
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of metabolites of liquiritin in rats by UHPLC-Q-TOF-MS/MS: metabolic profiling and pathway comparison in vitro and in vivo†
Xia Zhang,
Caijuan Liang,
Jintuo Yin,
Yupeng Sun and
Lantong Zhang *
*
Department of Pharmaceutical Analysis, School of Pharmacy, Hebei Medical University, Shijiazhuang 050017, P. R. China. E-mail: zhanglantong@263.net; Fax: +86-311-86266419; Tel: +86-311-86266419
First published on 27th March 2018
Abstract
Liquiritin (LQ), the main bioactive constituent of licorice, is a common flavoring and sweetening agent in food products and has a wide range of pharmacological properties, including antidepressant-like, neuroprotective, anti-cancer and anti-inflammatory properties. This study investigated the metabolic pathways of LQ in vitro (rat liver microsomes) and in vivo (rat model) using ultra high-performance liquid chromatography coupled with hybrid triple quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS/MS). Moreover, supplementary tools such as key product ions (KPIs) were employed to search for and identify compounds. As a result, 56 in vivo metabolites and 15 in vitro metabolites were structurally characterized. Oxidation, reduction, hydrolysis, methylation, acetylation, and sulfate and glucuronide conjugation were determined to be the major metabolic pathways of LQ, and there were differences in LQ metabolism in vitro and in vivo. In addition, the in vitro and in vivo metabolic pathways were compared in this study.
1. Introduction
Licorice root or Radix glycyrrhizae, a Chinese materia medica (CMM), is widely used to invigorate the spleen, replenish the Qi, dispel heat and remove toxic substances.1 In modern medical terms, the biologically active compounds in licorice are triterpene, saponins, flavonoids, polysaccharides and phenolic compounds,2 making licorice root a source of medicine and food. These compounds exhibit several important pharmacological activities, including anti-viral,3 anti-oxidant,4 anti-bacterial,5 anti-inflammatory6 and anti-HIV7 activities. Moreover, licorice and its extract are widely used in health foods because of the physiological activities of these substances.8 Currently, the use of licorice and its extract in grain products, oil products, meat products, beverages, candy, jelly, dried fruit, seeds, soy sauce, etc., are being extensively researched.9,10 Among the biologically active compounds in licorice, liquiritin (LQ, Fig. 1) is the dominant component and is considered to be the major active ingredient.11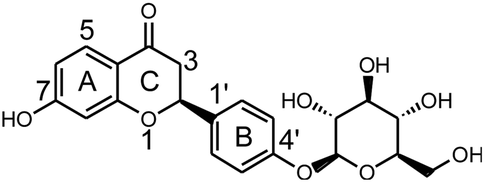 | ||
| Fig. 1 Chemical structure of LQ. | ||
Drug metabolism may lead to detoxification and/or activation reactions, and studies of drug metabolism can aid the identification of active compounds and explain the mechanisms of action of these compounds. It is well known that the liver plays a key role in the metabolism of orally administered drugs.12 The rat liver microsome system is often considered as a reasonable model in which to study drug metabolism. On the other hand, in vivo metabolic studies could comprehensively reveal the metabolic pathways of drugs.
In recent years, the use of liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) has been routinely used to detect and identify metabolites13,14 and has been used to study drug metabolism (flavones and flavonoids,15 phenylpropanoids,16–18 terpenes,19 alkaloids,20 saponins,21 stilbenes22 and traditional Chinese medicinal extracts23), pharmacokinetics and toxicokinetics of metabolites,24,25 and tissue distribution and excretion of metabolites.26 In addition, this method has applications in lipidomics,27 proteomics28 and metabolomics.29,30 A primary advantage of tandem mass spectrometry (MS/MS) is the ability of this method to detect a broad range of drugs with high sensitivity and specificity in a single analytical run.31–34 In addition, high-resolution mass spectrometry confirms structures by comparing the exact measured mass of a compound with the exact theoretical mass.35–37
To our knowledge, there has been one report of the metabolic profile of LQ China;38 however, this report was incomplete and only identified 7 metabolites. In this research contribution, a simple and rapid UHPLC-Q-TOF-MS/MS approach combined with pattern recognition analysis was first employed to rapidly screen and characterize metabolites of LQ in vitro and in vivo, which was the first systematic study of the metabolism of LQ in vitro and in vivo. The characterization of 56 in vivo metabolites and 15 in vitro metabolites was achieved by UHPLC-Q-TOF-MS/MS analysis based on the MS/MS spectra and clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values. In addition, the metabolic pathways of LQ were summarized. These results provide insight into the metabolic mechanism of LQ and lay the foundation for novel drug design.
P values. In addition, the metabolic pathways of LQ were summarized. These results provide insight into the metabolic mechanism of LQ and lay the foundation for novel drug design.
2. Materials and methods
2.1. Chemicals and materials
LQ (CAS No: 551-15-5) was purchased from Chengdu Desert Biotechnology Co., Ltd. (Chengdu, China). β-NADP (β-nicotinamine adenine dinucleotide phosphate), glucose-6-phosphate (G-6-P) and glucose-6-phosphatedehydrogenase (G-6-PD) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). MgCl2, UDPGA (uridine-5′-diphosphoglucuronic acid trisodium salt), Tris–HCl and alamethicin were purchased from BD Biosciences (Woburn, MA, USA). Phosphate-buffered saline (PBS) was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). HPLC-grade methanol was purchased from J.T. Baker Chemical Company (Phillipsburg, NJ, USA). Formic acid (HPLC grade) was provided by Diamond Technology (Dikma Technologies Inc., Lake Forest, CA, USA). Purified water was obtained from Hangzhou Wahaha Group Co., Ltd. (Hangzhou, China).2.2. Instrumentation and conditions
UHPLC-Q-TOF-MS/MS analysis was performed on a Shimadzu UHPLC system (Shimadzu Corp., Kyoto, Japan) coupled with a triple TOF™ 5600+ MS/MS system (AB Sciex, CA, USA). Information-dependent acquisition (IDA) was carried out. Chromatographic separation was conducted on a Poroshell 120 EC-C18 (2.1 × 100 mm, 2.7 μm) column with a SecurityGuard® UHPLC C18 pre-column (Agilent Corp, Santa Clara, CA, USA). The column temperature was maintained at 25 °C. The mobile phase was consisted of water containing 0.1% formic acid (A) and methanol (B). The gradient elution program was optimized for the separation, and the program was as follows: 0–2 min, 10–15% B; 2–15 min, 15–45% B; 15–20 min, 45–95% B; 20–25 min, 95–95% B. After maintaining the column at 95% solvent B for 5 min, the column was returned to its starting conditions over 1 min and was equilibrated in 10% solvent B for 5 min. The flow rate of the mobile phase was set to 0.3 mL min−1, and the injection volume was 3 μL.A Triple TOF™ 5600 system with DuoSpray™ ion sources (AB Sciex Triple TOF™ 5600+, Concord, Ontario, Canada) operating in the negative electrospray ionization mode was used for detection. The following MS/MS conditions were used: ion spray voltage, −4.5 kV; the turbo spray temperature, 550 °C; and declustering potential (DP), −60 V. Nitrogen was used as the nebulizer and auxiliary gas. Furthermore, the flows of the nebulizer gas (gas 1), heater gas (gas 2) and curtain gas were set to 55, 55 and 35 L min−1, respectively. The collision energy (CE) was set to −35 eV, and the collision energy spread (CES) was 15 eV.
Metabolite identification was performed with MetabolitePilot 1.5 (AB Sciex, CA, USA) based on accurate measurements of m/z values and on the processing of the data obtained from the XIC (extracted ion chromatography), MDF (mass defect filtering), PIF (product ion filtering) and NLF (neutral loss filtering) screening of putative metabolites. In addition, elemental compositions and chemical formulas were calculated.
2.3. Animals and drug administration
Thirty male Sprague-Dawley (SD) rats, 10–12 weeks in age and weighing 200–230 g, were obtained from the Experimental Animal Center of Hebei (Shijiazhuang, China). Rats were housed under standard temperature, humidity and light conditions. The animals were kept in an environmentally controlled breeding room for 7 days and fasted for 12 h before experiments. LQ was dissolved in a 0.5% carboxymethyl cellulose sodium (CMC-Na) aqueous solution. Thirty male SD rats were divided into six groups of five rats per group, which included experimental blood, urine and feces, and bile groups as well as blank blood, urine and feces, and bile groups. The prepared LQ suspension was orally administered to 15 rats from the experimental blood, urine and feces, and bile groups at a dose of 120 mg kg−1, and 0.5% CMC-Na aqueous solution was orally administered to 15 rats from the blank blood, urine and feces, and bile groups. All experiments were conducted in accordance with the guides of Animal Care and Use Committee at Hebei Medical University. This study was also performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of National Tissue Engineering Center (Shanghai, China).Plasma sample (five SD rats) collection was performed as follows: blood was taken from the canthi of the rats 0.17, 0.50, 0.75, 1, 2, 4, 6, 9, 12 and 24 h after administration. After centrifugation at 1400 × g for 5 min (Hunan Xiangyi Laboratory Instrument Development Co. Ltd., Hunan, China), the supernatant was collected, and all plasma samples were combined. Blank plasma was collected in the same manner from rats (five) administered 0.5% CMC-Na aqueous solution.
