
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of an autonomously concatenated hybridization chain reaction for signal amplification and intracellular imaging†
Jie
Wei
 a,
Xue
Gong
a,
Qing
Wang
a,
Min
Pan
a,
Xiaoqing
Liu
a,
Jing
Liu
b,
Fan
Xia
c and
Fuan
Wang
*a
a,
Xue
Gong
a,
Qing
Wang
a,
Min
Pan
a,
Xiaoqing
Liu
a,
Jing
Liu
b,
Fan
Xia
c and
Fuan
Wang
*a
aKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, P. R. China. E-mail: fuanwang@whu.edu.cn
bDepartment of Gastroenterology, Zhongnan Hospital of Wuhan University, Hubei Clinical Center & Key Lab of Intestinal & Colorectal Diseases, Wuhan, P. R. China
cDepartment of Urology, Union Hospital, Tongji Medical College, Hubei Key Laboratory of Bioinorganic Chemistry & Materia Medica, School of Chemistry and Chemical Engineering, Department of Epidemiology and Biostatistics, School of Public Health, Huazhong University of Science and Technology (HUST), Wuhan 430074, P. R. China
First published on 23rd October 2017
Abstract
Biomolecular self-assembly has spurred substantial research efforts for the development of low-cost point-of-care diagnostics. Herein, we introduce an isothermal enzyme-free concatenated hybridization chain reaction (C-HCR), in which the output of the upstream hybridization chain reaction (HCR-1) layer acts as an intermediate input to activate the downstream hybridization chain reaction (HCR-2) layer. The initiator motivates HCR-1 through the autonomous cross-opening of two functional DNA hairpins, yielding polymeric dsDNA nanowires composed of numerous tandem triggers T as output of the primary sensing event. The reconstituted amplicon T then initiates HCR-2 and transduces the analyte recognition into an amplified readout, originating from the synergistic effect between HCR-1 and HCR-2 layers. Native gel electrophoresis, atom force microscopy (AFM) and fluorescence spectra revealed that C-HCR mediated the formation of frond-like branched dsDNA nanowires and the generation of an amplified FRET signal. As a versatile and robust amplification strategy, the unpreceded C-HCR can discriminate DNA analyte from its mutants with high accuracy and specificity. By incorporating an auxiliary sensing module, the integrated C-HCR amplifier was further adapted for highly sensitive and selective detection of microRNA (miRNA), as a result of the hierarchical and sequential hybridization chain reactions, in human serum and even living cells through an easy-to-integrate “plug-and-play” procedure. In addition, the C-HCR amplifier was successfully implemented for intracellular miRNA imaging by acquiring an accurate and precise signal localization inside living cells, which was especially suitable for the ex situ and in situ amplified detection of trace amounts of analyte. The C-HCR amplification provides a comprehensive and smart toolbox for highly sensitive detection of various biomarkers and thus should hold great promise in clinical diagnosis and assessment. The infinite layer of multilayered C-HCR is anticipated to further strengthen the amplification capacity and reliability (anti-invasion performance) of intracellular imaging approach, which is of great significance for its bioanalytical applications.
Introduction
The isothermal amplified nucleic acid detection has attracted substantial research interests,1–3 and numerous isothermal amplification means have been developed and applied in homogeneous diagnostics, forensics, medicine, and agriculture.4–6 These include nucleic acid sequence-based amplification (NASBA),7,8 signal-mediated amplification of RNA technology (SMART),9,10 rolling circle amplification (RCA),11,12 strand displacement amplification (SDA),13,14 and loop-mediated isothermal amplification (LAMP).15,16 Isothermal amplification provides a rapid and efficient tool at a constant temperature without thermocycling required in traditional polymerase chain reaction (PCR).17–19 This method can be performed under simple conditions (e.g., water bath), which makes its applications more feasible on cell surfaces and even inside living cells. Unfortunately, these enzyme-mediated amplification schemes have the limitations of operating under conditions that might otherwise inhibit proteins. Recently, catalytic nucleic acids (DNAzymes)20–22 have been engineered and adapted to construct autocatalytic or cross-catalytic nucleic acid circuits for homogeneously amplified detection of nucleic acids or small molecules.23–26 However, the ease and accessibility of the DNAzyme substrates need to be solved before their universal applications can be extensively explored.Nonenzymatic nucleic acid circuits, which are merely composed of hybridization and strand-displacement reactions, have also been applied for isothermal signal amplification purpose. For example, the hybridization chain reaction (HCR),27–29 entropy-driven catalysis,30,31 and catalyzed hairpin assembly (CHA)32–34 have been adapted to analytical applications. HCR involves the analyte-triggered cross-opening of two DNA hairpins into long dsDNA copolymers.27 It has been successfully utilized for detecting various nucleic acid analytes.35–38 HCR has been further developed with a significant amplification capability by its conjugation with DNAzyme- or enzyme-mediated amplicons.39–42 The coupled HCR–DNAzyme system is based on the tailoring of two hairpin structures that involve hybridization chain reaction only upon exposure to target DNA, yielding RNA-cleaving or Hemin/G-quadruplex DNAzyme nanowires that provide catalytic reporter units for the sensing events.39–41 More efforts have been devoted to the reporter element of the amplification means, while rare interests are spent on nonenzymatic nucleic acid circuits themselves. More recently, a cascaded HCR–CHA circuit has been successfully developed for the amplified nucleic acid detection.43–45
An in-depth understanding of the comprehensive and complicated roles of microRNAs (miRNAs) requires the development of powerful intracellular imaging tools for visualizing the localization of endogenous miRNAs with low expression levels under in vivo conditions.46–48 Until now, the in situ intracellular miRNAs imaging still remains a challenge for the following issues that need to be addressed before their extensive applications can be envisaged. First, the short nature of miRNAs needs high affinity probes or significant signal amplification methods to acquire detectable readout signals. Second, the cellular diffusibility of miRNAs brings tremendous deflection to the reliable and accurate localization of miRNAs inside living cells after these requisite incubation and washing steps of intracellular imaging experiments. Third, the varied expression levels of miRNAs in different cells requires the development of a versatile and robust signal amplification strategy. In addition, to carry out intracellular imaging implementations in living cells, two molecular concerns have to be considered and resolved: efficient intracellular delivery of imaging probes into cells, and considerably improved resistance of imaging probes to intracellular endo- and exo-nucleases. Various transfection approaches have thus been developed for highly efficient cytosolic delivery of DNA probes into cells.49–51 Chemical modifications of the nucleic acid probes offers a considerably improved thermostability and nuclease resistance for molecular imaging at physiological condition.50–52 With these approaches, the probes could be efficiently transferred into living cells for accurate and amplified sensing without interference from surrounding complex cellular components.
Here, we introduce a programmable paradigm for constructing an isothermal enzyme-free signal amplification platform that is based on the unpreceded concatenated hybridization chain reaction (C-HCR). A novel two-layered concatenated HCR-1/HCR-2 circuit with synergistic amplification performance was constructed as the simplest model system for homogeneous assay. The amplicon product of the upstream HCR (HCR-1) layer is used as a transmission trigger for downstream HCR (HCR-2) transduction amplifier, leading to the construction of a branched long dsDNA copolymeric nanostructure. We start the robust amplification strategy with HCR-1, where the initiator (I) motivates the successive cross-opening of two functional DNA hairpins, yielding copolymeric DNA nanowires consisting of the amplicon trigger (T) as a result of the primary recognition event. The reconstitute T then initiates downstream HCR-2 and transduces analyte recognition into an amplified fluorescence readout signal. Furthermore, C-HCR is inherently modular and scalable, requiring only the design of base-pairing between strands. Thus, a previously optimized C-HCR amplifier can be immediately adopted to new detection systems without redesigning the main circuitry and reporter constructs. This enables us to utilize C-HCR as a versatile and convenient sensing platform for highly sensitive and selective detection of other clinically important analytes, e.g. miRNAs, through a convenient and universal ‘plug-and-play’ approach. The sequential hybridization behaviors of C-HCR amplifier provide an efficient signal amplification in living cells, achieving a simple and accurate C-HCR imaging system. Furthermore, the low cellular diffusibility of the high-molecular-weight branched dsDNA copolymers, as compared with the high cellular diffusibility of DNA hairpin probes, offers a high signal-to-noise ratio and an excellent localization contrast in intracellular imaging research, facilitating a precise and accurate positioning of the analyte. The C-HCR scheme can be easily engineered to generate a sophisticated hierarchical structure of programmable and multiple order, which is infeasible for traditional HCR or CHA/HCR systems. In particular, the use of C-HCR transducer in living cell detection applications may lead to concomitant improvements in the convenience and programmability of isothermal enzyme-free reactions for intracellular biomarkers of interest, leading to an accurate in situ diagnoses and effective treatment of key diseases.
