
A novel colorimetric and near-infrared fluorescent probe for hydrogen peroxide imaging in vitro and in vivo†
Peng Wang*,
Ke Wang,
Dan Chen,
Yibo Mao and
Yueqing Gu*
Department of Biomedical Engineering, School of Engineering, China Pharmaceutical University, 210009 Nanjing, China. E-mail: wangpeng159seu@hotmail.com
First published on 30th September 2015
Abstract
A novel NIR fluorescent probe (DCM-B2) based on dicyanomethylene-4H-pyran was synthesized for the detection of H2O2. This colorimetric fluorescent probe displays a fluorescence turn-on response following the conversion of the aryl boronate unit to phenol in the presence of H2O2. It could offer good performance in terms of sensitivity, selectivity, and low cytotoxicity. Furthermore, bioimaging investigations indicated that this probe was cell permeable and suitable for monitoring H2O2 in vitro and in vivo.
Introduction
To date, reactive oxygen species (ROS) have received considerable attention. ROS as a diverse group of small molecules with different reactivity, sources of production, and, ultimately, biological functions, are important contributors to the pathogenesis of major chronic diseases including cancer, diabetes and atherosclerosis. Moreover, some of these molecules play major roles in environmental, radiation and space biology.1–3Among different ROS, the role of H2O2 as a second messenger, in regulating fundamental biological processes, has been identified not long ago and is increasingly supported by new data.4 H2O2 is a non-polar molecule, which can diffuse relatively readily across biological membranes and exert its effect in multiple cellular compartments. Due to its low reactivity, H2O2 also has a relatively long half-life, a feature necessary to carry out long-distance effects across the cell. Escalated levels of H2O2 could be highly harmful, causing oxidative stress through the oxidation of biomolecules, and leading to cellular damage that may become irreversible and cause cell death. It has been reported that when cells are dysfunctional, H2O2 can accumulate and cause oxidative damage to cellular proteins, nucleic acids, and lipid molecules, thereby leading to aging and age-related diseases ranging from neurodegeneration to diabetes.5–7
Taking advantage of their high sensitivity, noninvasiveness and high spatiotemporal resolution for visualizing bioactive species in a biological system, fluorescent probes are the preferred methods to elucidate the mechanisms of these species. Fluorescent H2O2 probes, designed to detect this oxygen metabolite with high selectivity, are powerful tools for real-time, noninvasive monitoring of H2O2 chemistry in biological specimens.
Recently, a few metal-mediated fluorescent probes have been developed for the detection of H2O2.8 However, the above-mentioned fluorescent probes are not applicable for use in biological systems due to a slow response time or incompatibility with biological milieus. Compared to most other conventional fluorescent probes, those that rely on near-infrared (NIR) fluorescence possess unique advantages for tracing molecular processes in vitro and in vivo.9 NIR photons can penetrate tissue more deeply and avoid background noise. Therefore, it would be desirable to utilize NIR fluorophores as the signaling subunit in probes. At present, most of the fluorophores used for H2O2 probes, such as xanthenones,10 naphthalenes,11 and Peroxy Crimson,12 suffer from short wavelength emission. There is still a lack of probes for detecting H2O2 in vivo and in situ, which is most probably ascribed to the poor photostability of fluorophores. Moreover, the other challenges to detect H2O2 using fluorescence probes include chemoselectivity, selectivity of H2O2 over other ROS, and bioorthogonality, i.e. not interfering with intrinsic cellular biochemistry.
To solve the above problems, we invested effort into developing novel NIR fluorescent probes for H2O2 detection. As donor–π–acceptor (D–π–A) structured chromophores, dicyanomethylene-4H-pyran (DCM) derivatives have attracted considerable attention owing to their attractive features such as controllable emission wavelength in the NIR region via the tuning electron donor ability, large Stokes shift from the ultrafast intramolecular charge transfer (ICT), and high photostability.13–15 In addition, some studies established that H2O2 can react with aryl boronates to achieve selectivity over other ROS. And the reaction of H2O2 with boronates is faster than those of the corresponding alkyl peroxides, making the reaction of free H2O2 selective over lipid-derived peroxides.16 This reaction may provide a promising opportunity for H2O2 detection chemically.
In the current study, novel NIR probes based on DCM were designed for the visualization of H2O2 in cells and in vivo. As shown in Scheme 1, DCM-B1 was constructed by introducing a boronate-based self-immolative linker to DCM-OH, which has been successfully utilized in cell imaging.17 Meanwhile DCM-B2 was designed by installation of a boronic ester group at the 4′-position of the benzene ring, which can react with H2O2 to release DCM-OH.