Urine and feces (five SD rats) collection was performed as follows: urine and feces were collected during the 0–4 h, 4–8 h, 8–12 h, 12–24 h, 24–36 h, 36–48 h, 48–60 h and 60–72 h periods after administration, and all the urine and feces samples were combined. Rats (five) administered 0.5% CMC-Na aqueous solution were subjected to the same process to collect blank urine and feces samples.
Bile (five SD rats) collection was performed as follows: rats were administered urethane-containing physiological saline solution (1.5–2 g kg−1) after gavage, and then, bile duct cannulation. Then, bile samples were collected during 0–1 h, 1–3 h, 3–5 h, 5–8 h, 8–12 h, 12–20 h and 20–24 h periods after administration. Finally, all bile samples were consolidated. Rats (five) administered 0.5% CMC-Na aqueous solution were subjected to the same process to collect a blank bile sample.
Three milliliters of blood, urine and bile samples were taken, and the protein in the samples was precipitated by methanol. Then, the supernatant was concentrated to dryness under reduced pressure at 25 °C using a Heidolph Laborota 4001 rotatory evaporator (Heidolph Instruments, GmbH & Co., Schwabach, Germany). The dried samples were dissolved in 300 μL of methanol in an ultrasonic bath for 5 min, and then, the samples were centrifuged for 10 min at 10000 × g. Then, the supernatant was injected into the UHPLC-Q-TOF-MS/MS system for further analysis.
Methanol (20 mL) was added to the feces sample (2.0 g), and then, the sample was ultrasonicated for 45 min (Kun Shan Ultrasonic Instruments Co., Kunshan, China). After the mixture was centrifuged at 10000 × g for 10 min, the supernatant was collected and blow-dried in a nitrogen atmosphere. The residue was dissolved in 400 μL of methanol and centrifuged at 10000 × g for 10 min. The supernatant (3 μL) was injected into the chromatographic instrument.
All the bio-samples were placed in the −80 °C freezer for storage.
2.4. Microsomal incubation
000 × g for 20 min at 4 °C, the precipitate was discarded. After additional centrifugation at 100000 × g for 60 min at 4 °C, the supernatant was discarded. The precipitate was washed with 4 times as much cold Tris–HCl solution. After centrifugation at 100000 × g for 60 min at 4 °C, the precipitation obtained was resuspended using 4 times as much Tris–HCl solution to obtain liver microsomes. Finally, the liver microsomes were placed at −80 °C in the freezer for storage until further use. In addition, the protein concentration of the liver microsome suspension was determined by the Lowry method.40000 × g for 10 min, the organic phase was collected and evaporated under nitrogen gas. Residues were redissolved in 100 μL of methanol, and an aliquot (3 μL) was injected into the chromatographic system for analysis. The blank sample was incubated without LQ, while the control sample was incubated without the NADPH-generating system by following the method described above.3. Results and discussion
A total of 56 in vivo metabolites and 15 in vitro metabolites were detected in the experimental conditions used. The metabolites detected in vivo and in vitro are listed in Tables 1 and 2, respectively, and the structures of the metabolites are shown in Fig. 2. The XIC and MS/MS data of the metabolites detected in vivo and in vitro are presented in Fig. 3 and S1,† respectively.| Metabolites ID | Composition shift | Formula | m/z | Error (ppm) | tR (min) | Score (%) | MS/MS fragments | clogP |
Blood | Urine | Bile | Feces |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a + Detected, − undetected, a, b and c – possible metabolites. | ||||||||||||
| M1 | Hydrolysis + tera-oxidation | C15H12O8 | 319.0477 | 1.6 | 7.95 | 88.5 | 273.1616, 255.1246, 239.0917, 221.0782, 195.1014, 177.0920, 151.0475, 79.9581 | 0.509837 | − | + | + | + |
| M2 | Hydrolysis + tera-oxidation | C15H12O8 | 319.0477 | 2.6 | 9.54 | 88.5 | 273.1699, 255.1614, 239.0926, 221.0815, 195.1019, 177.0911, 151.0476, 79.9588 | 0.569225 | − | + | + | + |
| M3 | Hydrolysis + tera-oxidation | C15H12O8 | 319.0482 | 1.0 | 13.20 | 92.8 | 273.1696, 255.1595, 239.0923, 221.0814, 195.1021, 177.0916, 151.0475, 79.9589 | 0.629837 | − | + | + | + |
| M4 | Hydrolysis + tri-oxidation | C15H12O7 | 303.0533 | 1.4 | 8.98 | 84.6 | 285.1333, 259.1542, 255.1213, 241.1432, 223.0969, 217.1069, 205.0863, 163.0757, 153.0922, 137.0967 | 1.16623 | − | + | + | + |
| M5 | Hydrolysis + tri-oxidation | C15H12O7 | 303.0536 | 4.6 | 11.42 | 83.4 | 285.1340, 259.1548, 255.1233, 241.1436, 223.0967, 217.1073, 205.0858, 163.0755, 153.0916, 137.0965 | 1.22684 | − | + | + | + |
| M6 | Hydrolysis + Tri-oxidation | C15H12O7 | 303.0532 | 3.2 | 11.94 | 90.1 | 285.1342, 259.1540, 255.1299, 241.1430, 223.0975, 217.1075, 205.0865, 163.0750, 153.0920, 137.0972 | 1.28623 | − | + | + | + |
| M7 | Oxidation | C21H22O10 | 433.1115 | −0.9 | 9.01 | 83.1 | 271.0596, 243.0645, 227.0692, 164.0107, 136.0151, 109.0290 | 0.300687 | + | + | + | + |
| M8 | Oxidation | C21H22O10 | 433.1111 | −2.7 | 12.61 | 85.3 | 271.0593, 243.0646, 227.0695, 164.0103, 136.0152, 109.0289 | 0.500687 | + | + | + | + |
| M9 | Oxidation | C21H22O10 | 433.1115 | −1.9 | 15.65 | 80.4 | 271.0598, 243.0650, 227.0705, 164.0098, 136.0142, 109.0294 | 0.558399 | + | + | + | + |
| M10 | Oxidation | C21H22O10 | 433.1114 | −2.1 | 16.45 | 83.4 | 271.0594, 243.0652, 227.0689, 164.0099, 136.0154, 109.0285 | 0.948399 | + | + | + | + |
| M11 | Oxidation + ketone formation | C21H20O11 | 447.0909 | −2.4 | 9.11 | 75.0 | 271.0600, 175.0238, 135.0443, 113.0242 | −0.178159 | − | + | + | − |
| M12 | Oxidation + ketone formation | C21H20O11 | 447.0891 | −4.4 | 10.84 | 78.4 | 271.0607, 175.0235, 135.0445, 113.0248 | 0.0218411 | − | + | + | − |
| M13 | Oxidation + ketone formation | C21H20O11 | 447.0903 | −4.8 | 11.67 | 74.4 | 271.0603, 175.0240, 135.0444, 113.0250 | 0.0795532 | − | + | + | − |
| M14 | Oxidation + ketone formation | C21H20O11 | 447.0909 | −4.3 | 13.12 | 76.7 | 271.0601, 175.0232, 135.0446, 113.0255 | 0.469553 | − | + | + | − |