Results and discussion
The isothermal C-HCR is composed of two successive HCRs, the upstream HCR-1 and downstream transducer HCR-2. The execution of HCR-2 is dependent upon the formation of a particular DNA nanostructure, an assembled HCR-1 nanowire, as schematically shown in Fig. 1. The upstream HCR-1 system consists of two hairpin nucleic acid structures H1 and H2. H1 includes the sequence a–b (red) that is complementary to the initiator (I). H2 includes the domain b–c* (green) that is complementary to the sequence c–b* (red) of hairpin H1. H2 is further elongated with domains d and e at its 5′- and 3′-ends, respectively. In the presence of initiator I, hairpin H1 opens by a toehold-mediated strand displacement mechanism, giving rise to the formation of hybrid I–H1. The resulting single-stranded domain c–b* (red) opens hairpin H2 by hybridizing with domain b–c*, yielding an intermediate structure I–H1·H2 that includes the same exposed domain b*–a* (green) as initiator I. This results in an autonomous cross-opening of hairpins H1 and H2 that brings the separated segments d and e into close proximity, leading to the assembly of dsDNA nanowires analogous to alternating copolymers and the concomitant formation of tandem adjacent regions d and e. Thus, the I-stimulated HCR-1 leads to the formation of chains of the successively reconstituted d–e (blue) colocalized structure T. In the subsequent transducer downstream HCR-2 with H3, H4, H5 and H6, segments e and d act as a toehold and a branch-migration domain, respectively. The downstream HCR-2 system consists of four DNA hairpins H3, H4, H5 and H6. H3 includes the sequence e*–d* (pink) that can recognize and hybridize with the colocalized structure T. H4 includes domain d*–f* (purple) that is complementary to the sequence f–d (pink) of hairpin H3 while H5 includes domain g*–d* (pink) that is complementary to the sequence d–g (purple) of hairpin H4. H6 consists of sequence d*–h* (purple) that is complementary to the sequence h–d of hairpin H5. H6 also includes sequence d–e (purple) that is an analog sequence of the colocalized structure T. In addition, H3 is functionalized at its 3′-end with a fluorescence acceptor (TAMRA) while H5 is functionalized at its 5′-end with a fluorescence donor (FAM). Upon the formation of the tandem repeated structure T through the aforementioned upstream HCR-1 system, segment e docks to the toehold e* of H3, leading to a toehold-mediated strand displacement that opens TAMRA-labelled H3 with the formation of an intermediate T–H3 structure. The exposed domain f of T–H3 acts as a toehold to bind H4 to yield intermediate structure T–H3·H4. Then H4 is opened via branch migration, releasing hidden single-stranded region d-g, facilitating its hybridization with FAM-labelled H5 and producing an intermediate T–H3·H4·H5 hybrid. This brings the two fluorophores (FAM and TAMRA) into close proximity and enables the Förster resonance energy transfer (FRET) process (Fig. S1, ESI†). The single-stranded sequence h–d of the opened H5 again binds and unfolds H6 to construct an intermediate T–H3·H4·H5·H6 hybrid, restoring an exposed single-stranded sequence d–e in H6 with an analog sequence of the colocalized structure T. Thus the exposed sequence of H6 again opens H3 and leads to HCR-2-involved multiple assembly of H3, H4, H5 and H6 into long dsDNA copolymers on the backbone of HCR-1-derived nanowires (detailed see Fig. S2 of ESI†). | ||
| Fig. 1 Scheme of the isothermal concatenated hybridization chain reaction (C-HCR) circuit. | ||
In short, the upstream HCR-1-triggered repeating multiple cross-hybridization of H1 and H2 leads to the formation of a tandem DNA structure that offers an accessible stage for a downstream HCR-2-motivated cyclic sequential hybridization between H3, H4, H5 and H6, accelerating the construction of a frond-like dsDNA copolymer structure (Fig. S3†). Each H1–H2 pair hybridization event produces one HCR-2 copolymeric dsDNA nanowire and each H3–H4–H5–H6 hybridization event leads to one FRET signal generation. The initial I-triggered cross-opening of H1 and H2 (upstream HCR-1) produces a long nicked concatemer dsDNA nanowire carrying numerous T for triggering downstream HCR-2. The subsequent alternating hybridization of H3, H4, H5 and H6 with T yields the long DNA concatemer carrying a large number of adjacent FAM and TAMRA fluorophore pair, producing a remarkable FRET signal. The isothermal C-HCR is kinetically impeded and stays metastable without initiator, due to the closed formation of the hairpin stems. The synergistic effect of HCR-1 and HCR-2 thus generates significantly amplified readout signal for highly sensitive detection of the analyte. A homogeneous FRET-based mechanism provides a feasible approach for precise detection.
In order to eliminate the frustrating false-positive signal from C-HCR, all these hairpin species, key components of C-HCR, should not interact with each other until a specific analyte is introduced. The unique programmable nature of the loop-stem hairpin structures paves the way for accurate analyte assay. In a primary design, the single-stranded tether sequences e and d of H2 were theoretically (by Mfold software)53 and experimentally optimized to avoid a possible signal leakage from upstream HCR-1 to downstream HCR-2 (Table S1, ESI†). It is necessary to protect the toehold or/and binding regions of output sequences of upstream HCR-1 in order to prevent their possible interactions with downstream HCR-2 before exposure. It was found that caging partial sequence d into the stem region of H2 could dramatically decrease the signal leakage from upstream HCR-1 to downstream HCR-2 while maintain the high performance of the present C-HCR amplifier (Fig. S4†). The proof-of-concept demonstration of the proposed C-HCR-mediated signal amplification strategy was first examined. As shown in Fig. 2(A), the C-HCR mixture of H1 + H2 + H3 + H4 + H5 + H6 shows no fluorescence change (curve a), indicating these hairpins are metastable and no obvious signal leakage (spontaneous cascaded hybridization chain reactions) can happen. A T-analog structure (TI/TII/TIII) encoding with sequence e–d was designed for specifically triggering HCR-2 scheme (Fig. S2†). An apparent decrease of fluorescence intensity was observed, curve b of Fig. 2(A), basically due to the implementation of downstream HCR-2 (trigger-mediated successive opening of HCR-2 mixture of H3 + H4 + H5 + H6) as discussed previously. Such phenomena also imply that H1 and H2, the indispensable components of upstream HCR-1, have no side effect on downstream HCR-2 process of C-HCR circuit. However, when the same amount of initiator I (compared with trigger T) was incubated with the intact C-HCR mixture as described above, a dramatically decreased fluorescence was observed and it leveled off after ca. 2 h, curve c of Fig. 2(A). Accordingly, the resulting fluorescence spectra were monitored and recorded after 2 h (Fig. S5†). This tremendous decreased fluorescence of I-triggered C-HCR is attributed to the upstream HCR-1 that produced a long tandem repeated dsDNA copolymer, consisting of numerous colocalized trigger T, and the subsequent T-mediated downstream HCR-2 that dominates the cyclic H3–H4–H5–H6 hybridization and the amplified FRET signal generation. It should be noted that it is a characteristic HCR system (HCR-2 circuit) when the C-HCR mixture was incubated with trigger T only since then the downstream HCR-2 is merely activated to transduce the readout signal and the upstream HCR-1 is not involved in this process. The present C-HCR strategy showed an enormous fluorescence response over conventional HCR, indicating an enhanced signal amplification efficacy of the C-HCR amplifier over traditional HCR scheme. These results shown here clearly demonstrate the successful implementation of C-HCR amplifier and the significant signal enhancement capacity of our proposed sensing platform. Control experiments show that no significant fluorescence changes were observed by subtracting H1 or H2 from upstream HCR-1 or by subtracting H4 or H6 from downstream HCR-2 (Fig. S6†). This clearly indicates that C-HCR leads to the sequential and successive cross-opening of the corresponding hairpin mixtures and the effective FRET generation. Evidently, the C-HCR is only activated in the presence of both upstream HCR-1- and downstream HCR-2-involved hairpin mixtures. Noted that the H6-excluded C-HCR system represents a characteristic traditional HCR through the stoichiometric FRET readout of HCR-1 scheme (Fig. S6†), which shows a much lower signal amplification efficacy as compared with the present C-HCR amplifier. Target signaling occurs in a multiple reaction ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N) in the conventional HCR, whereas one target yields quadratic signal amplification (1:N2) in the newly developed C-HCR amplifier, facilitating the amplified detection of trace amount of target.
