
Oxidation- and thermo-responsive poly(N-isopropylacrylamide-co-2-hydroxyethyl acrylate) hydrogels cross-linked via diselenides for controlled drug delivery†
Xinfeng Chengab,
Yong Jin*cd,
Tongbing Sunab,
Rui Qiab,
Baozhu Fanab and
Hanping Licd
aChengdu Institute of Organic Chemistry, Chinese Academy of Science, Center of Polymer Science and Technology, Chengdu 610041, People's Republic of China
bUniversity of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, People's Republic of China
cNational Engineering Laboratory for Clean Technology of Leather Manufacture, Sichuan University, Chengdu 610065, People's Republic of China. E-mail: jinyong@cioc.ac.cn; Tel: +86-28-85214963
dKey Laboratory of Leather Chemistry and Engineering (Sichuan University), Ministry of Education, Chengdu 610065, People's Republic of China
First published on 5th December 2014
Abstract
A novel diselenide crosslinked poly(N-isopropylacrylamide-co-2-hydroxyethyl acrylate) hydrogel was successfully synthesized and characterized. The resultant hydrogel showed a swelling–shrinkage behavior as a function of external temperature because of the unique hydrophilic–hydrophobic transition of poly(N-isopropylacrylamide) (PNIPAM) segments. In addition to the temperature-dependent response, the oxidation-induced gel-to-sol transition of the hydrogel was also observed. The salicylic acid (SA) loaded hydrogels were prepared in order to investigate their stimuli-responsive release behaviors. The cumulative release profile of the SA-loaded hydrogels showed a thermo-induced slow sustained drug release and an oxidation-induced quick burst release, exhibiting temperature/oxidation dual-stimuli-responsive drug release. This novel type of hydrogels may hold great promise for controlled drug delivery systems.
1. Introduction
Stimulus-responsive hydrogels (SRHs) have attracted tremendous interest over the past decades due to their potential application in biomedical and pharmaceutical fields.1–4 These intelligent materials are assumed to be promising carriers for drug delivery due their abilities to sense changes in a specific environment and stimulate structural and/or morphological responses when the environmental factors change.5,6 For example, these SRHs can undergo sluggish or abrupt changes in response to physical changes (temperature,7–10 light,11,12 electric13 and magnetic fields14) or (bio)chemical stimulus (ionic strength,15,16 pH9,10 or chemical agents17–19). The responses can be very diverse, such as swelling, shrinking, sol–gel transition or degradation of the hydrogels.2,20,21Among the above-mentioned stimuli, temperature is the most common physical one used in biomedical applications, so temperature-sensitive hydrogels have been extensively studied as controlled drug delivery systems.22–24 Particularly, as one of the most popular temperature-responsive polymers, poly(N-isopropylacrylamide) (PNIPAM) has been widely utilized as an ideal basic building block for the temperature-sensitive hydrogels because of its excellent thermo-sensitivity.25,26 In an aqueous solution, PNIPAM can undergo a reversible phase transition around the lower critical solution temperature (LCST) depending on the hydrophilic–hydrophobic interactions between polymer and water molecules.27 As for PNIPAM based hydrogels, this unique phase transition endows them with reversible swelling–shrinkage abilities, which is very useful for the encapsulation and controlled release of hydrophilic drugs.28,29 There have been a large number of papers focusing on the preparation of thermo-responsive PNIPAM-based hydrogels for the application in controlled drug delivery systems.7,10,18,30,31
While, a challenge existing in most of the above mentioned chemically crosslinked PNIPAM hydrogels is that the reversible swelling and shrinking of the hydrogels lead to a long-term and incomplete drug release owing to their slow biodegradation by enzymes in vivo. To resolve this problem, various decomposable crosslinkers, such as ester,32 peptide33,34 and other labile bonds,35 have been incorporated in the synthesis of hydrogels. The resultant crosslinked hydrogels are degradable to water-soluble polymers, exhibiting a gel-to-sol transition accompanied by a quick and complete drug release. Disulfides represent an excellent choice for a degradable crosslinker. The disulfide bond (S–S) can be cleaved in aqueous media by redox agents such as dithiothreitol (DTT), glutathione (GSH) and reactive oxygen species (ROS).36 Various studies have shown that the enhanced drug release behavior was observed for the disulfide crosslinked hydrogels by the addition of a redox agent.19,37–40 Thus, the incorporation of disulfide bonds in polymer networks makes it possible to achieve controlled drug release of hydrogels at different redox conditions. Notably, a series of disulfide crosslinked hydrogels being sensitive to both temperature and redox have been synthesized.41–44 The dual-stimuli-responsive systems offer a very useful tool to optimize the control of drug release. That is to say, both thermo-induced long-term release and redox-induced short-term release can be combined together to control the drug release of the dual-responsive hydrogels.