| M15 | Hydrolysis + oxidation | C15H12O5 | 271.0635 | −0.6 | 11.52 | 84.1 | 253.1385, 235.1150, 227.0705, 209.1546, 191.1430, 183.0113, 151.1109, 145.0525, 119.0500 | 1.93694 | − | + | + | + |
| M16 | Hydrolysis + oxidation | C15H12O5 | 271.0610 | −2.5 | 13.66 | 90.3 | 253.1437, 235.1322, 227.1286, 209.1168, 191.1421, 183.1393, 151.0022, 145.0499, 119.0497 | 2.05485 | − | + | + | + |
| M17 | Hydrolysis + oxidation | C15H12O5 | 271.0605 | −1.4 | 13.88 | 74.8 | 253.1437, 235.1332, 227.1643, 209.1540, 191.1436, 183.1380, 151.0033, 145.0292, 119.0505 | 2.44485 | − | + | + | + |
| M18 | Hydrolysis + tri-oxidation + desaturation | C15H10O7 | 301.1284 | 4.0 | 12.89 | 75.1 | 283.1184, 257.1402, 255.0857, 241.1068, 239.1275, 221.1159, 211.0960, 169.0858, 118.9799 | 1.67912 | + | + | − | − |
| M19 | Hydrolysis + oxidation + desaturation | C15H10O5 | 269.0464 | 0.1 | 14.64 | 75.1 | 241.0512, 225.0547, 135.0085, 133.0286 | 2.90529 | − | + | + | + |
| M20a | Hydrolysis + oxidation + methylation | C16H14O5 | 285.0791 | −2.8 | 16.21 | 82.8 | 262.0798, 255.0729, 254.0721, 183.0107, 119.0495, 96.9603, 79.9575 | 2.50762 | − | + | + | + |
| M20b | 2.96762 | |||||||||||
| M20c | 3.03085 | |||||||||||
| M21 | Oxidation + methylation | C22H24O10 | 447.1278 | −4.2 | 17.00 | 83.9 | 385.1283, 285.1180, 271.0971, 255.0731, 175.0243, 165.0551, 113.0248 | 1.01117 | + | + | + | + |
| M22 | Oxidation + methylation | C22H24O10 | 447.1273 | −3.2 | 17.65 | 84.7 | 385.1281, 285.1150, 271.0967, 255.0739, 175.0241, 165.0554, 113.0246 | 1.47117 | + | + | + | + |
| M23 | Desaturation | C21H20O9 | 415.1780 | −4.1 | 17.03 | 82.6 | 253.0132, 252.0214, 223.0244, 142.9952, 112.9901 | 1.06205 | − | + | + | + |
| M24 | Hydrolysis + desaturation | C15H10O4 | 253.0509 | 0.1 | 17.58 | 91.4 | 135.0082, 133.0291, 117.0346 | 2.5753 | − | + | + | + |
| M25 | Hydrolysis + methylation | C16H14O4 | 269.0780 | −4.4 | 12.40 | 82.6 | 253.0637, 191.0387, 96.9607, 79.9585 | 2.83559 | − | + | + | + |
| M26 | Hydrolysis + methylation | C16H14O4 | 269.0781 | −3.9 | 13.87 | 87.4 | 253.0639, 191.0392, 96.9600, 79.9579 | 3.11994 | − | + | + | + |
| M27 | Hydrolysis | C15H12O4 | 255.0661 | −1.2 | 16.06 | 92.3 | 135.0090, 119.0510, 91.0204 | 2.53394 | + | + | + | + |
| M28 | Hydrolysis + bis-glucuronide conjugation | C27H28O16 | 607.1310 | −4.5 | 7.33 | 80.8 | 431.0975, 255.0655, 175.0240, 113.0248 | −1.70101 | − | + | + | + |
| M29 | Hydrolysis + oxidation + sulfate + glucuronide conjugation | C21H20O14S | 527.0471 | −4.8 | 7.55 | 81.1 | 447.0917, 351.0165, 271.0599, 193.0343, 175.0237, 135.0081, 113.0242 | −1.91668 | − | + | + | − |
| M30 | Hydrolysis + oxidation + sulfate + glucuronide conjugation | C21H20O14S | 527.0471 | −3.6 | 8.32 | 75.2 | 447.2034, 351.0510, 271.0601, 193.0340, 175.0233, 135.0068, 113.0240 | −1.61368 | − | + | + | − |
| M31 | Hydrolysis + oxidation + sulfate + glucuronide conjugation | C21H20O14S | 527.0480 | −4.1 | 10.20 | 78.1 | 447.0939, 351.0181, 271.0610, 193.0339, 175.0239, 135.0084, 113.0245 | −1.15368 | − | + | + | − |
| M32 | Glucuronide conjugation | C27H30O15 | 593.1479 | −1.6 | 7.63 | 83.7 | 417.1176, 255.0647, 175.0238, 135.0082, 119.0503, 113.0242 | −1.22216 | + | + | + | − |
| M33 | Glucuronide conjugation | C27H30O15 | 593.1476 | −2.1 | 12.01 | 83.3 | 417.1143, 255.0658, 175.0254, 135.0101, 119.0517, 113.0253 | −1.01791 | + | + | + | − |
| M34 | Hydrolysis + bis-sulfate conjugation | C15H12O10S2 | 415.1517 | −2.8 | 8.05 | 89.8 | 335.0230, 255.0658, 135.0080, 119.0494 | −0.870411 | − | + | + | − |
| M35 | Sulfate conjugation | C21H22O12S | 497.0759 | −3.0 | 8.70 | 86.6 | 417.1185, 283.0806, 255.0655, 206.9858, 167.0074, 135.0074, 113.0241 | −0.806862 | − | + | + | + |
| M36 | Hydrolysis + sulfate + glucuronide conjugation | C21H20O13S | 511.0541 | −1.3 | 8.92 | 74.2 | 431.0970, 335.0220, 255.0654, 238.9310, 175.0244, 135.0084, 119.0506, 113.0242 | −1.28571 | + | + | + | + |
| M37 | Tri-oxidation + glucuronide conjugation | C27H30O18 | 641.1338 | −0.7 | 9.25 | 79.1 | 337.0366, 303.0870, 281.1053, 255.0655, 135.0080, 119.0504 | −2.78222 | − | − | − | + |
| M38 | Tri-oxidation + glucuronide conjugation | C27H30O18 | 641.1397 | −0.4 | 12.14 | 76.0 | 337.0390, 303.0881, 281.1069, 255.0665, 135.0081, 119.0498 | −2.64222 | − | − | − | + |
| M39 | Oxidation + glucuronide conjugation | C27H30O16 | 609.1423 | −1.2 | 9.67 | 82.6 | 433.1126, 255.0806, 175.0239, 151.0395, 113.0243 | −1.55013 | − | + | − | − |
| M40 | Oxidation + glucuronide conjugation | C27H30O16 | 609.1430 | −3.2 | 12.55 | 81.2 | 433.1146, 255.0812, 175.0237, 151.0395, 113.0240 | −1.09013 | − | + | − | − |
| M41 | Hydrolysis + oxidation + sulfate conjugation | C15H12O8S | 351.0164 | −4.5 | 10.67 | 80.8 | 271.0614, 151.0039, 119.0507 | 0.361621 | − | + | + | + |
| M42 | Hydrolysis + oxidation + sulfate conjugation | C15H12O8S | 351.0163 | −4.8 | 11.45 | 79.8 | 271.0610, 151.0040, 119.0509 | 0.821621 | − | + | + | + |
| M43 | Hydrolysis + oxidation + sulfate conjugation | C15H12O8S | 351.0164 | −4.7 | 12.00 | 93.3 | 271.0609, 151.0040, 119.0507 | 0.884852 | − | + | + | + |
| M44 | Hydrolysis + glucuronide conjugation | C21H20O10 | 431.0976 | −2.6 | 11.17 | 85.6 | 255.0658, 175.0245, 135.0086, 119.0503, 113.0247 | 0.27429 | + | + | + | + |
| M45 | Hydrolysis + glucuronide conjugation | C21H20O10 | 431.0976 | −1.5 | 11.97 | 94.0 | 255.0662, 175.0253, 135.0089, 119.0502, 113.0251 | 0.55864 | + | + | + | + |
| M46 | Hydrolysis + desaturation + glucuronide conjugation | C21H18O10 | 429.0828 | 0.2 | 12.13 | 82.6 | 253.0505, 113.0235 | 0.390001 | − | + | + | − |
| M47 | Hydrolysis + desaturation + glucuronide conjugation | C21H18O10 | 429.0824 | −0.7 | 12.78 | 85.7 | 253.0495, 113.0230 | 0.583201 | − | + | + | − |
| M48 | Oxidation + sulfate conjugation | C21H22O13S | 513.0667 | −2.1 | 12.54 | 93.4 | 433.0514, 431.0986, 337.0368, 255.0641, 175.0226, 151.0390, 113.0244 | −1.13483 | + | + | + | + |
