DOI:
10.1039/C4RA12888E
(Paper)
RSC Adv., 2015,
5, 4368-4375
Biindole-based double D–π–A branched organic dyes for efficient dye-sensitized solar cells†
Received
22nd October 2014
, Accepted 8th December 2014
First published on 8th December 2014
Abstract
A novel class of double D–π–A branched organic dyes based on 2,2′-disubstituted-1H,1′H-3,3′-biindole moiety have been synthesized, characterized and applied as photosensitizers for dye-sensitized solar cells. Their photophysical, electrochemical and photovoltaic properties are further investigated. These type of organic dyes contain two cyanoacrylic acid moieties as electron acceptors/anchoring groups and different electron-rich conjugated linkers such as furan (JY11), thiophene (JY12) and 2,2′-bithiophene (JY13) as π-bridges. The superiority of the cross X-shaped structure of these double D–π–A branched organic dyes is the suppression of the intermolecular interactions and the guarantee of fast electron injection into the TiO2 semiconductor in the dye-sensitized solar cells. A highest power conversion efficiency of 6.54% was achieved for JY13-based cell with an iodine electrolyte under simulated AM 1.5 G solar irradiation (100 mW cm−2).
Introduction
Since the breakthrough work by Grätzel and co-workers, dye-sensitized solar cells (DSSCs) have attracted extensive attention during the past two decades due to their significant potential as low-cost and environmental friendly devices for efficient sunlight-to-electricity conversion.1 As one of the most important components of DSSCs, sensitizers have shown great influence on light-harvesting, charge transfer and power conversion efficiency (PCE).2 Recently, several zinc-porphyrin sensitizers, together with CoII/CoIII electrolyte, have been designed and successfully applied in DSSC devices to give over 12% PCEs under standard conditions.3 However, the practical application of porphyrin sensitizer would be inevitably confined by its complex synthetic process and low yield. Alternative metal-free organic dye sensitizers have thus attracted considerable attention for the facile structure tuning, easy synthesis process and low cost.4
Donor–π-bridge–acceptor (D–π–A) structure has been commonly involved in most metal-free organic dyes for DSSCs.5 With an aim of the expansion of the absorption spectra, many single D–π–A dyes have been designed with a long and rod-like configuration, which may facilitate undesirable dye aggregation and charge recombination.6 The intermolecular π–π stacking of dye molecules often cause self-quenching of excited states and hence inefficient electron injection. Because photovoltage is related to the conduction band level of TiO2 and the charge recombination rate in DSSCs, such interfacial charge carrier recombination may lead to reduction of photovoltage. Therefore, the suppression of dye aggregation is very important for minimizing the charge recombination and improving photovoltage.7 To control the intermolecular π–π stacking of organic dyes and related charge recombination processes, several kinds of coadsorbents, such as chenodeoxycholic acid (CDCA), have been