Similar to the disulfide bonds (S–S), the diselenide bonds (Se–Se) also show a cleavable behavior under redox conditions. Especially, the Se–Se bond is more easily cleaved and reduced to selenol in a reducing environment and oxidized to seleninic acid in the presence of oxidants because Se–Se bonds have a lower bond energy (172 kJ mol−1) relative to S–S bonds (240 kJ mol−1).45,46 Therefore, diselenide bond (Se–Se) can be a promising candidate for stimuli-responsive cleavable linkages, which has gained more and more attention during the past few years. For instance, Xu et al. reported a series of diselenide-containing amphiphilic block copolymers with repeating diselenides on the hydrophobic chains, which can self-assemble into micelles, and then disassemble and release the payloads under redox conditions.47,48 Meanwhile, Jin's group has also reported two single-diselenide labeled block copolymers and studied their self-assembly properties and enhanced release behaviors of encapsulated molecules under oxidation condition.49,50 Moreover, exploiting oxidative microenvironments in the body as triggers for drug delivery systems have attracted great interest.51 Reactive oxygen species (ROS) including H2O2, superoxide and hydroxide radical play an important role in cell signaling pathways. The overproduced ROS from cells in disease state may disrupt cellular homeostasis, giving rise to pathological conditions.52 Biomaterials that are sensitive to ROS can be strategically used to specifically release therapeutics agents to regions undergoing oxidative stress. For instance, Hubbell et al. developed a poly(propylene sulfide) (PPS) based amphiphilic diblock copolymer PEG-b-PPS, which could self-assemble into micelles and then underwent H2O2-triggered disassembly and drug release behaviors due to the oxidation-induced solubility switch.53 Fréchet et al. fabricated a ROS-responsive aryl-boronic esters modified dextran microspheres.54 These resulting nanoparticles were very sensitive to oxidants, which could undergo a H2O2-triggered degradation of the dextran particles and a rapid release of the payloads. Considering the aforementioned solubility switch or degradation behaviors of the diselenide-containing polymers under oxidation conditions, they could be an important part of the promising oxidation-sensitive biomaterials for biomedical application.
Stimulated by the good features of diselenides mentioned above, it can be anticipated that the incorporation of cleavable Se–Se crosslinkers into the hydrogels may result in an oxidation-responsive degradable hydrogel carriers, which can provide an accelerated dissociation of the carriers and more complete release of encapsulated drugs upon oxidation stimulus. Compared with disulfide crosslinked hydrogels, diselenide crosslinked hydrogels may exhibit enhanced degradable ability under a relatively milder physiological conditions serving as a fine-controlled drug delivery system. However, until now, literature on oxidation-responsive diselenide crosslinked hydrogels is scarcely investigated. Therefore, it is of great importance to construct such diselenide crosslinked hydrogels and explore their potential as functional carriers for controlled drug delivery.
Herein, we report an easy method to synthesize a dual stimuli-responsive hydrogel by cross-linking copolymers of N-isopropylacrylamide (NIPAM) and 2-hydroxyethyl acrylate (HEA) with a diselenide-bearing cross-linking agent. The resultant poly(NIPAM-co-HEA) hydrogels possess diselenide linkages, so they could degrade into water soluble polymer in the presence of oxidizing agents. Also, these hydrogels have PNIPAM segments, which offer them swelling–shrinkage ability in response to temperature change. Utilizing H2O2 as an oxidizing agent or increasing the temperature, the dual-response and drug release behaviors of the diselenide cross-linked hydrogels to oxidizing agents or temperature were studied in detail. The temperature and oxidation induced release behaviors indicate the promising application of these materials as controlled drug delivery vehicles (Scheme 1).
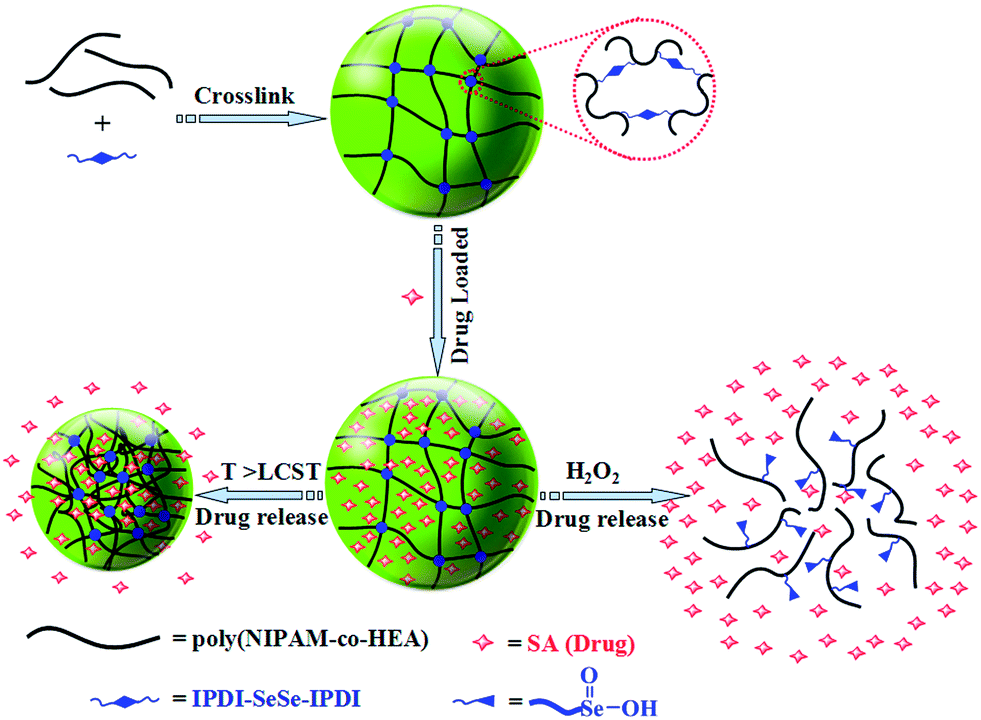 | ||
| Scheme 1 Schematic of the thermo- and oxidation-responsive morphology transformation of diselenide crosslinked poly(NIPAM-co-HEA) hydrogels. | ||
2. Experimental
2.1 Materials
N-Isopropylacrylamide (NIPAM) (Tokyo Chemical Industrial Co., Japan) was purified by recrystallization in a mixture of toluene and hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v) and dried in vacuum. 2-Hydroxyethyl acrylate (HEA) (Aldrich) was passed through a basic alumina column to remove the inhibitor before use. 2,2′-Azo-bis-isobutyrylnitrile (AIBN) was purchased from Aldrich Chemical and used after recrystallization from ethanol. Sodium boro-hydride (NaBH4), selenium powder, tetrahydrofuran (THF), dibutyltin dilaurate (DBTDL), hydrogen peroxide (H2O2), Salicylic Acid (SA) were analytical grade products purchased from Aladdin Reagents. 3-Isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate (IPDI, Analytical grade), supplied by Shanghai Chemical Reagent Corporation (Shanghai, China), was used without further purification. All other reagents were of analytical grade and used as received.