:N) in the conventional HCR, whereas one target yields quadratic signal amplification (1:N2) in the newly developed C-HCR amplifier, facilitating the amplified detection of trace amount of target.
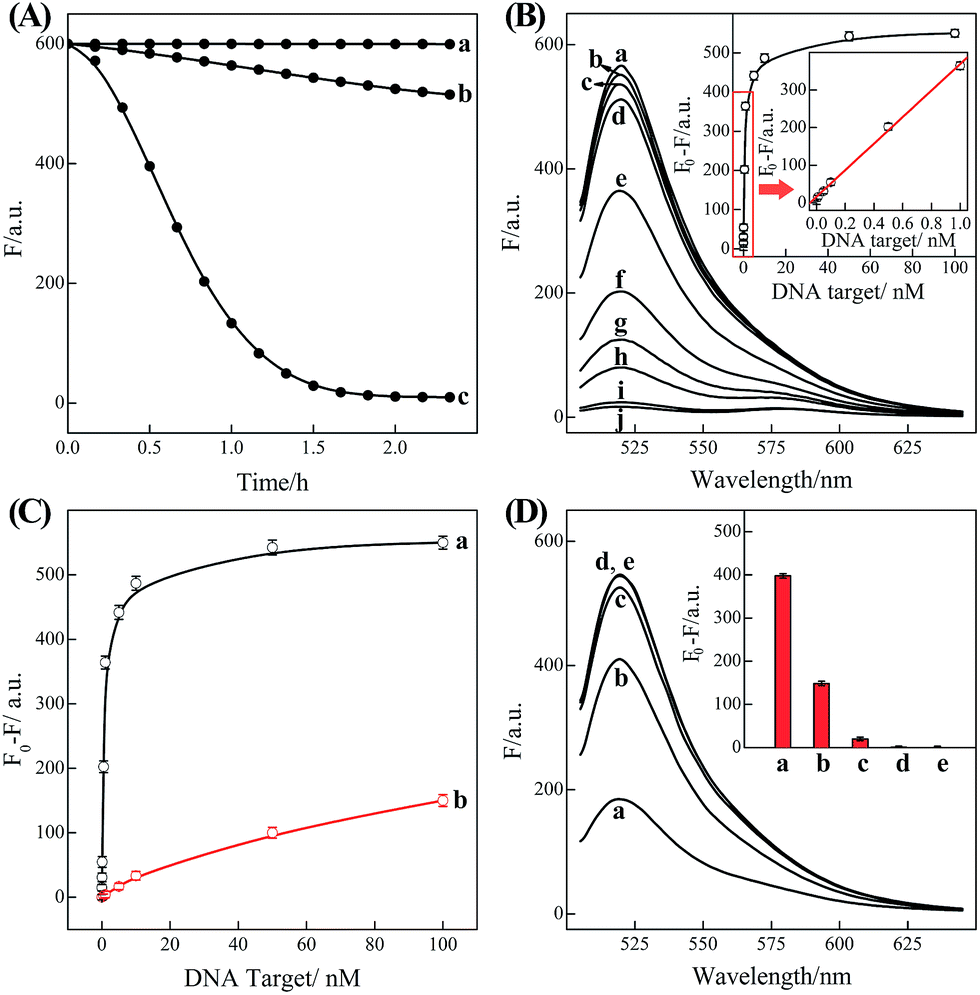 | ||
| Fig. 2 (A) Time-dependent fluorescence changes of the C-HCR circuit shown in Fig. 1 without analyte (a) and upon analyzing 50 nM T (b) or 50 nM I (c). (B) Fluorescence spectra generated by the C-HCR circuit upon analyzing different concentrations of I: (a) 0, (b) 1 × 10−11, (c) 5 × 10−11, (d) 1 × 10−10, (e) 5 × 10−10, (f) 1 × 10−9, (g) 5 × 10−9, (h) 1 × 10−8, (i) 5 × 10−8, and (j) 1 × 10−7 M. Inset: resulting calibration curve. (C) Calibration curves of C-HCR (a) and the conventional HCR (b) systems upon analyzing different concentrations of I. (D) Fluorescence spectra generated by the C-HCR system upon analysis of different analytes: (a) I, 10 nM, (b) IA, 10 nM, (c) IB, 10 nM, (d) IC, 10 nM, (e) no analyte. Inset: summary of the results of fluorescence spectra at λ = 520 nm. The respective system was carried out in reaction buffer for a fixed time interval of 2 h. F0 represents the original fluorescence intensity. Error bars were derived from n = 5 experiments. | ||
Based on these aforementioned convincing results, it is evident the current C-HCR circuit as illustrated in Fig. 1 can be utilized for the amplified detection of initiator DNA. Accordingly, the C-HCR system was applied for analyzing different concentrations of initiator I, by recording the fluorescence spectra after a fixed time interval of 2 h (Fig. 2(B)). A substantial decrease of fluorescence intensities was observed as the concentration of initiator I increased, consistent with the enhanced formation of long tandem DNA nanowires and the efficient generation of FRET signals. From the derived calibration curve (Fig. 2(B) inset), the detection limit corresponds to 3 pM for the homogeneous C-HCR strategy according to the 3σ calculation method. The performance of the H6-excluded C-HCR system (as conventional HCR control) was also examined for analyzing the same initiator of varied concentrations under the same reaction conditions (Fig. 2(C)). A dramatically lower signal response was observed for conventional HCR control, which was reasonable since the amplification efficacy of the C-HCR circuit (1/N2) was overwhelmingly enhanced over that of the traditional HCR (1/N) system.
The C-HCR-amplified detection platform shown in Fig. 1 is not only sensitive, but also selective. To evaluate this property, initiator I and the sequences of the one-, two-, and three-base mutations IA, IB, and IC were examined to elucidate the selectivity of the present C-HCR amplifier (for detailed kinetics analysis, see Fig. S7 of ESI†). Fig. 2(D) shows the fluorescence spectra generated by the analytical platform shown in Fig. 1 upon analyzing 10 nM of initiator I, and its one-, two-, and three-base mutants IA, IB and IC, respectively. Fig. 2(D), curve e shows the fluorescence spectra (background fluorescence) of the C-HCR sensing platform in the absence of the analyte. It is obvious that the two-base and three-base mismatched analytes IB (curve c) and IC (curve d) show fluorescence intensities that are almost identical to the background signal of the system. Also, the single-base mismatched analyte IA (curve b) can be easily discriminated from initiator I (curve a), implying that the system reveals high selectivity. These results clearly demonstrate the high selectivity of the present C-HCR strategy.