| M49 | Oxidation + sulfate conjugation | C21H22O13S | 513.0677 | −1.1 | 13.24 | 91.6 | 433.0558, 431.0976, 337.0365, 255.0665, 175.0247, 151.0393, 113.0241 | −0.674831 | + | + | + | + |
| M50 | Hydrolysis + sulfate conjugation | C15H12O7S | 335.0216 | −4.4 | 12.49 | 84.3 | 255.0656, 135.0083, 119.0500 | 0.689589 | + | + | + | + |
| M51 | Hydrolysis + sulfate conjugation | C15H12O7S | 335.0217 | −4.1 | 17.30 | 92.0 | 255.0660, 135.0088, 119.0502 | 0.97394 | + | + | + | + |
| M52 | Ketone formation | C21H18O11 | 445.1883 | −2.1 | 12.53 | 89.4 | 269.0443, 113.0239, 104.9542 | 0.924951 | + | + | + | − |
| M53 | Loss of O + bis-ketone formation | C21H18O9 | 413.0854 | −1.8 | 13.24 | 83.4 | 369.0968, 255.0660, 135.0088, 119.0505 | 0.29614 | − | + | − | − |
| M54 | Loss of O + bis-ketone formation | C21H18O9 | 413.0853 | −2.1 | 14.22 | 74.4 | 369.0983, 255.0666, 135.0087, 119.0501 | 0.34904 | − | + | − | − |
| M55 | Acetylation | C23H24O10 | 459.1297 | 0.0 | 16.56 | 75.3 | 417.1198, 399.1064, 255.0645, 186.9290, 135.0070, 119.0489 | 0.769139 | + | + | − | − |
| M56 | Methylation | C22H24O9 | 431.0992 | 2.7 | 18.33 | 77.4 | 369.0977, 255.0666, 175.0244, 135.0086, 113.0248 | 1.33914 | + | + | + | + |
| Metabolites ID | Composition shift | Formula | m/z | Error (ppm) | tR (min) | Score (%) | MS/MS fragments | clogP |
Blanks | Controls | Samples |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a + Detected, − undetected, a and b – possible metabolites. | |||||||||||
| N1 | Oxidation | C21H22O10 | 433.1110 | −2.0 | 8.62 | 79.1 | 271.0592, 243.0645, 109.0289 | 0.300687 | − | − | + |
| N2 | Oxidation | C21H22O10 | 433.1111 | −2.8 | 9.01 | 79.0 | 271.0595, 243.0647, 109.0282 | 0.500687 | − | − | + |
| N3 | Oxidation | C21H22O10 | 433.1112 | −2.6 | 9.51 | 82.5 | 271.0596, 243.0645, 109.0292 | 0.558399 | − | − | + |
| N4 | Oxidation | C21H22O10 | 433.1109 | −2.2 | 14.03 | 84.4 | 271.0589, 243.0676, 109.0290 | 0.948399 | − | − | + |
| N5 | Hydrolysis + oxidation | C15H12O5 | 271.0596 | −1.1 | 11.12 | 89.7 | 253.0474, 151.0016, 135.0439, 119.0487, 91.0191 | 1.93694 | − | − | + |
| N6 | Hydrolysis + oxidation | C15H12O5 | 271.0595 | −1.3 | 13.60 | 81.1 | 253.0486, 151.0024, 135.0444, 119.0495, 91.0136 | 2.05485 | − | − | + |
| N7 | Hydrolysis + oxidation | C15H12O5 | 271.0592 | −1.5 | 13.83 | 73.4 | 253.0480, 151.0021, 135.0435, 119.0480, 91.0131 | 2.44485 | − | − | + |
| N8 | Desaturation | C21H20O9 | 415.1011 | −1.7 | 12.63 | 76.2 | 253.0518, 252.0421, 223.0455, 142.99500, 112.9840 | 1.06205 | − | − | + |
| N9 | Hydrolysis | C15H12O4 | 255.0650 | −3.4 | 16.03 | 96.8 | 135.0083, 119.0500, 91.0193 | 2.53394 | − | − | + |
| N10 | Hydrolysis + desaturation + oxidation | C15H10O5 | 269.0437 | −3.9 | 16.38 | 79.2 | 135.0090, 133.0287 | 2.90529 | − | − | + |
| N11 | Hydrolysis + desaturation | C15H10O4 | 253.0496 | −2.4 | 17.58 | 85.6 | 135.0077, 133.0284, 117.0341, 91.0187 | 2.5753 | − | − | + |
| N12a | Glucuronide conjugation | C27H30O15 | 593.1565 | 3.7 | 7.62 | 74.4 | 417.1208, 255.0659, 175.0241, 135.0078, 117.0187, 113.0241 | −1.22216 | − | − | + |
| N12b | −1.01791 | ||||||||||
| N13 | Hydrolysis + glucuronide conjugation | C21H20O10 | 431.0989 | 1.3 | 11.19 | 86.4 | 255.0662, 226.9648, 175.0239, 135.0080, 119.0495, 113.0242 | 0.27429 | − | − | + |
| N14 | Hydrolysis + glucuronide conjugation | C21H20O10 | 431.0985 | 0.2 | 11.93 | 82.8 | 255.0666, 226.9703, 175.0233, 135.0079, 119.0496, 113.0229 | 0.55864 | − | − | + |
| N15 | Glucose conjuagtion | C27H32O14 | 579.1752 | 3.9 | 12.45 | 89.3 | 417.1273, 402.9939, 255.0669, 238.9306, 135.0085, 119.0492 | −0.743309 | − | − | + |
 | ||
| Fig. 2 Chemical structures of all metabolites of LQ detected in vitro and in vivo (a, b and c—possible chemical structure). | ||
 | ||
| Fig. 3 Extracted ion chromatograms of all metabolites of LQ detected in vitro and in vivo. (A1–2 in rat blood sample, B1–7 in rat urine sample, C in rat bile sample, D in rat feces sample, E1–4 in rat liver microsomes). Additional graph: metabolites detected by KPIs in rat blood, urine, bile and feces samples. | ||
3.1. Analytical strategy and metabolite analysis
In this study, a method of metabolite identification was developed that was based on a Triple TOF™ instrument with a multiple mass defect filter (MMDF) combined with dynamic background subtraction (DBS)-dependent on-line data acquisition and multiple post-acquisition data mining technologies. First, on-line data and accurate MS/MS data were acquired using a full-scan, unique and effective MMDF and a DBS-dependent data acquisition method.41 Then, post-acquisition data mining was performed using various data-mining tools such as XIC, MDF, PIF and NLF.42 Furthermore, structures of the metabolites of LQ were elucidated based on accurate mass measurements, knowledge of relevant drug bio-transformation, previously investigated fragmentation patterns of LQ, and MS/MS spectra of metabolites. Peakview 1.2 software was used to identify possible metabolites by comparing the extracted ion chromatograms and base peak chromatograms of the sample group with those of the blank group.43 Finally, the useful clogP parameter, which was calculated using the ChemDraw Ultra 12.0 program, was introduced to distinguish between structural isomers. Generally, compounds with the larger values have longer retention times in reversed-phase liquid chromatography systems.44,45
All chemical constituents in TMM can be categorized into different families based on structural types. Thus, groups of compounds with identical carbon skeletons may yield similar fragmentation patterns and then generate the same characteristic fragment ions when subjected to collision-induced dissociation (CID) for mass spectrometry. Accordingly, a core supplementary tool in this approach is to use key product ions (KPIs) as markers for compounds detection and identification.46–48 In this study, a KPI at m/z 255.0651 could be generated from common substructures as standards and was selected as a diagnostic ion for detecting relevant analogues of these substructures (shown in Fig. 3).