:5 v/v) and dried in vacuum. 2-Hydroxyethyl acrylate (HEA) (Aldrich) was passed through a basic alumina column to remove the inhibitor before use. 2,2′-Azo-bis-isobutyrylnitrile (AIBN) was purchased from Aldrich Chemical and used after recrystallization from ethanol. Sodium boro-hydride (NaBH4), selenium powder, tetrahydrofuran (THF), dibutyltin dilaurate (DBTDL), hydrogen peroxide (H2O2), Salicylic Acid (SA) were analytical grade products purchased from Aladdin Reagents. 3-Isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate (IPDI, Analytical grade), supplied by Shanghai Chemical Reagent Corporation (Shanghai, China), was used without further purification. All other reagents were of analytical grade and used as received.
2.2 Instrumentation
The 1H-NMR and 13C NMR spectra were recorded on a JEOL JNM-ECA 300 (300 MHz) spectrometer. Gel permeation chromatography (GPC) measurement was performed on a HLC-8320 GPC using polystyrene as a standard and THF as an eluent. Fourier transform infrared (FTIR) spectra were recorded on ThermoFisher Nicolet 6700 spectrophotometer in KBr pellets. X-ray photo-electron spectroscopy (XPS) measurements were performed on ESCA Lab220i-XL equipment with Si wafers as the substrates. A Hitachi U-2010 spectrophotometer was used to record the transmittance and absorbance of samples. Digital photographs were taken by a Canon Power Shot A3000 IS digital camera.2.3 Synthesis of poly(N-isopropylacrylamide-co-2-hydroxyethyl acrylate) (PNH) by free radical copolymerization of NIPAM and HEA
Poly(N-isopropylacrylamide-co-2-hydroxyethyl acrylate) copolymer (PNH) was synthesized with a feed ratio of NIPAM/HEA at 8:2 by the free radical polymerization. Initially, NIPAM (5.09 g; 45 mM), HEA (1.31 g; 11.25 mM) and 20 mg of AIBN (initiator) were dissolved in 30 mL THF. The solution was degassed by N2 for 30 min, and then polymerization reaction was carried out at 70 °C under N2 flow. The synthetic process was described in Scheme 2a. After 20 h, the reaction was terminated and the products were precipitated with diethyl ether. The products were purified by repeated precipitation in diethyl ether from THF, followed by vacuum-dried for 24 h. Finally, a white powder of PNH with a yield of 93% was obtained.
 | ||
| Scheme 2 Synthetic routes of the copolymer poly(NIPAM-co-HEA) (PNH) (a) cross-linker IPDI-SeSe-IPDI (b), and hydrogel PNH–SeSe (c). | ||
2.4 Synthesis of diselenide crosslinker IPDI-SeSe-IPDI (CL)
Scheme 2b shows the synthetic routs of diselenide crosslinker IPDI-SeSe-IPDI (CL). 0.5 g (6.3 mmol) of selenium was added to 25 mL of water containing 0.5 g dissolved sodium borohydride (13.2 mmol) with magnetic stirring at room temperature. After the initial vigorous reaction had subsided (10 min), an additional 0.5 g of selenium (6.3 mmol) was added. The mixture was stirred for 15 min and then warmed briefly on a steam bath to complete the dissolution of the selenium. A brownish red aqueous solution of Na2Se2 was obtained.The Na2Se2 solution was removed to a 100 mL flask which was sealed with a rubber plug, and then a solution of 1.575 g (12.6 mmol) 2-bromoethanol in 20 mL THF was injected into it under N2 flow. The reaction was performed at 50 °C for 6 h and the obtained solution was extracted three times with 20 mL of CH2Cl2 and dried with anhydrous Na2SO4. Then the product was purified by column chromatography with a 1:1 (volume/volume) mixture of CH2Cl2 and ethyl acetate as eluent. A yellow transparent liquid of di(1-hydroxyethylene) diselenide (DHE-SeSe) was obtained. 1H NMR (400 M Hz, CDCl3): δ (ppm) 3.95 (4H, t, HOCH2), 3.10 (4H, t, SeSeCH2).
0.223 g (0.9 mmol) di(1-hydroxyethylene) diselenide (DHE-SeSe) and 10 mg of DBTDL were dissolved in 5 mL anhydrous THF in a 20 mL flask and sealed with a rubber plug. The flask was then degassed by N2 for 30 min. A solution of 0.400 g (1.8 mmol) IPDI in 5 mL anhydrous THF was injected into the flask under N2 flow. The system was placed at room temperature to react for 24 h with stirring. The solvent was then removed by rotary evaporation under reduced pressure and the residual liquid was dissolved by acetone and cyclohexane. Finally, a yellow solid of IPDI-SeSe-IPDI (CL) with a yield of 95% was obtained after cryogenic drying.