Native electrophoretic experiment were utilized to prove the whole working principle in Fig. 1. To verify whether the C-HCR proceeded as designed, each layer of the C-HCR circuit, upstream HCR-1 and downstream HCR-2, was first validated separately. As indicated in Fig. 3(A), the bands of monomer hairpins became weakened and even vanished while many bright bands with a maximum size of tens of thousands of base-pairs were obtained for upstream HCR-1 or downstream HCR-2 when their respective hairpin mixtures were incubated with their corresponding trigger DNAs. Gel electrophoresis was further utilized to confirm the performance of the C-HCR circuit. We can observe that no new band emerged for C-HCR hairpin mixture, while many bright bands appeared by introducing their respective trigger DNAs into their corresponding hairpin mixtures. These results demonstrate that the upstream HCR-1, downstream HCR-2 or concatenated HCR-1/HCR-2 (C-HCR) can only be triggered by their respective initiators, yielding dsDNA supramolecular structures composed of hundreds of the corresponding hairpin components. This evidences the production of high-molecular-weight dsDNA products and indicates the potential of C-HCR for amplified bioanalysis. These gel electrophoresis results are in good agreement with those of fluorescence experiments shown in Fig. 2(A) and S2.† Furthermore, atomic force microscopy (AFM) was utilized for characterizing the C-HCR-involved branched dsDNA copolymers. Initiator-triggered HCR-1 results in tandem colocalized triggers for downstream HCR-2 that mediates the assembly of branched dsDNA nanochains on the backbone of upstream HCR-1-generated copolymeric dsDNA nanostructures. This leads to the formation of frond-like branched dsDNA nanowires. Fig. 3(B) shows AFM image of the C-HCR-motivated dsDNA nanochains (more see Fig. S8, ESI†). As expected, micrometer-long branched DNA copolymeric nanowires are observed with the height of ∼1.5 nm (Fig. 3(B) inset), corresponding to the height of a characteristic dsDNA. Some of the chains form bundles, which presumably originates from cross-interaction of the exposed single stranded DNAs (ssDNAs) associated with different chains. Only tiny spots of hairpin monomers without any assembled product were observed for C-HCR system without initiator (as a negative control, Fig. 3(C)). As expected, AFM imaging of the same initiator-triggered HCR-1 shows long linear dsDNA structures (as a positive control, Fig. 3(D)), validating the underlying reaction mechanism of our proposed C-HCR system.
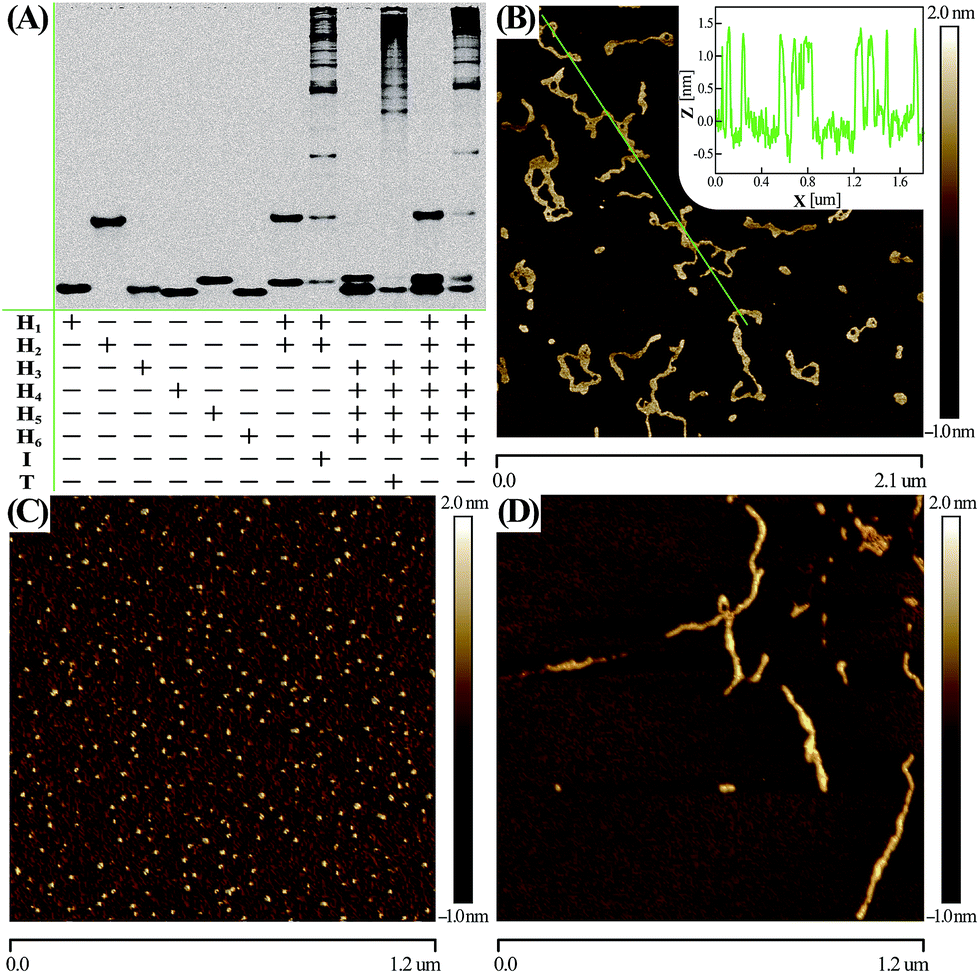 | ||
| Fig. 3 (A) Native gel electrophoresis characterization of C-HCR and its individual upstream HCR-1 and downstream HCR-2 systems. The “+” and “−” denote the presence and absence of the corresponding DNA components, respectively. (B) AFM image and cross-section analysis of the resulting C-HCR-motivated dsDNA branched nanowires. (C) AFM characterization of the C-HCR system without initiator. (D) AFM image of HCR-1-motivated linear dsDNA nanowires. For detailed experimental conditions see Experimental section. | ||
It should be noted that the isothermal C-HCR amplifier represents an optimized sensing platform. The C-HCR strategy can be easily adapted to analyze other nucleic acid targets without further significant optimization of the established system. This was exemplified with the analysis of microRNA (miRNA) by using microRNA-21 (miR-21) as the model target. Abundant miR-21 is overexpressed in a wide range of human cancers54–56 and has been recognized as an important oncogenic miRNA.57 As shown in Fig. 4, a “helper” hairpin H7 was introduced into C-HCR amplifier to recognize miR-21, and upon opening, it activated the sequential cross-opening of the C-HCR hairpins mixture, giving rise to the assembly of highly branched dsDNA nanowires. Fig. S9(A)† depicts the time-dependent fluorescence changes upon analysis of different concentrations of miR-21 and Fig. 5(A) depicts the resulting fluorescence spectra after a fixed time interval of 2 h. From the derived calibration curve (Fig. 5(A) inset), the system enabled the sensitive detection of miR-21 with a detection limit that corresponded to 3 pM, which is comparable to and even lower than most of the enzyme-free sensing platforms (Table S3, ESI†). This indicates that the C-HCR scheme can be used as a general amplification module with the aid of a sensing module (foreign helper DNA), and is specifically suitable for amplified detection of low abundant analyte in a simple ‘plug-and-play’ format.