3.2. Mass fragmentation behavior of LQ
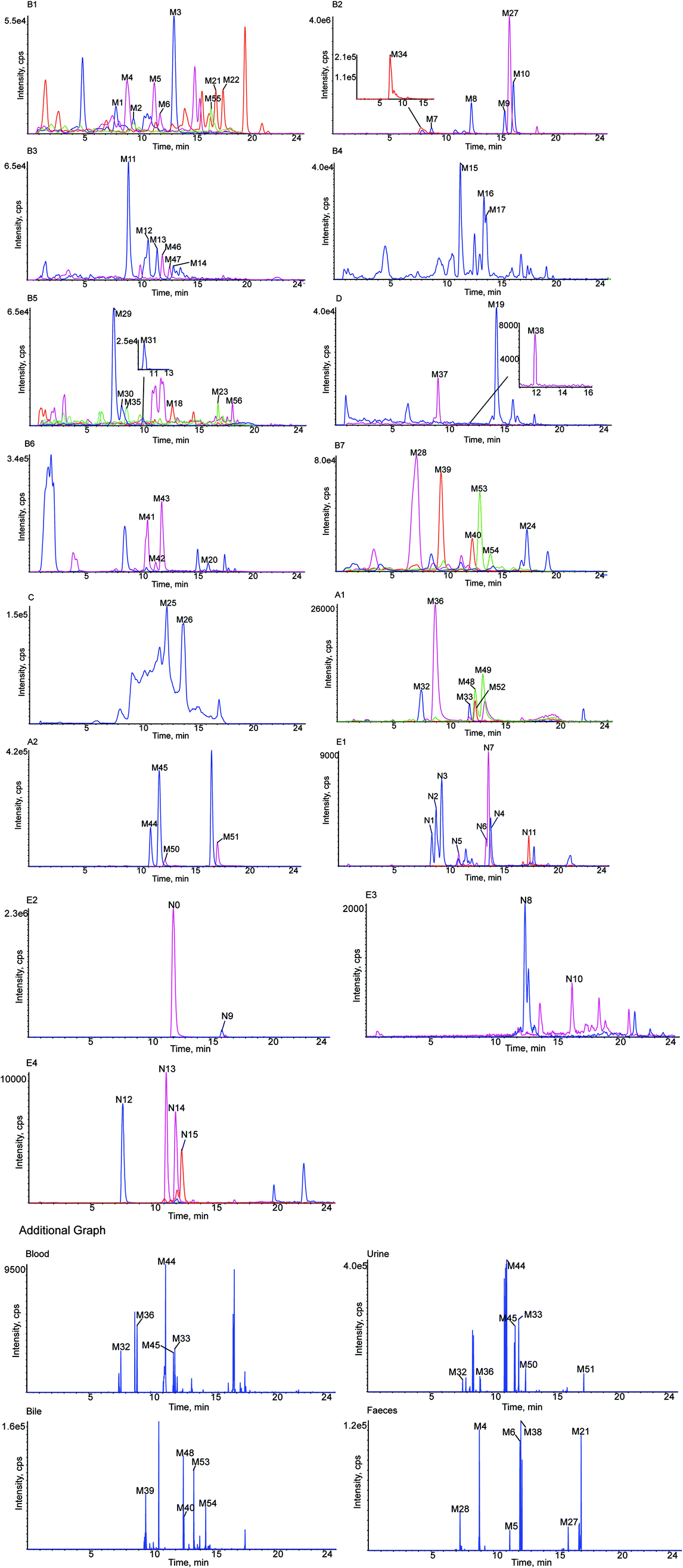
The retention time of LQ was 12.01 min, and LQ produced a molecular ion [M − H]− at m/z 417.1176 under the experimental conditions. Moreover, major secondary fragment ions at m/z 255.0651 [M–H–C6H10O5]−, 135.0079 (RDA reaction (retro Diels−Alder reaction)), 119.0496 (RDA reaction) and 91.0190 (RDA reaction) were detected. The proposed MS/MS fragmentation behavior and fragmentation pathways of LQ are shown in Fig. 4. The molecular weight and elemental composition of LQ were used as a baseline for comparison with some metabolites.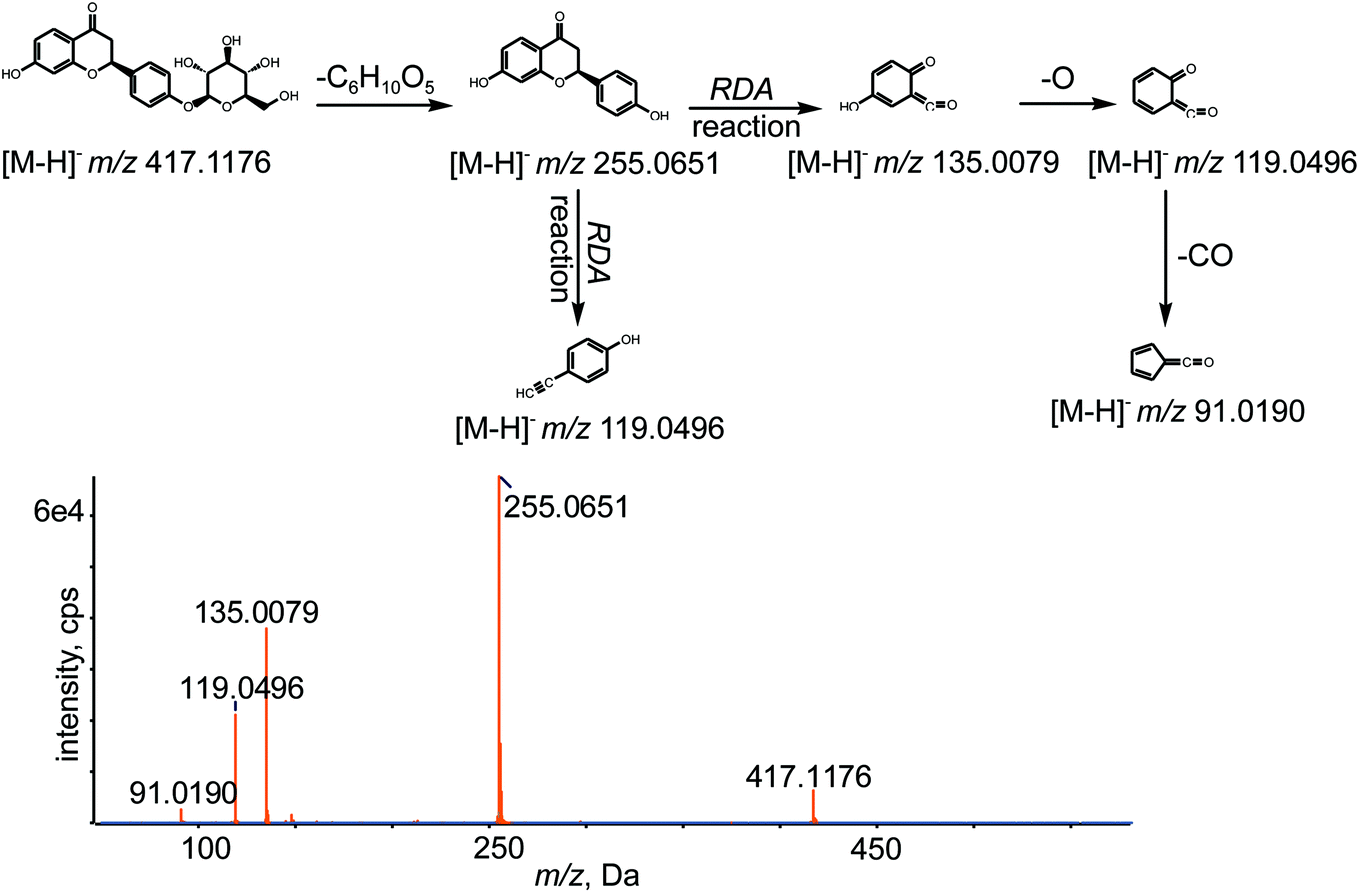 | ||
| Fig. 4 MS/MS spectrum of LQ and its predominant fragmentation pathways. | ||
3.3. Identification of in vivo phase I metabolites
Metabolites M1, M2 and M3. M1–M3 were eluted at 7.95, 9.54 and 13.20 min, respectively, with deprotonated molecular ions [M − H]− at m/z 319.0477, 319.0477 and 319.0482, respectively, which were 98 Da less than the value obtained for LQ and corresponded to C15H12O8. The fragment ions at m/z 273.1696, 255.1595, 239.0923, 221.0814 and 151.0475 were produced from M1, M2 and M3 by the loss of CO, H2O, O and by RDA reaction. Among these fragments, the fragment ion at m/z 255.1595 was hydrolyzed LQ. M1, M2 and M3 were oxidation metabolites of LQ and were assigned based on clog
P values of 0.509837, 0.569225 and 0.629837 by ChemDraw 12.0 software.
Metabolites M7, M8, M9 and M10. M7–M10, eluted at 9.01, 12.61, 15.65 and 16.45 min, respectively, were characterized as deprotonated molecular ions [M − H]− at m/z 433.1115, 433.1111, 433.1115 and 433.1114, respectively, which were 16 Da more than the value obtained for the parent drug LQ. This finding suggested that M7–M10 were oxidation metabolites of LQ and that the chemical formula of M7–M10 was C21H22O10. The observed diagnostic fragment ions at m/z 271.0596, 243.0645, 227.0692, 136.0151 and 109.0290 were generated by the loss of C6H10O5, CO and O and by RDA reaction. The clog
P values of M7–M10 were 0.300687, 0.500687, 0.558399 and 0.948399, respectively. Therefore, the four compounds were immediately identified on the basis of the retention times and clogP values.