2.5 Preparation of diselenide cross-linked poly(NIPAM-co-HEA) hydrogels
Poly(NIPAM-co-HEA) hydrogels were synthesized through the coupling reaction between –OH-pending copolymer and –NCO-containing IPDI-SeSe-IPDI crosslinker (Scheme 2c). The typical procedure was followed. Poly(NIPAM-co-HEA) (3.28 g), IPDI-SeSe-IPDI (0.2 g, 0.6 g or 1.8 g) and DBTDL (10 mg) were dissolved in THF (10 mL). The mixture was deoxygenated by three freeze–pump–thaw cycles and then maintained at room temperature without stirring. When the mixture became an immobile yellow bulk gel in the inverted cuvette, the gelation time was recorded. After 24 hours, the resultant hydrogels were cut into discs for further purification by dialysis against THF/water (1/1, volume/volume) for 2 days and water for 3 days. The final dry gels were obtained by the lyophilization in a freeze drier for 48 h.2.6 Phase transition of poly(NIPAM-co-HEA) copolymers
The turbidities of the aqueous solutions of poly(NIPAM-co-HEA) copolymers (0.5 wt%) were measured at 500 nm by using a UV-vis spectrometer (Hitachi U-2010) equipped with a temperature controller. The heating rate was 0.5 °C min−1. At each temperature, the solution was equilibrated for 10 min. The lower critical solution temperature (LCST) was determined as the temperature at 50% of the initial transmittance.55,56 In addition, the turbidity of PNIPAM homopolymer was also measured for a comparison.2.7 Thermo-responsive swelling–shrinkage behavior of poly(NIPAM-co-HEA) hydrogels
To measure the swell–shrinkage behaviors of hydrogels, the dried gel discs (16 mm diameter, 1.8 mm thickness) were firstly immersed into 20 mL of distilled water at 25 °C until the equilibrium degree of swelling and a constant weight was achieved, which usually took up to two days. Then the fully swollen gel samples were quickly immersed into 20 mL of distilled water at 40 °C until the equilibrium degree of deswelling and another constant weight was achieved. The swelling ratio (SR) of the hydrogels was determined using the following equation:| SR = (Wt − W0)/W0 |
2.8 Oxidation responsive behavior of poly(NIPAM-co-HEA) hydrogels
The oxidation responsive behavior of poly(NIPAM-co-HEA) hydrogels was studied by using H2O2 as an oxidizing agent to cleave the diselenide linkages of hydrogels. The dried gel discs (16 mm diameter, 1.8 mm thickness) were immersed into 0.5 wt% H2O2 solution at room temperature. Then a digital camera was used to record the gel-to-sol transition of hydrogels.2.9 Drug loading and release of Salicylic Acid (SA) of poly(NIPAM-co-HEA) hydrogels
Salicylic Acid (SA) was chosen as the model drug to test the drug loading and release behaviors of hydrogels. The dry poly(NIPAM-co-HEA) hydrogel discs (250 mg) were swollen in SA aqueous solution (1 mg mL−1, 20 mL) at 20 °C for 48 h. After swelling equilibrium was reached, the swollen hydrogels were taken out and then rinsed thoroughly with distilled water. Then the residual SA solution and the distilled water used to rinse the drug-loaded hydrogels were combined together and diluted to 50 mL in a volumetric flask. The amount of SA left in the loading medium was determined by a UV/Vis spectrometer (Hitachi U-2010) at a wavelength of 296 nm. The loading amount (mg) of SA for 250 mg hydrogels was determined. The drug loading capacities (DLC) and drug loading efficiency (DLE) were calculated by the following equations:| DLC(%) = WSL/(WHG + WSL) × 100 |
| DLE(%) = WSL/WS0 × 100 |
The drug release was carried out at different concentrations of H2O2 or temperatures. For the measurement of temperature-induced drug release, the drug-loaded hydrogel was kept in distilled water at the temperature of 25 °C or 40 °C. For the measurement of oxidation-induced drug release, the drug-loaded hydrogel was kept in 0.25 wt% or 0.5 wt% H2O2 solutions at the temperature of 25 °C. At each of determined time intervals, 2 mL solution was taken out for UV-vis (Hitachi U-2010) measurement at a wavelength of 296 nm and its concentration of SA was determined using the calibration built on the absorbance of SA solutions with known concentrations. At each sampling, 2 mL fresh solution was added to keep the constant volume of medium. Each release experiment was conducted in triplicate.
3. Results and discussion
3.1 Synthesis of the copolymers and hydrogels
| χNIPAM = 4I1.1/(6I3.7∼4.1 + 3I1.1) |
 | ||
| Fig. 1 1H NMR spectra of PNH (a) and IPDI-SeSe-IPDI (b) in CDCl3. | ||
To further prove the copolymers composed of NIPAM and HEA units, the structures were also confirmed by FT-IR spectra analysis. As depicted in Fig. 2a, a broad band at 3437 cm−1 is assigned to the stretching vibrations of –OH groups of HEA units. The band at 3302 cm−1 is ascribed to the N–H stretching vibrations of NIPAM units. 1734 cm−1 and 1648 cm−1 are contributed to the vibration of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups from –COO– of HEA units and –CONH– of NIPAM units, respectively. Notably, the absorption band at 1734 cm−1 was strengthened with the increase of the molar content of HEA units in the copolymers (Fig. S2†). All the results clearly confirmed that the PNH copolymers composed of NIPAM and HEA units were successfully prepared. Moreover, the molecular weight and its distribution of the copolymer PNH were also characterized by GPC (Fig. S3†), which were determined to be 1.7544 × 104 (Mw) and 1.92 (PDI), respectively.