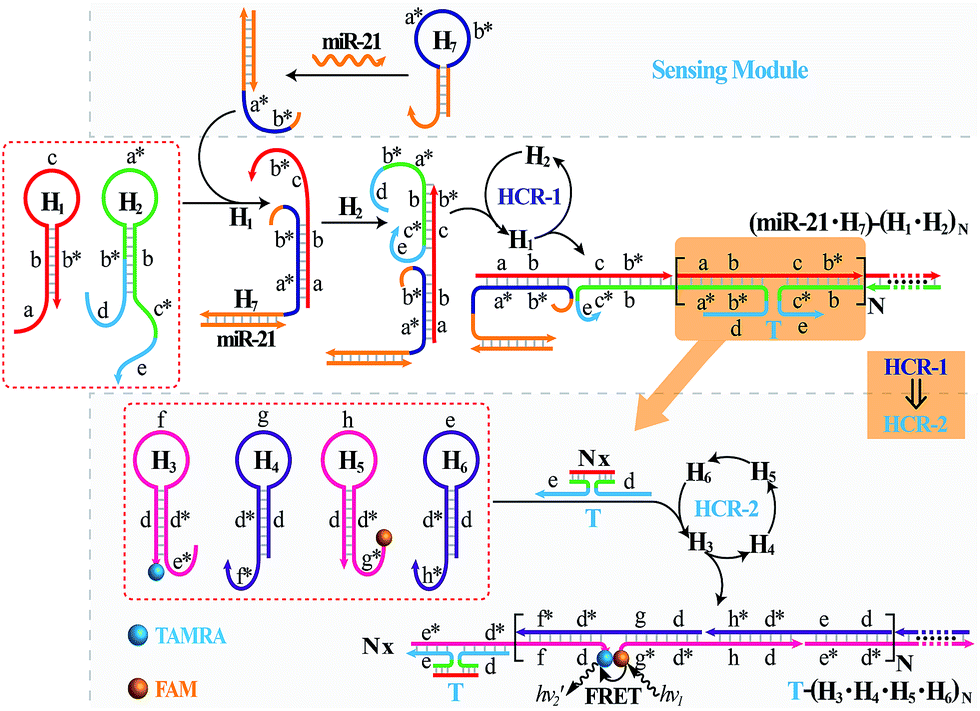 | ||
| Fig. 4 Schematic representation of the general analyte-sensing platform by introducing a sensing module consisting of a foreign helper DNA H7 into the well-established C-HCR circuit. | ||
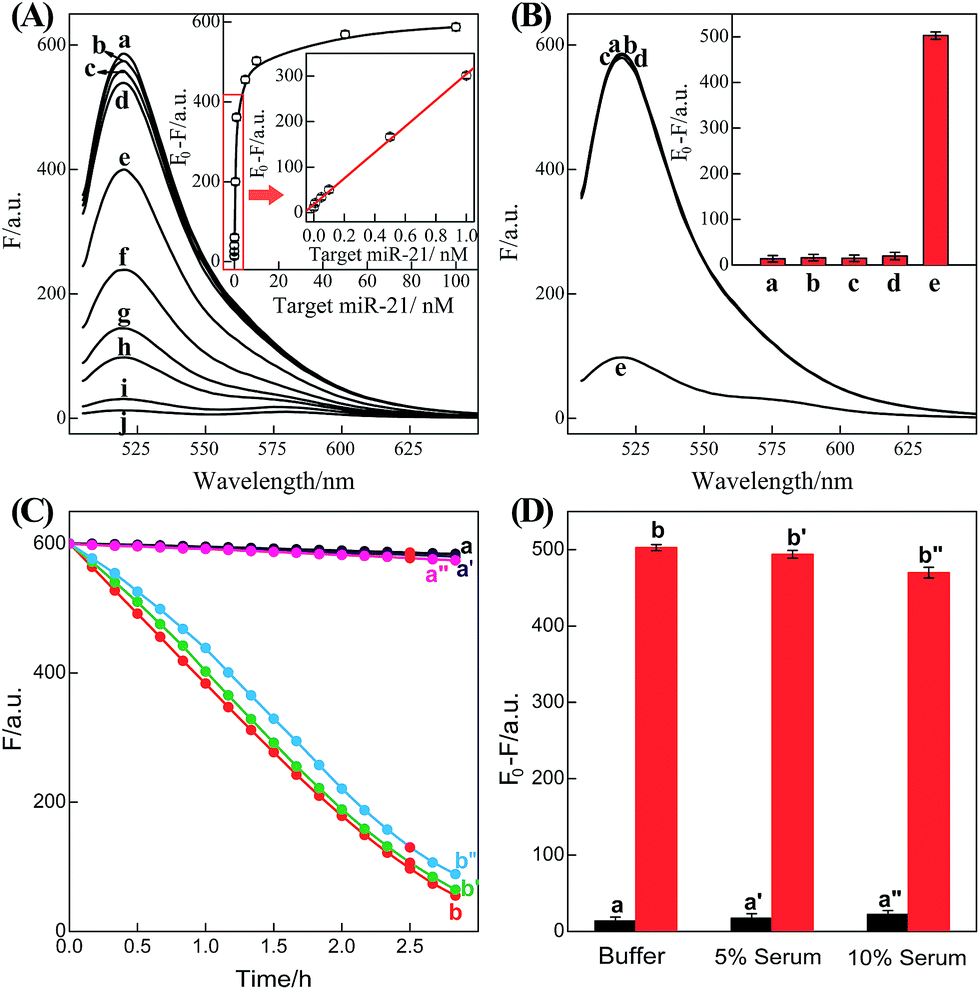 | ||
| Fig. 5 (A) Fluorescence spectra generated by the updated C-HCR-amplifier outlined in Fig. 4 upon analyzing different concentrations of miR-21: (a) 0, (b) 1 × 10−11, (c) 5 × 10−11, (d) 1 × 10−10, (e) 5 × 10−10, (f) 1 × 10−9, (g) 5 × 10−9, (h) 1 × 10−8, (i) 5 × 10−8, and (j) 1 × 10−7 M. Inset: resulting calibration curve. (B) Fluorescence spectra generated by the updated C-HCR system upon analyzing different analytes: (a) no analyte, (b) β-actin mRNA, 10 nM, (c) let-7a, 10 nM, (d) son DNA, 10 nM, and (e) miR-21, 10 nM. Inset: summary of the fluorescence spectra at λ = 520 nm. (C) Time-dependent fluorescence changes (at λ = 520 nm) of C-HCR system upon analyzing miR-21 in different serum solutions: (a) 0 M miR-21 in buffer, (b) 10 nM miR-21 in buffer, (a′) 0 nM miR-21 in 5% serum, (b′) 10 nM miR-21 in 5% serum, (a′′) 0 nM miR-21 in 10% serum, and (b′′) 10 nM miR-21 in 10% serum. (D) Summary of the fluorescence intensity changes (at λ = 520 nm) as shown in (C). The system consisting of H1 + H2 + H3 + H4 + H5 + H6 mixture (200 nM each) and a “helper” hairpin H7 (50 nM) was carried out for a fixed time interval of 2 h. F0 represents the original fluorescence intensity. Error bars were derived from n = 5 experiments. | ||
To evaluate the specificity of the present microRNA detection system, we challenged the updated C-HCR circuit with a series of interfering nucleic acids: β-actin mRNA, son DNA and let-7a miRNA. Fig. S9(B)† depicts the time-dependent fluorescence changes upon analyzing 10 nM miR-21 and its control nucleic acids: β-actin mRNA, son DNA and let-7a miRNA. Fig. 5(B) shows the fluorescence spectra generated by the sensing platform shown in Fig. 4 after a fixed time interval of 2 h. One may realize that the fluorescence intensities generated by the different interfering nucleic acids are very close to the background signal of the system (in the absence of analyte). This comparison clearly demonstrates that the C-HCR amplifier reveals high selectivity against other control nucleic acids and can offer potential applications to discriminate target miRNA from biological interference sequences. We have further examined the possibility to implement the homogeneous C-HCR sensing platform under biological conditions, e.g., serum samples. Human serum samples were diluted with buffer and analyzed by the C-HCR amplifier. Fig. 5(C) outlines the time-dependent fluorescence changes upon analyzing 10 nM miR-21 in 5% and 10% human serum samples. One may realize that target can be detected without obvious interference in real samples. These results show good agreement with that of the serum-free sensing environment (Fig. 5(D)), indicating an acceptable accuracy of the C-HCR system for analytes quantification in complex biological fluids.