Metabolites M11, M12, M13 and M14. M11–M14 were eluted at 9.11, 10.84, 11.67 and 13.12 min, respectively, with deprotonated molecular ions [M − H]− at m/z 447.0909, 447.0891, 447.0903 and 447.0909, respectively, which were 30 Da more than the value obtained for LQ. The major fragment ions detected at m/z 271.0600, 135.0443 and 113.0242 were generated by the loss of C6H8O6 and by RDA reaction, which implied that M11–M14 were oxidation products of LQ and that the chemical formula was C21H20O11. In addition, M11, M12, M13 and M14 were assigned based on the clog
P values of M11, M12, M13 and M14, which were −0.178159, 0.0218411, 0.0795532 and 0.469553, respectively.
Metabolite M18. M18 was detected at a retention time of 12.89 min, with a deprotonated molecular ion [M − H]− at m/z 301.1284, 46 Da higher than that of M0 losing C6H10O5, which suggested that M18 was oxidation metabolite. The fragment ions at m/z 283.1184, 255.0857, 239.1275, 169.0858 and 118.9799 were gained by loss of H2O, CO, O and by RDA reaction. According to the fragmentation, the chemical formula of M18 was C15H10O7.
Metabolite M19. M19 was detected at a retention time of 14.64 min with a deprotonated molecular ion [M − H]− at m/z 269.0464, which was generated by the loss of two oxygens from M18. Typical fragment ions at m/z 241.0512, 225.0547, 135.0085 and 133.0286 were detected due to successive loss of CO and O and due to RDA reaction. Thus, M19 was deduced to be C15H10O5.
Metabolite M20. M20 had a retention time of 16.21 min and was detected at m/z 285.0791 ([M − H]−), which was 30 Da more than the value obtained for hydrolyzed LQ, which implied that oxidation and methylation reactions had occurred. Representative fragment ions were observed at m/z 255.0729 and 119.0495 due to loss of CH2O and due to RDA reaction, which suggested that the formula of M20 was C16H14O5.
Metabolite M23. A peak was eluted at a retention time of 17.03 min. The MS/MS spectrum of M23 showed a deprotonated molecular ion [M − H]− at m/z 415.1780, which was 2 Da less than the value obtained for M0, which confirmed the molecular formula to be C21H20O9. In addition, the MS/MS spectrum of M23 showed a number of characteristic fragment ions at m/z 253.0132 [M–C6H10O5–H]−, 142.9952 (RDA reaction) and 112.9901 (RDA reaction).
Metabolite M24. M24 was detected at 17.58 min and showed a deprotonated molecular ion [M − H]− at m/z 253.0509, which was 164 Da lower than the value obtained for M0, which suggested that the loss of two hydrogens from hydrolyzed M0. Meanwhile, representative fragment ions at m/z 135.0082, 133.0291 and 117.0346 were obtained due to RDA reaction. The formula was identified as C15H10O4.38
Metabolites M25, M26. M25 and M26 displayed molecular ions [M − H]− at m/z 269.0780 and 269.0781, respectively, and had retention times of 12.40 and 13.87 min, respectively. The masses of the deprotonated M25 and M26 were 14 Da higher than the mass of hydrolyzed LQ, which suggested that a methylation reaction occurred on LQ after the loss of C6H10O5. The MS/MS spectra showed a characteristic fragment ion at m/z 253.0637 [M–O–H]−, which implied that the formula of M25 and M26 was C16H14O4. Then, M25 and M26 were identified based on their clog
P values of 2.83559 and 3.11994, respectively.
Metabolite M27. The metabolite M27 exhibited a deprotonated ion [M − H]− at m/z 255.0661, which was 162 Da lower than the value obtained for M0, confirming the occurrence of hydrolysis. In addition, typical fragment ions at m/z 135.0090, 119.0510 and 91.0204 were produced by RDA reaction. The formula of M27 was deduced to be C15H12O4.38
3.4. Identification of in vivo phase II metabolites
P values of M29, M30 and M31 were −1.91668, −1.61368 and −1.15368, respectively, and M29, M30 and M31 were identified based on this information.P values of −1.22216 and −1.01791, respectively.P values of M44 and M45 were 0.27429 and 0.55864, respectively. Hence, M44 and M45 were identified on the basis of their retention times and clogP values.P values of 0.390001 and 0.583201, respectively.P values of M50 and M51 were 0.689589 and 0.97394, respectively. Hence, these metabolites were identified based on the retention times and clogP values.P values of 0.29614and 0.34904, respectively.3.5. Identification and characterization of in vitro metabolites
In this incubation system, fifteen metabolites were detected, including eleven phase I and four phase II metabolites. Furthermore, metabolites N1–N14 were identified in the in vivo metabolic study. However, N15 was not found in vivo bio-samples. The structures of all the metabolites are shown in Fig. 2.Metabolites N1, N2, M3 and N4. N1–N4, with retention times of 8.62, 9.01, 9.51 and 14.03 min, exhibited sharp peaks of deprotonated molecular ions [M − H]− at m/z 433.1110, 433.1111, 433.1112 and 433.1109, respectively, which were 16 Da more than the value obtained for LQ, suggesting that N1–N4 were oxidation metabolites and corresponded to the molecular formula C21H22O10. In addition, typical fragment ions at m/z 271.0592, 243.0645 and 109.0289 were generated by the loss of C6H10O5 and CO and by RDA reaction. Moreover, N1–N4 were identified based on the clog
P values of N1–N4 at 0.300687, 0.500687, 0.558399 and 0.948399, respectively.
Metabolite N8. N8 was detected at 12.63 min and exhibited a sharp peak for a deprotonated molecular ion [M − H]− at m/z 415.1011, which was 2 Da less than the value obtained for M0. Fragment ions at m/z 253.0518 [M–C6H10O5–H]− suggested that a desaturation reaction had occurred, which was consistent with a molecular formula of C21H20O9.
Metabolite N9. N9 was detected at 16.03 min. N9 exhibited a sharp peak for a deprotonated molecular ion [M − H]− at m/z 255.0650, which was 162 Da lower than the value obtained for LQ, implying that a hydrolysis had occurred. The secondary fragment ions at m/z 135.0083 and 119.0500, caused by RDA reaction, confirmed that the molecular formula of N9 was C15H12O4.38
Metabolite N10. N10 exhibited a deprotonated molecular ion [M − H]− at m/z 269.0437, which was 14 Da more than the value obtained for N9, indicating that the molecular formula of N10 was C15H10O5. In addition, N10 had a retention time of 16.38 min, and the important fragment ions were at m/z 135.0090 (RDA reaction) and 133.0287 (RDA reaction).
Metabolite N11. N11 showed a deprotonated molecular ion [M − H]− at m/z 253.0496, which was 2 Da less than the value obtained for N9, suggesting that a desaturation reaction occurred for N9, and the molecular formula of N11 was predicted to be C15H10O4.38 In addition, the secondary fragment ions at m/z 135.0077, 133.0284 and 117.0341, caused by RDA reaction, were consistent with the chemical structure of N11. Moreover, the retention time of N11 was 17.58 min.
Metabolite N12. N12 presented a deprotonated molecular ion [M − H]− at m/z 593.1565, which was 176 Da higher than the value obtained for LQ, suggesting that glucuronide conjugation had occurred. In addition, the molecular formula was determined to be C27H30O15.38 Moreover, the MS/MS spectrum exhibited a series of typical fragment ions at m/z 417.1208 [M–C6H8O6–H]−, 255.0659 [M–C6H8O6–C6H10O5–H]−, 135.0078 (RDA reaction) and 117.0187 (RDA reaction). In addition, N12 was eluted at 7.62 min.
