
Hydrogen energy future with formic acid: a renewable chemical hydrogen storage system
Ashish Kumar
Singh
*a,
Suryabhan
Singh
*b and
Abhinav
Kumar
*c
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: ashish.bhuchem@gmail.com; Tel: +91 9450209554
bDepartment of Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore 560012, India. E-mail: sbs.bhu@gmail.com; Tel: +91 9453269249
cDepartment of Chemistry, University of Lucknow, Lucknow 226007, India. E-mail: abhinavmarshal@gmail.com; Tel: +91 9451891030
First published on 15th October 2015
Abstract
Formic acid, the simplest carboxylic acid, is found in nature or can be easily synthesized in the laboratory (major by-product of some second generation biorefinery processes); it is also an important chemical due to its myriad applications in pharmaceuticals and industry. In recent years, formic acid has been used as an important fuel either without reformation (in direct formic acid fuel cells, DFAFCs) or with reformation (as a potential chemical hydrogen storage material). Owing to the better efficiency of DFAFCs compared to several other PEMFCs and reversible hydrogen storage systems, formic acid could serve as one of the better fuels for portable devices, vehicles and other energy-related applications in the future. This perspective is focused on recent developments in the use of formic acid as a reversible source for hydrogen storage. Recent developments in this direction will likely give access to a variety of low-cost and highly efficient rechargeable hydrogen fuel cells within the next few years by the use of suitable homogeneous metal complex/heterogeneous metal nanoparticle-based catalysts under ambient reaction conditions. The production of formic acid from atmospheric CO2 (a greenhouse gas) will decrease the CO2 content and may be helpful in reducing global warming.
 Ashish Kumar Singh | Dr. Ashish Kumar Singh was born in Varanasi, U. P., India in 1985. He received his PhD degree in Inorganic Chemistry from Banaras Hindu University, Varanasi, India in 2011. He then worked with Prof. Qiang Xu (AIST) as a JSPS postdoctoral fellow between 2011 and 2013. He is currently working as a DSK postdoctoral fellow in the group of Prof. B. R. Jagirdar at IPC, IISc. He is currently interested in the development of homo-/heterogeneous catalysts for the activation of small molecules for chemical hydrogen storage. |
 Suryabhan Singh | Dr. Suryabhan Singh was born in Varanasi, U. P., India in 1982. He received his MSc degree in Chemistry in 2007 and his PhD degree in Inorganic Chemistry under the supervision of Prof. S. Bhattacharya from Banaras Hindu University, Varanasi, India in 2013. He is currently working as a DSK postdoctoral fellow in the group of Prof. S. Natarajan at SSCU, IISc. His research work is focused on the development of metal–organic frameworks for catalytic and gas storage applications. |
 Abhinav Kumar | Dr. Abhinav Kumar was born in Varanasi, U. P., India in 1979. He received his PhD degree in Inorganic Chemistry under the supervision of Prof. N. Singh from Banaras Hindu University, Varanasi, India in 2009. He has been working as an Assistant Professor at the Department of Chemistry, University of Lucknow, India since 2009. His research work is focused on the development of organometallic/coordination compounds for photovoltaic and material applications. |
1. Introduction
It is well known that non-renewable fossil fuels can be sustained only for next few decades. In the future, there will be a strong demand for safe and renewable energy carriers for transportation and other energy-related applications. Several people are actively working in different fields such as solar energy conversion, lithium ion batteries, geothermal power and nuclear energy, which could deal with the energy problem for a long time. Hydrogen is a promising and widely considered option as an alternative energy feedstock, however, despite extensive efforts by researchers to use hydrogen as a possible energy source, its storage and transportation is a major hurdle to its direct use.1–12 To use hydrogen as a clean fuel and to overcome the hurdles in its safe and efficient storage, various advanced research approaches for the development of new materials that can store and deliver hydrogen at acceptable rates have been discovered.1–12 Based on the methods, it can be mainly divided into two ways – either physical or chemical storage.1–13For the physical storage method, hydrogen is stored in its diatomic molecular form either in a closed container at high pressure and low temperature, such as using high-pressure tanks or cryo-compression,14,15 or getting it adsorbed on high surface area materials viz. various carbon materials,16–19 zeolites,20,21 clathrate hydrates,22,23 or recently the most developed or attractive materials – metal–organic frameworks.12,24–27 However, the hydrogenation/dehydrogenation energy levels of these materials have a large energy gap, hence, they are usually less energy efficient. For the chemical storage method, hydrogen is stored in the chemically bonded form instead of its molecular form. Usually, some suitable molecules having a higher hydrogen content are selected which could release hydrogen efficiently under ambient conditions either via a catalytic or a non-catalytic process. Examples of these molecules include sodium borohydride, ammonia borane, formic acid, hydrous hydrazine, metal hydrides, metal borohydrides, metal amidoborates, etc.2–10 Among them, formic acid is widely explored as a possible fuel for fuel cells because of its properties such as being non-toxic (although neat formic acid is corrosive and its vapour is harmful), liquid at room temperature, high density (1.22 g cm−3) and normal handling conditions (8.4–100.8 °C).2–11
Formic acid is most commonly found in nature in the bites and stings of insects28 and is the major by-product of petroleum refining (naphtha partial oxidation, methanol carbonylation/methyl formate hydrolysis), biomass processing, and several industrial organic syntheses. Hydrogen is easily produced from electrolysis of water.29 Scientists have been able to produce formic acid by the hydrogenation of CO2 present in the atmosphere or separated from industrial waste30 using suitable catalysts.31–34
Formic acid (FA) is considered as one of the most promising materials for hydrogen storage today. Despite the fact that the hydrogen content (4.4 wt%) in FA is less than the target set by the US Department of Energy for 2012 (ref. 35), it surpasses that of most other state-of-the-art storage materials utilized today owing to its simplicity and useable/net capacity.3 Useable or net capacity is defined as the effective hydrogen content that can be recovered in the form of H2 from a chemical hydrogen storage system.36 The produced H2 can be utilized for clean electricity production at low temperature with the production of merely H2O. In fact, the gravimetric energy density of formic acid is 7 times superior to that of commercial lithium ion batteries.37,38 Besides formic acid, hydrogen generation from other liquid organic molecules, also named as liquid organic hydrogen carriers (LOHCs),39,40 has been extensively studied e.g. methanol,41,42 carbazole, cycloalkanes, etc. However, these systems exhibit diverse problems for use as hydrogen storage materials, such as toxicity, cost, limited stability, low dehydrogenation kinetics, and low efficiency of the regeneration processes.
Formic acid has also been considered as a fuel in direct formic acid fuel cells (DFAFCs).43 Chemical processes in DFAFCs involve two-electron direct oxidation of formic acid at the anode and two-electron reduction of O2 at the cathode (eqn (1)–(3)). However, in addition to the fuel crossover and catalyst deactivation, DFAFCs suffer from more specific detrimental effects: CO from the dehydration of FA through an undesired route (eqn (4)) poisons the catalysts (20 ppm already destroys the fuel cell). Anode:
| HCOOH → CO2 + 2H+ + 2e− E0 ~ −0.25 V | (1) |
| 1/2O2 + 2H+ + 2e− → H2O E0 = 1.23 V | (2) |
| HCOOH + 1/2O2 → CO2 + H2O E0Cell ~ 1.48 V | (3) |
| HCOOH → −COads + H2O | (4) |
Sometimes the catalyst's active phase is prone to FA corrosion and FA's hydrophilicity can dehydrate the proton exchange membrane (PEM) and cause increased cell resistance.
In recent years, immense progress has been made and many examples have been reported on the efficient use of formic acid as a renewable source of energy with great efforts from researchers all over the world.1–10 This perspective provides an overview of catalytic hydrogen release and regeneration/production of formic acid. The first part is focused on the decomposition of formic acid for chemical hydrogen storage using a suitable homo-/heterogeneous catalyst. The second part particularly deals with the production and regeneration of formic acid from various processes such as biomass conversion and hydrogenation of CO2. The third part gives an overview of the recent developments in the practical set up for formic acid-based “Rechargeable Hydrogen Batteries”. In the last section, we will briefly discuss the current research progress, expected improvements and future outlook in this research area.
2. Formic acid decomposition
The challenge of producing, storing and transporting hydrogen affordably has kept fuel cells from becoming popular. Instead of transporting hydrogen gas, it is more practical to have a hydrogen-containing material or a chemical hydrogen storage material that can be broken down under ambient conditions to generate H2 gas whenever required.Formic acid, containing 4.4 wt% hydrogen, can be decomposed by following two principal pathways (eqn (1) and (2)), in which the process producing CO2 and H2 (5) is the desired reaction and that producing CO and H2O (6) is the undesired side reaction.3–10
| HCOOH → H2 + CO2 ΔG = −32.9 kJ mol−1 | (5) |
| HCOOH → H2O + CO ΔG = −28.5 kJ mol−1 | (6) |
CO-free decomposition of formic acid through pathway 1 is crucial for formic acid-based hydrogen storage.3–10 The combination of carbon dioxide and formic acid as a hydrogen storage system might act as an elegant and simple concept wherein selective decomposition of formic acid to H2 and CO2 and recycling of CO2 by reduction in the presence of H2 to formic acid can be achieved. Meanwhile, the abundance of CO2 on the earth makes it a cheap and readily available chemical. In this case, decreasing CO2 emissions by reduction using H2 makes CO2 itself as a hydrogen carrier. Tremendous research has been done in search of suitable catalysts (homo-/heterogeneous) for selective decomposition of FA. However, achievement of complete selectivity for the decomposition of formic acid through the desired pathway is still a challenging task.
2.1 Homogeneous catalysts
Over the last few years, there has been a remarkable increase in research activities in search of high-performance homogeneous catalysts for hydrogen release from formic acid. Various groups have highlighted the performances of these homogeneous catalysts in excellent review articles.3–10 A pioneering study by Coffey in 1967 (ref. 44) described the use of soluble Pt, Ru and Ir phosphine complexes for selective decomposition of formic acid to H2 and CO2. Among all the complexes, iridium complex IrH2Cl(PPh3)3 gave the highest rate of decomposition. Rh(C6H4PPh2)(PPh3)2, an organometallic complex, is active for the decomposition of formic acid.45 A platinum dihydride complex catalyzed the reversible formation of carbon dioxide and hydrogen from formic acid. The process was somewhat dependent on the choice of solvent and was promoted by the addition of a small amount of sodium formate.46 King and Bhattacharyya observed that nitrate ions promoted the formic acid decomposition reaction catalyzed by a rhodium(III) catalyst.47 The reactivity of a hydride and equivalent halide complexes of molybdenum was studied for formic acid decomposition. It was observed that the use of the hydride is important for catalysis, as the equivalent halide complexes were inactive.48 Puddephatt and co-workers have studied the detailed mechanism of the formic acid decomposition process over a binuclear, diphosphine-bridged, diruthenium catalyst [Ru2(μ-CO)(CO)4(μ-dppm)2] and characterized the intermediates of the FA decomposition process using X-ray crystallography.49,50 This complex catalyzes the reversible formation/decomposition of FA.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4; amine to HCO2H) or N,N-dimethyl-n-hexylamine (4:5), fast hydrogen generation was observed with [RuCl2(benzene)]2/dppe (TON = 1644 and 1469 h−1, respectively).
:4; amine to HCO2H) or N,N-dimethyl-n-hexylamine (4:5), fast hydrogen generation was observed with [RuCl2(benzene)]2/dppe (TON = 1644 and 1469 h−1, respectively).
A series of amine-functionalized ionic liquids (ILs) were prepared and used for hydrogen generation by the selective catalytic decomposition of formic acid in the presence of the [{RuCl2(p-cymene)}2] catalyst.54,55 Amongst the investigated ILs, the 1-(2-diisopropylaminoethyl)-3-methylimidazolium chloride–sodium formate (iPr2NEMimCl–HCOONa) system exhibited high activity (TOF > 600 h−1) at 333 K. However, this process was not recyclable. Wasserscheid and co-workers reported an outstandingly simple, active and recyclable ionic liquid-based system for the catalytic decomposition of formic acid. The most efficient catalytic system was RuCl3 dissolved in 1-ethyl-2,3-dimethylimidazolium acetate. This catalyst system converted formic acid to hydrogen and carbon dioxide selectively and was recyclable for at least nine cycles without deactivation or change in selectivity.56 Decisively, this simple catalytic system exhibits TOFs of 150 h−1 at 353 K and 850 h−1 at 393 K.
Laurenczy and co-workers carried out the decomposition of FA/sodium formate solution using hydrophilic ruthenium-based catalysts, generated from the highly water-soluble ligand meta-trisulfonated triphenylphosphine (TPPTS) with either [Ru(H2O)6]2+ or, more conveniently, commercially available RuCl3.57,58 This catalyst system could operate over a wide range of pressure, under mild conditions, and at a controllable rate without CO contamination. Later, they immobilized this catalyst on various supports using ion exchange, coordination or physical absorption methods, to get advantage of recycling, especially for dilute formic acid solutions, or for mobile, portable applications of heterogenized catalysts.59
Wills and co-workers investigated the activity of FA dehydrogenation using several RuII and RuIII catalyst precursors ([Ru2Cl2(DMSO)4], [RuCl2(NH3)6], RuCl3 and [Ru2(HCO2)2(CO)4]) in triethylamine at 393 K, explicitly without adding phosphine ligands.60 As can be expected for such a high temperature, FA decomposition activities are exceptionally high (up to ca. 1.8 × 104 h−1). Unfortunately, the CO concentration consistently exceeded 200 ppm for all the catalysts tested. The authors suggested the formation of [Ru2(HCO2)2(CO)4] as the active species common to all the precursors under these reaction conditions. It is interesting to note that during reuse, all the catalysts exhibited a slight enhancement in activity with each run, indicating ongoing formation of the active catalyst species. Later, the same group used their [Ru2Cl2(DMSO)4]/NEt3 system in an attempt to continuously decompose formic acid at a rate approaching the catalyst's maximum activity without acid accumulation in the system.61 Of the two concepts tested – one temperature-based and the other using an impedance probe – the latter gave promising results, even though the gas flow decreased slightly over several days.
Homogeneous Ru catalysts based on polydentate tripodal ligands 1,1,1-tris-(diphenylphosphinomethyl)ethane (triphos) and tris-[2-(diphenylphosphino)ethyl]amine (NP3), which can either be prepared in situ from suitable Ru(III) precursors or as molecular complexes, exhibited moderate to good activity for the selective dehydrogenation of formic acid to CO2 and H2.62 The complex [Ru(κ3-triphos)(MeCN)3](OTf)2 showed superior performance with a TON of 10000 after 6 h using 0.01 mol% catalyst and allowed recycling up to eight times (0.1 mol% catalyst) with a total TON of 8000 after ca. 14 h of continuous reaction at 353 K in the presence of n-octyldimethylamine (OctNMe2). Preliminary mechanistic NMR studies using in situ generated [Ru(κ3-triphos)(MeCN)3](PF6)2 and the molecular complex [Ru(κ4-NP3)Cl2] demonstrated that the NP3 ligand helps to stabilise Ru-hydrido species, hence there is a subtle difference in activity due to the ligand effects. Later, to clarify the mechanism of the catalytic dehydrogenation of formic acid (whether a metal-centered inner-sphere or a ligand-centered outer-sphere pathway) using the aforementioned complexes, an integrated experimental–theoretical study has been performed (Fig. 1).63 The mechanism depends on the choice of polydentate phosphines, i.e., triphos vs. NP3, and the nature and number of ancillary ligands (Cl vs. MeCN), resulting in a different number of vacant coordination sites for the activation of catalysts to their active forms. From the mechanistic study, it was concluded that Ru-hydrido vs. Ru-formato species are pivotal to bring about the efficient release of H2 and CO2 following either a metal-centered (inner-sphere) or a ligand-centered (outer-sphere) pathway, respectively.
 | ||
| Fig. 1 Schematic reaction pathway for formic acid dehydrogenation using [Ru(κ4-NP3)Cl2]. Reprinted with permission from ref. 63. Copyright 2013 American Chemical Society. | ||
Enthaler et al. performed a preliminary study on the ruthenium-catalysed decomposition of formic acid to yield hydrogen by using a ruthenium complex-modified polyformamidine network as a solid catalyst.64 The polyformamidine acted as a dual support for [RuCl2(p-cymene)]2 or [RuCl2(p-cymene)]2/PPh3: on the one hand, as a ligand and on the other hand, as a base for the activation of formic acid. It is noteworthy that the polyformamidine supported [RuCl2(p-cymene)]2/PPh3 catalyst system has a higher activity (TON = 325 for 3 h) than the unsupported [RuCl2(p-cymene)]2/PPh3 with NEt3 addition (TON = 169 for 3 h).
Fukuzumi et al. reported the dehydrogenation of FA, catalyzed by a water-soluble Rh catalyst, [RhIII(Cp*)(bpy)(H2O)](SO4) (Cp* = pentamethylcyclopentadienyl, bpy = 2,2′-bipyridine) in aqueous solution at room temperature.65 Similarly, an iridium catalyst [IrIII(Cp*)(dhbpy)(H2O)](SO4) (dhbpy = 4,4′-dihydroxy-2,2′-bipyridine) was reported by Himeda for CO-free FA dehydrogenation.66 High catalytic activity (TOF = 14000 h−1 at 363 K) without deterioration of the catalyst in continuous runs was observed for this catalytic system. They also demonstrated that the heteronuclear iridium–ruthenium complex [IrIII(Cp*)(H2O)(bpm)RuII(bpy)2](SO4)2 {bpm = 2,2′-bipyrimidine} is a highly active catalyst for hydrogen generation in an aqueous solution under ambient conditions, giving a TOF of about 426 h−1.67
In 2013, a new bisMETAMORPhos (MMP) ligand and its iridium complex (Ir-MMP), in which the ligand is in the dianionic state, were reported by Reek and co-workers (Scheme 1).68 The anionic form of the ligand MMP functions as an internal base, hence this catalyst system is active for FA dehydrogenation in “base-free” conditions. Base-free dehydrogenation of formic acid is important as it can act as a convenient carrier for hydrogen storage, because it increases the hydrogen content from 2.3 wt% in typical HCOOH/NEt3 5:2 mixtures to 4.4 wt% in pure HCOOH. The Ir-MMP catalyst not only operates under such base-free conditions, but also produces clean, CO-free dihydrogen and is very robust and active (base free, TOF of 3092 h−1 in toluene). As such, the cooperative catalysis concept, employing a bifunctional ligand, may hold promise for the efficient and effective (storage and) release of H2 ideally generated from a renewable source, such as solar energy.