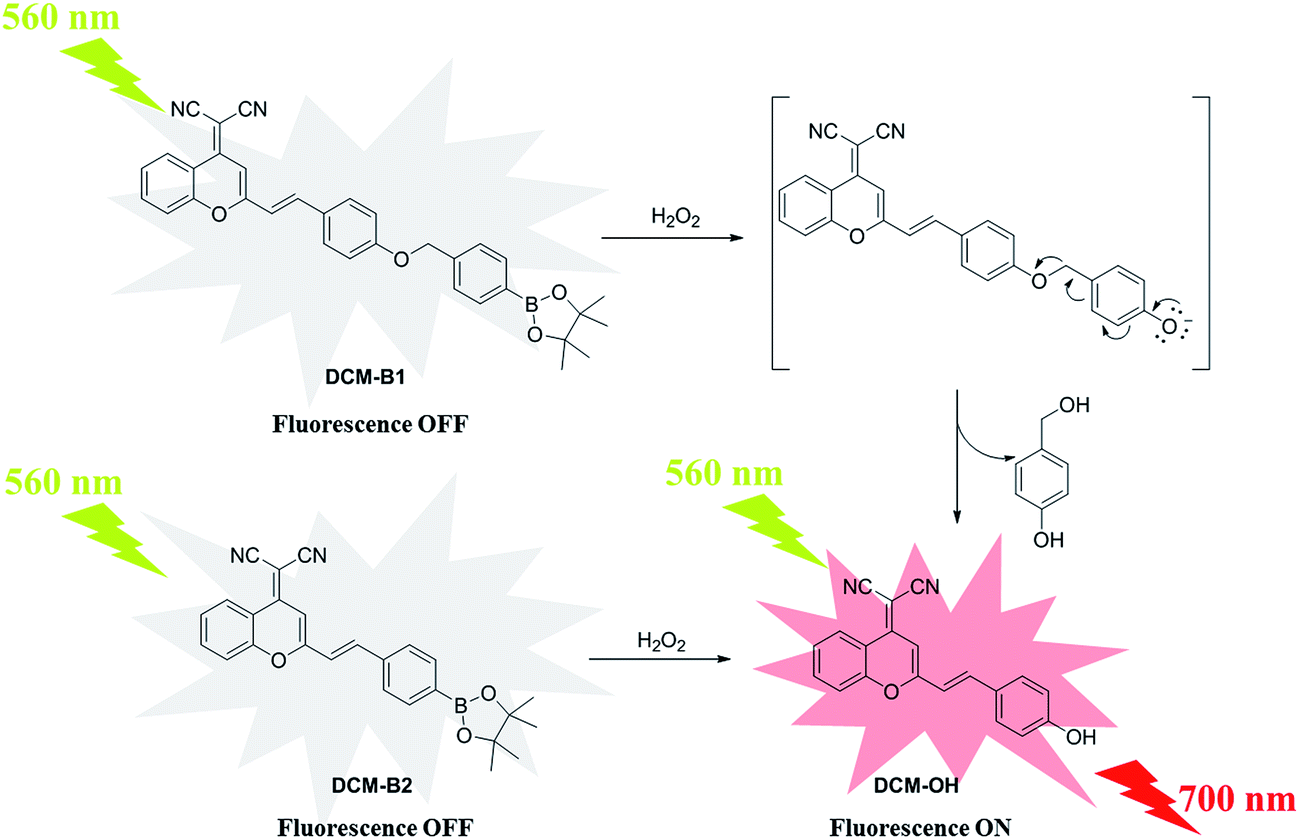 | ||
| Scheme 1 The structure and reaction mechanism of the probes DCM-B1 and DCM-B2. | ||
Experimental
General
All solvents and other reagents were of commercial quality and used without further purification. A UV-vis spectrophotometer (JH 754PC, Shanghai, China) was used for the absorption measurements. A PerkinElmer LS55 was utilized for fluorescence spectra detection. Laser confocal fluorescence microscopy (FluoView™, FV1000, Olympus, Japan) was used for cell imaging. IR spectra were measured using a Bruker Tensor-27 FRIR spectrometer using a KBr pellet. 1H-NMR and 13C-NMR spectra were taken on a Bruker Advance 300 MHz spectrometer where δ values are in ppm relative to TMS. Mass data (ESI) were recorded using quadruple mass spectrometry. For the H2O2 selectivity experiments, H2O2, TBHP and hypochlorite (NaClO) were delivered in 30%, 70% and 5% aqueous solutions, respectively. Superoxide (O2−) was added as solid KO2. HO˙ and t-BuO˙ were generated by the reaction of 1 mM Fe2+ with 100 mM H2O2 or 100 mM tert-butyl hydroperoxide (TBHP), respectively. NO was added using NO gas. Singlet oxygen (1O2) was generated from the thermodissociable endoperoxide of disodium 3,3′-(1,4-naphthalene)bispropionate.Synthesis of DCM-OH
2-(2-Methyl-4H-chromen-4-ylidene)malononitrile (1.0 mmol) and 4-hydroxybenzaldehyde (1.0 mmol) were dissolved in 10 mL anhydrous ethyl alcohol. Then piperidine (4.0 mmol) was added and heated to reflux for 5 h. The mixture was cooled to room temperature and filtered to obtain red solid DCM-OH in 80% yield. 1H-NMR (300 MHz, DMSO-d6): −δ 10.12 (s, 1H), 8.66 (d, J = 7.4 Hz, 1H), 7.86 (s, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.57 (m, 4H), 7.18 (d, J = 15.9 Hz, 1H), 6.86 (m, 3H). 13C-NMR (75 MHz, DMSO-d6): δ 159.9, 158.7, 152.6, 151.9, 139.1, 135.1, 130.3, 126.0, 125.9, 124.5, 118.9, 117.3, 117.0, 116.0, 115.8, 105.6, 59.1. ESI-MS: 311.0 [M − H]−.Synthesis of DCM-B1
The compound 2-(4-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.1 mmol) and DCM-OH (1.0 mmol) were dissolved in 10 mL CH3CN. Then K2CO3 (2.0 mmol) was added under nitrogen atmosphere, and was heated to 70 °C for 3 h. After cooling, the solid was removed by filtration and washed with CH3CN. The solution was concentrated on a rotary evaporator. The resultant crude material was recrystallized by ethyl alcohol to afford the compound DCM-B1 in 70% yield. 