O groups from –COO– of HEA units and –CONH– of NIPAM units, respectively. Notably, the absorption band at 1734 cm−1 was strengthened with the increase of the molar content of HEA units in the copolymers (Fig. S2†). All the results clearly confirmed that the PNH copolymers composed of NIPAM and HEA units were successfully prepared. Moreover, the molecular weight and its distribution of the copolymer PNH were also characterized by GPC (Fig. S3†), which were determined to be 1.7544 × 104 (Mw) and 1.92 (PDI), respectively.
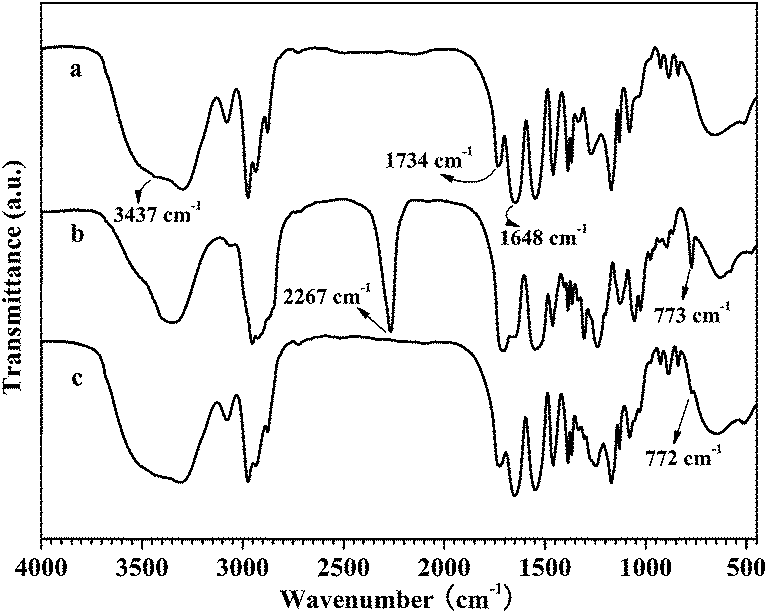 | ||
| Fig. 2 FT-IR spectra of copolymer PNH (a), crosslinker IPDI-SeSe-IPDI (b) and gel PNH–SeSe (c). | ||
Then poly(NIPAM-co-HEA) hydrogels (PNH–SeSe) were obtained through the group coupling reaction between the pendent –OH groups of copolymers and the terminal –NCO groups of IPDI-SeSe-IPDI crosslinkers at room temperature (Scheme 2c). As seen in Fig. 3, a dramatic sol-to-gel transition was observed for the PNH–SeSe gels with a feed ratio of crosslinker/copolymer at 0.6 g/3.28 g (denoted as Gel 1). Generally, by changing the feed ratio of IPDI-SeSe-IPDI crosslinkers to PNH copolymers, an accelerated gelation process was represented for the higher feed ratio of 1.8 g/3.28 g (denoted as Gel 2), while no formation of bulk gel was observed for the lower feed ratio of 0.2 g/3.28 g (Table S2 and Fig. S5†). FT-IR analyses confirmed the structures of the hydrogels PNH–SeSe. The FTIR spectrum of PNH–SeSe hydrogel (Gel 1) was shown in Fig. 2 together with PNH copolymer and IPDI-SeSe-IPDI crosslinker for comparison. Compared with the crosslinker IPDI-SeSe-IPDI, it was clearly to see that the absorption peak of –NCO group at 2267 cm−1 was disappeared in the spectrum of PNH–SeSe, informing that the –NCO groups from crosslinkers was completely reacted with –OH groups from copolymers to form a crosslinked network. In addition, a weaken characteristic band at 772 cm−1 originated from the vibration of C–Se bond from IPDI-SeSe-IPDI crosslinkers was observed in the spectrum of PNH–SeSe gels due to the lower ratio of crosslinkers in the PNH–SeSe gels. All the above features indicate that the diselenide crosslinked PNH hydrogels were successfully prepared.
 | ||
| Fig. 3 The sol–gel transition of mixing solution containing PNH copolymers and IPDI-SeSe-IPDI crosslinkers. | ||
3.2 Thermo- and oxidation-responsive characteristics of hydrogels
Stimuli-responsive characteristics of the resulting PNH–SeSe hydrogels were studied under physical and chemical conditions. Owing to the incorporation of PNIPAM segments in the PNH–SeSe hydrogels, the hydrogels exhibit a thermo-sensitive swelling–shrinkage characteristic due to the unique hydrophilic–hydrophobic transition of PNIPAM segments. As temperature changes, the PNIPAM segments undergo a reversible hydrophilic–hydrophobic transition at its lower critical solution temperature (LCST), which leaded the copolymer to be soluble in water below LCST or insoluble above LCST. The copolymer PNH was found to display a transparence to turbidity transition with the increase of temperature (Fig. S6†). The LCST of the copolymer PNH was determinated to be 34.7 °C, which was defined as the temperature at 50% of the initial transmittance. This value is higher than that of PNIPAM homopolymer (30.3 °C), due to the fact that the copolymer PNH containing HEA units becomes more hydrophilic and the consequent overall hydrogen bonding ability of the copolymers is increased, which leads to a higher transition temperature.The thermo-induced swelling–shrinking behaviors of the hydrogels were studied in distilled water at 25 and 40 °C, respectively. At 25 °C, all the hydrogels exhibited temperature-responsive swelling behaviors (Fig. S7†). The swelling ratio (SR) of PNH–SeSe hydrogel was calculated to be 150% for Gel 1 composed of 15 wt% crosslinker (Fig. S8†). As expected, the Gel 2 with more crosslinker content (35 wt%) exhibited a lower SR (58%) in comparison with Gel 1 at 25 °C. In addition, all the hydrogels displayed similar thermo-induced shrinking behaviors at 40 °C. As shown in Fig. 4, when increasing the temperature of the swollen hydrogel to 40 °C, the hydrogel underwent a typical shrinking process, resulting in the water to be squeezed out of the bulk gel during the volume phase transition. At 40 °C, the SRs were decreased to be 121% for Gel 1 and 15% for Gel 2.