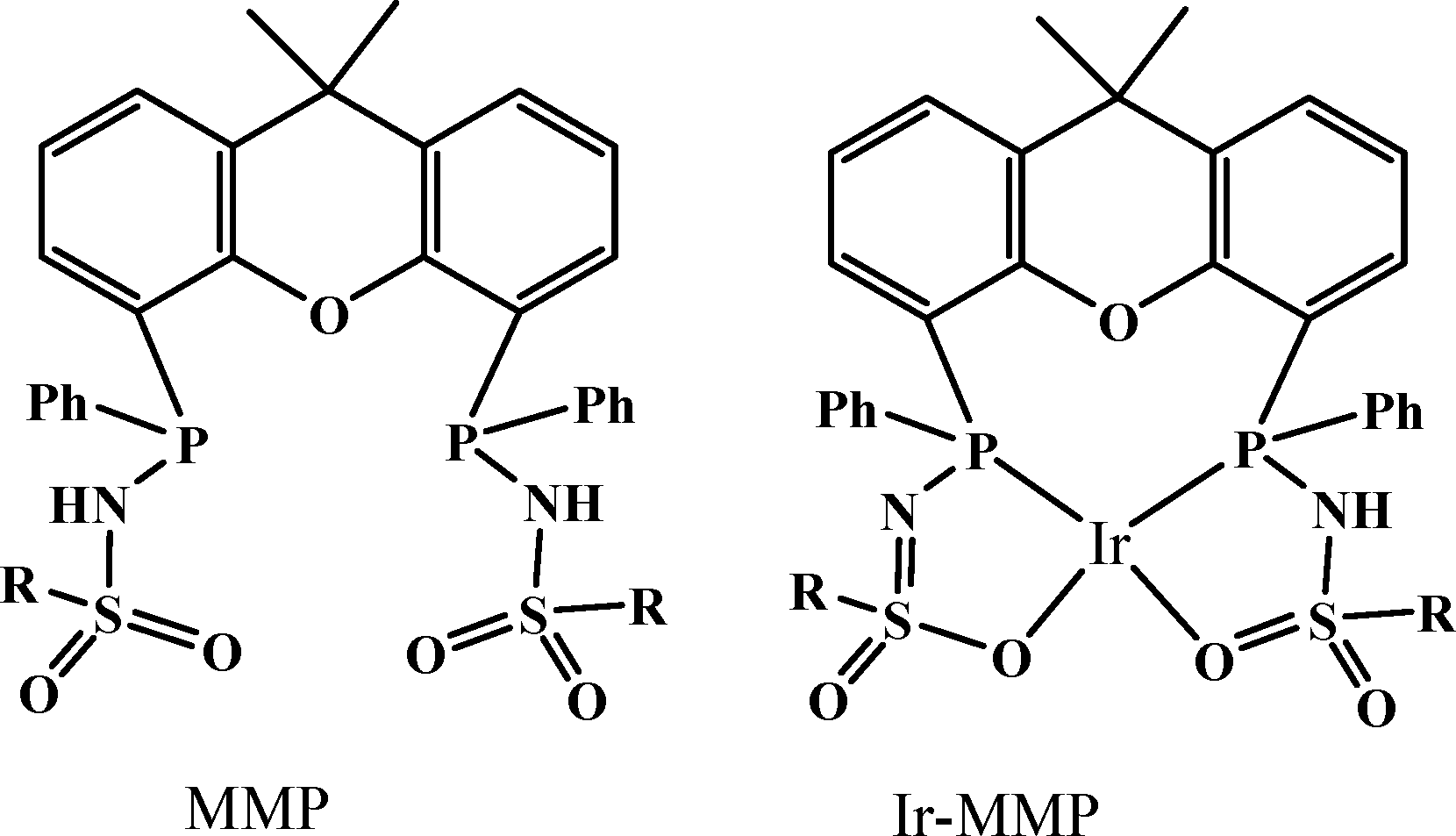 | ||
| Scheme 1 Structure of bisMETAMORPhos (MMP) ligand and its Ir complex (Ir-MMP). (Where R = 4-t-butylphenyl). | ||
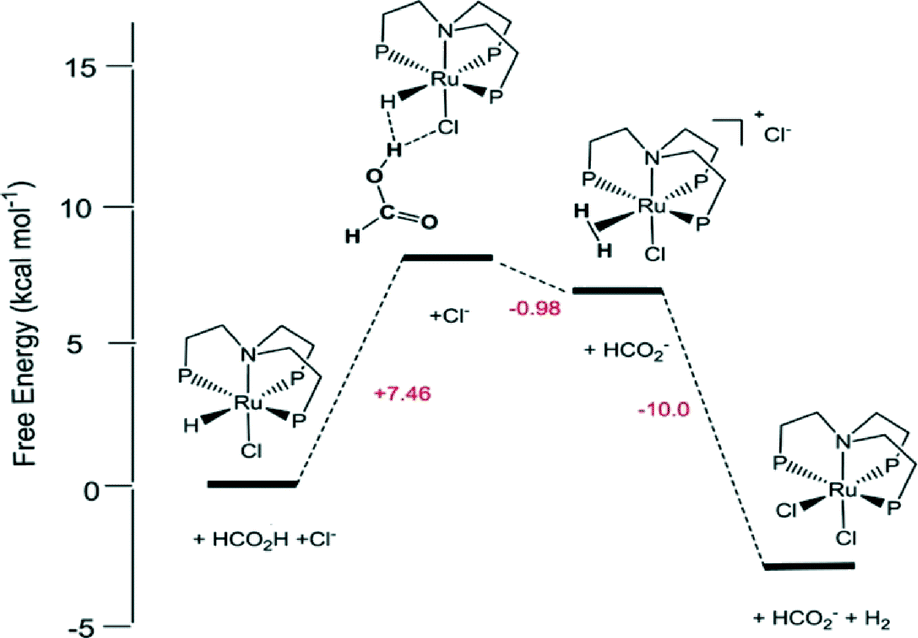
Later, they demonstrated another in situ generated iron-based system as a highly active catalyst (TON up to 100000 and TOF nearly 10000 h−1) for the CO-free decomposition of formic acid in a common organic solvent (propylene carbonate) without any further additives or light (Fig. 2 and Table 1).70 The catalyst can be formed in situ from Fe(BF4)2·6H2O and the tetradentate phosphine ligand tris[(2-diphenylphosphino)ethyl] phosphine (PP3) under the reaction conditions or can be added to the reaction mixture in a pre-synthesized form as [FeH(PP3)]+. During the process, the iron cation is permanently coordinated to four phosphorus centers of PP3, while the remaining two coordination sites are occupied by the HCOOH substrate and/or product-derived species during the catalytic cycle. Spectroscopic studies and density functional theory calculations suggested two possible pathways for H2 release from HCOOH, both of which went through a common Fe-hydride species, [FeH(PP3)]+.70
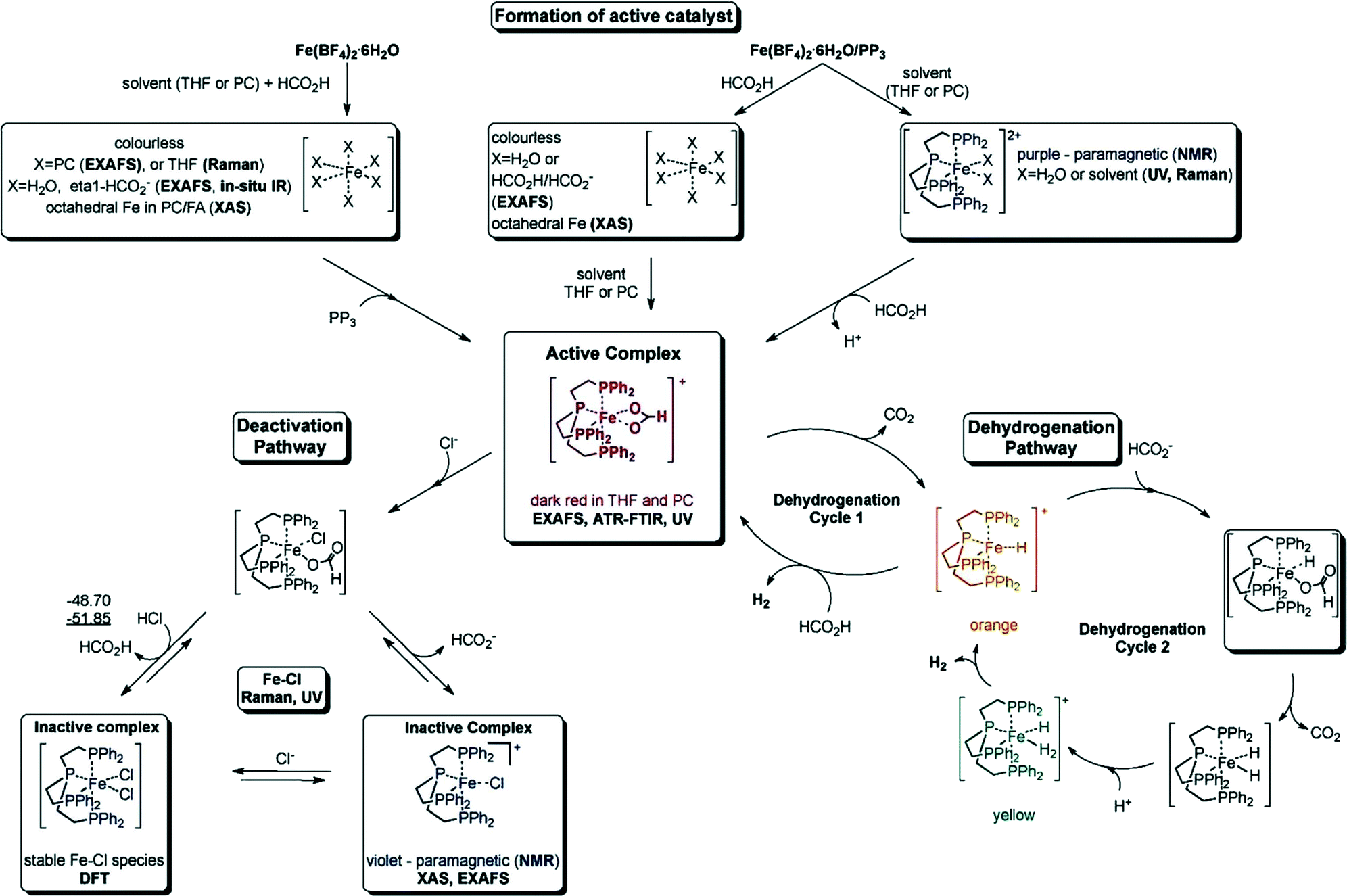 | ||
| Fig. 2 Summary of the activation and deactivation pathways of the Fe(BF4)2·6H2O/PP3 catalyst system as well as the proposed species formed based on the results of spectroscopic analyses. Free enthalpies calculated at the B3PW91/6-31G (including polarization functions except those of hydrogen) and B3PW/6-311G (including polarization functions also for hydrogen; value underlined) levels of theory. The relative enthalpies of the reaction of the interconversion are given in kJ mol−1. It is assumed that iron retains the formal oxidation state +2. Reprinted with permission from ref. 71. Copyright 2014 Wiley-VCH. | ||
| Metal precursor | T (K) | V 2h (mL) | TON2h | Yield (%) | CO (ppm) | Ref. |
|---|---|---|---|---|---|---|
| V 3h (mL) | TON3h | |||||
|
a Reaction conditions: 5.3 μmol of metal precursor (100 ppm) plus 10.6 μmol of PP3 (1) were added to 2 mL of FA and 5 mL of PC at 313 or 333 K. The volume of gas was measured with an automatic gas burette and analyzed by GC (H2/CO2, 1:1).
b Traces of H2 detected.
c No H2 detected. All values given are corrected by the value of a blank reaction without a catalyst as the reference. Values are an average of at least two experiments and have an error within 10%.
|
||||||
| Fe(BF4)2·6H2O | 313 | 333 | 1279 | 100 | <1 | 70 |
| 505 | 1942 | |||||
| [Fe(acac)2] | 313 | 315 | 1217 | 100 | <1 | 71 |
| 486 | 1879 | |||||
| [Fe(acac)3] | 313 | 324 | 1253 | 100 | <1 | 71 |
| 503 | 1943 | |||||
| Fe(ClO4)2·xH2O | 313 | 258 | 997 | 100 | <1 | 71 |
| 388 | 1500 | |||||
| Fe(ClO4)3·xH2O | 313 | 240 | 928 | 100 | <1 | 71 |
| 367 | 1418 | |||||
| Fe(OAc)2 | 313 | 245 | 945 | 98 | 70 | 71 |
| 489 | 1889 | |||||
| [Fe3(CO)12] | 333 | 84 | 325 | — | 1120 | 71 |
| 131 | 505 | |||||
| [Fe(CO)3COT] | 333 | 7.8 | 30 | — | 450 | 71 |
| 33 | 128 | |||||
| FeCl2b | 333 | 0.4 | 1.4 | — | <10 | 71 |
| 0.8 | 3.0 | |||||
| FeCl3c | 333 | 0 | — | — | <10 | 71 |
| 0 | — | |||||
| Co(BF4)2·6H2O | 333 | 27 | 103 | — | <10 | 71 |
| 51 | 197 | |||||
| [Mn(acac)2]b | 333 | 0.15 | 0.6 | — | <10 | 71 |
| 0.23 | 0.9 | |||||
| Fe(BF4)2·6H2O | 333 | 1583 | 6119 | 89 | <1 | 71 |
| 2101 | 8117 | |||||
Recently, they have extensively studied the iron-catalyzed dehydrogenation of formic acid both experimentally and mechanistically.71 The active catalysts were generated in situ from different cationic FeII/FeIII precursors and PP3 (Table 1). These catalysts are active at amine-free and ambient conditions, and the activity of these catalysts was highly dependent on the nature of solvent used, the presence of halide ions, the water content, and the ligand-to-metal ratio. The optimal catalytic performance was achieved by using [FeH(PP3)]BF4/PP3 in propylene carbonate in the presence of traces of water. With the exception of fluoride, the presence of halide ions in solution inhibited the catalytic activity. The in situ transmission FTIR measurements revealed the formation of an active iron formate species (evidenced by the band observed at 1543 cm−1), which could be correlated with the evolution of gas. This active species was deactivated in the presence of chloride ions due to the formation of a chloro species.
Milstein and co-workers reported the iron-based PNP pincer complex [(tBu-PNP)Fe(H)2(CO)] (tBu-PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) as an efficient and selective catalyst for the decomposition of formic acid to carbon dioxide and hydrogen at 313 K in the presence of trialkylamines with turnover numbers of up to 100000.72 From experimental studies, it is observed that the catalytic process proceeds via protonation of the iron dihydride catalyst, followed by dihydrogen liberation which led to an unsaturated species that is transformed into a hydrido–formate complex. Regeneration of the iron dihydride catalyst is finally achieved by CO2 elimination. This step is predicted to proceed through a novel, non-classical intramolecular β-H elimination according to DFT calculations.
In another recent work, Bielinski et al. reported a homogeneous iron catalyst that gives approximately 1000000 turnovers for FA dehydrogenation, when used with a Lewis acid co-catalyst.73 To date, this is the highest turnover number reported for a first-row transition metal catalyst. Based on the preliminary studies, they suggested that the Lewis acid assisted in the decarboxylation of a key iron formate intermediate, as shown in Scheme 2. They have studied the promotion of catalysis by different Lewis acids and observed that the highest TON and TOF were achieved with alkali or alkaline earth metal salt co-catalysts. Importantly, the enhancement of activity is associated with the chemical affinity for carboxylate. The best results were obtained for LiBF4 containing an alkali metal ion (Li+) and a weakly coordinating ligand (BF4−).
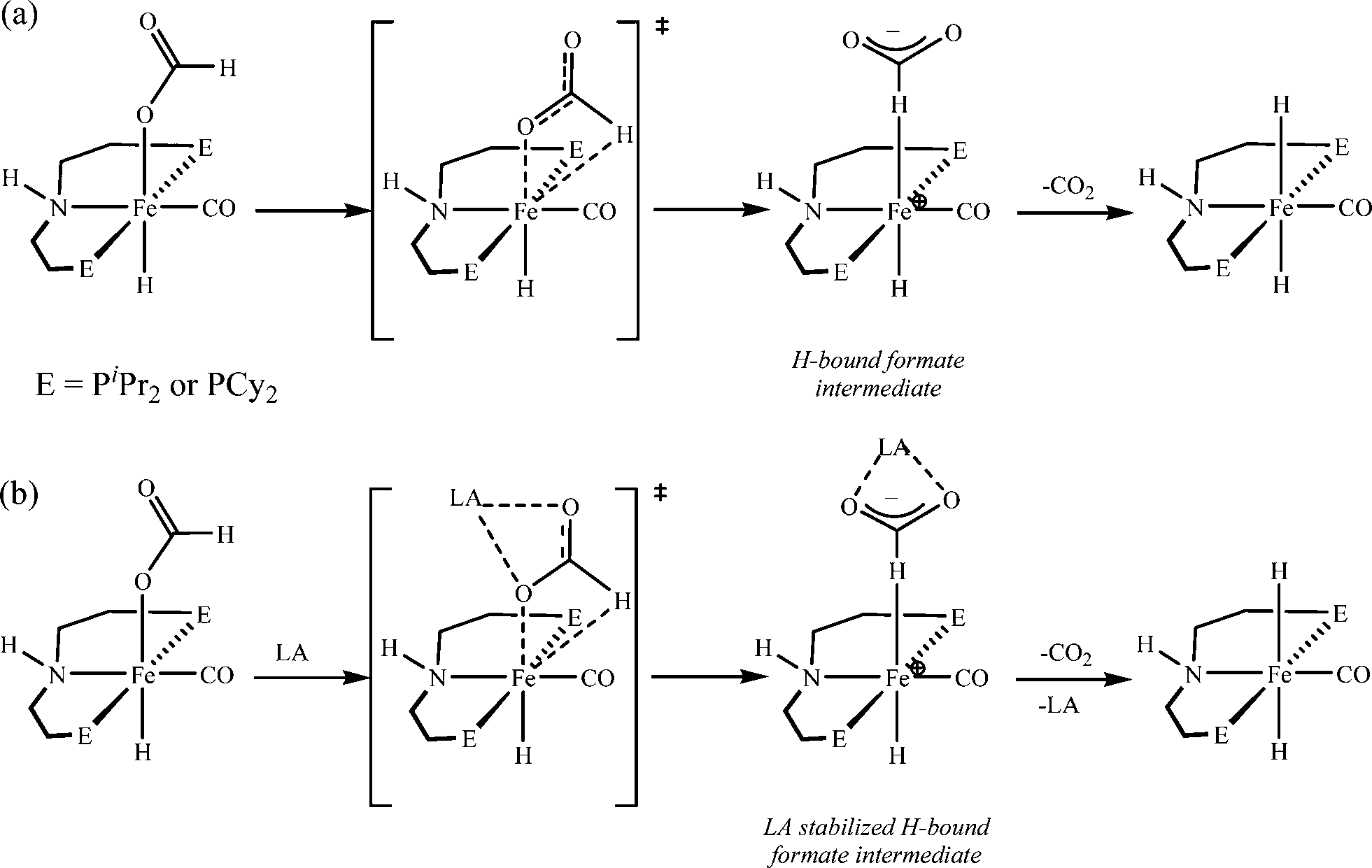 | ||
| Scheme 2 Proposed pathway for decarboxylation of Fe catalysts in the absence (a) and presence (b) of a Lewis acid. Adopted with permission from ref. 73. Copyright 2014 American Chemical Society. | ||
Complete FA dehydrogenation was achieved using 0.01 mol% catalyst and 10 mol% Lewis acid (NaCl, NaBF4, or LiBF4). Among them, LiBF4 gave the fastest time for completion of reaction. In fact, with catalyst loadings as low as 0.0001 mol%, a TON of 983642 and a TOF of 196728 h−1 were obtained. From this study, they concluded that this Lewis acid promotion was general for other catalysts used for FA dehydrogenation, as well as the reverse process, i.e. CO2 hydrogenation.
Myers and Berben reported the first molecular aluminium complexes of the bis(imino)pyridine ligand, (PhI2P2−)Al(THF)X (where X = H or CH3) for the selective dehydrogenation of formic acid to H2 and CO2 with an initial turnover frequency of 5200 turnovers per hour.74 Low-temperature reactions show that the reaction of the Al-hydride complex with HCOOH afforded a complex that was protonated three times: twice on the PhI2P2− ligand and once to liberate H2 or CH4 from the Al-hydride or Al-methyl, respectively. In the absence of protons, insertion of CO2 into the Al-hydride bond is facile and produces an Al-formate. Upon addition of protons, liberation of CO2 from the Al-formate complex affords an Al-hydride. Deuterium labelling studies and the solvent dependence of the reaction indicated that outer sphere β-hydride abstraction supported by metal–ligand cooperative hydrogen bonding is the likely mechanism for C–H bond cleavage.
Based on previous reports, the most commonly observed mechanism (it may not be applicable for all the catalyst systems) for the catalytic decomposition of formic acid using homogeneous catalysts could be given in Scheme 3. These reactions have the following steps: (1) deprotonation of formic acid to formate either by itself or by addition of base, (2) formation of a metal formate complex, (3) the most important and rate-determining step is β-hydride elimination of CO2, and finally (4) elimination of CO2 and H2.