1H-NMR (300 MHz, DMSO-d6): δ 8.72 (d, J = 8.2 Hz, 1H), 7.91 (t, J = 7.8 Hz, 1H), 7.78–7.67 (m, 6H), 7.59 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 7.6 Hz, 2H), 7.34 (d, J = 16.0 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.96 (s, 1H), 5.22 (s, 2H), 1.30 (s, 12H). 13C-NMR (75 MHz, DMSO-d6): δ 160.1, 158.5, 152.8, 151.9, 140.0, 138.5, 135.3, 134.5, 130.0, 127.8, 126.8, 126.0, 124.6, 119.0, 117.2, 117.2, 117.1, 115.9, 115.4, 106.0, 83.6, 69.2, 59.5, 24.6. IR (KBr): 2974, 2211, 2198, 1628, 1592, 1501, 1478, 1410, 1357, 1321, 1211, 1170, 1140, 1087, 977, 854, 813, 742, 651 cm−1.Synthesis of DCM-B2
2-(2-Methyl-4H-chromen-4-ylidene)malononitrile (1.0 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (1.0 mmol) were dissolved in 10 mL anhydrous ethyl alcohol. Then piperidine (4.0 mmol) was added and heated to reflux for 5 h. The mixture was cooled to room temperature and filtered to obtain yellow solid DCM-B2 in 78% yield. 1H-NMR (300 MHz, CDCl3): δ 8.93 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.75 (t, J = 7.8 Hz, 1H), 7.62 (m, 4H), 7.46 (t, J = 7.7 Hz, 1H), 6.90 (t, J = 8.0 Hz, 2H), 1.36 (s, 12H). 13C-NMR (300 MHz, DMSO-d6): δ 157.5, 152.6, 151.8, 137.8, 137.4, 135.2, 134.7, 127.2, 125.9, 124.5, 120.7, 118.8, 116.8, 116.7, 115.4, 107.0, 83.6, 24.5. IR (KBr): 3067, 3028, 2975, 2209, 1634, 1500, 1460, 1327, 1262, 1089, 1016, 765, 740, 651, 618 cm−1.Cell culture and confocal fluorescence imaging
The human cell line MCF-7 (breast cancer cells) was purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Hyclone), 100 μg mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. One day before imaging, cells were seeded in laser scanning confocal microscope (LSCM) culture dishes at a density of 5 × 105 cells per well. The dishes were subsequently incubated at 37 °C in a humidified atmosphere containing 5% CO2. Then the cells were incubated with 10 μM DCM-B2 for 30 min. Subsequently, 100 μM H2O2 was added and incubated at 37 °C for 30 min. The cells were washed three times with Dulbecco’s PBS (pH 7.0) to remove free compound before analysis. MCF-7 cells only incubated with 10 μM DCM-B2 for 30 min acted as a control. Confocal luminescence images of MCF-7 cells were carried out on an Olympus FV1000 laser scanning confocal microscope.Cytotoxic assay