The modular and effective amplification features of the homogeneous C-HCR system not only facilitate the accurate quantification of analyte in diluted serum, but also enable in situ intracellular imaging and monitoring of the cellular analyte in living cells. We then applied the aforementioned miR-21-targeting C-HCR system to investigate the miR-21 expression levels in different mammalian cells by confocal laser scanning microscopy (CLSM). To ensure the biostability of hairpin probes in cell culture medium, the DNA hairpin probes were synthesized with phosphorothioate bonds for C-HCR imaging (detailed see Table S2†). Moreover, to avoid undesired interfere from the complex intracellular environment, here the fluorescence emission ratio of acceptor to donor (FA/FD) was adopted as the detecting FRET signal to analyze microRNA in living cells (Fig. 6(A)). Statistical histogram analysis of the relative fluorescence intensity (FA/FD) of the C-HCR imaging system was shown in Fig. 6(B). MCF-7 cells, known to express relatively high levels of miR-21, were first chosen as a model system for studying the capacity of C-HCR-mediated intracellular imaging in living cells. The aforementioned miR-21-targeting C-HCR system was transfected into MCF-7 cells via lipofectamine 3000, and incubated with cells at 37 °C for 2 h. An intense FRET signal was observed, indicating a high miR-21 expression level in live MCF-7 cells, and the C-HCR platform offered distinct localization signals in each cell (sample a of Fig. 6(A), (B), and S10†). Meanwhile, an amplified efficiency of the present C-HCR-imaging system was further demonstrated by using control experiments that expelled one of the non-fluorophore-labelled hairpin probes from the C-HCR imaging system. No significant FRET signal was observed for H1-, H2- or H4-excluded C-HCR system (Fig. S11†) while a slight change of the FRET signal was revealed for conventional HCR imaging scheme (H6-excluded C-HCR imaging strategy), indicating the profound amplification nature of the present C-HCR imaging platform inside living cells (sample b of Fig. 6(A) and (B)). It shows a good agreement with that of fluorescence experiments (Fig. S6†). To evaluate the effectiveness of the C-HCR imaging system in cells with different miR-21 expression profiles, the present strategy was then carried out on different cell types and controls. Scarcely no FRET signal was observed when the miR-21 expression was knocked down by introducing an anti-miRNA antisense inhibitor oligonucleotide into MCF-7 cells (sample c of Fig. 6(A) and (B)), indicating that the C-HCR system can discriminate microRNA of different expression levels in living cancer cells. To further verify the robustness of our C-HCR imaging system for monitoring low expression levels of miRNA target, HeLa cells, exhibiting a relatively low miR-21 expression, were analyzed and indeed showed a comparably reduced generation of FRET signal (sample d of Fig. 6(A) and (B)). Clearly, the C-HCR imaging system could easily distinguish different cells with distinct miRNA expression levels. Moreover, the FRET efficiency of the C-HCR intracellular imaging system was measured and acquired by a conventional acceptor-photo-bleaching technique (Fig. S12†). A mean FRET efficiency of 0.62 was obtained from the C-HCR-imaging system by confocal laser scanning microscopy measurements (Fig. 6(C)). These results clearly demonstrate that the C-HCR imaging system indeed offers a programmable and efficient amplification toolbox for analyzing miRNA of interest in living cells, in real-time. It should be noted that the molecularly engineered C-HCR system, in principle, can improve the concomitant signal amplification and accurate localization of the target analyte as compared with the traditional HCR (Fig. S11†) and CHA-HCR systems. The distinct diffusion efficiency between the C-HCR monomers and C-HCR polymers leads to the construction of a compact hierarchical DNA nanostructure and to the distinct FRET signal generation. The inherited and infinite layer features of multilayered C-HCR are anticipated to further strengthen the programmable and robust construction of hierarchical DNA nanostructures of higher orders. With further molecular engineering and the aid of multiplexed fluorophore-labels, this homogeneous C-HCR amplifier is anticipated to realize simultaneous and quantitative imaging of multiple RNA targets in living cells.49,58 Given the fact that other powerful affinity probes, e.g., aptamers, could be easily adapted into the present C-HCR sensing strategy, the C-HCR imaging system is envisaged to play an important role in monitoring other small metabolites and cellular proteins targets inside living cells. The capability to visualize and localize lowly expressed endogenous RNA of interest in living cells may offer new possibilities for early cancer diagnosis and therapeutic intervention.
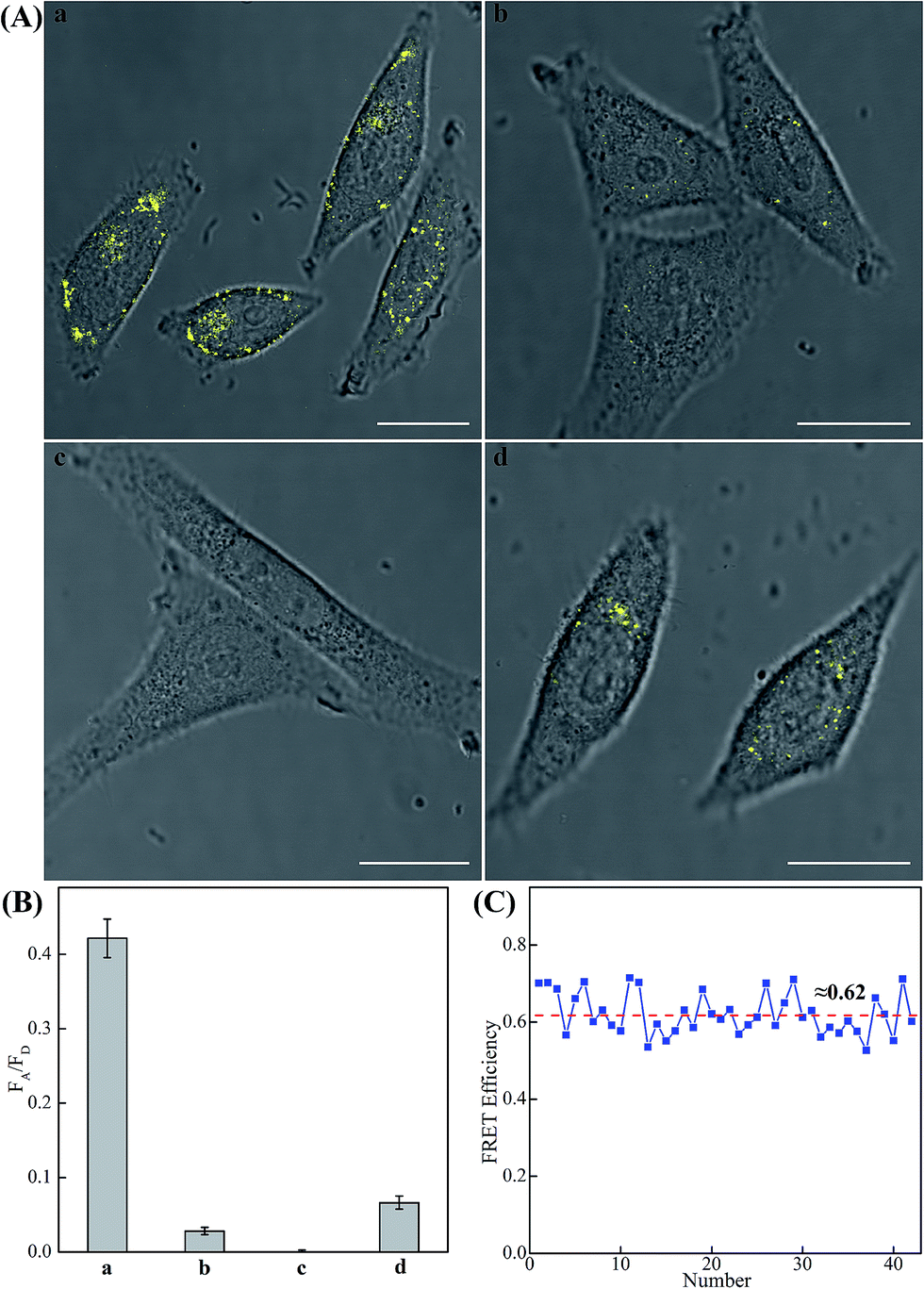 | ||
| Fig. 6 (A) Living cell analysis of miR-21 based on C-HCR or HCR strategy and FRET transduction (in the form of FA/FD). Confocal laser scanning microscopy (CLSM) imaging of miR-21 in (a) routine MCF-7 live cells by C-HCR amplifier, (b) routine MCF-7 live cells by conventional HCR amplifier (H6-excluded C-HCR), (c) MCF-7 live cells treated with a chemically modified miR-21 inhibitor by C-HCR amplifier, and (d) HeLa live cells by C-HCR amplifier. All of the aforementioned living cells were transfected and incubated with miR-21-targeting C-HCR mixture at 37 °C for 2 h. All scale bars correspond to 20 μm. (B) Statistical histogram analysis of the relative fluorescence intensity (FA/FD) of the above four cell samples through C-HCR imaging system. (C) Determination of the FRET efficiency of the C-HCR-imaging system collected from a large number of MCF-7 living cells. | ||
Conclusions
We have developed an isothermal enzyme-free concatenated hybridization chain reaction (C-HCR) strategy for constructing a robust and versatile signal amplification platform and for engineering an in situ intracellular imaging system. Initiator-induced upstream HCR-1 results in dsDNA copolymer consisting of tandem colocalized trigger DNAs for downstream HCR-2 that assembles polymeric dsDNA nanowires on the backbone of upstream HCR-1 nanochains. The novel C-HCR scheme mediates the formation of frond-like branched dsDNA nanowires and continually brings two fluorophores into close proximity, leading to the generation of an amplified FRET signal. By taking advantage of the signal amplification features of synergistic HCRs, the C-HCR system enables the amplified detection of target with substantial signal enhancement capacity. It can also discriminate target DNA from its one-, two-, and three-base mutation sequences. The homogeneous C-HCR system can be utilized as a general sensing platform and easily be adapt for analyzing other biologically important analytes by introducing a sensing module consisting of a foreign helper DNA. This easy-to-integrate ‘plug-and-play’ amplification scheme was further applied for highly sensitive and selective detection of miR-21 down to 3 pM in buffer, as well as in human serum. In addition, the molecularly engineered C-HCR was successfully adapted for highly efficient and accurate miRNA imaging in living cells. The C-HCR imaging system is proved to enable a precise signal localization at single cell level, resulting in an enzyme-free amplified detection of lowly expressed, short RNA targets in real time. The C-HCR amplifier can be easily adapted as a general imaging system for other endogenous targets of interest by simply reconfiguring and engineering the existing C-HCR sensing module consisting of a foreigner recognition probe. The inheritance of C-HCR sequential hybridization features contribute to the amplified detection of any desired endogenous RNAs in living cells in situ. The flexible and programmable nature of homogeneous C-HCR circuit provides a versatile and robust toolbox to probe different native RNAs in living cells in real time, which we believe can be implemented for potential bioengineering and synthetic biology applications.Experimental