Metabolite N15. The molecular formula was determined to be C27H32O14 based on the deprotonated molecular ion [M − H]− at m/z 579.1752, which was 162 Da higher than the value obtained for LQ. N15 was eluted at 12.45 min and had characteristic fragment ions at m/z 417.1273 [M–C6H10O5–H]−, 255.0669 [M–2C6H10O5–H]−, 135.0085 (RDA reaction) and 119.0492 (RDA reaction), which implied that N15 was a glucose metabolite. Other metabolites were showed in ESI.†
3.6. Metabolic pathways of LQ
Based on the elemental compositions of the metabolites, the accurate MS/MS spectra, the chemical structures of the metabolites and the fragment ions of the metabolites, the metabolic pathway of LQ could be tentatively proposed. In total, 56 in vivo metabolites and 15 in vitro metabolites were characterized. Based on these results, LQ mainly underwent oxidation, reduction, hydrolysis, methylation, acetylation, glucuronide and sulfate conjugation. Based on the in vivo metabolic data, sulfate and glucuronide conjugation reactions were the major bio-transformations. However, based on the in vitro metabolic data, the metabolic pathways of LQ were concentrated in phase I. The proposed metabolic pathways of LQ in vitro and in vivo are shown in Fig. 5.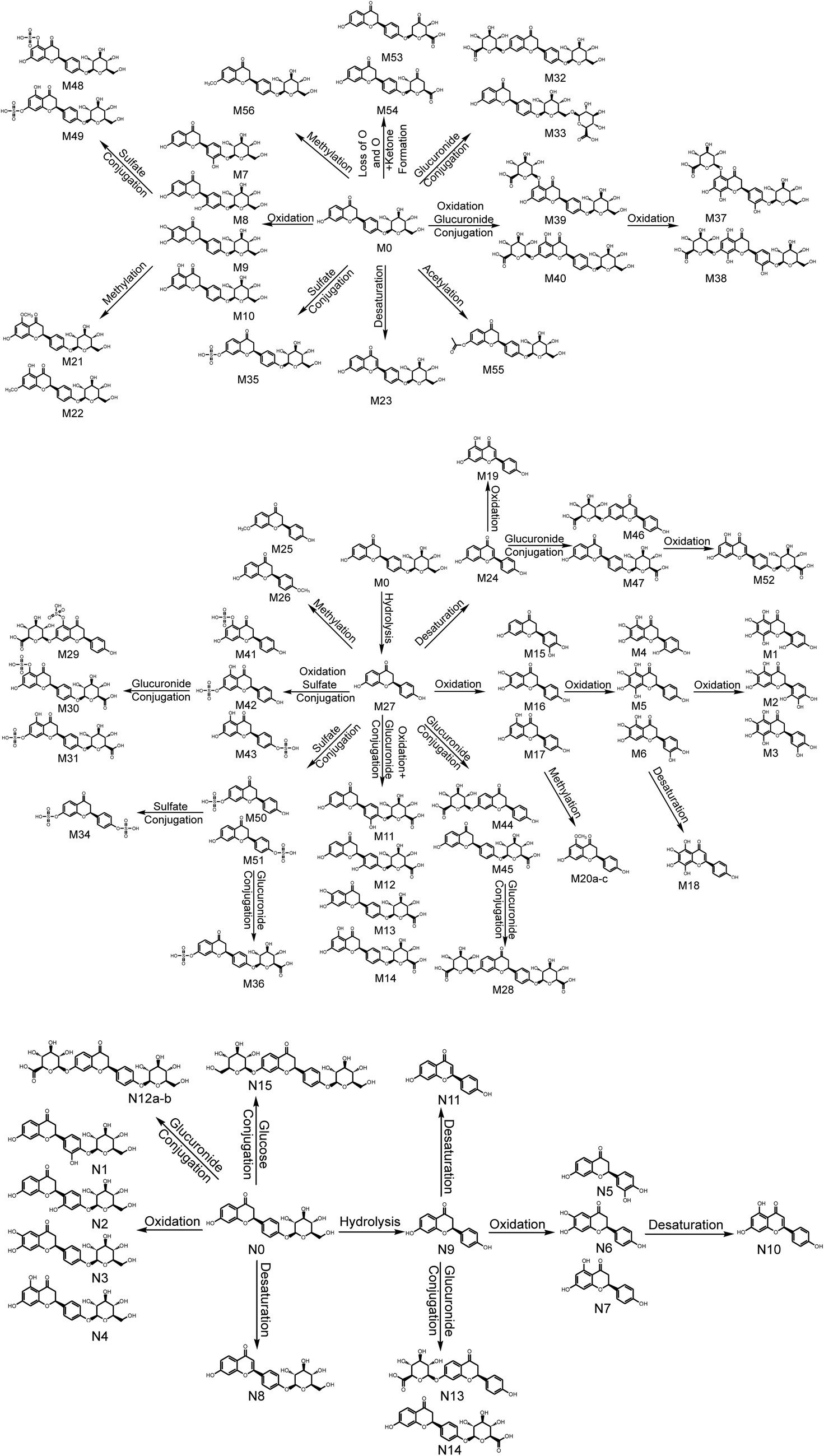 | ||
| Fig. 5 Metabolic profile and proposed metabolic pathways of LQ in vitro and in vivo (a, b, c—possible chemical structure). | ||
3.7. Comparison of metabolic pathways in vitro and in vivo
Drug metabolism plays an important role in different areas of the pharmaceutical industry and in drug development and toxicology. The in vivo approach is quantitative and very effective in studies of drug metabolism.49,50 The in vitro method is generally suitable for targeted studies and is often predictive of real hazards and risks.51 In this study, in vitro (rat liver microsomes) and in vivo (blood, urine, feces and bile in rats) metabolic profiles were investigated by UHPLC-Q-TOF-MS/MS. Consequently, a total of 56 in vivo metabolites and 15 in vitro metabolites were screened and characterized. The in vitro and in vivo metabolisms were concentrated in phase I and II, respectively. The metabolites detected in vitro were all detected in vivo as well. However, the isomer of N12 was detected in vivo, and N15 was not found in vivo. It was speculated that oxidation of N15 had occurred in vivo. Therefore, the isomer of N12 was observed in vivo, which suggested that the metabolism in vivo was more complex and elusive than that in vitro.3.8. Comparison of metabolites identified in this study and in a previous study
A study of the metabolic pathways of LQ has been reported in China, which only detected 7 metabolites (M24, M27, M32 (N12), M35, M36, M44 and M45 (N13 and N14), and M51). However, in this study, 56 metabolites were detected in vivo and 15 metabolites were detected in vitro, including the 7 metabolites identified in the previous study. Moreover, the metabolic sites of two metabolites were not determined in the previous study, and there were some differences between this study and the previous study. In this manuscript, M32, M33 and M50, M51 were detected as two pairs of isomers; however, these metabolites were detected as a single chromatographic peak in the previous study. Moreover, in this study, M32 and M33 were both not detected in vitro (N12), and no isomers of M36 were detected in vivo. Furthermore, in this manuscript, the metabolic sites of M44 and M45 were identified based on the important clogP values, which were the same as those of N13 and N14 in vitro. In addition, in this study, metabolite N15 was not detected in vivo; therefore, it was speculated that N15 had undergone oxidation and that metabolites M32 and M33 were found in vivo.
4. Conclusion
In conclusion, the UHPLC-Q-TOF-MS/MS method was first established to screen and identify the metabolites of LQ in vitro and in vivo. Based on the preceding data acquisition and mining strategy, twenty, fifty-four, forty-eight, thirty-seven and fifteen metabolites were detected in rat blood, urine, bile, feces and liver microsomes, respectively. The metabolic pathways of LQ were determined to be oxidation, reduction, hydrolysis, methylation, acetylation, and sulfate and glucuronide conjugation; these findings filled in the gaps remaining from the previous incomplete study. Key product ion (KPI) was used to aid the detection of metabolites of LQ. The results lay the foundation for active screening studies. In addition, this study demonstrated a powerful strategy for rapid screening and identifying metabolites and chemical constituents of traditional Chinese medicines.Conflicts of interest
All the authors have declared no conflict of interest.Acknowledgements
The work received financial support from the National Natural Science Foundation of China (No. 81473180).References
- M. N. Asl and H. Hosseinzadeh, Phytother. Res., 2008, 22, 709–724 CrossRef CAS PubMed.
- B. Q. Fu, H. Li, X. R. Wang, F. S. C. Lee and S. F. Cui, J. Agric. Food Chem., 2005, 53, 7408–7414 CrossRef CAS PubMed.
- J. Cinatl, B. Morgenstern, G. Bauer, P. Chandra, H. Rabenau and H. W. Doerr, Lancet, 2003, 361, 2045–2046 CrossRef CAS.
- T. Yokozawa, Z. W. Liu and C. P. Chen, Phytomedicine, 2000, 6, 439–445 CrossRef CAS PubMed.
- J. He, L. Chen, D. Heber, W. Y. Shi and Q. Y. Lu, J. Nat. Prod., 2006, 69, 121–124 CrossRef CAS PubMed.
- G. Khaksa, M. E. Zolfaghari, A. R. Dehpour and T. Samadian, Planta Med., 1996, 62, 326–328 CrossRef CAS PubMed.
- F. Rauchensteiner, Y. Matsumura, Y. Yamamoto, S. Yamaji and T. Tani, J. Pharm. Biomed. Anal., 2005, 38, 594–600 CrossRef CAS PubMed.
- Y. X. Gao, B. F. Cheng, J. J. Lian, D. D. Guo, J. W. Qin, Y. B. Zhang, H. J. Yang, M. Wang, L. Wang and Z. W. Feng, J. Funct. Foods, 2017, 33, 142–148 CrossRef CAS.