MCF-7 cells were seeded in a 96-well plate (1 × 104 cells per well). After cultivation for 24 h, DCM-B2 (it was dissolved in DMSO first, then it was added into the cell culture medium) of different concentrations was added into the wells (n = 6) and incubated for 48 h. Then a stock solution of MTT (20 μL; 5 mg mL−1) was added into each well. After a 4 h incubation at 37 °C, the MTT solution was replaced with 150 μL DMSO in each well. The absorbance in each well was measured at 570 nm with a multi-well plate reader. Cell viability was calculated using the following formula: cell viability = (mean absorbance of test wells − mean absorbance of medium control wells)/(mean absorbance of untreated wells − mean absorbance of medium control well) × 100%.Fluorescence imaging in living mice
Athymic nude mice were purchased from Charles River Laboratories (Shanghai, China) for in vivo imaging investigation. All animal experiments were carried out in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China (Document no. 55, 2001) and the guidelines for the Care and Use of Laboratory Animals of China Pharmaceutical University. Athymic nude mice, 5–10 g, were selected and divided into two groups. The mice were given an s.p. (skin-pop) injection of the probe DCM-B2 (100 μM, in DMSO/saline = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v) in the back. Then one group of mice were injected with 2 mM hydrogen peroxide at the same region. The other group was given saline as the control. Images were taken after incubation for 30 min by using a NIR fluorescence imaging system. This home-built imaging system was reported in our previous works.18–20 The NIR system contains an excitation laser (λ = 660 nm), a high sensitivity NIR CCD camera (PIXIS 512B, Princeton Instrumentation) and a 700 nm long pass filter for capturing fluorescence emission from the tissue.
:9, v/v) in the back. Then one group of mice were injected with 2 mM hydrogen peroxide at the same region. The other group was given saline as the control. Images were taken after incubation for 30 min by using a NIR fluorescence imaging system. This home-built imaging system was reported in our previous works.18–20 The NIR system contains an excitation laser (λ = 660 nm), a high sensitivity NIR CCD camera (PIXIS 512B, Princeton Instrumentation) and a 700 nm long pass filter for capturing fluorescence emission from the tissue.
Results and discussion
Synthesis of DCM-B1 and DCM-B2