 | ||
| Fig. 4 Dual thermo- and oxidation-responsive characteristics of PNH–SeSe hydrogels. | ||
In addition, the oxidation-responsive behavior of the PNH–SeSe hydrogel was also studied. It is known that the diselenide bond tends to be cleaved under redox conditions. So we assumed that the resulting diselenide-crosslinked hydrogels could be also cleaved and degraded upon addition of a reducing or oxidizing agent. In order to confirm this assumption, the small molecule HOCH2CH2SeSeCH2CH2OH was chosen as a model compound and H2O2 was selected as an oxidizing agent for the oxidation reaction test.57,58 The oxidation product was characterized by XPS experiments. The XPS data (Fig. S9†) demonstrates that the binding energy of Se 3d5 shifts from 56.7 eV to 60.6 eV, suggesting that a higher valence of Se that is quite close to seleninic acid group59 was obtained after the oxidation of 0.5 wt% H2O2 solution. The oxidation reaction equation of the model compound HOCH2CH2SeSeCH2CH2OH is as follows:
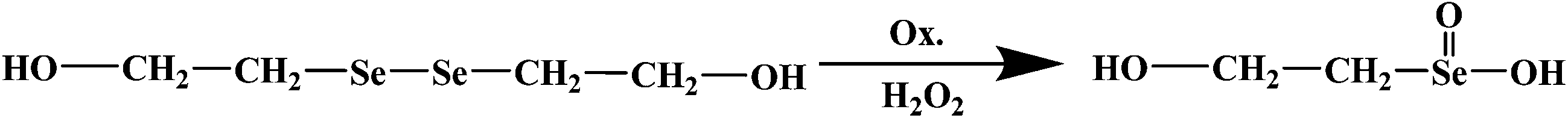
Therefore, H2O2 was employed as the oxidant to study the oxidation-responsive cleavage of the PNH–SeSe hydrogels. The cleavage reaction was implemented using 2 mL of a solution of 0.5 wt% H2O2 under a nitrogen atmosphere. Within few hours, the bulk gel was destroyed and then transformed into a clear solution, as depicted in Fig. 4. The oxidative cleavage of the diselenide linkages in the cross-linked networks resulted in a degradation into soluble linear polymer chains with pendant seleninic acid groups (Scheme 1).
3.3 In vitro stimuli-responsive drug release of Salicylic Acid (SA)
Due to the thermo-responsive swelling–shrinking and oxidation-responsive gel-to-sol transition behaviors of the diselenide-crosslinked hydrogels, it is possible to use the PNH–SeSe hydrogels as drug-carriers to encapsulate and release some functional molecules, such as the hydrophilic drugs. For this purpose, the controlled release experiment under thermo- and oxidation stimuli was conducted. Salicylic Acid (SA) was chosen as a simple model drug in loading and release experiment. It has strong UV-vis absorption at 296 nm, so its concentration in water can be easily determined by using its calibration curve (Fig. S10†). The release behaviors were monitored through the increase of UV absorption intensity of the dialysis extracting solution. The Gel 1 with lower crosslinking density (15 wt% crosslinker content) was chosen as an instructive example to examine the drug release behavior of the PNH–SeSe hydrogels. The drug loading capacities (DLC) and drug loading efficiency (DLE) of Gel 1 were calculated to be 3.1% and 48%, respectively. | ||
| Fig. 5 In vitro SA release from the Gel 1 at 25 and 40 °C, respectively. | ||
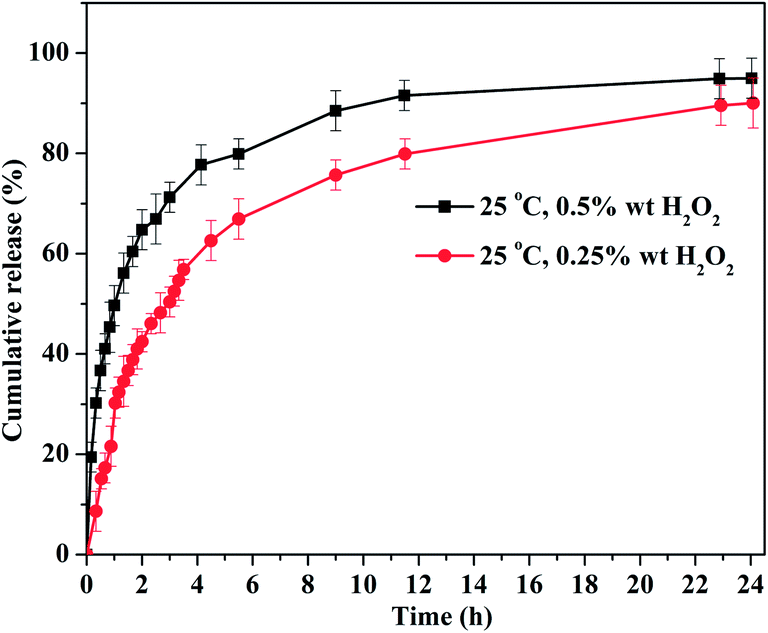 | ||
| Fig. 6 In vitro SA release from the Gel 1 upon exposure to different concentrations of H2O2 solution at 25 °C. | ||
Overall, by increasing the temperature or concentration of oxidizing agent, the hydrophilic–hydrophobic balance between PNH and water molecules, or the crosslinking network is destroyed, resulting in a phase transition or degradation of hydrogels, corresponding to a relative slow sustained or quick burst drug release behavior. According to the above drug release features of the PNH–SeSe hydrogel, single temperature-induced drug release cannot provide high drug release ratio because its cross-linked network still exists under this condition. While, treatment with H2O2 leads to the expected structural loss and dissociation of the bulk gels, along with an approximate complete release of the encapsulated drugs. These preliminary in vitro studies have proved that the release of encapsulated molecules can be controlled and adjusted by external temperature and H2O2 stimuli, indicating that this new kind of dual-responsive hydrogels could be serving as promising drug carriers for controlled drug release. Considering the complexity of human physiological environment, the in vitro tests here could not be mimicking the true environmental factors observed in vivo very well, such as the release medium, the concentration of H2O2, etc. Therefore, further insight into this stimulus-dose-dependent degradation and responsive release of drugs in vivo are being under investigation.