- H. J. Kwon, H. H. Kim, Y. B. Ryu, J. H. Kim, H. J. Jeong, S. W. Lee, J. S. Chang, K. O. Cho, M. C. Rho, S. J. Park and W. S. Lee, Bioorg. Med. Chem., 2010, 18, 7668–7674 CrossRef CAS PubMed.
- P. Montoro, M. Maldini, M. Russo, S. Postorino, S. Piacente and C. Pizza, J. Pharm. Biomed. Anal., 2011, 54, 535–544 CrossRef CAS PubMed.
- X. Y. Meng, S. B. Yang, Z. F. Pi, F. R. Song, H. Y. Jiang and Z. Q. Liu, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 1538–1549 CAS.
- C. F. Xue, S. Jiang, J. M. Guo, D. W. Qian, J. A. Duan and E. X. Shang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3901–3908 CrossRef CAS PubMed.
- C. Y. Li, L. W. Qi and P. Li, J. Pharm. Biomed. Anal., 2011, 55, 146–160 CrossRef CAS PubMed.
- G. G. Tan, M. Liu, X. Dong, S. Wu, L. Fan, Y. B. Qiao, Y. F. Chai and H. Wu, J. Pharm. Biomed. Anal., 2014, 96, 187–196 CrossRef CAS PubMed.
- Y. Z. Zhang, F. Xu, J. Dong, J. Liang, Y. Hashi, M. Y. Shang, D. H. Yang, X. Wang and S. Q. Cai, J. Pharm. Biomed. Anal., 2012, 70, 425–439 CrossRef CAS PubMed.
- M. P. Gonthier, C. Remesy, A. Scalbert, V. Cheynier, J. M. Souquet, K. Poutanen and A. M. Aura, Biomed. Pharmacother., 2006, 60, 536–540 CrossRef CAS PubMed.
- A. H. Yang, J. X. Chen, Y. T. Ma, L. L. Wang, Y. W. Fan and X. He, J. Pharm. Biomed. Anal., 2017, 141, 200–209 CrossRef CAS PubMed.
- M. Y. Liu, S. H. Zhao, Z. Q. Wang, Y. F. Wang, T. Liu, S. Li, C. C. Wang, H. T. Wang and P. F. Tu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 949–950, 115–126 CrossRef CAS PubMed.
- H. Han, W. L. Zeng, C. Y. He, S. W. A. Bligh, Q. Liu, L. Yang and Z. T. Wang, J. Mass Spectrom., 2014, 49, 1108–1116 CrossRef CAS PubMed.
- K. Wang, L. W. Chai, X. C. Feng, Z. B. Liu, H. X. Liu, L. Q. Ding and F. Qiu, J. Pharm. Biomed. Anal., 2017, 139, 73–86 CrossRef CAS PubMed.
- X. D. Cheng and M. G. Wei, Molecules, 2014, 19, 18881–18896 CrossRef PubMed.
- D. G. Wang, T. J. Hang, C. Y. Wu and W. Y. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 829, 97–106 CrossRef CAS PubMed.
- H. L. Ma, Y. Liu, X. Mai, Y. J. Liao, K. Zhang, B. Liu, X. Xie and Q. L. Du, J. Pharm. Biomed. Anal., 2016, 125, 194–204 CrossRef CAS PubMed.
- K. Xiong, T. T. Gao, T. Zhang, Z. T. Wang and H. Han, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1065–1066, 1–7 CrossRef CAS PubMed.
- C. P. Yang, M. H. Liu, W. Zou, X. L. Guan, L. Lai and W. W. Su, J. Asian Nat. Prod. Res., 2012, 14, 68–75 CrossRef CAS PubMed.
- C. R. Deng, C. Y. Gao, X. H. Tian, B. Chao, F. Wang, Y. Zhang, J. T. Zou and D. C. Liu, J. Funct. Foods, 2017, 35, 332–340 CrossRef CAS.
- Y. Y. Zhao, S. P. Wu, S. M. Liu, Y. M. Zhang and R. C. Lin, Chem.-Biol. Interact., 2014, 220, 181–192 CrossRef CAS PubMed.
- Y. Y. Zhao and R. C. Lin, Chem.-Biol. Interact., 2014, 215, 7–16 CrossRef CAS PubMed.
- Y. Y. Zhao, L. Zhang, F. Y. Long, X. L. Cheng, X. Bai, F. Wei and R. C. Lin, Chem.-Biol. Interact., 2013, 201, 31–38 CrossRef CAS PubMed.
- Y. Y. Zhao, J. Liu, X. L. Cheng, X. Bai and R. C. Lin, Clin. Chim. Acta, 2012, 413, 642–649 CrossRef CAS PubMed.
- X. Wang, S. S. Johansen, M. K. K. Nielsen and K. Linnet, Drug Test. Anal., 2017, 9, 1137–1151 CrossRef CAS PubMed.
- J. M. Wang, Z. Yang and J. Lechago, Biomed. Chromatogr., 2013, 27, 1463–1480 CrossRef CAS PubMed.
- S. Hegstad, S. Hermansson, I. Betner, O. Spigset and B. M. H. Falch, J. Chromatogr. B, 2014, 947–948, 83–95 CrossRef CAS PubMed.
- J. J. Pitt, Clin. Biochem. Rev., 2009, 30, 19–34 Search PubMed.
- A. H. Wu, R. Gerona, P. Armenian, D. French, M. Petrie and K. L. Lynch, Clin. Toxicol., 2012, 50, 733–742 CrossRef CAS PubMed.
- H. H. Maurer, Ther. Drug Monit., 2010, 32, 324–327 CrossRef CAS PubMed.
- H. H. Maurer and M. R. Meyer, Arch. Toxicol., 2016, 90, 2161–2172 CrossRef CAS PubMed.
- S. Q. Dong, H. R. Fan, Q. S. Li, G. L. Wei, W. H. Liu and D. Y. Si, Chin. Tradit. Herbal Drugs, 2014, 45, 2499–2505 CAS.
- W. T. Liang, H. B. Huang and T. Liu, Pharm. Today, 2012, 22, 13–16 CAS.
- H. B. Li, Z. R. Li, Z. L. Wang and L. Y. Sun, Prog. Biochem. Biophys., 1993, 20, 402–403 CAS.
- Y. H. Ma, W. W. Xie, T. T. Tian, Y. R. Jin, H. J. Xu, K. R. Zhang and Y. F. Du, Anal. Biochem., 2016, 511, 61–73 CrossRef CAS PubMed.
- P. P. Jia, Y. Q. Zhang, Q. Y. Zhang, Y. P. Sun, H. T. Yang, H. Shi, X. X. Zhang and L. T. Zhang, Biomed. Chromatogr., 2016, 30, 1498–1505 CrossRef CAS PubMed.
- M. Y. Liu, S. H. Zhao, Z. Q. Wang, Y. F. Wang, T. Liu, S. Li, C. C. Wang, H. T. Wang and P. F. Tu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 949–950, 115–126 CrossRef CAS PubMed.
- T. T. Tian, Y. R. Jin, Y. H. Ma, W. W. Xie, H. J. Xu, K. R. Zhang, L. T. Zhang and Y. F. Du, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1006, 80–92 CrossRef CAS PubMed.
- J. Liang, F. Xu, Y. Z. Zhang, S. Huang, X. Y. Zang, X. Zhao, L. Zhang, M. Y. Shang, D. H. Yang, X. Wang and S. Q. Cai, J. Pharm. Biomed. Anal., 2013, 83, 108–121 CrossRef CAS PubMed.
- J. F. Chen, Y. L. Song, X. Y. Guo, P. F. Tu and Y. Jiang, Analyst, 2014, 139, 6474–6485 RSC.
- L. L. Wang, M. M. Sang, E. W. Liu, P. O. Banahene, Y. Zhang, T. Wang, L. F. Han and X. M. Gao, J. Pharm. Biomed. Anal., 2017, 140, 45–61 CrossRef CAS PubMed.
- K. W. Luo, Q. S. Shi and F. Feng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1040, 260–272 CrossRef CAS PubMed.
- S. J. Fenwick and J. P. Scarth, Drug Test. Anal., 2011, 3, 705–716 CrossRef CAS PubMed.
- J. P. Scarth, H. A. Spencer, S. E. Timbers, S. C. Hudson and L. L. Hillyer, Drug Test. Anal., 2010, 2, 1–10 CAS.
- I. Wilk-Zasadna, C. Bernasconi, O. Pelkonen and S. Coecke, Toxicology, 2015, 332, 8–19 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information regarding other metabolites found in vitro and in vivo. Fig. S1 MS/MS spectra of all metabolites of LQ detected in vitro and in vivo. See DOI: 10.1039/c7ra13760e |
| This journal is © The Royal Society of Chemistry 2018 |