According to the synthetic route shown in Scheme 2, DCM-B1 was prepared in two reaction steps and was obtained as a yellow solid in a reasonable yield.21,22 Similarly, DCM-B2 was obtained in good yield by the reaction between 2-(2-methyl-4H-chromen-4-ylidene)malononitrile and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehydes. The structures of DCM-B1 and DCM-B2 were characterized using NMR and mass spectrometry from which satisfactory results corresponding to its structure were obtained. Both of them are soluble in common organic solvents such as DMSO, acetonitrile and dichloromethane, but slightly soluble in water.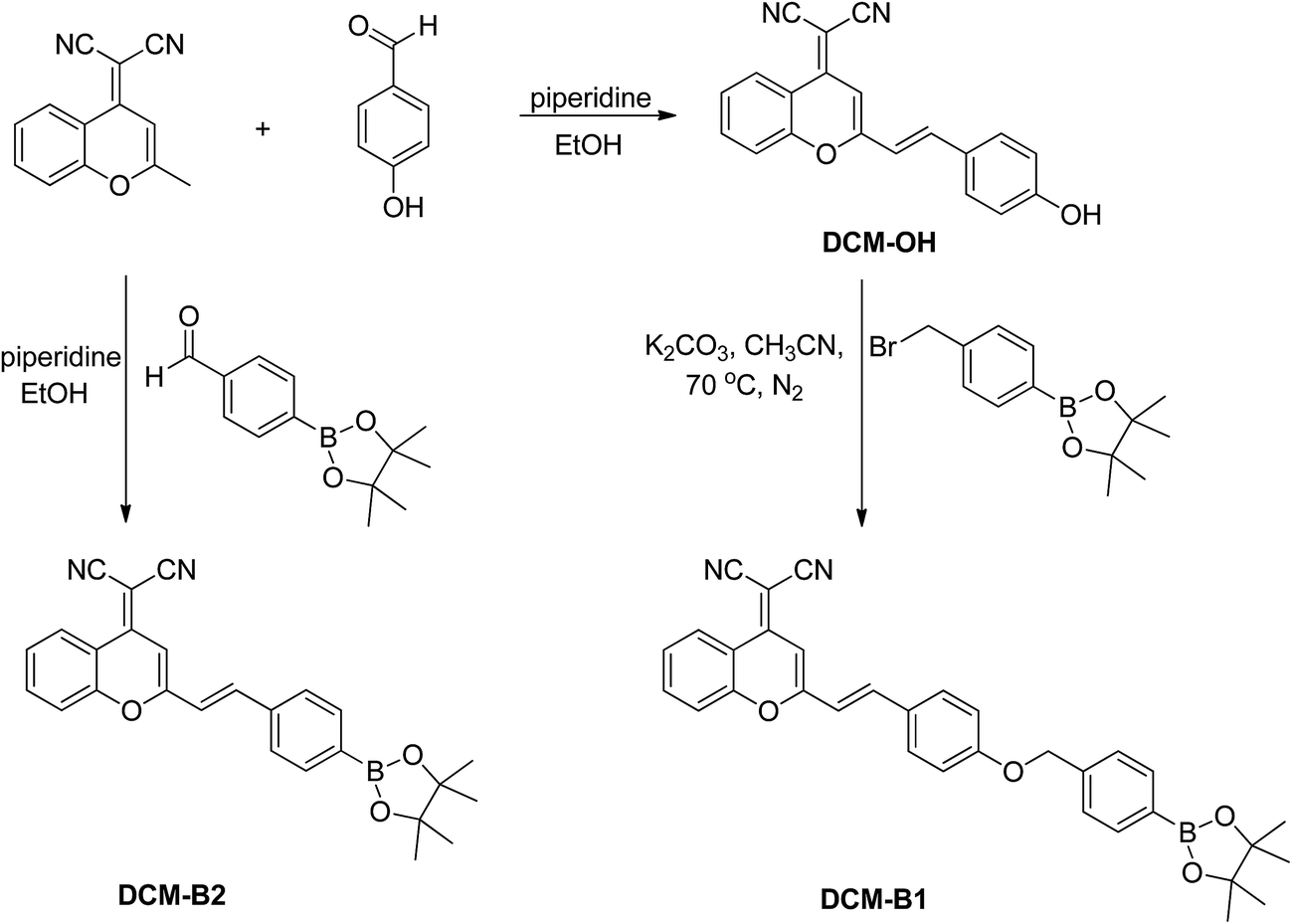 | ||
| Scheme 2 The synthetic route of probes DCM-B1 and DCM-B2. | ||
UV-vis and fluorescence responses
We evaluated the optical properties of DCM-B1 and DCM-B2 in PBS buffer (20 mM, 50% DMSO, pH 7.4). DCM-B1 features one prominent absorption band in the visible region centered at 450 nm and one weak absorption band at around 550 nm (Fig. S1†). DCM-B1 shows weak fluorescence with an emission maximum at 560 nm. Similarly, DCM-B2 has two absorption maxima at around 420 nm and 450 nm, with a corresponding weak emission band centered at 560 nm (Fig. 1). After treatment of DCM-B1 and DCM-B2 with H2O2 for 30 min, a new absorption peak appeared at 560 nm for these two probes. Notably, the red-shift of 110 nm in the absorption is very large with respect to that of other H2O2 chemosensors, indicating the capacity of DCM-B1 and DCM-B2 for colorimetric detection of H2O2 even with the naked eye. | ||
| Fig. 1 Absorption spectra of the probe DCM-B2 (5 μM) before (blue line) and after reacting with H2O2 (100 μM, red line). | ||
Probe DCM-B1 and DCM-B2 showed almost no fluorescence emission upon excitation at 560 nm. Addition of H2O2 resulted in a marked increase in red fluorescence for DCM-B1 and DCM-B2 (Fig. 2a and S2†). With increasing concentrations of H2O2, the fluorescence titration curve showed a steady and smooth enhancement. Reaction of DCM-B1 with H2O2 triggered an almost 30-fold fluorescence turn-on, whereas H2O2 elicited a 50-fold increase in fluorescence for DCM-B2. Absorption and emission spectra, along with mass spectrometry data, establish that the H2O2-mediated boronate deprotection of DCM-B1 and DCM-B2 generate DCM-OH as the fluorescent product. Upon addition of H2O2, the fluorescence intensity of DCM-B1 and DCM-B2 enhanced apparently with a maximum at 700 nm, indicating both of these two probes were suitable for application in cells and in vivo. These results indicated that DCM-B1 and DCM-B2 were turn-on type fluorescent probes for H2O2 detection.