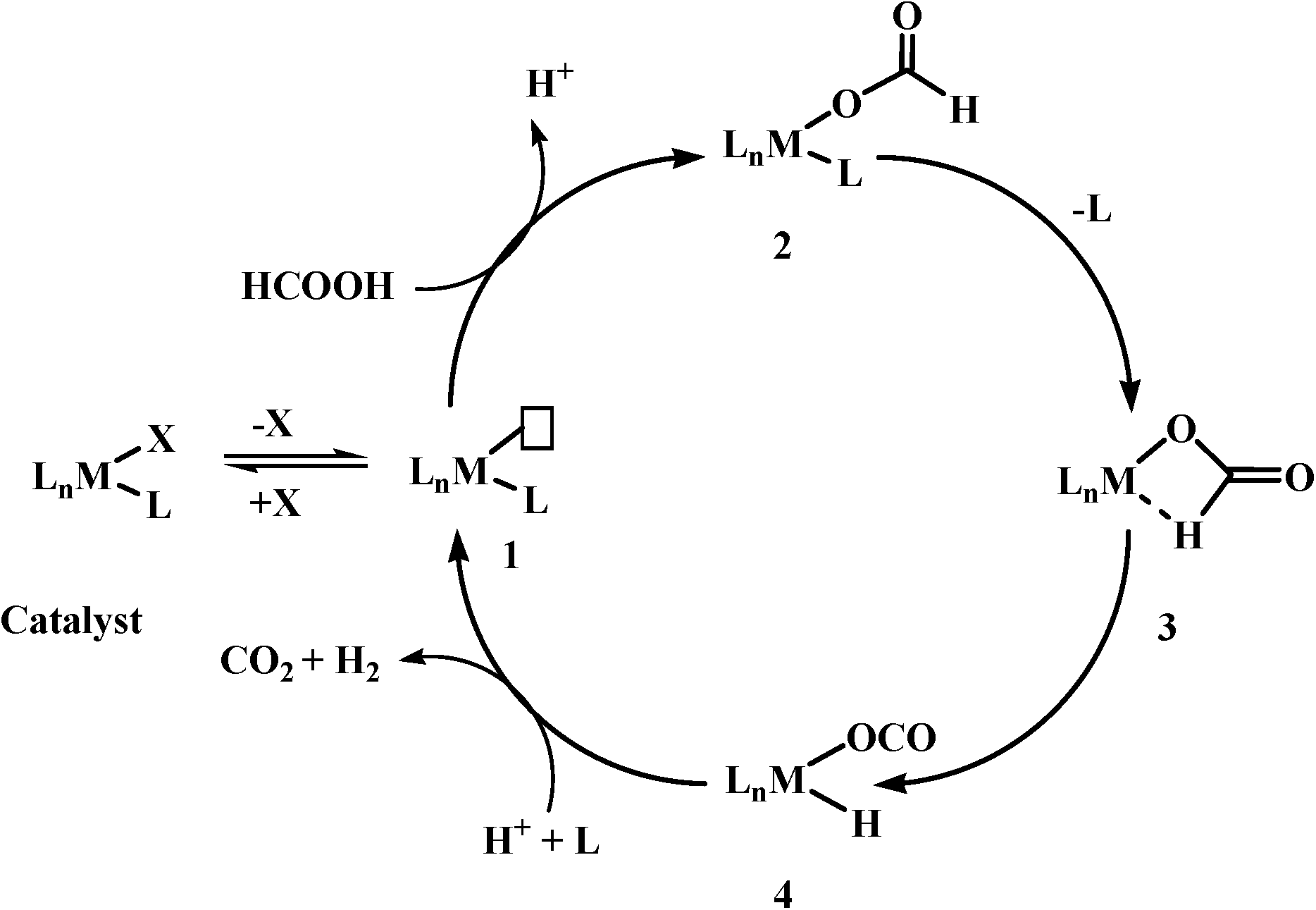 | ||
| Scheme 3 A general mechanism for catalytic decomposition of formic acid. | ||
As β-hydride elimination is the rate-determining step, the catalyst with the tendency to stabilize structure 3 has better activity for formic acid dehydrogenation. This goal could be achieved not only by modifying the metal centre/geometry of metal centre/ligand geometry of homogeneous catalysts, but also by using different kinds of bases as well as other additives, e.g. Lewis acids, solvents (ionic liquids), etc. In some cases, base-free dehydrogenation was achieved because of the presence of basic sites in the catalyst itself or basic ionic liquid. Further, the search for suitable reaction conditions and homogeneous catalysts based on non-noble metals for selective dehydrogenation of formic acid is still in progress.
2.2 Heterogeneous catalysts
Initially, the decomposition of formic acid using heterogeneous catalysts has been mainly performed in the gas phase10 using a metal,75–78 metal oxides79–84 and metal supported on oxides or carbon catalysts.85–93 A gas phase reaction requires the introduction of an inert carrier gas to dilute formic acid below its saturated vapor pressure or heating above the normal boiling point of formic acid (>373 K). However, researchers had also studied hydrogen generation from formic acid decomposition in the liquid phase or at low temperature. Table 2 summarizes selected heterogeneous catalysts for the decomposition of aqueous formic acid to hydrogen and carbon dioxide. Most of the heterogeneous catalysts are supported mono-/bi-/trimetallic nanoparticles with Pd as the major component.| Catalyst | TOF (h−1) | T (K) | Ref. |
|---|---|---|---|
| GN = graphene nanosheets, Fc BR = functionalized basic resin, CB = carbon black, N–mrGO = nitrogen-doped mildly reduced graphene oxide, CN = nitrogen doped carbon, mpg-C3N4 = mesoporous graphitic carbon nitride. | |||
| PdAu/C | 27 | 365 | 94 |
| PdAu/C–CeO2 | 227 | 365 | 94 |
| PdAu@Au | — | 365 | 95 |
| Pd–Au–Dy/C | 269 | 365 | 98 |
| Pd–Au–Eu/C | 387 | 365 | 98 |
| Pd–Au–Ho/C | 224 | 365 | 98 |
| PtRuBiOx | 312 | 353 | 99 |
| Ag@Pd/C (core–shell) | 125 | 293 | 100 |
| 626 | 363 | 100 | |
| Pd/C | 64 | 298 | 102 |
| Au@Pd/N–mrGO | 89.1 | 298 | 103 |
| Ag0.1Pd0.9/rGO | 105.2 | 298 | 104 |
| Co0.30Au035Pd0.35 | 80 | 298 | 105 |
| Ni0.40Au0.15Pd0.45/C | 12.4 | 298 | 106 |
| CoAuPd/DNA–rGO | 85 | 298 | 107 |
| Pd@SiO2 or Pd/SiO2 | — | 363 | 108 |
| Pd@CN (N-doped carbon) | 49.8 | 288 | 109 |
| Pd/–N(CH3)2 Fc BR | 820 | 348 | 110 |
| Pd/MSC-30 | 2623 | 323 | 111 |
| PdAu/MIL-101 | — | 363 | 112 |
| AgPd(Ag80Pd20)/MIL-101 | 848 | 353 | 113 |
| Pd–NH2-MIL-125 | 214 | 305 | 114 |
| PdNi@Pd/GNs–CB | 577 | 300 | 115 |
| PdNi/GNs–CB | 529 | 300 | 115 |
| Au/Al2O3 | — | 353 | 89 |
| Au@SiO2 | — | 363 | 116 |
| Au/ZrO2 | 1590 | 323 | 117 |
| 1% Pt/C–NF | 0.09 (s−1) | 373 | 118 |
| Pd–Ni–Ag/C | 85 | 323 | 119 |
| Pd/C | 304 | 298 | 120 |
| Pd–B/C | 1184 | 298 | 120 |
| Pd/mpg-C3N4 | 144 | 298 | 121 |
Xing and co-workers found that Pd–Au/C and Pd–Au@Au/C have a unique characteristic of evolving high-quality hydrogen from the catalytic decomposition of liquid formic acid in the presence of sodium formate (HCOONa) as a base at a convenient temperature.94,95 The order of catalytic activity based on TOF values is Pd/C < Pd–Cu/C < Pd–Ag/C < Pd–Au/C. The higher catalytic activities of bimetallic Pd–Ag/C and Pd–Au/C catalysts were attributed to the higher tolerance of Ag and Au to CO poisoning. The catalytic activity was further improved by the addition of CeO2(H2O)x because CeO2 produces cationic palladium species, which showed high activity in CO oxidation96 and methanol decomposition.97 Another reason was that CeO2(H2O)x on the Pd surface can induce the decomposition of formic acid in a more efficient route, in which fewer poisoning intermediates would be produced.94
Later, they reported a novel Pd–Au bimetallic catalyst with a Pd–Au@Au core–shell nanostructure supported on carbon. This was synthesized by a simultaneous reduction method without using any stabilizer and was successfully used as a catalyst for hydrogen generation by formic acid decomposition. The catalyst exhibited high activity, selectivity and stability at a low temperature and the catalytic performance was much better than that of the corresponding monometallic catalysts. Especially, the reformed gas from formic acid decomposition contained only 30 ppm of CO and can be used directly to feed the fuel cells.95 Inspired by the promotion effect of CeO2 on the catalytic activities of Pd–Au/C and Pd–Ag/C catalysts, they extended their investigations on other rare earth elements (Dy, Eu, and Ho) and obtained TOFs of 269 ± 202, 387 ± 292, 224 ± 73, and 45 ± 11 h−1 for Pd–Au–Dy/C, Pd–Au–Eu/C, Pd–Au–Ho/C and Pd–Au/C catalysts, respectively, at 365 K.98 In addition, these catalysts were active even at room temperature temporarily and above 325 K steadily. All the rare earth metal oxides that promoted the catalytic activity of Pd–Au/C catalysts showed lower activation energies for decomposition of formic acid than Pd–Au/C and Pd–Au–Eu/C (84.2 ± 7.4 kJ mol−1). The metal/metal oxide catalysts composed of platinum, ruthenium and bismuth, denoted as PtRuBiOx, catalyzed the selective dehydrogenation of FA in water at nearly ambient temperature.99 The observed activation energy was 37.3 kJ mol−1 and the TOF was estimated to be 312 h−1 in the first hour of the decomposition.
In an effort, Tsang and co-workers prepared various core–shell nanoparticles having an inner core of a metal element and an external shell of palladium.100,101 Among all metals tested, the highest activity towards FA decomposition at room temperature is obtained for Ag@Pd NPs (diameter 8 nm) with the thinnest continuous Pd shell (1–2 atomic layers), and the corresponding Ag/Pd alloy and pure Pd catalysts showed very low activity.100,101 The turnover frequencies per surface Pd site were comparable to homogeneous catalysts: 125 h−1 at 293 K and 252 h−1 at 323 K. At 293 K, an equimolar mixture of hydrogen and CO2 was continuously produced without any trace of CO; on the other hand, CO was detected at temperatures higher than 323 K. Furthermore, theoretical calculations showed a strong correlation between the catalytic activity and the work function of the metal core: the largest net difference with the work function of the Pd shell led to the highest adsorption energy by charge transfer from the core to the shell, and hence to the best possible activity of the resulting bimetallic structure for formic acid decomposition. The very short range of this so-called “ligand” electronic effect between the two metals explained why the highest performance was achieved for the thinnest Pd layer. The nanomaterial interface definitely plays a key role in catalysis.
Yan and co-workers have extensively studied the catalytic dehydrogenation of formic acid using mono-/bi-/trimetallic nanoparticle-based catalysts supported on different carbon materials.102 A low cost Pd/C catalyst, synthesized in situ with citric acid, has been reported for highly efficient CO-free hydrogen generation from formic acid/sodium formate aqueous solution at room temperature (Fig. 3).102 The presence of citric acid during the formation and growth of the Pd nanoparticles on carbon drastically enhanced the catalytic properties of the resulting Pd/C at room temperature (conversion efficiency 85% in 160 min and turnover frequency 64 molH2 molcatalyst−1 h−1).
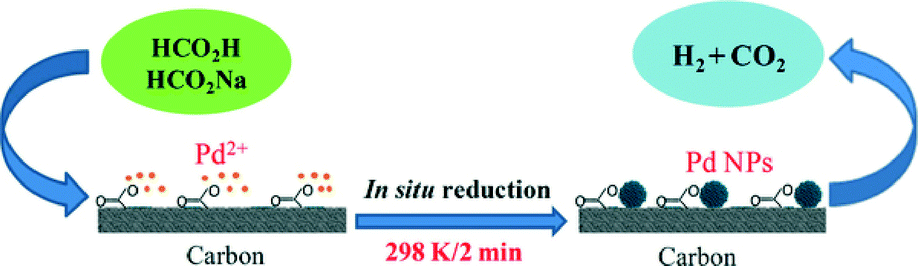 | ||
| Fig. 3 Schematic presentation of Pd nanoparticle formation over carbon surface. Reprinted with permission from ref. 102. Copyright 2012 Nature Publishing group. | ||
Ultrafine (1.8 nm) and well dispersed Au@Pd core–shell nanoclusters on nitrogen-doped mildly reduced graphene oxide (Au@Pd/N–mrGO), synthesized by a green and facile strategy, without any surfactant and additional reducing agent, exhibited much greater activity than its alloy or monometallic counterparts towards hydrogen generation from formic acid aqueous solution without using any additive at room temperature.103 During the synthesis, N–mrGO acts as both a reducing agent and a support by taking advantage of its moderate reducing and high dispersing capabilities. In another report, they found that Ag0.1Pd0.9 nanoparticles assembled on reduced graphene oxide (Ag0.1Pd0.9/rGO) could be used as an efficient catalyst for formic acid dehydrogenation reaction with 100% H2 selectivity and exceedingly high activity at room temperature under ambient conditions.104
Yan and co-workers also reported the Co0.30Au035Pd0.35 nano-alloy supported on carbon as a stable, low-cost, and highly efficient catalyst for CO-free hydrogen generation from formic acid dehydrogenation at room temperature.105 The elevated stability of Co0 in the protective nano-alloy structure makes its first application in FA dehydrogenation successful. More interestingly, the prepared Co–Au–Pd/C catalyst with a lower consumption of noble metals exhibits 100% H2 selectivity, the highest activity, and excellent stability toward H2 generation from FA without any additive at 298 K. The catalytic activities of Co0.30Au0.35Pd0.35/C together with its mono-metallic (Pd/C, Au/C, and Co/C) and bi-metallic (Au0.50Pd0.50/C, Co0.30Pd0.70/C, and Co0.30Au0.70/C) counterparts for H2 generation from FA decomposition at 298 K in ambient atmosphere are presented in Fig. 4. The as-prepared Co0.30Au0.35Pd0.35/C exhibited a much better activity than the mono- and bi-metallic catalysts synthesized by the same method. In a similar way, carbon supported highly homogeneous trimetallic NiAuPd alloy nanoparticles (Ni0.40Au0.15Pd0.45/C) have also been employed as a catalyst for the selective dehydrogenation of formic acid.106 This catalyst also exhibited high activity and 100% selectivity for dehydrogenation of FA without any additives at room temperature. The catalytic activity of Ni0.40Au0.15Pd0.45/C was much higher than its monometallic (Pd/C, Ni/C, Au/C) bimetallic counterparts (Ni0.40Pd0.60/C, Au0.25Pd0.75/C, Ni0.40Au0.60/C) as well as physical mixtures of Ni/C, Au/C and Pd/C (Ni:Au:Pd = 0.40:0.15:0.45) under similar reaction conditions.
 | ||
| Fig. 4 Gas generation by decomposition of FA (0.5 M, 10 mL) versus time in the presence of a) Co0.30Au0.35Pd0.35/C, b) Co/C, c) Au/C, d) Co0.30Au0.70/C, e) Pd/C, f) Co0.30Pd0.70/C and g) Au0.50Pd0.50/C (nmetal/nFA = 0.02) at 298 K in ambient atmosphere. Reprinted with permission from ref. 105. Copyright 2013 Wiley-VCH. | ||
They also reported a DNA-directed and facile approach to synthesize the Co–Au–Pd/DNA–rGO composite (Co:Au:Pd = 1:1:1).107 The catalytic performance of the Co–Au–Pd/DNA–rGO composite toward the dehydrogenation of FA without any additives at room temperature was compared with the Co–Au–Pd/rGO composite and Co–Au–Pd NPs. The Co–Au–Pd/DNA–rGO composite exhibited the highest activity compared to the other catalysts and the order of activity is Co–Au–Pd < Co–Au–Pd/rGO < Co–Au–Pd/DNA–rGO. Additionally, no gas was generated from aqueous FA solution in the presence of DNA or the DNA–GO composite, thereby suggesting that DNA and DNA–GO serve as a template and a support, respectively, for the growth of Co–Au–Pd NPs, and not as catalysts for FA dehydrogenation.
We have prepared a high-performance palladium silica nanosphere (20–35 nm) catalyst using Pd(NH3)4Cl2 as a precursor in a polyoxyethylene-nonylphenyl ether/cyclohexane reversed micelle system followed by NaBH4 reduction.108 Pd NPs supported on silica nanospheres (Pd/SiO2) were prepared by the conventional impregnation of the Pd(NH3)4Cl2 precursor to silica nanospheres, which were prepared using a similar reversed micelle system without a Pd precursor, followed by NaBH4 reduction. The as-synthesized Pd@SiO2 and Pd/SiO2 catalysts have high catalytic activities as compared to Pd NPs supported on commercial silica at convenient temperature. Remarkably, it was observed that the interactions of the Pd nanoparticles with the surface groups of the silica support are important for the catalytic performance. The strong cooperative effects from surface molecular groups of silica on the metal surface, that is, a strong metal–molecular support interaction (SMMSI), might bring new opportunities in the development of high-performance heterogeneous catalysts from inactive or less active metal catalyst systems.
Cai et al. synthesized Pd nanoparticles immobilized on mesoporous carbon nitride (Pd@CN) as a highly efficient Mott–Schottky photocatalyst for dehydrogenation of formic acid.109 The exceptional catalytic performance of this catalyst was due to the enhanced electron enrichment of the Pd nanoparticles through charge transfer operating at the interface of the Mott–Schottky contact. Under similar operating conditions, the catalytic performance of Pd@CN is orders of magnitude higher than that of similar Pd@carbon catalysts. Nanostructured carbon nitride acted both as a stabilizer and a semiconducting support for coupling of metal NPs to form the required rectifying Mott–Schottky nano-heterojunctions.
A basic resin bearing –N(CH3)2 functional group within its macro-reticular structure served as an efficient organic support for the active Pd nanoparticles responsible for the production of high-quality H2via formic acid (HCOOH) decomposition at convenient temperature (Fig. 5).110 Physicochemical characterization as well as the kinetic isotope effect (KIE) revealed that not only the formation of small Pd NPs but also the cooperative action by the –N(CH3)2 groups within the resins play crucial roles in achieving efficient catalytic performances. In addition to advantages such as simple workup procedure, additive-free, and superior catalytic activity compared with conventional inorganic supports, this catalytic system can suppress unfavorable CO formation of <5 ppm, which makes it an ideal hydrogen vector in terms of potential industrial application for PEMFCs. Moreover, the basic resin support also provided a Pd–Ag nanocatalyst from an aqueous solution of a mixture of PdCl2 and AgNO3. The catalytic activities for H2 production from formic acid decomposition were strongly dependent on the presence of Ag atoms and the Pd–Ag catalyst was found to perform significantly better than the pure Pd and Ag catalysts.
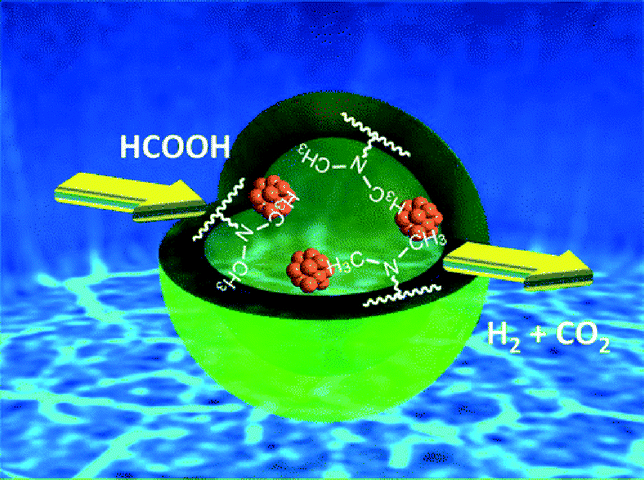 | ||
| Fig. 5 Model of Pd nanoparticles supported on basic resin bearing –N(CH3)2 functional groups within its macroreticular structure. Reprinted with permission from ref. 110. Copyright 2013 American Chemical Society. | ||
Highly dispersed Pd nanoparticles (NPs) deposited on nanoporous carbon MSC-30 have been synthesized using a sodium hydroxide-assisted reduction approach.111 The use of NaOH during the formation and growth of the particles resulted in well-dispersed ultrafine Pd NPs on MSC-30. The combination of metal–support interaction and high dispersion of NPs significantly enhanced the performance of the resulting catalyst. This catalyst exhibited 100% H2 selectivity and very high activity (TOF = 2623 h−1) for heterogeneously catalyzed decomposition of FA at 323 K. The activity of the prepared catalyst was comparable to those acquired from the most active homogeneous catalysts as well as heterogeneous catalysts under ambient conditions.
Xu and co-workers reported the synthesis of bimetallic Au–Pd NPs immobilized in the mesoporous metal–organic framework (MOF) MIL-101 (pore size = 2.9–3.4 nm and window size = 1.2–1.4 nm) as efficient catalysts for decomposition of formic acid towards generation of hydrogen.112 Because of its ordered pore structure, small window and hybrid pore surface, MIL-101 facilitates the encapsulation of metal NPs and the adsorption of formic acid inside the pores. MIL-101 has coordinatively unsaturated Cr3+ centres. In order to improve the interactions between the metal precursors and the MIL-101 support, ED-MIL-101 has been prepared by grafting MIL-101 with the electron-rich functional group ethylenediamine (ED). ED-MIL-101 exhibited improved immobilization of small metal NPs. Among the resulting bimetallic Au–Pd NPs immobilized in the MOFs, Au–Pd/MIL-101 and Au–Pd/ED-MIL-101 represent the first highly active MOF-immobilized metal catalysts for the complete conversion of formic acid to hydrogen at a convenient temperature.
Dai et al. investigated the activity of bimetallic Ag–Pd nanoparticles immobilized into MIL-101 for catalytic dehydrogenation of formic acid.113 Among all the AgPd@MIL-101 catalysts with different compositions of Ag and Pd (Ag@MIL-101, Ag78Pd22@MIL-101, Ag63Pd37@MIL-101, Ag48Pd52@MIL-101, Ag35Pd65@MIL-101, Ag20Pd80@MIL-101 and Pd@MIL-101), the Ag20Pd80@MIL-101 catalyst exhibited the highest catalytic activity for the conversion of formic acid to high-quality hydrogen at 353 K with a TOF value of 848 h−1, which is among the highest values reported at 353 K (Fig. 6).