physically co-adsorbed on the TiO2 surface to prevent the intermolecular aggregation of dyes. Another effective strategy is introducing long alkyl chains into the dye skeleton, which would diminish the charge recombination between dyes and I3− ions in the electrolyte.8 Employing starburst dyes with bulky multidonor, as well as double D–π–A branched dyes with “X” or “H” shape, has also proved to be effective methods to prevent intermolecular aggregation, retard charge recombination rate and therefore enhance the photovoltage.9 It was also found that the dye with two D–π–A segments had significantly larger amount of D–π–A segment adsorbed on TiO2 compared to the single D–π–A dye, which can more effectively suppress the dark current in addition to better light-harvesting and higher photocurrent.10
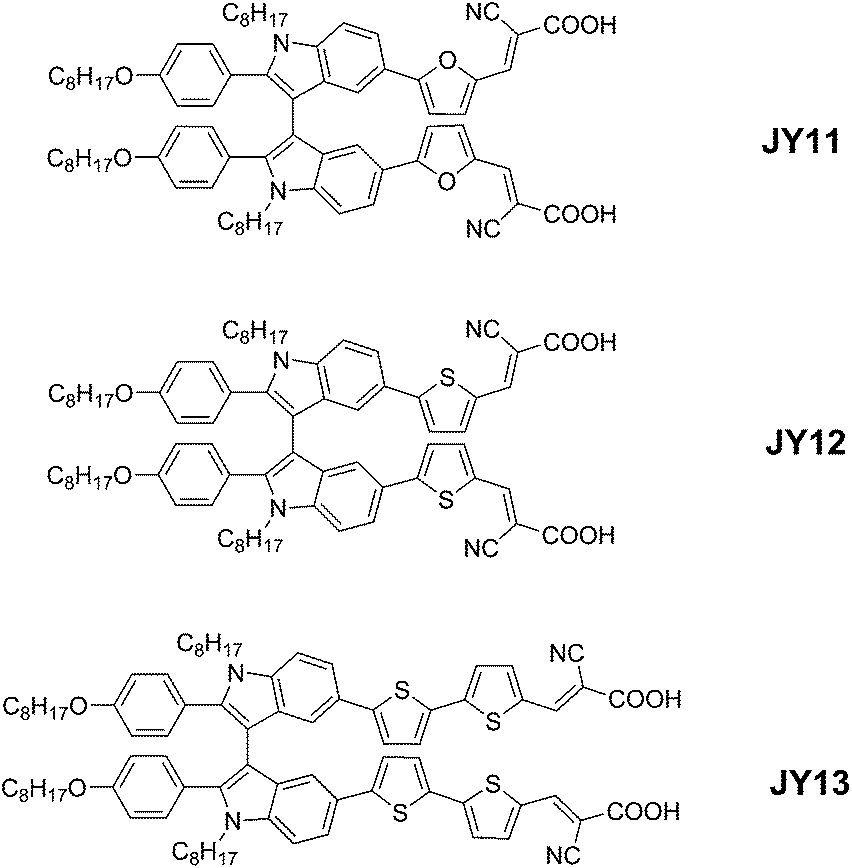
Indole derivatives are important well-known heteroaromatic compounds in diverse natural products and also considered as potential building blocks for functional electroactive materials. Recently, we have incorporated unique discotic triindole to build organic dyes with D–π–A configuration for the first time and an efficiency of 6.1% has been achieved with CDCA as coadsorbent.11 Because of the highly π-conjugated planar structure, the triindole-based dyes tend to aggregate when anchored on TiO2 films, which is unfavorable for retarding charge recombination and improving power conversion efficiency. In this report, we present a new extension of double D–π–A branched dyes based on indole derivatives, with the aim of further expanding the possibility of improving the optical and energetic properties of the sensitizers. To the best of our knowledge, the construction of organic dyes with electron-rich 3,3′-biindole core for DSSCs has not been reported yet. So we elaborate the double D–π–A branched dyes that consist of a 1,1′-dioctyl-2,2′-bis(4-(octyloxy)phenyl)-3,3′-biindole moiety acting as the electron donor and two cyanoacrylic acid acting as the electron acceptors/anchoring groups. Aromatic π-bridge such as furan, thiophene and 2,2′-bithiophene have been selected to act as the π-conjugated linkers and resulted in sensitizers JY11, JY12 and JY13, respectively. The molecular structures of these dyes are shown in Fig. 1.
 |
| | Fig. 1 Chemical structures of the dyes JY11–13. | |
Results and discussion
Molecular design and synthesis
The syntheses of the sensitizers JY11–13 were achieved in four major steps through a synthetic route displayed in Scheme 1. 2-[(2-Aminophenyl)ethynyl]phenylamine (compound 1) was prepared according to literature procedure.12 An acid-catalyzed cascade reaction of compound 1 with 4-octyloxybenzaldehyde afforded 2,2′-disubstituted-1H,1′H-3,3′-biindole (compound 2). Alkylation reaction of compound 2 with 1-bromooctane yielded compound 3. After a bromination of intermediate 3 with NBS and a successive Suzuki cross-coupling with substituted aromatic aldehydes, the π-extended biindole bearing aldehydes 4a–c were prepared. Finally, the aldehydes 4a–c were treated with cyanoacetic acid by Knoevenagel reactions to give the dyes JY11–13.