 | ||
| Fig. 2 (a) Emission spectra of probe DCM-B1 in the presence of different equivalents of H2O2 (0, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, 10.0, 15.0, and 20.0 eq., 30 min) excited at 560 nm; (b) a linear correlation between emission intensity and concentration of H2O2. | ||
To further confirm the sensitivity of the probe, the fluorescence of the probes was measured by adding diverse concentrations of H2O2. Fig. 2b suggested that the fluorescence intensity of probe DCM-B1 and DCM-B2 increased following the increased concentration of H2O2 within a certain range. There was a good linearity between relative fluorescent intensity at 700 nm and the concentration of H2O2 ranging from 0–100 μM (Fig. 2b and S3†). The regression equation was F700 nm = 4.1993 [H2O2] (μM) + 26.319 (r = 0.9959). The detection limit of H2O2 was calculated from the equation DL = 3σ/S, where σ is the standard deviation of a blank measurement and S is the slope between intensity versus sample concentration.23 The detection limit of DCM-B1 toward H2O2 was calculated to be 7.9 × 10−8 M, suggesting that DCM-B1 was highly sensitive to H2O2. Similarly, the regression equation of DCM-B2 was F700 nm = 4.7851 [H2O2] (μM) + 21.624 (r = 0.9937). The detection limit of DCM-B2 for H2O2 was 3.9 × 10−8 M, indicating that DCM-B2 had greater sensitivity than DCM-B1. These results indicated that the probe DCM-B2 can be used to quantify the concentration of H2O2.
The time-dependent fluorescence changes of DCM-B1 and DCM-B2 were also investigated (Fig. 3). As shown in Fig. 3c, the response time of DCM-B2 was shorter than that of DCM-B1, which indicated that the reaction rate between DCM-B2 and H2O2 was faster than DCM-B1. It might be due to the attack of the boronic ester group of DCM-B2 by H2O2 to release DCM-OH directly, while DCM-B1 needs a self-immolative process.
 | ||
| Fig. 3 (a) The change in the emission spectra of DCM-B1 (5 μM) with time after addition of 100 μM H2O2; (b) the change in the emission spectra of DCM-B2 (5 μM) with time after addition of 100 μM H2O2; (c) time-dependent fluorescence changes of DCM-B1 and DCM-B2 (5 μM) upon addition of H2O2 (100 μM). | ||
H2O2 selectivity
For an excellent probe, high selectivity is a very important parameter. We next examined the selectivity of DCM-B1 and DCM-B2 towards H2O2 across a series of other related reactive oxygen species in biological systems. DCM-B1 and DCM-B2 were respectively incubated with various biologically relevant species including the hydroxyl radical (OH˙), t-butoxy radical (t-BuO˙), superoxide anion (O2−), t-butyl hydroperoxide (TBHP), hypochlorite (ClO−), singlet oxygen (1O2), and nitric oxide (NO). As a result, only H2O2 induced a remarkable fluorescence enhancement with an excitation at 560 nm. Fig. 4 shows that the addition of H2O2 presented a 30-fold increase in fluorescence intensity compared to the control at 30 min. However, there was little to no increase in intensity when the probe reacted with other ROS for over an hour. These results indicated that DCM-B2 has reasonable activity and selectivity to identify H2O2 in a complex biological environment. Although DCM-B1 also has good selectivity to identify H2O2 (Fig. S4†), DCM-B2 has a better detection limit, faster reaction rate, and lower background noise than DCM-B1. Hence, the following experiments were focused on the probe DCM-B2 for visualization of H2O2 in cells and in vivo. | ||
| Fig. 4 Fluorescence intensity of 5 μM DCM-B2 in response to the test species in a PBS buffer solution (20 mM, 50% DMSO, pH 7.4) at 700 nm excited at 560 nm. Bars represent fluorescence intensity 0, 10, 30 and 60 min after addition of various compounds excited at 560 nm. | ||
Fluorescence imaging and cytotoxicity assay
Next, we investigated the ability of DCM-B2 to detect H2O2 in living cells. Initially, MCF-7 cells were incubated with 10 μM DCM-B2 for 30 min at 37 °C. With the control, the cells exhibited almost no fluorescence (Fig. 5a). Bright-field measurements after treatment with DCM-B2 confirmed that the cells were viable throughout the imaging experiment (Fig. 5b and c). In contrast, when incubated with 100 μM H2O2 for 30 min after treatment with DCM-B2, the cells displayed obvious fluorescence (Fig. 5e). An overlay of the confocal fluorescence and bright-field images demonstrated that fluorescence was evident (Fig. 5f). Therefore, these results indicated that DCM-B2 is cell membrane-permeable and available for detection of H2O2 in living cells. | ||
| Fig. 5 Fluorescence and bright-field images of MCF-7 cells. (a) Bright-field image of cells incubated with DCM-B2 (10 μM) for 30 min; (b) fluorescence image of cells shown in panel (a); (c) overlay image of (a) and (b); (d) bright-field image of cells pretreated with DCM-B2 (10 μM) and then incubated with H2O2 (μM) for 30 min; (e) fluorescence image of cells shown in panel (d); (f) overlay image of (d) and (e). Excitation was provided at 568 nm. | ||
Moreover, the cytotoxicity of DCM-B1 was evaluated using a cell viability assay. MCF-7 cells were treated with different concentrations of DCM-B1 (from 10 to 100 μM) for 24 h, and then cell viability was evaluated using a MTT assay. The results showed that cell viability was over 85% even though 100 μM DCM-B2 was added for 24 h, indicating that the fluorescence probe DCM-B2 had low cytotoxicity (Fig. S5†). The above experimental results proved that DCM-B2 could offer a good performance in terms of sensitivity, selectivity, and low cytotoxicity.