4. Conclusions
A novel dual-stimuli-responsive hydrogel was prepared by cross-linking copolymers of N-isopropylacrylamide (NIPAM) and 2-hydroxyethyl acrylate (HEA) with a diselenide-bearing cross-linking agent. The obtained hydrogel showed a smart response to the external stimuli of temperature and oxidation. The elevation of environment temperature will directly result in the formation of shrinking structures of hydrogel due to the LCST phase transition of PNIPAM segments. Moreover, the diselenide cross-linked hydrogels can be dissolved into polymer solutions by adding the oxidizing agent (H2O2). The Salicylic Acid (SA) loaded hydrogels were prepared in order to investigate their stimuli-responsive release. The cumulative release profile of the SA-loaded hydrogels showed a relatively low level of drug release (48 wt% in 24 h) at 40 °C and a quick release (over 90 wt% in 24 h) with oxidizing environment, exhibiting temperature/oxidation dual-stimuli-responsive drug release. The hydrogels possess many favorable merits of drug carriers, such as satisfactory drug loading capacity and controlled drug release in response to the external temperature and/or oxidation. These results confirmed their great potential as novel drug carriers and controlled delivery systems.Acknowledgements
This work was financially supported by the National High-tech Research and Development Projects (863) (2013AA06A306), the National Natural Science Foundation of China (20874102 and 21474065), and the Sichuan Province Science and Technology Support Projects (2010FZ0093).References
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- D. Y. Ko, U. P. Shinde, B. Yeon and B. Jeong, Prog. Polym. Sci., 2013, 38, 672–701 CrossRef CAS PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jüngst, W. E. Hennink and W. J. A. Dhert, et al., Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS PubMed.
- A. Doring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- Y. Liu, Z. C. Li and D. H. Liang, Soft Matter, 2012, 8, 4517–4523 RSC.
- L.-W. Xia, R. Xie, X.-J. Ju, W. Wang, Q. Chen and L.-Y. Chu, Nat. Commun., 2013, 4, 2226 Search PubMed.
- A. Nemethy, K. Solti, L. Kiss, B. Gyarmati, M. A. Deli and E. Csanyi, et al., Eur. Polym. J., 2013, 49, 2392–2403 CrossRef CAS PubMed.
- X. Gao, Y. Cao, X. Song, Z. Zhang, C. Xiao and C. He, et al., J. Mater. Chem. B, 2013, 1, 5578–5587 RSC.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS PubMed.
- B. Kim, H. S. Lee, J. Kim and S. H. Kim, Chem. Commun., 2013, 49, 1865–1867 RSC.
- A. Servant, C. Bussy, K. Al-Jamal and K. Kostarelos, J. Mater. Chem. B, 2013, 1, 4593–4600 RSC.
- Y. Li, G. Huang, X. Zhang, B. Li, Y. Chen and T. Lu, et al., Adv. Funct. Mater., 2013, 23, 660–672 CrossRef CAS.
- M. Dadsetan, Z. Liu, M. Pumberger, C. V. Giraldo, T. Ruesink and L. Lu, et al., Biomaterials, 2010, 31, 8051–8062 CrossRef CAS PubMed.
- S. Zhou, A. Bismarck and J. H. G. Steinke, J. Mater. Chem. B, 2013, 1, 4736–4745 RSC.
- M. F. Maitz, U. Freudenberg, M. V. Tsurkan, M. Fischer, T. Beyrich and C. Werner, Nat. Commun., 2013, 4, 2168 Search PubMed.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang and P. C. Wang, et al., J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- C. Legros, M.-C. De Pauw-Gillet, K. C. Tam, S. Lecommmandoux and D. Taton, Polym. Chem., 2013, 4, 4801–4808 RSC.
- S. C. Lee, I. K. Kwon and K. Park, Adv. Drug Delivery Rev., 2013, 65, 17–20 CrossRef CAS PubMed.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- Z. Zhang, X. Y. Gao, A. P. Zhang, X. W. Wu, L. Chen and C. L. He, et al., Macromol. Chem. Phys., 2012, 213, 713–719 CrossRef CAS.
- C. W. Zhao, X. L. Zhuang, P. He, C. S. Xiao, C. L. He and J. R. Sun, et al., Polymer, 2009, 50, 4308–4316 CrossRef CAS PubMed.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Macromolecules, 2011, 44, 6629–6636 CrossRef CAS.
- S. Sun, J. Hu, H. Tang and P. Wu, J. Phys. Chem. B, 2010, 114, 9761–9770 CrossRef CAS PubMed.