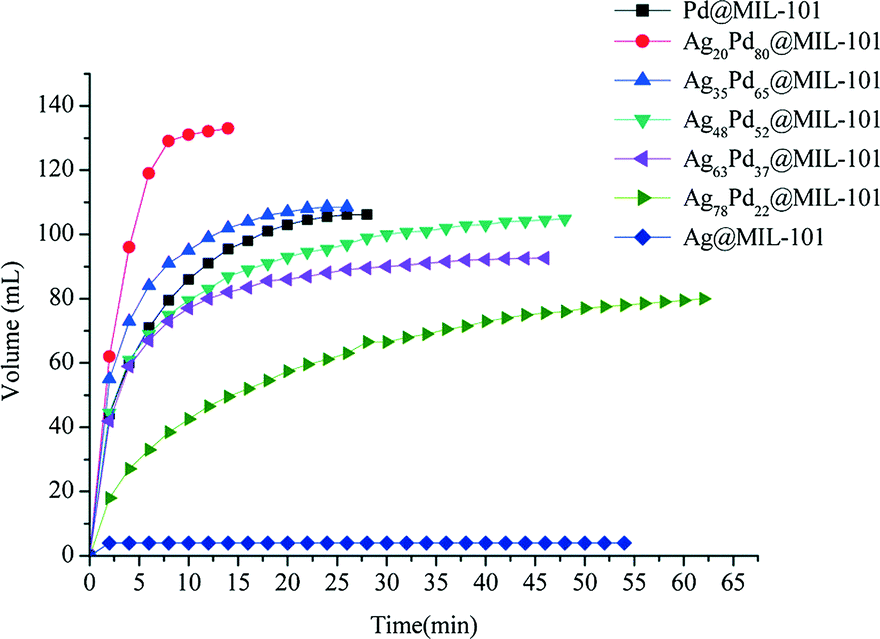 | ||
| Fig. 6 Hydrogen generation from HCOOH in the presence of different catalysts: Ag20Pd80@MIL-101; Ag35Pd65@MIL-101; Ag48Pd52@MIL-101; Ag63Pd37@MIL-101; Ag78Pd22@MIL-101; Ag@MIL-101; Pd@MIL-101 at 353 K. Reprinted with permission from ref. 113. Copyright 2013 Royal Society of Chemistry. | ||
In another report, Yamashita and co-workers used the photocatalytically active metal–organic framework MIL-125 and its amine-functionalized equivalent NH2–MIL-125 to immobilize Pd NPs by photo-assisted and ion exchange deposition methods.114 The Pd–NH2–MIL-125 catalyst showed high catalytic activity for H2 generation from FA at ambient temperature (TOF = 214 h−1 at 305 K) in comparison with Pd–MIL-125 and other titanium-based porous materials. The main factors responsible for the high catalytic performance were the basic functionalization of the MOF and small NP sizes. In addition, the photo-assisted deposition method was found to be a more effective method for producing small and highly dispersed NPs within the MOF structure.
Zhang and co-workers synthesized well-dispersed Pd–Ni nanocatalysts grown on a graphene nanosheet–carbon black (GNs–CB) composite support to combine the advantages of GNs and CB.115 Unexpectedly, Pd–Ni NCs loaded on GNs–CB exhibited higher catalytic performance for FA decomposition at room temperature in aqueous media than on Pd or Ni alone. Furthermore, Pd is employed to replace the surface Ni of Pd–Ni NCs toward a novel Pd–Ni@Pd catalyst to further improve the catalytic activity and stability. The use of GNs–CB as a new kind of carbon support to disperse, anchor, and further promote nanocatalysts with active components would undoubtedly assist long-term endeavours to further optimize and enhance the catalytic efficacy of catalysts in the development of FA as a hydrogen-storage material.
There are few reports for formic acid decomposition by Au NP-based heterogeneous catalysts. The first report on Au NP-based heterogeneous catalysts for formic acid dehydrogenation in the gas phase at ambient temperatures has been reported by Ojeda and Iglesia in 2009.89 Well-dispersed Au species immobilized on Al2O3 catalyzed the decomposition of HCOOH with metal-time yields (rates per Au atom) larger than Pt clusters supported on Al2O3 at 353 K. HCOOH dehydrogenation proceeds via either a H-assisted formate decomposition mechanism or sequential cleavage of O–H and C–H bonds and H-atom recombination on isolated sites.
The first work on liquid phase dehydrogenation of formic acid using a monometallic Au NP-based catalyst was reported by Xu and co-workers.116 Monometallic gold nanoparticles encapsulated in amine functionalized silica nanospheres acted as a high-performance catalyst for hydrogen generation from aqueous formic acid. It was observed that the unsupported or silica-supported gold NPs without the amine functional group are inactive for FA dehydrogenation reaction. Hence, the presence of amine in the silica sphere is important for the activity of the gold nanoparticles due to the strong metal–molecular support interaction (SMMSI).
Bi et al. demonstrated the mild and selective dehydrogenation of FA/amine mixtures using ultradispersed sub-nanometric gold NPs on acid-tolerant ZrO2 as catalysts.117 The catalytic reactions proceed efficiently and selectively under ambient conditions, without the generation of any unwanted by-product, such as CO, and with high TOFs/TONs. A unimolecular mechanism involving unique amine-assisted formate decomposition at the Au–ZrO2 interface is supported by the exclusive formation of HD and the primary kinetic isotope effects measured from HCOOD or DCOOH dehydrogenation (Fig. 7). The exceptional activity of sub-nanometric Au toward FA activation promises a new area of gold research by fine-tuning of the dispersed Au clusters at the sub-nano level.
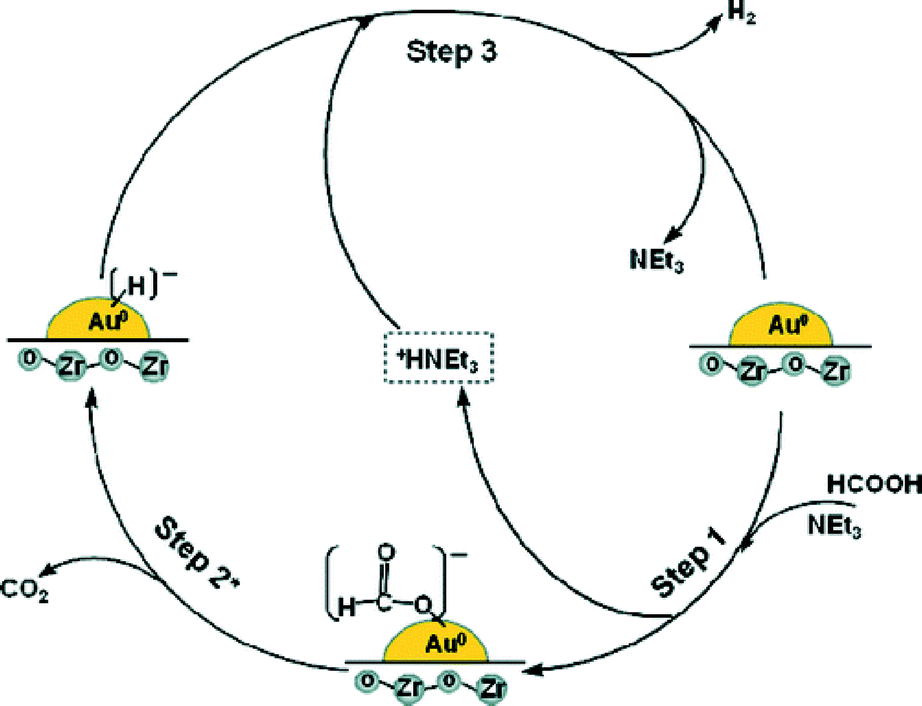 | ||
| Fig. 7 Possible reaction pathway for hydrogen evolution from the 5:2 FA–NEt3 azeotropic mixture system over the Au/ZrO2 NC catalyst. Reprinted with permission from ref. 117. Copyright 2012 American Chemical Society. | ||
The activities of Pt catalysts on carbon nanofibers with different nitrogen contents were compared for hydrogen production by formic acid decomposition.118 The catalysts contained a fraction of Pt clusters with a mean size of 1.0–2.3 nm and possibly a considerable fraction of Pt clusters with a diameter of less than 0.75 nm which were invisible by transmission electron microscopy (TEM) observation. The activities of N-doped catalysts with low Pt contents (≤1 wt%) were 10 times higher than the activities of undoped catalysts. The N-doped catalysts demonstrated an improved selectivity to hydrogen and an increased resistance to CO inhibition. However, they were inactive for ethylene hydrogenation. These results are explained by the presence of electron-deficient, two-dimensional sub-nm sized Pt clusters stabilized by pyridyl nitrogen on vacancy sites. In accordance, the Pt-4f7/2 binding energies measured by X-ray photoelectron spectroscopy were 0.6 eV higher for the N-doped samples than for the undoped ones.
Yurderi et al. synthesized trimetallic Pd–Ni–Ag nanoparticles with different ratios of metals as well as its bi-metallic (Pd–Ni, Ni–Ag and Pd–Ag) and mono-metallic (Pd, Ni and Ag) counterparts supported on activated carbon by simple and reproducible wet impregnation followed by a simultaneous reduction method without using any stabilizer at room temperature.119 All the prepared composites were employed as heterogeneous catalysts in the catalytic decomposition of formic acid under mild reaction conditions. It was found that Pd–Ni–Ag/C can catalyze the dehydrogenation of formic acid with high selectivity (~100%) and activity (TOF = 85 h−1) at 323 K. More importantly, the exceptional stability of Pd–Ni–Ag nanoparticles against agglomeration, leaching and CO poisoning makes Pd–Ni–Ag/C a reusable catalyst for formic acid dehydrogenation. The Pd–Ni–Ag/C catalyst retained almost its inherent activity (>94%) even at 5th reuse in the decomposition of formic acid with high selectivity (~100%) under ambient conditions. The quantitative kinetic studies depending on the catalyst [Pd–Ni–Ag], substrate [FA] and promoter [SF] concentrations revealed that the Pd–Ni–Ag/C catalyzed dehydrogenation of FA is a first-order reaction with respect to the catalyst concentration, half-order with respect to the promoter concentration, and with respect to the substrate concentration it appeared to be zero-order when [FA] < 0.140 M and half-order at higher concentrations. The Pd–Ni–Ag/C catalyzed dehydrogenation of FA at different temperatures provided the activation parameters (Ea = 20.5 kJ mol−1, ΔH≠ = 17.8 kJ mol−1, ΔS≠ = −190 J mol−1 K−1) and suggested the associative mechanism for the Pd–Ni–Ag/C catalyzed dehydrogenation of FA.
Recently, a potential boron-doped Pd nanocatalyst (Pd–B/C) was reported to enhance hydrogen generation at room temperature from aqueous formic acid–formate solutions at a record high rate.120 Pd/C and Pd–B/C catalysts with a Pd loading of 5 wt% were synthesized through wet chemical reduction of NaBH4 and dimethylamine borane (DMAB), respectively. To exclude any boron doping, HCOONa was also used as the reducing agent to synthesize Pd/C–HCOONa. Catalytic studies suggested only a slightly higher gas production rate on Pd/C–NaBH4 than on Pd/C–HCOONa (Fig. 8). In contrast, the gas production rate was nearly doubled on Pd–B/C as compared to the other two catalysts at a given mass of Pd. The present Pd–B/C catalyst showed superior catalytic activity, as compared to the TOF values reported so far for various Pd-based catalysts at room temperature.
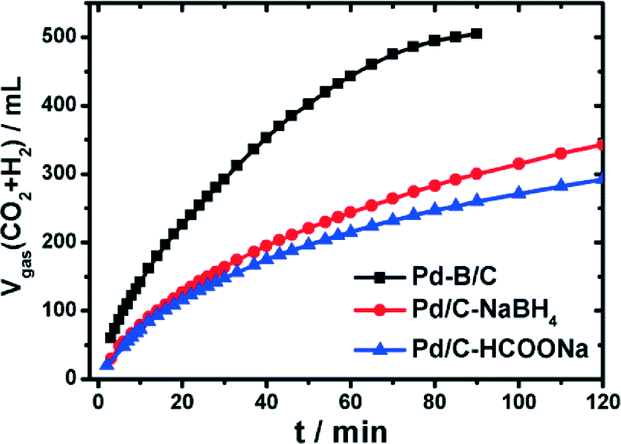 | ||
| Fig. 8 Time-course of reforming gas generation from 10 mL solution of 1.1 M FA + 0.8 M SF in the presence of 100 mg of Pd based/C (5 wt% Pd) catalysts at 303 K under ambient atmosphere. Reprinted with permission from ref. 120. Copyright 2014 American Chemical Society. | ||
Highly sensitive attenuated total reflection infrared spectroscopy (ATR-IR) was used to monitor dynamically the interfacial species on Pd–B/C or Pd/C in FA-SF solutions to provide a molecular-level understanding of the different activities of the examined catalysts. ATR-IR spectroscopy revealed that the superior activity of Pd–B/C is related to an apparently impeded COad (adsorbed CO) accumulation on the surface of the Pd–B/C catalyst. This work demonstrated that developing new anti-CO poisoning catalysts coupled with a sensitive interfacial analysis is an effective way toward rational design of cost-effective catalysts for better hydrogen energy exploitation.
Reversible, carbon dioxide-mediated chemical hydrogen storage was first demonstrated using a heterogeneous Pd catalyst supported on mesoporous graphitic carbon nitride (Pd/mpg-C3N4).121 The Pd nanoparticles were uniformly dispersed onto mpg-C3N4 with an average size of 1.7 nm without any agglomeration and further exhibited superior activity for the dehydrogenation of formic acid with a turnover frequency of 144 h−1 even in the absence of external bases at room temperature. From DFT studies, it was confirmed that basic sites located at the mpg-C3N4 support play synergetic roles in stabilizing the reduced Pd nanoparticles without any surfactant as well as in initiating H2 release by deprotonation of formic acid. These potential interactions were further confirmed by X-ray absorption near edge structure (XANES). Along with dehydrogenation, Pd/mpg-C3N4 was also proved to catalyze the regeneration of formic acid via CO2 hydrogenation. The governing factors for CO2 hydrogenation were further elucidated to increase the quantity of the desired formic acid with high selectivity.
Beller and co-workers carried out a detailed theoretical study of formic acid decomposition on various metal surfaces {such as Ni(111), Ni(211), Pd(111), Pd(211), Pt(111) and Pt(211)}, metal oxides (such as TiO2, MgO, ZnO and NiO) and molybdenum carbide (β-Mo2C(101)).122–125 A common pathway observed for the selective decomposition of formic acid to CO2 and H2 is shown in Scheme 4. The formate route (HCOOH → HCOO + H) plays the dominant role in formic acid decomposition, and the rate-determining step is the dissociation of formate into surface CO2 and H (HCOO → CO2 + H). The most stable adsorption configuration of formate (HCO2) has a bidentate bridging structure with its two oxygen atoms (C–O) at atop sites.
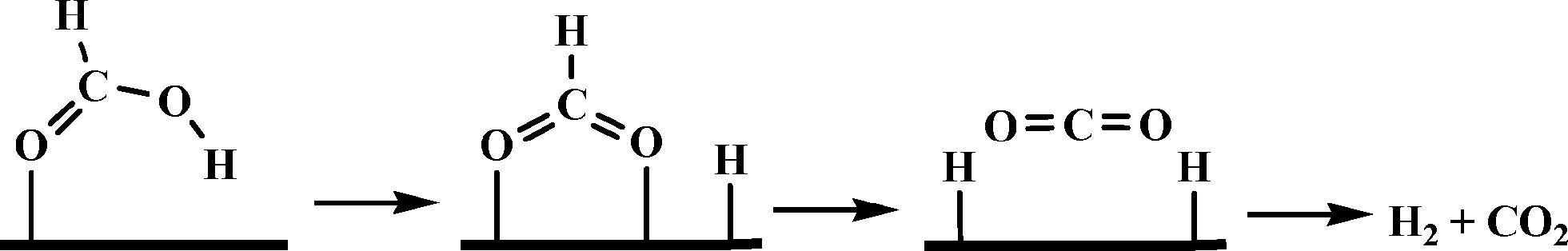 | ||
| Scheme 4 General mechanism of formic acid decomposition over various surfaces through formate route. | ||
Further work is required to develop an efficient system for hydrogen release from formic acid using a heterogeneous catalyst owing to selectivity problems induced by the high reaction temperatures for heterogeneous catalytic reactions.
3. Formic acid production
Formic acid is an important platform chemical for the synthesis of diverse organic substances. In nature, FA is found in the venom of ants28 and is present as a component of the atmosphere due primarily to forest emissions.126,127 Chemically, it can be prepared by various processes. The most common industrial process is the combination of methanol and carbon monoxide in the presence of a strong base at elevated pressure (40 atm) and temperature (353 K) for the preparation of methyl formate, followed by hydrolysis of methyl formate to formic acid.128| CH3OH + CO → CH3COOH | (7) |
| CH3COOH + H2O → HCO2H + CH3OH | (8) |
| CH3OH + CO → CH3OCHO | (9) |
| CH3OCHO + H2O → CH3OH + HCO2H | (10) |
| H2O + CO → HCO2H | (11) |
An issue is the separation of formic acid from the reaction mixture. Other methods for the synthesis of FA are as a by-product of acetic acid production, CO2 hydrogenation, oxidation of biomass, and biosynthesis by reduction of carbon dioxide catalyzed by the enzyme formate dehydrogenase.130,131 In this section, we will discuss in detail the different methods for formic acid production with major focus on CO2 hydrogenation. Carbon dioxide, a major by-product of FA decomposition either chemically or electrochemically and the last product of any organic compound burnt in air, is a major greenhouse gas. Utilization of this greenhouse gas as a source of formic acid is not only a smart step to establish FA as a renewable energy but may also help to reduce environmental deterioration.
3.1 Oxidation of biomass
Production of formic acid from biomass offers a mild and sustainable way to produce H2.132 Many researchers have studied the production of FA from biomasses (carbohydrates) using thermal cracking,133 SCW/H2O2,134 Fe2(SO4)3/O2 or CuSO4/O2 (ref. 135 and 136), OH−/H2O2 (ref. 137), etc. Wasserscheid and co-workers had performed a systematic study for the selective oxidation of carbohydrate-based biomass to formic acid and CO2 using a Keggin-type H5PV2Mo10O40 polyoxometalate as the catalyst under varying reaction conditions with different catalyst promoters.138 In their first report, several water-soluble carbohydrate-based biomasses had been fully and selectively converted to formic acid and CO2 by oxidation with molecular oxygen in aqueous solution under very mild conditions.138 Using glucose as a substrate, their catalyst loading and temperature optimization (353 K, 30 bar O2, 26 h) studies indicated that the characteristic FA yield at full conversion was 49 ± 2%. Interestingly, biogenic waste products, such as glycerol (from bio-diesel production) and xylan (from the hemicellulosic waste streams of paper manufacturing), were oxidized to FA in 40% and 33% yield, respectively. Using this catalyst, even complex biomass mixtures, such as popular wood sawdust, were transformed to formic acid, giving 19 wt% yields (11% based on the carbon atoms in the feedstock) under non-optimized conditions.Later, to optimize the reaction conditions for water-insoluble biomass conversion to FA and CO2, different additives (catalyst promoters) such as CaCl2, H3BO3, ZnCl2, LiCl, benzene/toluene/methane/trifluoromethane/camphor/xylene/chlorobenzene/p-toluenesulphonic acid, etc. were used with the polyoxometalate catalyst H5PV2Mo10O40 in water using oxygen (30 bar) as the oxidising agent at 363 K and the reaction was monitored for 24 h.139 Among all additives, p-toluenesulfonic acid acted as the best additive for the transformation of feedstock like wood, waste paper, or even cyanobacteria to formic acid and CO2 as the sole products. The produced FA can be separated from the aqueous reaction mixture by a simple extraction process and can be regarded as a hydrogen equivalent produced from biomass in a very robust process. The reaction achieved up to 53% yield of formic acid for xylan (hemicellulose) as the feedstock within 24 h at 363 K. Using this promoter, even third generation wet biomass such as algae could be efficiently converted to FA and CO2, offering a very interesting path to the energetic use of these side products for future biodiesel production plants from algae.
In another work, Wasserscheid and co-workers modified a Keggin-type polyoxometalate catalyst and synthesized a series of different heteropolyacids (HPAs) of the general type H3+n[PVnMo12−nO40] (n = 0–6) (Scheme 5).140 To confirm the reactivity, all the POM complexes were tested for the conversion of glucose to FA using oxygen (at 30 bar) as the oxidising agent. The higher V-substituted complexes (n = 2–6) showed superior activities to the lower V-substituted complexes (n = 0–1). Applying the optimized polyoxometalate catalyst system H8[PV5Mo7O40] (HPA-5), a total FA yield (with respect to carbon in the biogenic feedstock) of 60% for glucose within 8 h of reaction time and 28% for cellulose within 24 h of reaction time could be achieved at 363 K. The trend of the activity was explained on the basis of optical and electrochemical measurements which concluded that the formation of the pervanadyl species in the higher V-substituted system was responsible for the high activity of these HPA complexes. The mechanism comprises two alternative ways for the dissociation of HPA-n. On the one hand, before the oxidation reaction starts, the fully oxidized HPA-n with a reduction degree of m = 0 dissociates to free pervanadyl and residual degenerated lacunary species. On the other hand, the fully reduced HPA-n after substrate oxidation with a reduction degree of m = n dissociates to vanadyl (VO2+) and lacunary species.
 | ||
| Scheme 5 Dissociation/re-oxidation equilibria for HPA-n in aqueous solution. | ||
In an acidic solution, the free vanadyl (VO2+) ion cannot be oxidised by molecular oxygen. Instead, the V4+ formed in the reaction returns into the heteropolyanion either by an electron transfer or by association with the lacunary species (HPA-n-1). Consequently, V4+ is re-oxidised by oxygen inside HPA-n.