Materials
4-(2-Hydroxyethyl)piperazine-1 ethanesulfonic acid sodium salt (HEPES), (3-aminopropyl)trimethoxysilane (APTES), N,N-diisopropylethylamine (DIPEA), penicillin-streptomycin, sodium chloride and magnesium chloride were of analytical grade and were purchased from Sigma-Aldrich (MO, USA) unless otherwise indicated. The DNA marker, GelRed, fetal bovine serum (FBS) and Lipofectamine 3000 transfection Reagent were purchased from Invitrogen (Carlsbad, CA). Dulbecco's Modified Eagle Medium (DMEM) was purchased from HyClone (Logan, Utah, USA). MCF-7 cells and HeLa cells were obtained from Shanghai Institutes for Biological Sciences (SIBS). Trypsin was purchased from Genview (USA). Human serum samples were kindly donated by healthy volunteers. All oligonucleotides were custom-designed and then synthesized by Sangon Biotech. Co., Ltd. (Shanghai, China). They were HPLC-purified and freeze-dried by the company. They were used as provided and diluted in 20 mM phosphate buffer (PB, pH 7.0) to give stock solution of 100 μM. Tables S1 and S2† depicts the sequences of the oligonucleotides used in the study. All solutions were prepared using ultrapure water, which was obtained through a Millipore Milli-Q water purification system with an electric resistance >18.2 MΩ cm. Atomic force microscope (AFM) cantilever (SCANASYST-AIR) was purchased from Bruker (Camarilla, CA).Native polyacrylamide gel electrophoresis
For DNA copolymer samples used for gel electrophoresis assay, 20 nM of initiators (I for HCR-1 and C-HCR, T for HCR-2) were incubated with 200 nM of their corresponding hairpin mixtures (H1 + H2 for HCR-1, H3 + H4 + H5 + H6 for HCR-2, and H1 + H2 + H3 + H4 + H5 + H6 for C-HCR) in reaction buffer (10 mM HEPES, 1 M NaCl, 50 mM MgCl2, pH 7.2) for 2 h at 25 °C. Then each of the samples was mixed with loading buffer and loaded into the notches of the freshly prepared 12% native polyacrylamide gel. Electrophoresis was performed at a constant voltage of 120 V in 1 × TBE buffer (89 mM Tris, 89 mM boric acid, 2.0 mM EDTA, pH 8.3) for 3 h. The gel was then stained with GelRed and imaged by FluorChem FC3 (ProteinSimple, USA) under 365 nm UV irradiation.AFM characterization
For AFM characterization of the C-HCR-motivated branched dsDNA copolymers, the sample was prepared in reaction buffer (10 mM HEPES, 1 M NaCl, 50 mM MgCl2, pH 7.2) that contained H1 + H2 + H3 + H4 + H5 + H6 (200 nM each) and initiator I (20 nM). The HCR-1 mixture consisting of H1 + H2 (200 nM each) and initiator I (20 nM) was carried out in reaction buffer (10 mM HEPES, 1 M NaCl, 50 mM MgCl2, pH 7.2) for a fixed time interval of 2 h. Mica was modified to bear positive charges on its surface for sample loading and imaging. Briefly, the desiccator was filled with argon gas. APTES and DIPEA were separately placed on the bottom of the desiccator with a volume ratio of 3:1. The freshly cleaved mica was reacted with the evaporated chemicals in the desiccator for 2 h. The DNA sample was diluted and deposited on the modified mica for 15 min to allow its adsorption on the mica surface, followed by its rinsing with water and drying under a stream of nitrogen. The prepared sample was scanned in tapping mode by Multimode 8 Atomic Force Microscope with a NanoScope V controller (Bruker Inc.). The silicon tips used for AFM analysis were SCANASYST-AIR (tip radius: ∼2 nm; resonance frequency: ∼70 kHz; spring constant: ∼0.4 N m−1; length: 115 μm; width: 25 μm).
Construction of C-HCR for signal amplification
All the assays were prepared in reaction buffer (10 mM HEPES, 1 M NaCl, 50 mM MgCl2, pH 7.2). Each DNA hairpin (4 μM) was heated to 95 °C for 5 min and then allowed to cool to room temperature (25 °C) for at least 2 h before use. For sensitive detection of DNA by means of DNA-triggered C-HCR, DNA analyte was introduced into the H1 + H2 + H3 + H4 + H5 + H6 mixtures (200 nM each) to initiate the self-assembly process at 25 °C. For amplified detection of miR-21 by means of C-HCR-amplifier, miR-21 was introduced into the mixtures of “helper” hairpin H7 (50 nM) and H1 + H2 + H3 + H4 + H5 + H6 (200 nM each) to initiate the self-assembly process at 25 °C. Unless specifically indicated, all of the control experiments were performed without changing the concentration of DNA except subtracting the unwanted oligonucleotides from the mixtures. Subsequently, the time-dependent fluorescence measurements were performed and monitored spectroscopically by using a Cary Eclipse spectrometer (Varian Inc) at 25 °C. The emission spectra were acquired by exciting the samples at 490 nm, and the resulting fluorescence spectra were collected from 505 to 650 nm. The kinetically monitoring the fluorescence intensity changes were recorded at a fixed wavelength of 520 nm upon exciting the system at λ = 490 nm. F0 represents the original fluorescence intensity.Cell culture and transfection for C-HCR-imaging system
Human breast cancer cells (MCF-7) and cervical cancer cells (HeLa) were grown in Dulbecco's Modified Eagle Medium (DMEM) containing 10% FBS and 1% penicillin/streptomycin at 37 °C in 5% CO2 atmosphere. Cells were plated with a DMEM medium (1.0 mL) for 12 h in glass bottom culture dishes. The miR-21-targeting C-HCR mixture containing H7 (0.1 nmol) and H1 + H2 + H3 + H4 + H5 + H6 (0.2 nmol each) was prepared in Opti-MEM (200 μL), and then mixed with lipofectamine 3000 (6 μL) dispersed in Opti-MEM (200 μL) for 5 min. Then the plated cells were transfected with the ∼400 μL mixture supplied with 80 μL FBS at 37 °C for 2 h. Afterwards, the cultured cells were washed three times with phosphate buffered saline (PBS) and transferred to a freshly prepared DMEM medium (1.0 mL) containing 10% FBS for confocal laser scanning microscopy observations. For anti-miRNA antisense inhibiter oligonucleotide experiment, MCF-7 cell was transfected with inhibiter oligonucleotide (final concentration, 100 nM) for 1 h, followed by transfection and incubation with the C-HCR system as described above for 2 h.Confocal laser scanning microscopy (CLSM) characterization
All cellular fluorescence images were collected with Leica TCS-SP8 laser scanning confocal microscopy system. A series of FRET, donor and acceptor filters was arranged to detect the following signals: donor fluorescence, acceptor fluorescence from external excitation and FRET stimulation. An external 488 nm excitation with an accompanying emission ranging from 500 to 550 nm was selected for the green channel of fluorophore (FAM) donor. The external 488 nm FRET stimulation with an accompanying emission signal collection ranging from 570 to 630 nm was selected for the yellow channel of fluorophore (TAMRA) acceptor. An external 561 nm excitation with an accompanying emission ranging from 570 to 630 nm was chosen for the red channel of TAMRA fluorophore. All images were developed at 63.0 × 1.40 objective with oil. The FRET signal was denoted as the fluorescence emission ratio of acceptor to donor (FA/FD) in intracellular imaging systems upon an external 488 nm stimulation. The respective fluorescence (FA and FD) signals of different cells were acquired by integrating the fluorescence images of the corresponding samples. To achieve a reliable quantitative FRET readout, the background FRET signal, originating from solely FAM or TAMRA fluorophore, was subtracted from each of the samples. All FRET images of the living cells were normalized by using ImageJ and Fiji softwares.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Basic Research Program of China (973 Program, 2015CB932601), Hubei Provincial Natural Science Foundation of China (2015CFB503), Jiangsu Provincial Natural Science Foundation of China (BK20161248, BK20160381), National Natural Science Foundation of China (21503151, 81602610, 81472735) and 1000 Young Talent (to F. W. and X. L.) the Wuhan Youth Science and Technology Plan (2016070204010131) and the Foundation of Key Laboratory of Analytical Chemistry for Biology and Medicine (Wuhan University), Ministry of Education (No. ACBM2017005).Notes and references
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812–2841 CrossRef CAS PubMed.