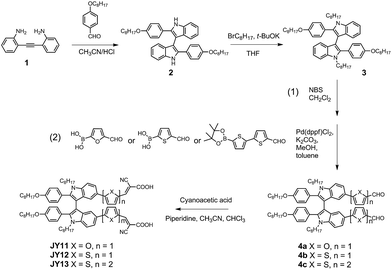 |
| | Scheme 1 Synthetic route to JY11, JY12 and JY13. | |
Photophysical and electrochemical properties
The photophysical characteristics of JY11, JY12 and JY13 were investigated by UV-vis absorption spectra. The absorption spectra of the three dyes in THF solutions and on TiO2 films are shown in Fig. 2. As can be seen from Fig. 2a, all dyes exhibit two distinct absorption bands in THF solutions. The absorption bands in the high-energy region (<370 nm) correspond to the localized aromatic π–π* electron transitions of the conjugated backbone, and the other relatively stronger absorption bands in the low-energy region (>370 nm) can be assigned to the intramolecular charge transfer (ICT) transitions from the electron donating part to the electron-withdrawing group.13 The maximum absorption peaks (λmax) for JY11, JY12 and JY13 are located at 468, 459 and 484 nm, respectively. Furthermore, it can be found that the presence of additional thiophene units in JY13 (ε = 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 170 M−1 cm−1) significantly red-shifts its ICT absorption band and results in great enhancement in the molar extinction coefficient relative to JY11 (ε = 51155 M−1 cm−1) and JY12 (ε = 48914 M−1 cm−1), illustrating the importance of extension of electron-rich groups on facilitating the donor–acceptor interactions in dipolar compounds and decreasing the HOMO–LUMO gap of the dyes. As shown in Fig. 2b, when adsorbed on TiO2, all these dyes showed slight blue-shift absorption peaks as compared to the corresponding spectra recorded in THF solutions. The blue-shift observed for these dyes is due to partial deprotonation of the carboxylic acid unit because of the interaction between the dye and titanium dioxide. The data of absorption and electrochemical properties of these dyes are summarized in Table 1.
170 M−1 cm−1) significantly red-shifts its ICT absorption band and results in great enhancement in the molar extinction coefficient relative to JY11 (ε = 51155 M−1 cm−1) and JY12 (ε = 48914 M−1 cm−1), illustrating the importance of extension of electron-rich groups on facilitating the donor–acceptor interactions in dipolar compounds and decreasing the HOMO–LUMO gap of the dyes. As shown in Fig. 2b, when adsorbed on TiO2, all these dyes showed slight blue-shift absorption peaks as compared to the corresponding spectra recorded in THF solutions. The blue-shift observed for these dyes is due to partial deprotonation of the carboxylic acid unit because of the interaction between the dye and titanium dioxide. The data of absorption and electrochemical properties of these dyes are summarized in Table 1.
 |
| | Fig. 2 UV-vis absorption spectra of these dyes (a) in THF solutions and (b) on TiO2 films. | |
Table 1 Photophysical, electrochemical data of JY11–13a
| Dye |
λmax/nm |
ε/M−1 cm−1 |
Eox/V |
E0–0/eV |
Ered/V |
HOMO/LUMO/eV |
| First oxidation potentials (vs. NHE) in benzonitrile were calibrated with ferrocene (0.63 V vs. NHE).14 E0–0 values (zeroth–zeroth transition energies) were estimated from the onset wavelength in absorption spectra in THF. Ered = Eox − E0–0. NHE vs. the vacuum level was set to 4.5 V.15 |
| JY11 |
468 |
51155 |
0.88 |
2.07 |
−1.19 |
−5.38/−3.31 |
| JY12 |
459 |
48914 |
0.89 |
2.09 |
−1.20 |
−5.39/−3.30 |
| JY13 |
484 |
85170 |
0.83 |
1.98 |
−1.15 |
−5.33/−3.35 |
To evaluate the possibility of electron injection from the excited state dye to the conduction band (CB) of TiO2 and dye regeneration on TiO2 electrode, cyclic voltammetry measurement was performed in benzonitrile solution with a 100 mV s−1 scan rate and calibrated against ferrocene, using 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. Cyclic voltammograms of the dyes are depicted in Fig. 3. As shown in Fig. 3, all three dyes exhibited two reversible oxidation couples. The highest occupied molecular orbitals (HOMOs) of JY11–13 corresponding to their first oxidation potentials (Eox) were 0.88, 0.89 and 0.83 V (vs. NHE), respectively. The band gap energies (E0–0) of the three dyes JY11–13 were 2.07, 2.09 and 1.98 eV, respectively, which were calculated from their onset wavelength in absorption spectra in THF solutions. The lowest unoccupied molecular orbitals (LUMOs) of JY11–13, which could be calculated from Eox − E0–0, were −1.19, −1.20 and −1.15 V (vs. NHE), respectively. Consequently, the LUMO levels of JY11–13 are all more negative than the CB of the TiO2 electrode (−0.5 V vs. NHE), indicating that electron injection from the LUMO orbital into the CB of TiO2 is energetically permitted. On the other hand, the HOMO levels of JY11–13 are sufficiently more positive than the I−/I3− redox couple (+0.4 V vs. NHE), ensuring the thermodynamic regeneration of the oxidized form of the dyes in a DSSC.