Li et al. also used the same catalyst and added H2SO4 as an additive to oxidize cellulose to obtain FA with 28% yield at 443 K and cellulose was completely converted in 9 h. The increase in temperature and acidity (by adding H2SO4) increased the FA yield, and the time was shortened to 9 h. Under modified reaction conditions, the conversion of glucose to formic acid could be achieved up to 52% yield within 3 h using 5 mol% H5PV2Mo10O40 at 373 K using air as the oxidant.141 Han et al. prepared three phosphovanadomolybdic acids with different contents of vanadium, namely H4PVMo11O40, H5PV2Mo10O40 and H6PMo9V3O40, and three Keggin-type HPA catalysts including two V-free HPAs (H3PW12O40 and H3PMo12O40) and one phosphovanadotungstic acid (H5PV2W12O40), and evaluated their catalytic performance for the conversion of cellulose.142 Although a relatively low temperature (~373 K) was used to convert the soluble carbohydrates, a higher reaction temperature (>423 K) was essential for the effective conversion of water-insoluble biomass (e.g. cellulose) with HPAs. H4PVMo11O40 acts as the most efficient catalyst capable of converting varieties of biomass-derived substrates to formic acid and acetic acid with high selectivity in aqueous medium and oxygen atmosphere. Under optimized reaction conditions, H4PVMo11O40 gave an exceptionally high yield of formic acid (67.8%) from cellulose.
Recently, Liu et al. observed that heteropolyanion-based ionic liquids with –SO3H functionalized cations and PMo11VO404− anions showed enhanced activity for catalysing cellulose conversion to FA in the presence of oxygen in water compared to H4PMo11VO40.143 Investigation of the possible pathway of FA production from cellulose indicated that heteropolyanion-based ionic liquids served as bifunctional catalysts with cations catalysing cellulose hydrolysis to glucose and anions catalysing glucose oxidation to FA. Optimization with different as-prepared ionic liquid catalysts resulted in high efficiency for FA production from cellulose conversion, giving a high FA yield of over 50% and a high FA concentration of ~10% in aqueous solution at 453 K and in an oxygen pressure of 1.0 MPa.
Marsh and co-workers developed a catalytic process for efficient production of FA from common carbohydrates via VO2+ formed by dissolving sodium metavanadate in acidic water. The hydrolysis reaction decomposed the polymerized structures of polysaccharides to produce monosaccharides, which were readily oxidized to FA under catalysis of VO2+. Typically, the molar yields of formic acid were 64.9% from cellulose and 63.5% from xylan (hemicellulose) at 433 K.144 Wang and co-workers used the cheaper and less toxic vanadium salt VOSO4 as a high-performance catalyst for the transformations of glucose and cellulose into either formic acid or lactic acid (LA) by simply tuning the reaction atmosphere from O2 to N2. The catalytic reaction proceeds through the isomerization of glucose to fructose, which subsequently underwent retro-aldol fragmentation to two trioses, that is, glyceraldehyde and 1,3-dihydroxyacetone. The isomerization of these trioses under N2 led to the formation of lactic acid. The oxidative cleavage of C–C bonds in the intermediates caused by the redox conversion of VO2+/VO2+ under aerobic conditions, giving formic acid and CO2. Addition of an alcohol suppresses the formation of CO2 and enhances the formic acid yield significantly to 70–75%.145
From all the observations by different groups, it can be concluded that biomass could be converted to formic acid by vanadyl species (VO2+/VO2+) or heteropolyacids (HPAs) containing vanadyl ions. These catalysts work as bifunctional catalysts for the conversion of polysaccharides to monosaccharides as well as the selective conversion of the generated monosaccharides to formic acid and CO2 under acidic conditions using air or oxygen as the oxidant.
Besides vanadyl-containing catalysts, some people reported the production of FA from biomass in the presence of other catalysts and some additives.146–148 Gao et al. successfully produced FA with 22% yield from pretreated cellulose under hydrothermal conditions at 483 K for 30 h.146 A high yield of 75% FA was produced by Jin et al. at 523 K using 120% H2O2 from glucose.147 Choudhary et al. designed a Cu catalyst suitable for the conversion of biomass to lactic acid (LA) and formic acid.148 They synthesized a magnesia supported copper catalyst by a hydrothermal methodology using CTAB as the capping agent (denoted as Cu–CTAB/MgO). Interestingly, this catalyst boosted the yields of LA and FA dramatically from sugars with decreasing reaction temperature from 523 K to 393 K. Glucose can be converted to lactic acid or formic acid depending on different additives. Lactic acid with a yield of 70% was produced in the presence of NaOH, while FA was produced in the presence of H2O2; both were achieved from glucose at 393 K in water using the Cu-CTAB/MgO catalyst, which could be recycled without any significant loss of activity. The catalyst was also found to exhibit excellent activity for the transformation of other sugars.
Despite all efforts, complete and selective conversion of biomass to formic acid and CO2 is still a challenging task and further work is required to replace the harsh reaction conditions with mild conditions.
3.2 CO2 hydrogenation
Energy is the most indispensable element of our daily lives, which comes mostly from fossil fuels. Apart from the limited source of these non-renewable fossil fuels, our dependency on them has caused serious environmental concerns, such as greenhouse gases (CO2) as well as other toxic pollutants. The best way to overcome this problem is to use CO2 to make high value chemicals. Extensive research has been carried out on the utilization of carbon dioxide to prepare high value chemicals such as formic acid, formaldehyde, methanol, methane, and other long chain organics, and in synthetic chemistry to obtain different products at ambient conditions.129,149 The conversion of CO2 to useful chemicals is of significant importance not only due to reductions in greenhouse gases but also to build an energy infrastructure that uses methanol or formic acid etc.70,150–153 Unlike conversion to formic acid, conversion of CO2 to formaldehyde, methanol or methane requires harsh conditions which may deactivate the catalysts; hence, these reduction processes are economically unfavourable.The hydrogenation of CO2 to formic acid is an important step to establish formic acid as a reversible hydrogen storage material. Several research works have been done in this field and those works are summarized in several excellent review articles as well as book chapters.10,32,70,149–161 Hence, we present here only the basic concept of the conversion process and summarize only some important recent research on this topic.
| CO2(g) + H2(g) → HCOOH (l) ΔG = 32.9 kJ mol−1 | (12) |
| CO2(aq) + H2(aq) → HCOOH (aq) ΔG = −4.0 kJ mol−1 | (13) |
| CO2(aq) + H2(aq) + NH3(aq) → HCOO− (aq) + NH4+(aq) ΔG = −9.5 kJ mol−1 | (14) |
Most of the complexes used to catalyze CO2 hydrogenation are based on group VIII transition metals such as Pd, Ni, Rh, Ru, and Ir. Among these catalysts, Rh, Ru, and most recently Ir complexes were found to be the most effective. Recently, the groups of Himeda and Fujita summarized the homogeneous hydrogenation of CO2 to formate and methanol as an alternative to photo- and electrochemical CO2 reduction with major emphasis on a half-sandwich iridium catalyst based on proton-responsive ligands.161
Yang et al. studied the reaction mechanisms for the hydrogenation of carbon dioxide catalyzed by PNP-ligated (PNP = 2,6-bis(di-isopropylphosphinomethyl)pyridine) metal pincer complexes, (PNP)MHn (M = Ir, Fe and Co), computationally by using density functional theory (DFT).163 Iridium is a recently reported high efficiency catalyst for the formation of formic acid from H2 and CO2.164 Fe and Co metal complexes could be used as low-cost non-noble metal catalysts for CO2 reduction.
Using the rhodium-phosphine complex, [RhCl(mtppms)3] (mtppms = monosulfonated triphenylphosphine), as a catalyst, free formic acid was produced in the hydrogenation of carbon dioxide dissolved in aqueous sodium formate solutions under H2 and CO2 pressure with a water-soluble concentration of sodium formate.165 The total gas pressure and the pressure ratio of H2 to CO2 were the most important factors for the production of HCOOH. Up to 0.13 M concentration of HCOOH was achieved, while there was negligible formic acid production in the absence of sodium formate.
Himeda et al. reported the interconversion between formic acid and H2/CO2 using half-sandwich rhodium and ruthenium catalysts with 4,4′-dihydroxy-2,2′-bipyridine (DHBP).166 In the hydrogenation of CO2/bicarbonate to formate under basic conditions, activations of the catalysts were caused by the electronic effect of oxyanions generated by the deprotonation of the hydroxyl group. The turnover frequencies of these catalysts increased 65- and 8-fold, respectively, compared to the corresponding unsubstituted bipyridine complexes. The rhodium–DHBP catalyst showed high activity without CO contamination in a relatively wide pH range. These catalytic systems are extremely useful for H2 storage materials by combining CO2 hydrogenation with formic acid decomposition.
Maenaka et al. synthesized the [C, N] cyclometalated organoiridium complex, [IrIII(Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoic acid-κC3)(H2O)]2SO4, as an efficient catalyst for interconversion between hydrogen and formic acid in water at ambient temperature and pressure for both directions depending on pH.167 The hydrogenation of carbon dioxide occurs in the presence of a catalytic amount of the Ir complex under an atmospheric pressure of H2 and CO2 in weakly basic water (pH 7.5) at room temperature, whereas formic acid efficiently decomposes to afford H2 and CO2 in acidic water (pH 2.8).
Wesselbaum et al. presented a new idea that allows the continuous-flow hydrogenation of supercritical CO2 (scCO2) with integrated product separation from an immobilized catalyst and a stabilizing base to produce pure formic acid in a single processing unit.168 The concept is based on a biphasic reaction system consisting of scCO2 as the mobile phase and an ionic liquid as the stationary phase containing the catalyst and a nonvolatile base (Scheme 6). The pure HCO2H that is free of any cross-contamination can be obtained without interrupting the working catalyst inside the reactor by this method.
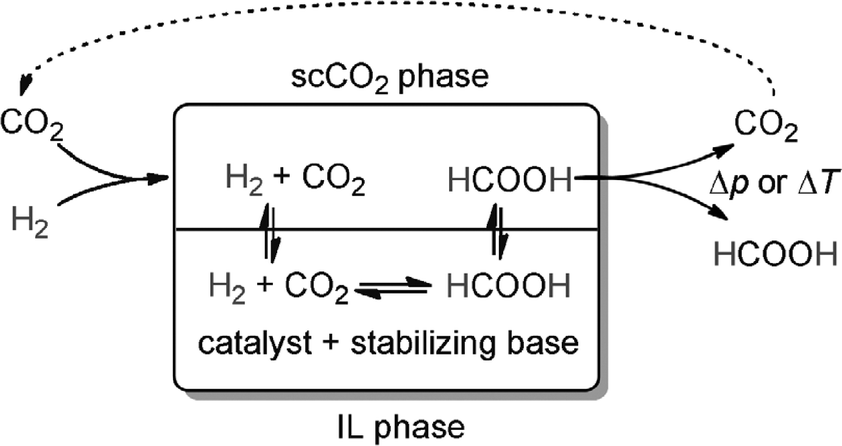 | ||
| Scheme 6 Process for the direct continuous-flow hydrogenation of CO2 to free formic acid based on a two-phase system with scCO2 as the extractive mobile phase and an ionic liquid (IL) as the stationary phase containing the catalyst and the stabilizing base. Reprinted with permission from ref. 168. Copyright 2012 Wiley-VCH. | ||
Huff et al. have synthesized the ruthenium pincer complex Ru(PNN)CO(H) (PNN = 6-(di-tert-butylphosphinomethylene)-2-(N,N-diethylaminomethyl)-1,6-dihydropyridine) which is an efficient catalyst for the hydrogenation of carbon dioxide to formate. Stoichiometric studies are presented that support the feasibility of individual steps in a proposed catalytic cycle for this transformation.169 They explored the influence of base and solvents on the catalytic performance. Under optimized conditions (using diglyme as the solvent and potassium carbonate as the base) up to 23000 turnovers of formate and a turnover frequency of up to 2200 h−1 were achieved.
The activity of homogeneous ruthenium(II)-1,2-bis(diphenylphosphino)ethane as a catalyst was studied for the equilibrium position in formic acid/amine–CO2 systems as a function of pressure and temperature under isochoric conditions.170 It was found that the Ru catalyst was active for both, that is, in formic acid cleavage producing pure hydrogen and CO2, and in carbon dioxide hydrogenation under basic conditions. High yields of formic acid dehydrogenation into H2 and CO2 are favoured by low gas pressures and/or high temperatures, and H2 uptake is possible at elevated H2–CO2 pressures.
Recently, the direct hydrogenation of CO2 into formic acid using a homogeneous ruthenium catalyst has been studied by Moret et al. in aqueous solution and in dimethyl sulphoxide (DMSO) without any additives.171 In water, at 313 K, 0.2 M formic acid can be obtained under 200 bar, however, in DMSO the same catalyst affords 1.9 M formic acid. In both solvents, the catalyst can be reused multiple times without a decrease in activity. Worldwide demand for formic acid continues to grow, especially in the context of a renewable energy hydrogen carrier, and its production from CO2 without a base, via direct catalytic carbon dioxide hydrogenation, is considerably more sustainable than the existing routes.
Drake et al. have investigated the role of various common inorganic salts in increasing the productivity for the hydrogenation of CO2 to formic acid in the presence of various catalysts.172 Among the various salts, KHCO3 acted as the best promoter for the hydrogenation of CO2. The selectivity for formic acid production catalyzed by RuCl2(PPh3)(p-cymene) could be increased by 84% to 140% depending on the amount of KHCO3. The effect was general for other noble-metal catalysts and for one of the most efficient non-noble-metal hydrogenation catalysts shown in Fig. 9. Mechanistic experiments revealed that the reaction likely proceeds via the formation of a metal–carbonate species.
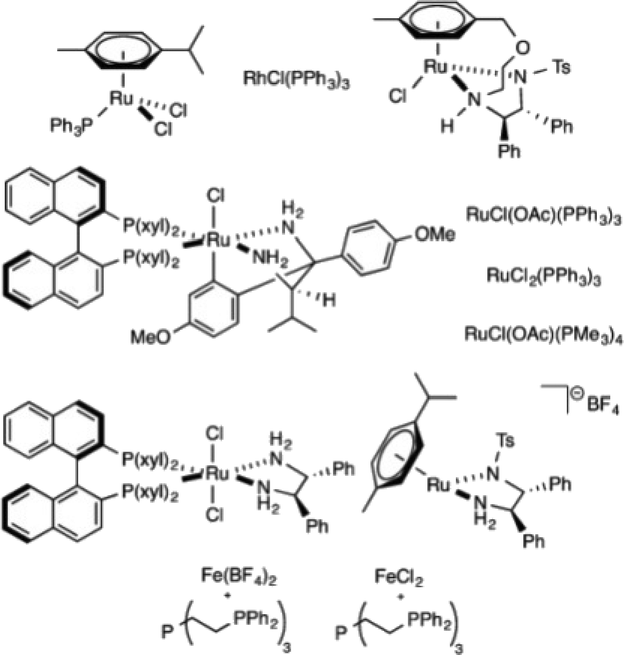 | ||
| Fig. 9 Catalysts used for hydrogenation of CO2. Reprinted with permission from ref. 172. Copyright 2013 American Chemical Society. | ||
Hsu et al. have reported the use of a Ru catalyst [(PNNP)(acetonitrile)RuII] for the efficient and reversible reduction of CO2 to formic acid in the presence of DBU as a base.173 At a catalyst loading of only 0.075 mol%, both the reduction using H2 pressure and the decomposition of the formic acid adduct under pressure-free conditions were established.