Fluorescent imaging in vivo
We further applied DCM-B2 for H2O2 imaging in nude mice. The backs of mice were injected with 100 μM DCM-B2 (100 μL in 1:9 DMSO/saline v/v), and 10 min later, they were injected with 2 mM H2O2 (100 μL in saline) in the same region. After 30 min, there was a remarkable increase in fluorescence in the injection region (Fig. 6b). As a control, the other group of mice were only injected with 100 μM DCM-B2 (100 μL in 1:9 DMSO/saline, v/v) and imaged after 40 min. The control showed only slight fluorescence (Fig. 6a). These results showed that the NIR fluorescent probe DCM-B2 could be successfully applied for deep imaging in live mice and effectively avoid organisms’ autofluorescence. The quantification of the mean fluorescence intensity of each group is shown in Fig. 6c. It is noteworthy that the total number of photons from the region of interest was 3-fold that of the control. The above experiments demonstrated that DCM-B2 achieved noninvasive imaging in living mice and was sensitive enough to visualize H2O2 in living animals.
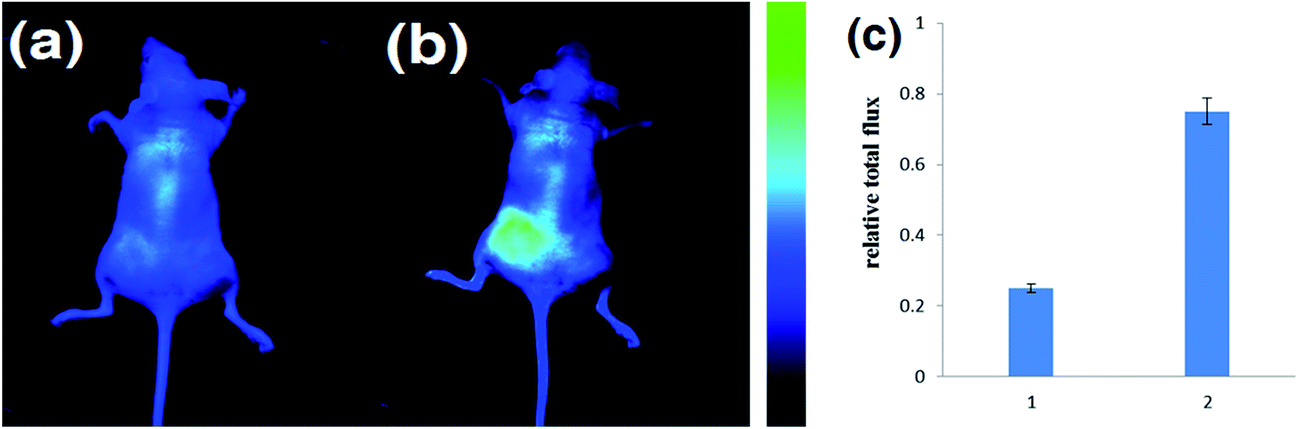 | ||
| Fig. 6 Fluorescence imaging of exogenous H2O2 activity using DCM-B2 in nude mice. (a) Mice injected s.p. with DCM-B2 (100 μL in 1:9 DMSO/saline v/v) for 40 min; (b) mice injected s.p. with DCM-B2 (100 μL in 1:9 DMSO/saline v/v), and then loaded with H2O2 (2 mM, 100 μL in saline) for 30 min; (c) quantification of the total photon flux from the region of interest for each group. Images constructed from a 700 nm fluorescence window, λex = 660 nm. | ||
Conclusions
In summary, novel NIR fluorescent probes DCM-B1 and DCM-B2 were designed and synthesized. These probes can detect H2O2 with a fluorescence turn-on effect, and the detection limits are 7.9 × 10−8 M and 3.9 × 10−8 M, respectively. Moreover, DCM-B2 exhibits high selectivity, good sensitivity and low cytotoxicity in the detection of H2O2. We confirm that DCM-B2 can detect H2O2 in mice without interference from background fluorescence. The probe DCM-B2 may present a promising tool to detect H2O2 during physiological and pathological processes.Acknowledgements
We are grateful for the financial support from the NSFC (National Nature Science Foundation of China, No. 81501529) and Fundamental Research Funds for the Central Universities (ZJ15011).Notes and references
- S. V. Chetyrkin, M. E. Mathis, A. J. L. Ham, D. L. Hachey, B. G. Hudson and P. A. Voziyan, J. Free Radicals Biol. Med., 2008, 44, 1276–1285 CrossRef CAS PubMed.