 |
| | Fig. 3 Cyclic voltammograms of the dyes recorded in benzonitrile solutions. | |
Computational analysis
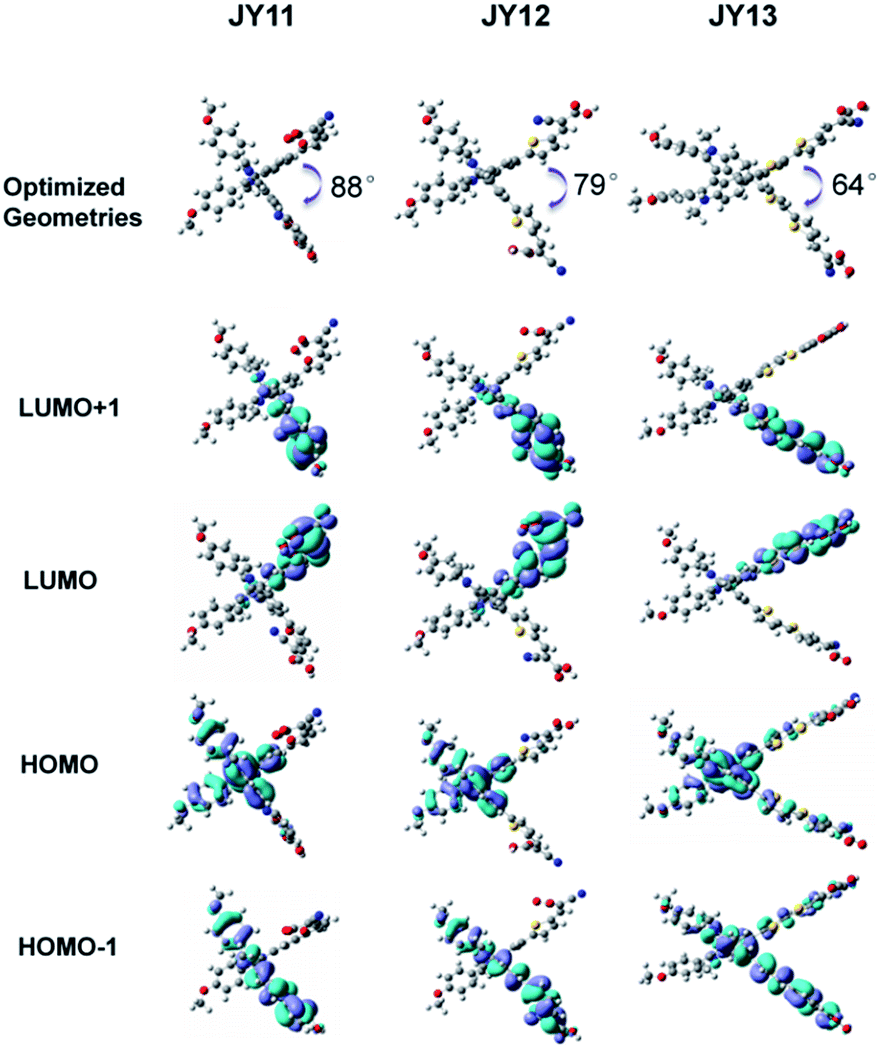
To deep investigate the geometrical structures and the frontier molecular orbital configurations of the three double D–π–A type dyes, density functional theory calculations were conducted with the Gaussian 03 program at the B3LYP/6-31G(d) level. All octyl groups have been replaced with methyl groups to save calculation time. The optimized geometries and frontier molecular orbital distributions of three dyes are shown in Fig. 4. Optimized geometry demonstrated all dyes present a “X” shape, which benefits the suppression of electron recombination and improvement of open-circuit voltage. Notably, the intersection angle of the two D–π–A branches of JY11 (88°) is more closer to vertical angle as compared to that of JY12 (79°) and JY13 (64°), which indicates a weaker intermolecular aggregation of JY11 and the cell based on JY11 may have a relative higher open-circuit voltage. As shown in Fig. 4, for the HOMOs of all dyes, the electron density is uniformly distributed on biindole core. For the HOMO−1 state, the electron density is almost distributed on single D–π–A branch. At the meantime, the LUMO and LUMO−1 of these dyes are very similar, and the electron density shows localized electron distributions through the each single cyanoacrylic acid unit and its adjacent π-spacer. Because the HOMO, HOMO−1, LUMO and LUMO+1 are more important than other orbitals because their electron transitions are very favorable for the photon-to-electron conversion and the electron may be easily transferred from the HOMO and HOMO−1 to the LUMO and LUMO+1 and then be injected into the semiconductor. Therefore, these electron distributions will preferentially ensure that