The homogeneously catalyzed hydrogenation of carbon dioxide to formic acid and its derivates has been well studied. Recently, an increase in product formation was achieved by further development of homogeneous catalysts. Currently, iridium-based catalysts offer the highest catalytic activity known in the hydrogenation of carbon dioxide. Here we summarized various catalyst systems for CO2 hydrogenation to formic acid (Table 3) as reported in a recent book chapter by Behr et al.38
| SN | Catalyst | Solvent | Base/additive | T (K) | P CO2/H2 (bar) | Time (h) | TON (h−1) | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|
| bipy = 2,2′-bipyridine, cod = cyclooctadiene, CYPO = Cy2PCH2CH2OCH3, DBF = dibutylformamide, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, dcpb = Cy2P(CH2)4PCy2, dcpe = 1,2-dicyclohexylphosphinoethane, dhbipy = 4,4′-dihydroxy-2,2′-bipyridine, DHphen = 4,7-dihydroxy-1,10-phenanthroline, dippe = 1,2-(diisopropylphosphino)ethane, dppb = 1,4-bis(diphenylphosphino)butane, dppbts = tetrasulfonated-1,4-bis(diphenylphosphino)butane, dppp = 1,3-bis(diphenylphosphino)propane, hfacac = hexafluoroacetylacetonate, mTPPMS = meta-monosulfonated triphenylphosphine, NBD = bicycle[2.2.1]hepta-2,5-diene, NHC = N-heterocyclic carbine, PP3 = [P(CH2CH2PPh2)3], PTA = 1,3,5-triaza-7-phosphaadamantane, TEtA = triethanolamine, Tp = hydrotris(pyrazolyl)borate, TPPMS = monosulfonated triphenylphosphine, TPPTS = trisulfonated triphenylphosphine. | ||||||||
| 1 | [Ru(Cl2bipy)2(H2O)2] [CF3SO3]2 | EtOH | NEt3 | 323 | 30/30 | 8 | 5000 | 625 |
| 2 | RuCl2(PMe3)4 | scCO2 | NEt3 | 323 | 120/80 | 47 | 7200 | n.a. |
| 3 | [RuCl(C6Me6)(DHphen)]Cl | H2O | KOH | 393 | 30/30 | 24 | 15400 |
642 |
| 4 | RuCl(OAc)(PMe3)4 | scCO2 | NEt3 | 323 | 120/70 | 0.3 | 31700 |
95000 |
| 5 | [RuCl2(mTPPMS)2]2 | H2O | NaHCO3 | 353 | 35/60 | 0.03 | 320 | 9600 |
| 6 | RuH2(PMe3)4 | scCO2 | NEt3 | 323 | 120/85 | 1 | 1400 | 1400 |
| 7 | Ru2(CO)5(dppm)2 | Acetone | NEt3 | RT | 38/38 | 21 | 2160 | 103 |
| 8 | RuCl3/PPh3 | EtOH | NEt3 | 333 | 60/60 | 5 | 200 | 40 |
| 9 | CpRu(CO)(μ-dppm)Mo(CO)2Cp | Benzene | NEt3 | 393 | 30/30 | 45 | 43 | 1 |
| 10 | TpRuH(PPh3)(CH3CN) | THF | NEt3 | 373 | 25/25 | 16 | 1815 | 113 |
| 11 | RuH2(PPh3)4 | Benzene | NEt3 | RT | 25/25 | 20 | 87 | 4 |
| 12 | RuH2(PPh3)4 | Benzene | Na2CO3 | 373 | 25/25 | 4 | 169 | 42 |
| 13 | [(C5H4(CH2)3NMe2)Ru(dppm)]BF4 | THF | None | 353 | 40/40 | 16 | 8 | 0.5 |
| 14 | [RuCl2(CO)2]n | H2O, iPrOH | NEt3 | 353 | 27/81 | 0.3 | 400 | 1300 |
| 15 | RuCl3(aq)/dppbts | H2O | HNMe2 | 333 | 25/25 | 15 | 4980 | n.a. |
| 16 | K[RuCl(EDTA-H)Cl]·2H2O | H2O | None | 313 | 17/3 | 0.5 | n.a. | 250 |
| 17 | RuCl2(PTA4) | H2O | NaHCO3 | 353 | 0/60 | n.a | n.a | 807 |
| 18 | [(Cl2bipy)2Ru(H2O)2][CF3SO3]2 | EtOH | NEt3 | 423 | 30/30 | 8 | 5000 | n.a. |
| 19 | [(NHC)2RuCl][PF6] | H2O | KOH | 473 | 20/20 | 75 | 23000 |
n.a. |
| 20 | [(C6Me6)Ru(dhbipy)] | H2O | KOH | 393 | 30/30 | 8 | 13620 |
4400 |
| 21 | [(dppm)Ru(H)(Cl)] | H2O | NaHCO3 | 343 | 0/50 | 2 | 1374 | 687 |
| 22 | [RuCl2(TPPMS)2]2 | H2O | NaHCO3 | 298 | 5/35 | 2 | 524 | 262 |
| 23 | [Cp*Ru(DHphen)Cl]Cl | H2O | KOH | 393 | 30/30 | 24 | 15400 |
3600 |
| 24 | [(η6-arene)Ru(η2-N, O–L)(H2O)]BF4 | H2O | NEt3 | 373 | 50/50 | 10 | 400 | n.a. |
| 25 | RuH2(PMe3)4 | scCO2 | NEt3 | 323 | 120/80 | 3 | 1900 | 630 |
| 26 | RuH2(PPh3)4 | DBF | TEtA | 323 | 30/30 | 1 | 698 | 698 |
| 27 | Rh(hfacac)(dcpb) | DMSO | NEt3 | 298 | 20/20 | 5 | n.a. | 1335 |
| 28 | RhCl(TPPTS)3 | H2O | NHMe2 | RT | 20/20 | 12 | 3439 | n.a. |
| 29 | [RhH(cod)]4/dppb | DMSO | NEt3 | RT | 20/20 | 18 | 2200 | 375 |
| 30 | RhCl(TPPTS)3 | H2O | NHMe2 | 354 | 20/20 | 0.5 | n.a. | 7260 |
| 31 | [RhCl(cod)]2/dippe | DMSO | NEt3 | 297 | 40 total | 18 | 205 | 11 |
| 32 | RhCl(PPh3)3 | Benzene | Na2CO3 | 373 | 55/60 | 3 | 173 | 58 |
| 33 | [Rh(η2-CYPO)2]BPh4 | MeOH | NEt3 | 328 | 25/25 | 7 | 1000 | n.a |
| 34 | RhCl3/PPh3 | H2O | NHMe2 | 323 | 10/10 | 10 | 2150 | 215 |
| 35 | [Rh(NBD)(PMe2Ph)3]BF4 | THF | None | 313 | 48/48 | 48 | 128 | 3 |
| 36 | RhCl(PPh3)3/PPh3 | MeOH | NEt3 | 298 | 40/20 | 20 | 2700 | 125 |
| 37 | RhCl3(aq)/dppbts | H2O | HNMe2 | 333 | 25/25 | 15 | 3910 | n.a. |
| 38 | [RhCl(cod)]2/dppb | DMSO | NEt3 | RT | 20/20 | 22 | 1150 | 52 |
| 39 | RhCl(TPPTS)3 | H2O | NEt3 | 296 | 20/20 | n.a. | n.a. | 1364 |
| 40 | [Cp*Rh(DHphen)Cl]Cl | H2O | KOH | 353 | 20/20 | 32 | 2400 | 270 |
| 41 | Rh(NO)(dcpe) | n.a. | DBU | 323 | 1.5/1.5 | 16 | 106 | n.a. |
| 42 | [RhCl(mTPPMS)3] | H2O | HCOONa | 323 | 50/50 | 20 | 65 | n.a. |
| 43 | [RhCl(mTPPMS)3] | H2O | CaCO3 | 323 | 20/80 | 24 | 300 | n.a. |
| 44 | [Cp*Ir(dhbipy)Cl]Cl | H2O | KOH | 393 | 30/30 | 57 | 42000 |
190000 |
| 45 | [Cp*Ir(DHphen)Cl]Cl | H2O | KOH | 393 | 30/30 | 48 | 222000 |
33000 |
| 46 | [Cp*IrCl(DHphen)]Cl | H2O | KOH | 393 | 30/30 | 10 | 21000 |
2100 |
| 47 | [(PNP)IrH3] | H2O | KOH | 393 | 30/30 | 48 | 3500000 |
73000 |
| 48 | [IrI2(AcO)(bis-NHC)] | H2O | KOH | 473 | 30/30 | 75 | 190000 |
n.a. |
| 49 | PdCl2 | H2O | KOH | 433 | n.a./110 | 3 | 1580 | 530 |
| 50 | PdCl2 | H2O | KOH | 513 | 40/106 | 3 | 340 | n.a. |
| 51 | Pd(dppe)2 | Benzene | NEt3 | 383 | 25/25 | 20 | 62 | 3 |
| 52 | Pd(dppe)2 | Benzene | NaOH | RT | 24/24 | 20 | 17 | 0.9 |
| 53 | PdCl2[P(C6H5)3]2 | Benzene | NEt3 | RT | 50/50 | n.a. | 15 | n.a. |
| 54 | FeCl3/dcpe | DMSO | DBU | 323 | 60/40 | 7.5 | 113 | 15.1 |
| 55 | Fe(BF4)2·6H2O, PP3 | MeOH | NaHCO3 | 353 | 0/60 | 20 | 610 | n.a. |
| 56 | Co(BF4)2·6H2O, PP3 | MeOH | NaHCO3 | 353 | 0/60 | 20 | 3877 | n.a |
| 57 | trans-[(tBu-PNP)Fe(H2)(CO)] | H2O | NaOH | 393 | 3.33/6.66 | 5 | 788 | n.a |
| 58 | NiCl2(dcpe) | DMSO | DBU | 323 | 160/40 | 216 | 4400 | 20 |
| 59 | Ni(dppe)2 | Benzene | NEt3 | RT | 25/25 | 20 | 7 | 0.4 |
| 60 | TiCl4/Mg | THF | None | RT | 1/1 | 24 | 15 | n.a. |
| 61 | MoCl3/dcpe | DMSO | DBU | 323 | 60/40 | 7.5 | 63 | 8 |
Some catalysts are found to be active for reversible CO2 hydrogenation as well as formic acid dehydrogenation by varying the reaction conditions.164,174 These systems hold promise for the development of a practical H2 storage technology based on the reversible catalytic hydrogenation of CO2. Nozaki and co-workers reported a highly active catalytic hydrogenation of carbon dioxide to form formic acid salts and the dehydrogenation of formic acid salt using a PNP-ligated iridium(III) trihydride complex (Fig. 10). The hydrogenation of carbon dioxide using this PNP-ligated iridium(III) trihydride complex afforded potassium formate in aqueous KOH solution with a TOF of 150000 h−1 and a TON of 3500000, both of the values being the highest ever reported.164,174 In the presence of triethanolamine as the base, both CO2 hydrogenation (TON 29000, TOF 14000 h−1 at 5.0 MPa at 297 K) and FA dehydrogenation (TON 4900, TOF 1000 h−1 at 333 K) proceed at an appreciable rate, showing that the iridium catalyst can be used for reversible hydrogen storage by changing the reaction conditions only.
 | ||
| Fig. 10 Plausible mechanism for hydrogenation of CO2 using iridium trihydride complex. Reprinted with permission from ref. 174. Copyright 2011 American Chemical Society. | ||
Pidko et al. reported the highly efficient reversible hydrogenation of carbon dioxide to formates using a ruthenium PNP-pincer catalyst.175 This catalyst has a very high activity for CO2 hydrogenation and FA dehydrogenation. If used in combination with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), the catalyst allows the control of hydrogen liberation activity in a narrow temperature interval. The H2 capacity is dependent on the base strength, and strong bases are required to generate a high acid-to-amine ratio at elevated temperature if the reaction times are short.
Besides the several works on noble-metal based catalysts, some non-precious-metal catalysts based on Fe,70,176–180 Co,181–183 Ni,177,184 Cu,185 Mo,177 and Ti (ref. 186) have also been investigated. Beller and co-workers reported the efficient, stable, and easy-to-synthesize non-noble metal complex iron(II)-fluoro-tris(2-(diphenylphosphino)phenyl)phosphino]tetrafluoroborate for the reduction of CO2 and bicarbonates.179 The catalytic hydrogenation of the bicarbonates proceeded in good yields with high catalyst productivity and activity (TON > 7500, TOF > 750 h−1).
Beller and co-workers successfully utilized the cationic Co(BF4)2·6H2O/PP3 catalyst system for the conversion of sodium bicarbonate/CO2 to sodium formate (TON 687 at 353 K and 3877 at 393 K).181 A cobalt-based catalyst system for the production of formate from CO2 and H2 under ambient conditions has been synthesized by Jeletic et al.182 The complex Co(dmpe)2H (dmpe = 1,2-bis(dimethylphosphino)ethane) catalyzes the hydrogenation of CO2, with a TOF of 3400 h−1 at room temperature and 1 atm of 1:1 CO2:H2 (74000 h−1 at 20 atm) in THF. These results highlighted the value of the fundamental thermodynamic properties for the rational design of catalysts. Himeda et al. reported new water-soluble pentamethylcyclopentadienyl cobalt(III) complexes with proton-responsive 4,4′- and 6,6′-dihydroxy-2,2′-bipyridine ligands as efficient catalysts for CO2 hydrogenation to formate in aqueous bicarbonate media at moderate temperature under a total pressure of 4–5 MPa (CO2:H2 1:1).183
Several possible mechanisms are conceivable for CO2 hydrogenation to formic acid which mainly involves the insertion of CO2 to the M–H bond followed by the elimination of FA. Th insertion of carbon dioxide into the M–H bond is identical for all mechanisms in which carbon dioxide forms a metal formate complex via η2-coordination and migration of hydrogen (Scheme 7).38
 | ||
| Scheme 7 General mechanism for the homogeneously catalyzed hydrogenation of CO2 to FA. | ||
Theoretically, it is established that the insertion can occur by M–H bond breaking based on the weak H–CO2 interaction.187–189 The elimination of formic acid can proceed via reductive elimination of formic acid, addition of hydrogen to the formate complex, direct hydrogenolysis of the M–O bond without previous oxidative addition to hydrogen or hydrolysis of the metal formate.38,162,190,191
Wiener et al. reported the reduction of carbonates to formic acid under mild reaction conditions using Pd/C as a catalyst.193 However, the reaction could not achieve completeness due to the chemical equilibrium between carbonate and formate. Preti et al. examined the activity of some heterogeneous catalysts, such as iron powder, RANEY® nickel and Co, Cu, Ru, Rh, Pd, Ag, Ir, Pt and Au black, for the formation of formic acid by CO2 hydrogenation.194 Au black catalyses the CO2 hydrogenation in the presence of neat NEt3 to form HCOOH/NEt3 adducts. Activity studies have been extended to commercially available aurolite (Mintek; 1 wt% Au/TiO2, extrudates). Formic acid can be separated from HCOOH/NEt3 with the help of a high-boiling point amine (n-C6H13)3N. When coupled with the catalytic HCOOH decomposition to CO-free hydrogen together with easily removable and reusable CO2, these findings complete the chemical loop for the long-sought-after hydrogen storage with CO2.
Hao et al. reported the preparation and application of heterogeneous supported ruthenium catalysts.195 The catalysts were active in the synthesis of formic acid from the hydrogenation of carbon dioxide. Abundant hydroxyl groups, which interact with the ruthenium components, play an important role in the catalytic reactions. Highly dispersed ruthenium hydroxide species enhance the hydrogenation of CO2, while crystalline RuO2 species, which are formed during the preparation of the catalyst, restrict the production of formic acid. The best activity of ruthenium hydroxide as a catalyst for the hydrogenation of CO2 to formic acid was achieved over a γ-Al2O3 supported 2.0 wt% ruthenium catalyst, which was prepared in a solution of pH 12.8 with NH3·H2O as a titration solvent.
A series of catalysts made of ruthenium loaded on γ-Al2O3 and γ-Al2O3 nanorods were studied for the hydrogenation of CO2 to formic acid by Preti et al.196 Among these catalysts, 2% Ru/Al (n) has the highest activity. The excellent catalytic activity is related to the highly dispersed ruthenium species on the surface of the support and abundant hydroxyl groups of the support.
A series of catalysts made of ruthenium loaded on γ-Al2O3 nanorods were prepared and their catalytic performances for the hydrogenation of CO2 to formic acid have been studied by Na Liu et al.197 The results revealed that the catalytic activity is determined by the structure of the supported ruthenium oxide species. Th optimal activity of the catalyst for the hydrogenation of CO2 to formic acid is achieved over a γ-Al2O3 nanorod supported 2.0 wt% ruthenium catalyst.
Peng et al. theoretically investigated the hydrogenation of CO2 to formic acid on transition metal surfaces because they exhibit unique reactivity for the hydrogenation reaction. They explored the potential of subsurface hydrogen for hydrogenating carbon dioxide on Ni(110).198 A heterogeneous, mesoporous silica-tethered iridium catalyst (Ir-PN/SBA-15) containing a bidentate iminophosphine ligand was reported for the hydrogenation of CO2 to formic acid in aqueous solution.199 This catalyst is recyclable and exhibits high activities for formic acid production under mild conditions [333 K, 4.0 MPa total pressure (H2/CO2 = 1:1)].
The reaction mechanisms of the hydrogenation of carbon dioxide to formic acid over a Cu-alkoxide-functionalized metal–organic framework (MOF) have been investigated theoretically.200 The reaction can proceed via two different pathways, namely, concerted and stepwise mechanisms. In the concerted mechanism, the hydrogenation of CO2 to formic acid occurs in a single step. It requires a high activation energy of 67.2 kcal mol−1. For the stepwise mechanism, the reaction begins with hydrogen atom abstraction by CO2 to form a formate intermediate. The intermediate then takes another hydrogen atom to form formic acid. The activation energies are calculated to be 24.2 and 18.3 kcal mol−1 for the first and second steps, respectively. Because of the smaller activation barriers associated with this pathway, it therefore seems to be more favoured than the concerted one.
Ruthenium complexes immobilized on amine-functionalized silica have been synthesized by an in situ synthetic approach.201 The catalysts exhibit high activity (TOF = 1384 h−1) with complete (100%) selectivity for CO2 hydrogenation to formic acid. Immobilization of homogeneous catalysts offers practical advantages such as easy separation and recycling. The combination of the basic ionic liquid 1-(N,N-dimethylaminoethyl)-2,3-dimethylimidazolium trifluoromethane-sulfonate ([mammim][OTf]) and a silica-supported ruthenium complex (“Si”–(CH2)3–NH(CSCH3)–RuCl3–PPh3) was found to catalyze CO2 hydrogenation to formic acid with satisfactory activity and selectivity.202 This immobilized catalyst catalyzes the hydrogenation of CO2, with a maximum TOF of 103 h−1 at a total pressure of 18 bar (H2:CO2 = 1:1) without loss of activity over 5 catalytic cycles.
A reversible heterogeneous Pd catalyst (>3 nm) supported on mesoporous graphitic carbon nitride (Pd/mpg-C3N4) for the interconversion of CO2 and formic acid has been synthesized by Lee et al. (Fig. 11).121 This catalyst catalyzes the formation of formic acid via CO2 hydrogenation along with dehydrogenation of formic acid with a turnover frequency of 144 h−1 even in the absence of external bases at room temperature. The basic sites of the support could also induce initial interaction with CO2 for the synthesis of FA. The governing factors for CO2 hydrogenation are further elucidated to increase the quantity of the desired formic acid with high selectivity.
 | ||
| Fig. 11 HRTEM analyses: (a and b) images of Pd/mpg-C3N4, (c) HADDF-STEM image of Pd/mpg-C3N4, and (d) particle size distribution of Pd/mpg-C3N4. Reprinted with permission from ref. 121. Copyright 2014 Royal Society of Chemistry. | ||
3.3. Photochemical and enzymatic reductions of CO2 to formic acid
CO2 fixation by photochemical and enzymatic reduction is an inexpensive, attractive and green method for the synthesis of organic compounds from CO2 as the starting material. Although several works have been done on CO2 fixation, the quantum yields and selectivity of the products based on the reduction of CO2 are still low.203–206 In artificial photocatalytic CO2 reduction, the reduction of CO2 to CO is more favourable than to formic acid or other products. In principle, the synthesis of formic acid by photocatalytic reduction of CO2 is mainly done by using the formate dehydrogenase (FDH) enzyme which catalyzes the reversible conversion of formic acid to CO2. NADH (nicotinamide adenine dinucleotide) dependent enzymes can be used to reduce the formed N,N′-alkyl-4,4′- or -2,2′-bipyridinium salt molecules as artificial substrates instead of NADH or NAD+. For example, FDH from Saccharomyces cerevisiae is a NADH-dependent enzyme and uses the various reduced forms of the bipyridinium salt molecules as substrates for the conversion of CO2 to formic acid.Willner and co-workers reported the enzymatic formic acid synthesis from HCO3− with FDH and the photoreduction of various 4,4′- or 2,2′-bipyridinium salts by a system containing [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and mercaptoethanol (RSH) as an electron donor.207–210 The efficiency of formic acid synthesis depends on the nature of the bipyridinium salts, and the quantum yield of formic acid from HCO3− is in the range of 0.5–1.6%. Schuchmann et al. reported the discovery of a bacterial hydrogen-dependent carbon dioxide reductase (HDCR) that directly uses H2 for the interconversion of CO2 to formate from the acetogenic bacterium Acetobacterium woodii.211 They targeted this bacterium because it grows with H2 + CO2 under ambient conditions using an ancient pathway for CO2 fixation and energy conservation. It lives under extreme energy limitation and directly uses H2 for CO2 reduction which would be beneficial for the cell.
Alissandratos et al. reported the biological conversion of CO2 and H2 into formate by utilising engineered whole-cell biocatalysts.212Escherichia coli. JM109(DE3) cells transformed for the overexpression of either native formate dehydrogenase (FDH), the FDH from Clostridium carboxidivorans, or genes from Pyrococcus furiosus and Methanobacterium thermoformicicum predicted to express FDH based on their similarity to known FDH genes were all able to produce levels of formate well above the background, when present with H2 and CO2.
Kodaka et al. reported the direct synthesis of formic acid from CO2 gas with the system consisting of triethanolamine (TEOA), [Ru(bpy)3]2+, various bipyridinium salts (1,1′-dialkyl-4,4′-bipyridinium salts or 1,1′-dialkyl-2,2′-bipyridinium salts) and FDH.213 The effects of the structure and redox potentials of various bipyridinium salts have been investigated for the synthesis of formic acid from CO2. After 7 h of irradiation, the formation of formic acid in the concentration range of 0.2–0.95 mM was achieved from a sample solution containing 0.5 M TEOA, 0.5 mM [Ru(bpy)3]2+, 3.0 mM bipyridinium salt, and 8 mg of FDH. The best activity for formic acid synthesis from CO2 was observed by using N,N′-trimethylene-2,2′-bipyridinium salt. The yield of formic acid depends on the redox potentials of the bipyridinium salts.