- M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. L. Qian, J. S. Lam, L. E. Ailles, M. Z. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman and M. F. Clarke, Nature, 2009, 458, 780-U123 CrossRef PubMed.
- P. D. Ray, B. W. Huang and Y. Tsuji, Cell Signal, 2012, 24, 981–990 CrossRef CAS PubMed.
- H. C. Guo, H. Aleyasin, B. C. Dickinson, R. E. Haskew-Layton and R. R. Ratan, Cell Biosci., 2014, 4, 64 CrossRef PubMed.
- Y. H. A. Liu and X. B. Liao, Curr. Org. Chem., 2013, 17, 654–669 CrossRef CAS.
- T. Finkel, M. Serrano and M. A. Blasco, Nature, 2007, 448, 767–774 CrossRef CAS PubMed.
- J. B. Pi, Y. S. Bai, Q. Zhang, V. Wong, L. M. Floering, K. Daniel, J. M. Reece, J. T. Deeney, M. E. Andersen, B. E. Corkey and S. Collins, Diabetes, 2007, 56, 1783–1791 CrossRef CAS PubMed.
- D. Song, J. M. Lim, S. Cho, S. J. Park, J. Cho, D. Kang, S. G. Rhee, Y. You and W. Nam, Chem. Commun., 2012, 48, 5449–5451 RSC.
- Z. Q. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS PubMed.
- C. Chung, D. Srikun, C. S. Lim, C. J. Chang and B. R. Cho, Chem. Commun., 2011, 47, 9618–9620 RSC.
- E. W. Miller, O. Tulyanthan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS PubMed.
- X. M. Wu, X. R. Sun, Z. Q. Guo, J. B. Tang, Y. Q. Shen, T. D. James, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2014, 136, 3579–3588 CrossRef CAS PubMed.
- Z. Q. Guo, W. H. Zhu and H. Tian, Chem. Commun., 2012, 48, 6073–6084 RSC.
- W. Zhu, X. M. Huang, Z. Q. Guo, X. M. Wu, H. H. Yu and H. Tian, Chem. Commun., 2012, 48, 1784–1786 RSC.
- A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed.
- D. H. Yu, Q. Zhang, S. S. Ding and G. Q. Feng, RSC Adv., 2014, 4, 46561–46567 RSC.
- H. Y. Chen, S. N. Wan, F. X. Zhu, C. Wang, S. S. Cui, C. L. Du, Y. X. Ma and Y. Q. Gu, Contrast Media Mol. Imaging, 2014, 9, 122–134 CrossRef CAS PubMed.
- H. Y. Chen, M. Zhang, H. B. Yang, W. X. Xu, Y. X. Ma and Y. Q. Gu, RSC Adv., 2014, 4, 8191–8199 RSC.
- X. Li, D. W. Deng, J. P. Xue, L. Z. Qu, S. Achilefu and Y. Q. Gu, Biosens. Bioelectron., 2014, 61, 512–518 CrossRef CAS PubMed.
- E. J. Na, K. H. Lee, Y. K. Kim and S. S. Yoon, Mol. Cryst. Liq. Cryst., 2012, 568, 8–14 CrossRef CAS PubMed.
- W. Sun, J. L. Fan, C. Hu, J. F. Cao, H. Zhang, X. Q. Xiong, J. Y. Wang, S. Cui, S. G. Sun and X. J. Peng, Chem. Commun., 2013, 49, 3890–3892 RSC.
- J. Zhou, Y. Li, J. N. Shen, Q. Li, R. Wang, Y. F. Xu and X. H. Qian, RSC Adv., 2014, 4, 51589–51592 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16827a |
| This journal is © The Royal Society of Chemistry 2015 |