electrons will be efficient transferred from the biindole core through the electron-rich π-space to the cyanoacrylic acid acceptor. From the analysis of the TD-DFT results in Table 2, we can assign the ICT absorption peaks of the three dyes as transitions from HOMO−1, HOMO−2 and HOMO−3 to LUMO and LUMO+1. The electronic transitions in the three dyes are positively contributed to electron injection since ICT transitions correspond to electron transfer from the whole molecule to the anchoring group. The order of calculated absorption peaks and absorption intensity of these dyes are also similarly ranked as those of experimental values.
 |
| | Fig. 4 The optimized geometries and frontier molecular orbital distributions of three dyes optimized by DFT calculations. | |
Table 2 Excitation wavelengths, orbital energies, oscillator strength (f) and assignment of the dyes in vacuum computed using B3LYP/6-31G(d)
| Dye |
λmax/nm |
Calculated energy/eV |
f |
Transition assignmenta |
| H = HOMO, L = LUMO. |
| JY11 |
431 |
2.88 |
0.62 |
H−3 → L (70%) |
| H−2 → L (9%) |
| JY12 |
428 |
2.90 |
0.60 |
H−1 → L+1 (33%) |
| H−3 → L (18%) |
| H−2 → L+1 (17%) |
| H−2 → L (15%) |
| JY13 |
489 |
2.54 |
1.41 |
H−1 → L+1 (41%) |
| H−2 → L (28%) |
DSSC performance
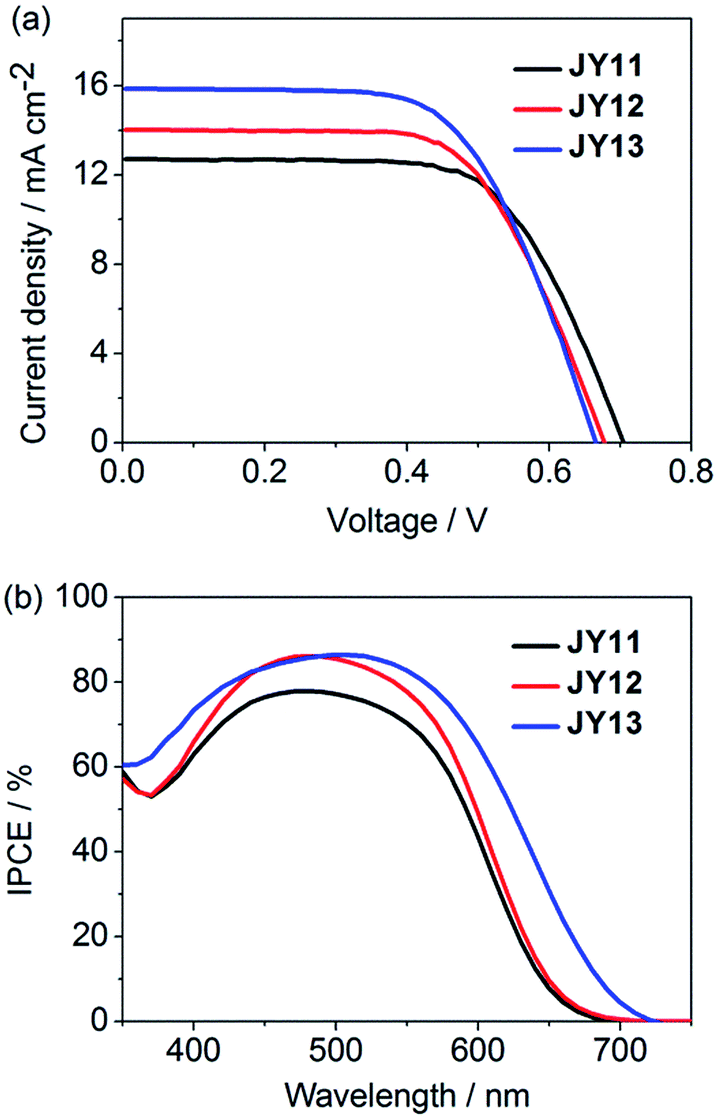
The photovoltaic characteristics of the DSSCs based on the three dyes under simulated AM 1.5G solar irradiation (100 mW cm−2) have been investigated, employing a liquid electrolyte composed of 0.3 M 1,2-dimethyl-3-propylimidazolium iodide (DMPII), 0.1 M LiI, 0.05 M I2 and 0.5 M 4-tert-butyl pyridine (TBP) in acetonitrile. The DSSCs performance parameters of JY11–13 and commercial N719 dye are displayed in Table 3. The photocurrent density–voltage (J–V) curves of JY11–13 are plotted in Fig. 5a. The JY12 dye exhibited a