Amao and Miyatani synthesized formic acid from HCO3− with FDH and the photoreduction of N,N′-dimethyl-4,4′-bipyridinium (MV2+).214–216 They used a water-soluble zinc porphyrin (zinc tetrakis(4-methylpyridyl)porphyrin, ZnTMPyP) as a photosensitizer. When the sample solution containing 0.3 M TEOA, 9.0 μM ZnTMPyP4+, 15 mM MV2+, 20 units of FDH and 1.0 mM NaHCO3 was irradiated, 60 μm of formic acid (estimated yield 6%) was produced after 3 h of irradiation. In another report, they also introduced a system for the synthesis of formic acid from CO2 gas with FDH, MV2+, NADPH as an electron relay and Mg chlorophyll-a (MgChl-a) as a visible light photosensitizer (Fig. 12).217 After 4 h of irradiation, the formation of 56 μM formic acid was achieved from the sample solution containing NADPH (3.0 mM), MgChl-a (9.0 μM), MV2+ (0.1 mM), FDH (5 units), and CO2 gas (saturated) at 303 K. Further, it has been observed that formic acid was not produced in the absence of any of the components.
 | ||
| Fig. 12 Photochemical CO2 reduction system that combines the photoreduction of NAD+ as an electron relay by the photosensitization of dye molecules (P) and CO2 reduction with FDH. Reprinted with permission from ref. 217. Copyright 2006 Elsevier. | ||
Sato and co-workers reported the immobilization of a molecular CO2 reduction catalyst onto a semiconductor surface for engineering hybrid photocatalytic materials.218–222 The ruthenium catalysts were grafted onto different p-type semiconductors such as N-doped Ta2O5, Zn doped InP, etc. via covalent linkages or via a polymerization process. It was reported that the robustness of a hybrid system depends strongly on the choice of grafting linkages. The [Ru(dpbpy)(bpy)(CO)2]/N–Ta2O5 (dpbpy = 4,4′-diphosphonate-2,2′-bipyridine, bpy = 2,2′-bipyridine) system with a phosphate linker displayed a 5 times higher rate of CO2 reduction to HCOOH (in acetonitrile solution using visible light) compared with the [Ru(dcbpy)(bpy)(CO)2]/N–Ta2O5 (dcbpy: 4,4′-dicarboxy-2,2′-bipyridine, bpy = 2,2′-bipyridine) system with a carbonate linker.218,221 By using a Deronzier's-type ruthenium bipyridine-based polymeric film catalyst, a robust system adapted for working in aqueous solution was achieved.221
Recently, Ishitani and co-workers investigated the photocatalytic reduction of CO2 in an aqueous solution using a supramolecular binuclear Ru(II)–Re(I) complex, of which the Ru(II) photosensitizer and Re(I) catalyst units are connected with a bridging ligand (Fig. 13).223 Sodium ascorbate was used as an electron donor. Formic acid is the main product of the photochemical reaction in aqueous solution (TON = 25, selectivity = 83%), whereas CO is the main product in DMF/TEOA.
 | ||
| Fig. 13 Reaction pathways of the photochemical reduction of CO2 to HCOOH using supramolecular Ru(II)–Re(I) bimetallic complex with sodium ascorbate in an aqueous solution. Adopted with permission from ref. 223. Copyright 2015 American Chemical Society. | ||
4. Practical set up for formic acid-based hydrogen storage
A sustainable cycle for energy storage based on formic acid and carbon dioxide can be designed as shown in Fig. 14.34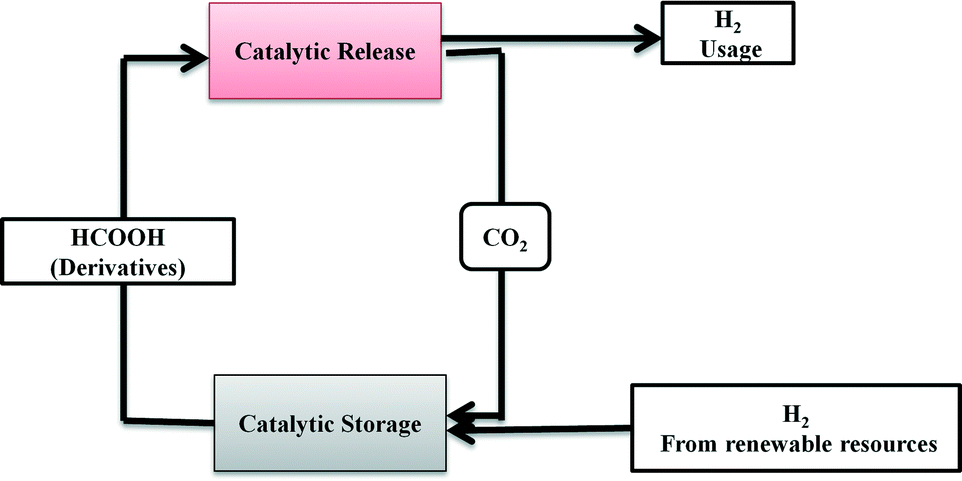 | ||
| Fig. 14 A catalytic cycle for hydrogen storage in formic acid. | ||
For energy storage, carbon dioxide is converted to formic acid or a formate derivative either electrochemically224,225 or by catalytic hydrogenation.32,156,226 The resulting material is a liquid at ambient conditions, either pure formic acid, an adduct containing formic acid, or an inorganic formate in aqueous solution, and can thus be handled, stored, and transported easily. On the other side of the cycle, energy is released either in a direct formic acid fuel cell, or by selective on-demand decomposition to carbon dioxide and hydrogen, which can be used directly in an appropriate hydrogen oxygen fuel cell.227–232 If pure hydrogen is required, the gases may also be separated using membrane techniques.233 The hydrogen content of pure formic acid is 43 g kg−1 or 52 g L−1; its release from formic acid is thermodynamically downhill by ΔG° = −32.8 kJ mol−1 at room temperature.156 As early as 1978, the electrochemical reduction of carbon dioxide to formic acid and its subsequent decomposition on Pd/C for energy storage were proposed by Williams et al.234 Later on, a system for solar energy conversion by reduction of aqueous carbonate was proposed by Halmann in 1983.235 In 1986, a similar concept was described by Wiener et al. They proposed the use of a Pd/C catalyst to decompose aqueous formate solutions to obtain hydrogen.236–238 However, both approaches have not led to any application. Two decades later, the research on the use of carbon dioxide for energy storage was resumed, which has been continued in recent years.6,34,61,239–242
To identify the main technological obstacles on the way to an efficient use of FA as a hydrogen source and storage material, a hydrogen generator based on the decomposition of FA over a homogeneous Ru(II) catalyst was designed and built by Laurenczy and co-workers (Fig. 15).6 The hydrogen generator successfully met the target power output of 1 kWel or roughly 30 L min−1 H2/CO2 assuming 50% fuel cell efficiency. The system was designed around a 5 L reaction vessel with electrical surface heating elements, containing 11.8 g of RuCl3·3H2O (0.045 mol) and 51.2 g of Na3TPPTS (Na3TPPTS sodium salt of meta-trisulfonated triphenylphosphine) (0.090 mol) catalyst in 1.5 L of aqueous HCOONa solution (1 M). The free reactor volume (~3.5 L) is used as a buffer in the case of rapid changes in the product flow rate set point as well as for gas–liquid separation and effectively prevents foam or spray from reaching the product gas outlet.
 | ||
| Fig. 15 A hydrogen generator designed and built in laboratories of Laurenczy for a power output of 1 kWel (30 L min−1 H2/CO2): (1) formic acid tank; (2) pump; (3) tube in tube heat exchanger; (4) reactor; (5) heat exchanger; (6 and 7) condensate separators; (8) mass flow controller. Adopted with permission from ref. 6. Copyright 2012 Royal Society of Chemistry. | ||
A standard mass flow controller is utilized to control the product flow rate, while a controlled formic acid feed flow is utilized to maintain the reaction pressure. To minimize water vapour entrainment in the product gas, the system also comprises a product gas cooler with a recycle condensate separator. As the control concept does not comprise the measurement of pH or formic acid concentration in the catalyst solution, the reaction has to run at a large catalyst excess for reasons of process safety. This setup has been used safely for over one year without the need to exchange or replenish the catalyst solution.
In 2013, Beller et al. designed Ru(II)/dppe as the catalyst system for continuous hydrogen production from formic acid (Fig. 16).243,244 The chemoselectivity is very high, resulting in very low CO contents of less than 10 ppm, which allows for its direct application for hydrogen generated in proton-exchange membrane (PEM) fuel cells. They presented the concept and insight into a setup for the continuous production of hydrogen from FA–amine adducts under mild conditions. This setup allows for a more secure system and a longer continuous hydrogen flow of up to 47 L h−1. The continuous setup consists of four main parts (Fig. 16): a high pressure reactor, an FA dosage system, a gas cleaning unit, and a quantification and analysis part for the resulting gas mixture. The central parts to control the dosage of FA are the pump and the liquid flow meter. The reactor, a stainless-steel autoclave, is connected to a condenser, a gas filter, and an active carbon column. These will remove any traces of discharged liquids or vapors. All components are commercially available. To produce hydrogen on demand, a certain amount of FA is dosed into the reactor containing the catalyst and the amine, and immediately, it is converted to hydrogen and carbon dioxide, in the same way that gasoline is consumed in an Otto engine. This allows for the use of the full hydrogen density of FA of 4.4 wt% (not including the surrounding system) because only the acid has to be stored in the tank.
 | ||
| Fig. 16 Schematic representation of the setup used for continuous hydrogen production. Reprinted with permission from ref. 243. Copyright 2013 Wiley-VCH. | ||
Beller et al. also developed a reversible “hydrogen battery” which allowed reversible fast hydrogen loading (even at RT) and unprecedented unloading (TON for H2 liberation of 800000).244 For this purpose, they used the defined [RuH2(dppm)2] complex instead of the in situ catalyst system. With this catalyst, hydrogenation of carbon dioxide and hydrogen release from the resulting formic acid were possible up to eight consecutive cycles at room temperature.
The group of Plietker reported a rechargeable H2 battery based on the principle of the Ru-catalyzed hydrogenation of CO2 to formic acid (charging process) and the Ru-catalyzed decomposition of formic acid to CO2 and H2 (discharging process).245 Both processes are driven by the same catalyst at elevated temperature either under pressure (charging process) or pressure-free conditions (discharging process). Up to five charging–discharging cycles were performed without a decrease in storage capacity. The resulting CO2/H2 mixture is free of CO and can be employed directly in fuel-cell technology (Fig. 17).
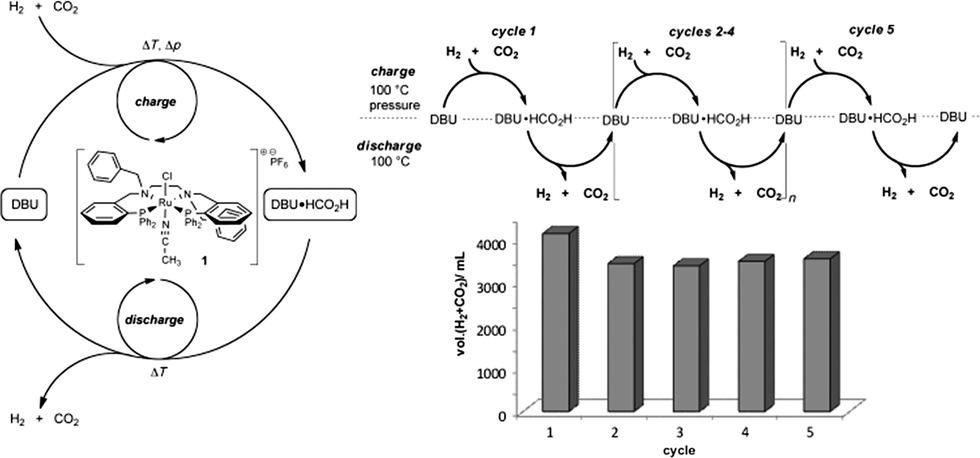 | ||
| Fig. 17 Schematic for charging and discharging process developed by Plietker et al. and charging and discharging cycles of the H2 battery. Reprinted with permission from ref. 245. Copyright 2013 Wiley-VCH. | ||
Some rechargeable hydrogen batteries were developed based on the selective hydrogenation of bicarbonates and the selective dehydrogenation of formates, instead of the hydrogenation of CO2 and the dehydrogenation of formic acid. Beller and co-workers have presented a ruthenium-based catalyst system ([{RuCl2(benzene)}2]/dppm (Ru/P = 1:6)) for the selective hydrogenation of bicarbonates and the selective dehydrogenation of formates (Fig. 18).246 It was demonstrated that the two reactions can be coupled leading to a reversible hydrogen-storage system. The hydrogenation of NaHCO3 to sodium formate was performed in 96% yield at 343 K in water/THF without additional CO2. The dehydrogenation of sodium formate was achieved with high conversion (>90%) at ambient temperature (303 K). In contrast to the dehydrogenation of formic acid/amine adducts, this process is amine-free.
 | ||
| Fig. 18 Reversible hydrogen storage with a bicarbonate/formate system: catalytic hydrogenation of bicarbonate and hydrogen release from formate with the same Ru/dppm in situ catalyst. Reaction conditions: dehydrogenation (DH): 40 mL of DMF, 10 mL of H2O, 303 K, 24–48 h; hydrogenation (H): 20 mL of H2O, 10 mL of THF, 80 bar H2, 24 h; sequence I: 71 μmol of [{RuCl2(benzene)}2], 0.43 mmol of dppm, 39.6 mmol of NaHCO2; sequence II: 10.4 μmol of [{RuCl2(benzene)}2], 41.6 μmol of dppm, 23.8 mmol of NaHCO3. Reprinted with permission from ref. 246. Copyright 2013 Wiley-VCH. | ||
Cao and co-workers developed the first heterogeneous catalytic system (1 wt% Pd/r-GO, Pd NPs supported on reduced graphene (rGO)) for the efficient reversible dehydrogenation of aqueous formates to bicarbonates.247 The r-GO nanosheets are a distinct and powerful support allowing for the generation of highly strained Pd NPs that enable facile and repetitive formate/bicarbonate interconversion under mild aqueous conditions. By controlling the temperature and the hydrogen pressure, the reversible catalytic transformations can be repeated nearly quantitatively six times with almost no loss of efficiency. This could lead to the construction of a simple, truly rechargeable HCOOK/KHCO3-based hydrogen storage device.
An ammonium bicarbonate/formate redox equilibrium system has also been demonstrated for reversible hydrogen storage and evolution using the same Pd/AC nanocatalyst by Lin and co-workers.248 The reaction temperature and H2 pressure are key factors in controlling the hydrogen storage–evolution equilibrium in this system. Up to 96% yield of ammonium formate was achieved when the hydrogenation reaction was carried out at room temperature at an initial hydrogen pressure of 2.75 MPa, whereas nearly 100% hydrogen yield was obtained from the dehydrogenation of ammonium formate at 353 K at an initial nitrogen pressure of 0.1 MPa. Compared to the homogeneous catalytic system, this heterogeneous system has the following advantages: no organic solvents or inorganic additives are needed; high volumetric energy density of hydrogen (stored in concentrated ammonium formate aqueous solutions) is achieved; the solid salts or their aqueous solutions can be easily transported and distributed; and the Pd/AC catalyst is stable, being more easily recycled and handled than homogeneous catalysts.
Further work on the design and testing of the hydrogen battery device based on a reversible catalytic system for formic acid dehydrogenation/hydrogenation of CO2 as well as dehydrogenation of formate/hydrogenation of bicarbonates is still under way.
5. Conclusions and perspectives
In this perspective, we have focused on the application of formic acid as a renewable hydrogen energy source for the future. In addition, we have tried to focus on formic acid production from various processes and, at the same time, the possibility of its regeneration from greenhouse gas CO2 has also been explored. We have attempted to present an up-to-date overview of this rapidly growing field of research. This subject area is very active and many papers are published every year (even during the preparation of this perspective) by chemists, physical and materials scientists. Therefore, it is difficult to take into account all the contributions to this field in a limited number of pages. We apologize if some significant contributions have been left out.In the first part, we have summarized the recent developments in formic acid decomposition reactions catalysed either by homogeneous or heterogeneous catalysts in the presence and absence of base and other additives. For improvement of catalytic performance, it is required to facilitate the activation of the C–H bond of the formate anion which is anticipated to be the rate-limiting step. Majority of homogeneous catalytic systems are based on Ru/Rh/Ir/Fe complexes, whereas Pd is the main constituent of heterogeneous catalysts.
Extensive work on the homogeneous catalytic decomposition of FA has been done by Beller and Leuranczy et al. Thanks to the people working in the field to prepare low-cost and efficient catalysts for hydrogen generation from formic acid. For homogeneous catalysts, β-hydride elimination is the rate-determining step of FA dehydrogenation. Thus, lowering the activation energy of β-hydride elimination is the key to success in enhancing the activity and selectivity of FA dehydrogenation. Scientists achieved this goal by modifying different parameters of the catalyst systems e.g. metal centre, design of ligands, reaction temperature, solvent medium, additives (phosphine/base/salts), etc. One of the recent approaches, as suggested by Chauvier et al., is to use a Lewis acid capable of converting the formate anion to CO2, with a low activation energy and negligible exergonicity.249
As obvious for most of the heterogeneous catalytic processes, the activity/selectivity of the heterogeneous catalytic FA dehydrogenation process also depends upon the sizes, morphologies, distributions, and surface states of NPs, capping agents, and catalyst supports, for bi-/tri-metallic NP counter elements. Generally, smaller size particles have better catalytic activity because of their large surface area but they easily aggregate due to their high surface energy and so induce low activity. Hence, a suitable capping agent or support is required for the dispersion of NPs to avoid the aggregation without severe deactivation of the surface of NPs by strong affiliation of the capping agent or support. In the FA dehydrogenation process, unsupported Pd catalysts are inactive due to the aggregation of NPs as well as poisoning of the surface by CO formed upon FA decomposition. Good activity with high selectivity for FA dehydrogenation was accomplished by immobilization of Pd NPs on/in carbon/graphene/silica/zirconia/ceria supports, etc. These supports not only facilitate the dispersion of metal NPs but also the catalytic CO oxidation at the metal–support interface, and thus prevent the deactivation of the catalyst by CO poisoning.
Similarly, in bimetallic NPs, the second component such as Au or Ag helps to diminish CO production by FA decomposition. As a result, the reaction proceeds via FA dehydrogenation with enhanced activity and selectivity. To further understand/explain the mechanism involved in formic acid decomposition on the surface, density functional theory approaches have also been adopted.
In the second part, we tried to cover various methods to prepare formic acid with special emphasis on biomass conversion to formic acid as well as CO2 hydrogenation by different methods. Wasserscheid and co-workers developed a method to obtain formic acid and CO2 selectively by different types of biomasses either water-soluble or insoluble. By varying the reaction conditions, such as pH, temperature or biomass substrate, they were successful in obtaining formic acid with up to 60% selectivity. We have also discussed CO2 reduction or hydrogenation to formic acid in detail. CO2 hydrogenation to FA depends on the initial pressure of the reactant (H2 and CO2), solvent, etc. Basic conditions favour the formation of FA in high yield owing to the neutralization of the acid product. Further, the presence of basic sites on supported/heterogeneous catalysts induces initial interaction with CO2 for the synthesis of FA. Better catalytic performance could be achieved by tuning such interactions.
Some catalyst systems are active for both dehydrogenation of formic acid and hydrogenation of CO2 by varying the reaction conditions only, thus providing an opportunity for the charge/discharge of hydrogen fuel cells similar to other rechargeable batteries. Beller and others developed few rechargeable hydrogen batteries based on these reversible catalyst systems, as summarized in the third part.
Besides the several advantages, the formic acid-based hydrogen storage system has only 4.4 wt% hydrogen content which gets further decreased by addition of amine as well as solvent to around 1%. To meet practical applications, further work is needed for the development of low-cost, more efficient, reversible catalyst systems. Moreover, the development of photocatalytic systems for both processes (fixation and evolution of hydrogen to/from formic acid), which employ sustainable/renewable energy resources, is highly desirable.
Acknowledgements
The authors thank the reviewers for their valuable suggestions. AK thanks DST for financial support. AKS and SS acknowledge the UGC for the DSK Postdoctoral Fellowship. The authors are thankful to those who have worked in this area; their names can be found in the references section. AKS is grateful to Prof. Q. Xu, AIST, Ikeda, Osaka, for his generous support and guidance during his postdoc in Japan.Notes and references
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- U. B. Demirci and P. Miele, Energy Environ. Sci., 2011, 4, 3334–3341 CAS.
- B. Loges, A. Boddien, F. Gartner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS.
- P. Makowski, A. Thomas, P. Kuhn and F. Goettman, Energy Environ. Sci., 2009, 2, 480–490 CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 CAS.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 CAS.
- E. Fujita, J. T. Muckerman and Y. Himeda, Biochim. Biophys. Acta, 2013, 1827, 1031–1038 CrossRef CAS PubMed.
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 CAS.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 668–681 RSC.
- F. Amaseder and G. Krainz, Liquid hydrogen storage systems developed and manufactured for the first time for customer cars. In: SAE paper 2006-01-0432, 2006 Search PubMed.
- S. M. Aceves, F. Espinosa-Loza, E. Ledesma-Orozco, T. O. Ross, A. H. Weisberg, T. C. Brunner and O. Kircher, Int. J. Hydrogen Energy, 2010, 35, 1219–1226 CrossRef CAS.
- B. Sakintuna and Y. Yürüm, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef CAS.
- A. C. Dillon, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377–379 CrossRef CAS.
- A. D. Lueking, R. T. Yang, N. M. Rodríguez and R. T. K. Baker, Langmuir, 2004, 20, 714–721 CrossRef CAS PubMed.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 CAS.
- J. Weitkamp, M. Fritz and S. Ernst, Int. J. Hydrogen Energy, 1995, 20, 967–970 CrossRef CAS.
- A. Zuttle, Mater. Today, 2003, 6, 24–33 CrossRef.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. Seo, H. Zeng, I. L. Moudrakovskl, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743–746 CrossRef CAS PubMed.
- Y. H. Hu and E. Ruckenstein, Angew. Chem., Int. Ed., 2006, 45, 2011–2013 CrossRef CAS PubMed.
- J. L. C. Roswell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef CAS PubMed.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- D. R. Hoffman, Ant venoms, Curr. Opin. Allergy Clin. Immunol., 2010, 10, 342–346 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. M. Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- Y. Yasaka, H. Yoshida, C. Wakai, N. Matubayasi and M. Nakahara, J. Phys. Chem. A, 2006, 110, 11082–11090 CrossRef CAS PubMed.
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81–88 RSC.
- Office of Energy Efficiency and Renewable Energy; The Freedom CAR and Fuel Partnership, Targets For Onboard Hydrogen Storage Systems For Light-duty Vehicles, September 2009, http://www1.eere.energy.gov.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC.
- A. Behr and K. Nowakowski, Adv. Inorg. Chem., 2014, 66, 223–258 CrossRef CAS.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 CAS.
- D. Teichmann, W. Arlt and P. Wasserscheid, Int. J. Hydrogen Energy, 2012, 37, 18118–18132 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- A. Monney, E. Barsch, P. Sponholz, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2014, 50, 707–709 RSC.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- R. S. Coffey, Chem. Commun., 1967, 923–924 Search PubMed.
- S. H. Strauss, K. H. Whitmire and D. F. Shriver, J. Organomet. Chem., 1979, 174, C59–C62 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 3529–3530 CrossRef CAS.