- W. Guo, C.-H. Lu, X.-J. Qi, R. Orbach, M. Fadeev and H.-H. Yang, et al., Angew. Chem., 2014, 126, 10298–10302 CrossRef.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- C. S. Brazel and N. A. Peppas, J. Controlled Release, 1996, 39, 57–64 CrossRef CAS.
- A. Gutowska, J. Seok Bark, I. Chan Kwon, Y. Han Bae, Y. Cha and S. Wan Kim, J. Controlled Release, 1997, 48, 141–148 CrossRef CAS.
- J. Zhang, R. Xie, S.-B. Zhang, C.-J. Cheng, X.-J. Ju and L.-Y. Chu, Polymer, 2009, 50, 2516–2525 CrossRef CAS PubMed.
- T.-T. Pan, W.-D. He, L.-Y. Li, W.-X. Jiang, C. He and J. Tao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2155–2164 CrossRef CAS.
- M. B. Runge, M. Dadsetan, J. Baltrusaitis, T. Ruesink, L. Lu and A. J. Windebank, et al., Biomacromolecules, 2010, 11, 2845–2853 CrossRef CAS PubMed.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS PubMed.
- Y. S. Jo, J. Gantz, J. A. Hubbel and M. P. Lutolf, Soft Matter, 2008, 5, 440–446 RSC.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- M. Huo, J.-Y. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- S.-C. Han, W.-D. He, J. Li, L.-Y. Li, X.-L. Sun and B.-Y. Zhang, et al., J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4074–4082 CrossRef CAS.
- H. Koo, G.-W. Jin, H. Kang, Y. Lee, H. Y. Nam and H.-S. Jang, et al., Int. J. Pharm., 2009, 374, 58–65 CrossRef CAS PubMed.
- M. Ejaz, H. Yu, Y. Yan, D. A. Blake, R. S. Ayyala and S. M. Grayson, Polymer, 2011, 52, 5262–5270 CrossRef CAS PubMed.
- B. Gyarmati, Á. Némethy and A. Szilágyi, RSC Adv., 2014, 4, 8764–8771 RSC.
- H. C. Chiu and C. H. Wang, Polym. J., 2000, 32, 574–582 CrossRef CAS.
- B.-Y. Zhang, W.-D. He, L.-Y. Li, X.-L. Sun, W.-T. Li and K.-R. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3604–3612 CrossRef CAS.
- W. Lv, S. Liu, W. Feng, J. Qi, G. Zhang and F. Zhang, et al., Macromol. Rapid Commun., 2011, 32, 1101–1107 CrossRef CAS PubMed.
- S. Aleksanian, Y. Wen, N. Chan and J. K. Oh, RSC Adv., 2014, 4, 3713–3721 RSC.
- R. Blom and A. Haaland, J. Mol. Struct., 1985, 128, 21–27 CrossRef CAS.
- N. K. Kildahl, J. Chem. Educ., 1995, 72, 423–424 CrossRef CAS.
- N. Ma, Y. Li, H. P. Xu, Z. Q. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442–443 CrossRef CAS PubMed.
- L. Wang, W. Cao, Y. Yi and H. Xu, Langmuir, 2014, 30, 5628–5636 CrossRef CAS PubMed.
- T. B. Sun, Y. Jin, R. Qi, S. J. Peng and B. Z. Fan, Polym. Chem., 2013, 4, 4017–4023 RSC.
- T. B. Sun, Y. Jin, R. Qi, S. J. Peng and B. Z. Fan, Macromol. Chem. Phys., 2013, 214, 2875–2881 CrossRef CAS.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H.-J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS PubMed.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- D. Velluto, D. Demurtas and J. A. Hubbell, Mol. Pharmaceutics, 2008, 5, 632–642 CrossRef CAS PubMed.
- K. E. Broaders, S. Grandhe and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed.
- X.-M. Liu, L.-S. Wang, L. Wang, J. Huang and C. He, Biomaterials, 2004, 25, 5659–5666 CrossRef CAS PubMed.
- Y. Zou, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5393–5405 CrossRef CAS.
- N. Ma, Y. Li, H. F. Ren, H. P. Xu, Z. B. Li and X. Zhang, Polym. Chem., 2010, 1, 1609–1614 RSC.
- H. F. Ren, Y. T. Wu, N. Ma, H. P. Xu and X. Zhang, Soft Matter, 2012, 8, 1460–1466 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectra, Appendix B. Chemical State Tables, Perkin-Elmer, Physical Electronics Division, Eden Prairie, MN 55344, 1992 Search PubMed.
- F. Puoci and M. Curcio, pH- and Temperature-Responsive Hydrogels in Drug Delivery, Smart Materials for Drug Delivery, The Royal Society of Chemistry, 2013, vol. 2, ch. 18, pp. 153–179 Search PubMed.
- Z. Tang, Y. Akiyama, M. Yamato and T. Okano, Biomaterials, 2010, 31, 7435–7443 CrossRef CAS PubMed.
- G. H. Zhang, Y. Fang, J. Shen, C. Mao and X. H. Huang, J. Biomater. Sci., Polym. Ed., 2012, 23, 1569–1578 CAS.
- P. Han, S. C. Li, W. Cao, Y. Li, Z. W. Sun and Z. Q. Wang, et al., J. Mater. Chem. B, 2013, 1, 740–743 RSC.
- J. X. Ding, C. S. Xiao, L. S. Yan, Z. H. Tang, X. L. Zhuang and X. S. Chen, et al., J. Controlled Release, 2011, 152, E11–E13 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13500h |
| This journal is © The Royal Society of Chemistry 2015 |