- R. Duan, X. Lou and F. Xia, Chem. Soc. Rev., 2016, 45, 1738–1749 RSC.
- S. Guo and E. Wang, Acc. Chem. Res., 2011, 44, 491–500 CrossRef CAS PubMed.
- C. Jung and A. D. Ellington, Acc. Chem. Res., 2014, 47, 1825–1835 CrossRef CAS PubMed.
- P. Gill and A. Ghaemi, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 224–243 CAS.
- L. Zhang, J. Zhu, S. Guo, T. Li, J. Li and E. Wang, J. Am. Chem. Soc., 2013, 135, 2403–2406 CrossRef CAS PubMed.
- B. Deiman, P. van Aarle and P. Sillekens, Mol. Biotechnol., 2002, 20, 163–179 CrossRef CAS PubMed.
- J. Compton, Nature, 1991, 350, 91–92 CrossRef CAS PubMed.
- M. J. Hall, S. D. Wharam, A. Weston, D. L. N. Cardy and W. H. Wilson, BioTechniques, 2002, 32, 604–611 CAS.
- S. D. Wharam, P. Marsh, J. S. Lloyd, T. D. Ray, G. A. Mock, R. Assenberg, J. E. McPhee, P. Brown, A. Weston and D. L. N. Cardy, Nucleic Acids Res., 2001, 29, e54 CrossRef CAS PubMed.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- P. M. Lizardi, X. Huang, Z. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowarski, M. Kotler and I. Willner, Angew. Chem., Int. Ed., 2006, 118, 7544–7548 CrossRef.
- K. Zhang, R. Deng, Y. Li, L. Zhang and J. Li, Chem. Sci., 2016, 7, 4951–4957 RSC.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- X. Fang, Y. Liu, J. Kong and X. Jiang, Anal. Chem., 2010, 82, 3002–3006 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS PubMed.
- F. Wang, C.-H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881–2941 CrossRef CAS PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- M. Deng, D. Zhang, Y. Zhou and X. Zhou, J. Am. Chem. Soc., 2008, 130, 13095–13102 CrossRef CAS PubMed.
- J. S. Hartig, I. Grüne, S. H. Najafi-Shoushtari and M. Famulok, J. Am. Chem. Soc., 2004, 126, 722–723 CrossRef CAS PubMed.
- F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295–299 CrossRef CAS PubMed.
- T. A. Lincoln and G. F. Joyce, Science, 2009, 323, 1229–1232 CrossRef CAS PubMed.
- F. Wang, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2012, 134, 5504–5507 CrossRef CAS PubMed.
- M. Levy and A. D. Ellington, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6416–6421 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS PubMed.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- G. Seelig, B. Yurke and E. Winfree, J. Am. Chem. Soc., 2006, 128, 12211–12220 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
- B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef CAS PubMed.
- X. Chen, J. Am. Chem. Soc., 2012, 134, 263–271 CrossRef CAS PubMed.
- Z. Wu, G. Q. Liu, X. L. Yang and J. H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef CAS PubMed.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
- F. Xuan and I. M. Hsing, J. Am. Chem. Soc., 2014, 136, 9810–9813 CrossRef CAS PubMed.
- J. Ren, J. Wang, L. Han, E. Wang and J. Wang, Chem. Commun., 2011, 47, 10563–10565 RSC.
- S. Shimron, F. Wang, R. Orbach and I. Willner, Anal. Chem., 2012, 84, 1042–1048 CrossRef CAS PubMed.
- L. Li, J. Feng, H. Liu, Q. Li, L. Tong and B. Tang, Chem. Sci., 2016, 7, 1940–1945 RSC.
- F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed.
- J. Huang, H. Wang, X. Yang, K. Quan, Y. Yang, L. Ying, N. Xie, M. Ou and K. Wang, Chem. Sci., 2016, 7, 3829–3835 RSC.
- B. Li, Y. Jiang, X. Chen and A. D. Ellington, J. Am. Chem. Soc., 2012, 134, 13918–13921 CrossRef CAS PubMed.
- Q. Wang, M. Pan, J. Wei, X. Liu and F. Wang, ACS Sens., 2017, 2, 932–939 CrossRef CAS PubMed.
- Y. Wei, W. Zhou, X. Li, Y. Chai, R. Yuan and Y. Xiang, Biosens. Bioelectron., 2016, 77, 416–420 CrossRef CAS PubMed.
- R. Deng, K. Zhang and J. Li, Acc. Chem. Res., 2017, 50, 1059–1068 CrossRef CAS PubMed.
- R. Deng, L. Tang, Q. Tian, Y. Wang, L. Lin and J. Li, Angew. Chem., Int. Ed., 2014, 53, 2389–2393 CrossRef CAS PubMed.
- R. Deng, K. Zhang, Y. Sun, X. Ren and J. Li, Chem. Sci., 2017, 8, 3668–3675 RSC.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS PubMed.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CrossRef CAS PubMed.
- C. C. Wu, S. Cansiz, L. Q. Zhang, I. T. Teng, L. P. Qiu, J. Li, Y. Liu, C. Zhou, R. Hu, T. Zhang, C. Cui, L. Cui and W. H. Tan, J. Am. Chem. Soc., 2015, 137, 4900–4903 CrossRef CAS PubMed.
- L. Wang, C. J. Yang, C. D. Medley, S. A. Benner and W. Tan, J. Am. Chem. Soc., 2005, 127, 15664–15665 CrossRef CAS PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebert, R. H. Mak, A. A. Ferrando, J. R. Downing, T. Jacks, H. R. Horvitz and T. R. Golub, Nature, 2005, 435, 834–838 CrossRef CAS PubMed.
- S. M. Hammond, Nat. Methods, 2006, 3, 12–13 CrossRef CAS PubMed.
- S. Alahari, MicroRNA in Cancer, 2013, VIII, 148 Search PubMed.
- P. J. Santangelo, B. Nix, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, e57 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DNA sequences, original C-HCR design, AFM characterization, control experiments and FRET efficiency for fluorescence imaging of miR-21 in MCF-7 cell. See DOI: 10.1039/c7sc03939e |
| This journal is © The Royal Society of Chemistry 2018 |