- R. B. King and N. K. Bhattacharyya, Inorg. Chim. Acta, 1995, 237, 65–69 CrossRef CAS.
- J. H. Shin, D. G. Churchill and G. Parkin, J. Organomet. Chem., 2002, 642, 9–15 CrossRef CAS.
- Y. Gao, J. Kuncheria, G. P. A. Yap and R. J. Puddephatt, Chem. Commun., 1998, 2365–2366 RSC.
- Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt and G. P. A. Yap, J. Chem. Soc., Dalton Trans., 2000, 3212–3217 RSC.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS PubMed.
- A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS PubMed.
- H. Junge, A. Boddien, F. Capitta, B. Loges, J. R. Noyes, S. Gladiali and M. Beller, Tetrahedron Lett., 2009, 50, 1603–1606 CrossRef CAS.
- X. Li, X. Ma, F. Shi and Y. Deng, ChemSusChem, 2010, 3, 71–74 CrossRef CAS PubMed.
- X. Li, F. Shi, X. Ma, L. Lu and Y. Deng, J. Fuel Chem. Technol., 2010, 38, 544–553 CrossRef CAS.
- M. E. M. Berger, D. Assenbaum, N. Taccardi, E. Spiecker and P. Wasserscheid, Green Chem., 2011, 13, 1411–1415 RSC.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed.
- C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem. – Eur. J., 2009, 15, 3752–3760 CrossRef CAS PubMed.
- W. Gan, P. J. Dyson and G. Laurenczy, React. Kinet. Catal. Lett., 2009, 98, 205–213 CrossRef CAS.
- D. J. Morris, G. J. Clarkson and M. Wills, Organometallics, 2009, 28, 4133–4140 CrossRef CAS.
- A. Majewski, D. J. Morris, K. Kendall and M. Wills, ChemSusChem, 2010, 3, 431–434 CrossRef CAS PubMed.
- I. Mellone, M. Peruzzini, L. Rosi, D. Mellmann, H. Junge, M. Beller and L. Gonsalvi, Dalton Trans., 2013, 42, 2495–2501 RSC.
- G. Manca, I. Mellone, F. Bertini, M. Peruzzini, L. Rosi, D. Mellmann, H. Junge, M. Beller, A. Ienco and L. Gonsalvi, Organometallics, 2013, 32, 7053–7064 CrossRef CAS.
- S. Enthaler, H. Junge, A. Fischer, A. Kammer, S. Krackl and J. D. Epping, Polym. Chem., 2013, 4, 2741–2746 RSC.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS PubMed.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed.
- S. Oldenhof, B. de Bruin, M. Lutz, M. A. Siegler, F. W. Patureau, J. I. van der Vlugt and J. N. H. Reek, Chem. – Eur. J., 2013, 19, 11507–11511 CrossRef CAS PubMed.
- A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS PubMed.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1734 CrossRef CAS PubMed.
- D. Mellmann, E. Barsch, M. Bauer, K. Grabow, A. Boddien, A. Kammer, P. Sponholz, U. Bentrup, R. Jackstell, H. Junge, G. Laurenczy, R. Ludwig and M. Beller, Chem. – Eur. J., 2014, 20, 13589–13602 CrossRef CAS PubMed.
- T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- K. Hirota, K. Kuwata and Y. Nakai, Bull. Chem. Soc. Jpn., 1958, 31, 861–864 CrossRef CAS.
- D. H. S. Ying and R. J. Madix, J. Catal., 1980, 61, 48–56 CrossRef CAS.
- Y. K. Sun, J. J. Vajo, C. Y. Chan and W. H. Weinberg, J. Vac. Sci. Technol., A, 1988, 6, 854–855 Search PubMed.
- X. D. Peng and M. A. Barteau, Catal. Lett., 1991, 7, 395–402 CrossRef.
- N. Aas, Y. Li and M. Bowker, J. Phys.: Condens. Matter, 1991, 3, S281–S286 CrossRef CAS.
- P. A. Dilara and J. M. Vohs, J. Phys. Chem., 1993, 97, 12919–12923 CrossRef CAS.
- V. A. Gercher and D. F. Cox, Surf. Sci., 1994, 312, 106–114 CrossRef CAS.
- J. Stubenrauch, E. Brosha and J. M. Vohs, Catal. Today, 1996, 28, 431–444 CrossRef CAS.
- R. Larsson, M. H. Jamroz and M. A. Borowiak, J. Mol. Catal. A: Chem., 1998, 129, 41–51 CrossRef CAS.
- A. Bandara, J. Kubota and A. Wada, J. Phys. Chem. B, 1997, 101, 361–368 CrossRef CAS.
- J. Rasko, T. Kecskes and J. Kiss, J. Catal., 2004, 224, 261–268 CrossRef CAS.
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71–81 CrossRef CAS.
- G. Jacobs, P. M. Patterson, U. M. Graham, A. C. Crawford and B. H. Davis, Int. J. Hydrogen Energy, 2005, 30, 1265–1276 CrossRef CAS.
- G. Jacobs, P. M. Patterson, U. M. Graham, A. C. Crawford, A. Dozier and B. H. Davis, J. Catal., 2005, 235, 79–91 CrossRef CAS.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed.
- D. A. Bulusheva, S. Beloshapkin and J. R. H. Ross, Catal. Today, 2010, 154, 7–12 CrossRef.
- F. Solymosi, Á. Koós, N. Liliom and I. Ugrai, J. Catal., 2011, 279, 213–219 CrossRef CAS.
- G. Halasi, G. Schubert and F. Solymosi, Catal. Lett., 2011, 142, 218–223 CrossRef.
- D. A. Bulushev, L. Jia, S. Beloshapkin and J. R. H. Ross, Chem. Commun., 2012, 48, 4184–4186 RSC.
- X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 3540–3542 RSC.
- Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122–5128 CrossRef CAS.
- G. Glaspell, L. Fuoco and M. S. El-Shall, J. Phys. Chem. B, 2005, 109, 17350–17355 CrossRef CAS PubMed.
- W.-J. Shen and Y. Matsumura, J. Mol. Catal. A: Chem., 2000, 153, 165–168 CrossRef CAS.
- X. Zhou, Y. Huang, C. Liu, J. Liao, T. Lu and W. Xing, ChemSusChem, 2010, 3, 1379–1382 CrossRef CAS PubMed.
- S. W. Ting, S. Cheng, K. Y. Tsang, N. van der Laak and K. Y. Chan, Chem. Commun., 2009, 7333–7335 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- K. Tedsree, C. W. A. Chan, S. Jones, Q. Cuan, W. K. Li, X. Q. Gong and S. C. E. Tsang, Science, 2011, 332, 224–228 CrossRef CAS PubMed.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang, Y. Ping and Q. Jiang, Sci. Rep., 2012, 2, 598 Search PubMed.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang, Y. Ping and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12721–12725 CAS.
- Y. Ping, J.-M. Yan, Z.-L. Wang, H.-L. Wang and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12188–12191 CAS.
- Z.-L. Wang, J.-M. Yan, Y. Ping, H.-L. Wang, W.-T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- Z.-L. Wang, Y. Ping, J.-M. Yan, H.-L. Wang and Q. Jiang, Int. J. Hydrogen Energy, 2014, 39, 4850–4856 CrossRef CAS.
- Z.-L. Wang, H.-L. Wang, J.-M. Yan, Y. Ping, O. Song-Il, S.-J. Li and Q. Jiang, Chem. Commun., 2014, 50, 2732–2734 RSC.
- M. Yadav, A. K. Singh, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 19146–19150 RSC.
- Y.-Y. Cai, X.-H. Li, Y.-N. Zhang, X. Wei, K.-X. Wang and J.-S. Chen, Angew. Chem., Int. Ed., 2013, 52, 11822–11825 CrossRef CAS PubMed.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114–1119 CrossRef CAS.
- Q.-L. Zhu, N. Tsumori and Q. Xu, Chem. Sci., 2014, 5, 195–199 RSC.
- X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- H. Dai, N. Cao, L. Yang, J. Su, W. Luo and G. Cheng, J. Mater. Chem. A, 2014, 2, 11060–11064 CAS.
- M. Martis, K. Mori, K. Fujiwara, W.-S. Ahn and H. Yamashita, J. Phys. Chem. C, 2013, 117, 22805–22810 CAS.
- Y.-L. Qin, J. Wang, F.-Z. Meng, L.-M. Wang and X.-B. Zhang, Chem. Commun., 2013, 49, 10028–10030 RSC.
- M. Yadav, T. Akita, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 12582–12586 RSC.
- Q.-Y. Bi, X.-L. Du, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS PubMed.
- L. Jia, D. A. Bulushev, O. Y. Podyacheva, A. I. Boronin, L. S. Kibis, E. Y. Gerasimov, S. Beloshapkin, I. A. Seryak, Z. R. Ismagilov and J. R. H. Ross, J. Catal., 2013, 307, 94–102 CrossRef CAS.
- M. Yurderi, A. Bulut, M. Zahmakiran and M. Kaya, Appl. Catal., B, 2014, 160–161, 514–524 CrossRef CAS.
- K. Jiang, K. Xu, S.-Z. Zou and W.-B. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- J. H. Lee, J. Ryu, J. Y. Kim, S.-W. Nam, J. H. Han, T.-H. Lim, S. Gautam, K. H. Chae and C. W. Yoon, J. Mater. Chem. A, 2014, 2, 9490–9495 CAS.
- Q. Luo, G. Feng, M. Beller and H. Jiao, J. Phys. Chem. C, 2012, 116, 4149–4156 CAS.
- Q. Luo, T. Wang, M. Beller and H. Jiao, J. Mol. Catal. A: Chem., 2013, 379, 169–177 CrossRef CAS.
- Q. Luo, T. Wang, M. Beller and H. Jiao, J. Power Sources, 2014, 246, 548–555 CrossRef CAS.
- Q. Luo, M. Beller and H. Jiao, J. Theor. Comput. Chem., 2013, 12, 1330001 CrossRef.
- http://en.wikipedia.org/wiki/Formic_acid .
- A new study confirms that nature is responsible for 90% of the Earth's atmospheric acidity – http://wattsupwiththat.com.
- W. Reutemann and H. Kieczka, Formic Acid, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931–1934 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121–125 CrossRef CAS.
- J. Song, H. Fan, J. Ma and B. Han, Green Chem., 2013, 15, 2619–2635 RSC.
- N. Taccardi, D. Assenbaum, M. E. M. Berger, A. Bosmann, F. Enzenberger, R. Wolfel, S. Neuendorf, V. Goeke, N. Schodel, H. J. Maass, H. Kistenmacher and P. Wasserscheid, Green Chem., 2010, 12, 1150–1156 RSC.
- D. Yu, M. Aihara and J. Antal, Energy Fuels, 1993, 7, 574–577 CrossRef CAS.
- G. D. Mcginnis, S. E. Prince, C. J. Biermann and J. T. Lowrimore, Carbohydr. Res., 1984, 128, 51–60 CrossRef CAS.
- G. D. Mcginnis, W. W. Wilson and C. E. Mullen, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 352–357 CrossRef CAS.
- F. M. Jin, J. Yun, G. M. Li, A. Kishita, K. Tohji and H. J. Enomoto, Green Chem., 2008, 10, 612–615 RSC.
- R. Wölfel, N. Taccardi, A. Bösmann and P. Wasserscheid, Green Chem., 2011, 13, 2759–2763 RSC.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962 CAS.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Green Chem., 2014, 16, 226–237 RSC.
- J. Li, D.-J. Ding, L. Deng, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS PubMed.
- J. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82 CrossRef CAS.
- J. Xu, H. Zhang, Y. Zhao, Z. Yang, B. Yu, H. Xu and Z. Liu, Green Chem., 2014, 16, 4931–4935 RSC.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang and Y. Wang, ChemSusChem, 2014, 7, 1557–1567 CrossRef CAS PubMed.
- P. Gao, G. Li, F. Yang, X.-N. Lv, H. Fan, L. Meng and X.-Q. Yu, Ind. Crops Prod., 2013, 48, 61–67 CrossRef CAS.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Green Chem., 2008, 10, 612–615 RSC.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., B, 2015, 162, 1–10 CrossRef CAS.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, 2006 Search PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- W. Leitner, E. Dinjus and F. Gassner, in Aqueous-Phase Organometallic Catalysis, Concepts and Applications, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1998, pp. 486–498 Search PubMed.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- P. G. Jessop, Homogeneous hydrogenation of carbon dioxide, in Handbook of Homogeneous Hydrogenation, ed. J. G. De Vries and C. J., Elsevier, Wiley-VCH, Weinheim, 2007, vol. 1, pp. 489–511 Search PubMed.
- C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- Recent advances in transition metal-catalysed homogeneous hydrogenation of carbon dioxide in aqueous media in Hydrogenation, Karamé, ed. W.-H. Wang and Y. Himeda, InTech, 2012, ISBN: 978-953-51-0785-9 Search PubMed.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00197.
- Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 5, 863–864 CrossRef.
- X. Yang, ACS Catal., 2011, 1, 849–854 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- G. Zhao and F. Joó, Catal. Commun., 2011, 14, 74–76 CrossRef CAS.
- Y. Himeda, S. Miyazawa and T. Hirose, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 CAS.
- S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- K. Sordakis, M. Beller and G. Laurenczy, ChemCatChem, 2014, 6, 96–99 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CAS.
- J. L. Drake, C. M. Manna and J. A. Byers, Organometallics, 2013, 32, 6891–6894 CrossRef CAS.
- S.-F. Hsu, S. Rommel, P. Eversfield, K. Muller, E. Klemm, W. R. Thiel and B. Plietker, Angew. Chem., Int. Ed., 2014, 53, 7074–7078 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS.
- G. A. Filonenko, R. V. Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- G. O. Evans and C. J. Newell, Inorg. Chim. Acta, 1978, 31, L387–L389 CrossRef CAS.
- C.-C. Tai, T. Chang, B. Roller and P. G. Jessop, Inorg. Chem., 2003, 42, 7340–7341 CrossRef CAS PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado and N. Hazari, Chem. Sci., 2015, 6, 4291–4299 RSC.
- C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem. – Eur. J., 2012, 18, 72–75 CrossRef CAS PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS PubMed.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS.
- C. M. Zall, J. C. Linehan and A. M. Appel, ACS Catal., 2015, 5, 5301–5305 CrossRef CAS.
- B. Jezowska-Trzebiatowska and P. Sobota, J. Organomet. Chem., 1974, 80, C27–C28 CrossRef CAS.
- Y. Musashi and S. Sakaki, J. Am. Chem. Soc., 2000, 122, 3867–3877 CrossRef CAS.
- A. Urakawa, M. Iannuzzi, J. Hutter and A. Baiker, Chem. – Eur. J., 2007, 13, 6828–6840 CrossRef CAS PubMed.
- C. Bo and A. Dedieu, Inorg. Chem., 1989, 28, 304–309 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- H. H. Karsch, Chem. Ber., 1977, 110, 2213–2221 CrossRef CAS.
- M. W. Farlow and H. Adkins, J. Am. Chem. Soc., 1935, 57, 2222–2223 CrossRef CAS.
- H. Wiener, J. Blum, H. Feilchenfeld, Y. Sasson and N. Zalmanov, J. Catal., 1988, 110, 184–190 CrossRef CAS.
- D. Preti, C. Resta, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2011, 50, 12551–12554 CrossRef CAS PubMed.
- C. Hao, S. Wang, M. Li, L. Kang and X. Ma, Catal. Today, 2011, 160, 184–190 CrossRef CAS.
- D. Preti, S. Squarcialupi and G. Fachinetti, ChemCatChem, 2012, 4, 469–471 CrossRef CAS.
- N. Liu, J. Lei, M. Y. Li and P. Wang, Adv. Mater. Res., 2014, 283, 881–883 Search PubMed.
- G. Peng, S. J. Sibener, G. C. Schatz and M. Mavrikakis, Surf. Sci., 2012, 606, 1050–1055 CrossRef CAS.
- Z. Xu, N. D. McNamara, G. T. Neumann, W. F. Schneider and J. C. Hicks, ChemCatChem, 2013, 5, 1769–1771 CrossRef CAS.
- T. Maihom, S. Wannakao, B. Boekfa and J. Limtrakul, J. Phys. Chem. C, 2013, 117, 17650–17658 CAS.
- Y. Zhang, J. Fei, Y. Yu and X. Zheng, Catal. Commun., 2004, 5, 643–646 CrossRef CAS.
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Zhiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS PubMed.
- Y. Amao, ChemCatChem, 2011, 3, 458–474 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Grazel, Nature, 1987, 327, 506–508 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- B. Aurian-Blajeni, M. Halmann and J. Manassen, J. Sol. Energy, 1980, 25, 165–170 CrossRef CAS.
- D. Mandler and I. Willner, Org. Biomol. Chem., 1988, 2, 997–1003 Search PubMed.
- I. Willner and D. Mandler, J. Am. Chem. Soc., 1989, 111, 1330–1336 CrossRef CAS.
- I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy and E. Katz, J. Am. Chem. Soc., 1994, 116, 1428–1441 CrossRef CAS.
- I. Willner, I. Willner and N. Lapidot, J. Am. Chem. Soc., 1990, 112, 6438–6439 CrossRef CAS.
- K. Schuchmann and V. Müller, Science, 2013, 342, 1382–1385 CrossRef CAS PubMed.
- A. Alissandratos, H.-K. Kim and C. J. Easton, Bioresour. Technol., 2014, 164, 7–11 CrossRef CAS PubMed.
- M. Kodaka and Y. Kubota, Org. Biomol. Chem., 1999, 2, 891–894 Search PubMed.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931–1934 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121–125 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Jpn. Pet. Inst., 2004, 47, 27–31 CrossRef CAS.
- I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173–176 CrossRef CAS.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664–12666 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800–1807 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- H. Arakawa, M. Aresta, J. Armor, M. Barteau, E. Beckman, A. Bell, J. Bercaw, C. Creutz, D. A. Dixon, D. Dixon, K. Domen, D. DuBois, J. Eckert, E. Fujita, D. Gibson, W. Goddard, D. Goodman, J. Keller, G. Kubas, H. Kung, J. Lyons, L. Manzer, T. Marks, K. Morokuma, K. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. Sachtler, L. Schmidt, A. Sen, G. Somorjai, P. Stair, B. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- F. A. de Bruijn, D. C. Papageorgopoulos, E. F. Sitters and G. J. M. Janssen, J. Power Sources, 2002, 110, 117–124 CrossRef CAS.
- R. K. Ahluwalia and X. Wang, J. Power Sources, 2008, 180, 122–131 CrossRef CAS.
- A. E. Russell, S. C. Ball, S. Maniguet and D. Thompsett, J. Power Sources, 2007, 171, 72–78 CrossRef CAS.
- S. Giddey, F. Ciacchi and S. Badwal, Ionics, 2005, 11, 1 CrossRef CAS.
- T. Tingelöf, L. Hedström, N. Holmström, P. Alvfors and G. Lindbergh, Int. J. Hydrogen Energy, 2008, 33, 2064–2072 CrossRef.
- J. Larminie and A. Dicks, Fuel cell systems explained, Wiley, Chichester, 2003 Search PubMed.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- R. Williams, R. S. Crandall and A. Bloom, Appl. Phys. Lett., 1978, 33, 381–383 CrossRef CAS.
- M. Halmann, M. Ulman and B. Aurian-Blajeni, J. Sol. Energy, 1983, 31, 429–431 CrossRef CAS.
- H. Wiener, Y. Sasson and J. Blum, J. Mol. Catal., 1986, 35, 277–284 CrossRef CAS.
- B. Zaidman, H. Wiener and Y. Sasson, Int. J. Hydrogen Energy, 1986, 11, 341–347 CrossRef CAS.
- H. Wiener, B. Zaidman and Y. Sasson, J. Sol. Energy, 1989, 43, 291–296 CrossRef CAS.
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed.
- F. Joo, ChemCatChem, 2014, 6, 3306–3308 CAS.
- S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351–1362 CrossRef CAS.
- Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 CAS.
- P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172–1176 CrossRef CAS PubMed.
- A. Boddien, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge, G. Laurenczy and M. Beller, Energy Environ. Sci., 2012, 5, 8907–8911 CAS.
- S.-F. Hsu, S. Rommel, P. Eversfield, K. Muller, E. Klemm, W. R. Thiel and B. Plietker, Angew. Chem., Int. Ed., 2014, 53, 7074–7078 CrossRef CAS PubMed.
- A. Boddien, F. Gärtner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS PubMed.
- Q.-Y. Bi, J.-D. Lin, Y.-M. Liu, X.-L. Du, J.-Q. Wang, H.-Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 13583–13587 CrossRef CAS PubMed.
- J. Su, L. Yang, M. Lu and H. Lin, ChemSusChem, 2015, 8, 813–816 CrossRef CAS PubMed.
- C. Chauvier, A. Tlili, C. D. N. Gomes, P. Thuéry and T. Cantat, Chem. Sci., 2015, 6, 2938–2942 RSC.
| This journal is © The Royal Society of Chemistry 2016 |