short-circuit photocurrent density (Jsc) of 14.0 mA cm−2, an open-circuit photovoltage (Voc) of 674 mV and a fill factor (FF) of 0.64, generating a PCE of 6.05%. The JY11 dye showed a 27 mV improved Voc benefitting from its nearly vertical intersection angle of the two D–π–A branches as compared to that of JY12, which endowed JY11 the potential of reduced intermolecular π–π stacking. Nevertheless, the JY11 dye displayed a lower Jsc of 12.7 mA cm−2 and a FF of 0.66, resulting in a slightly reduced PCE of 5.87%. Notably, JY13 exhibited a lowest Voc (663 mV) and a lowest FF of 0.62 with the extension of π-bridge compared to JY11 and JY12. But greatly benefited from its broader and more intense ICT band, JY13 finally displayed a relatively higher Jsc of 15.8 mA cm−2 and an increased PCE of 6.54%.
Table 3 Photovoltaic performance of JY11–13 with N719 as a referencea
| Dye |
Voc/mV |
Jsc/mA cm−2 |
FF |
PCE/% |
| Active area was 0.196 cm2 with a metal mask; used an electrolyte composed of 0.3 M 1,2-dimethyl-3-propylimidazolium iodide (DMPII), 0.1 M LiI, 0.05 M I2 and 0.5 M 4-tert-butyl pyridine in acetonitrile; applied the commercial N719 dye for comparison. |
| JY11 |
701 |
12.7 |
0.66 |
5.87 |
| JY12 |
674 |
14.0 |
0.64 |
6.05 |
| JY13 |
663 |
15.8 |
0.62 |
6.54 |
| N719 |
765 |
17.2 |
0.62 |
8.20 |
 |
| | Fig. 5 (a) The photocurrent density–voltage (J–V) curves and (b) the IPCE spectra of JY11–13. | |
In order to analyze the difference between the Jsc values of the three dyes, the incident photon-to-current conversion efficiencies (IPCE) as a function of incident wavelength for DSSCs based on these dyes are plotted in Fig. 5b. As shown in IPCE spectra, all of the dyes JY11–13 could efficiently convert the light to photocurrents in the UV-vis region and the onsets of the IPCE spectra for the three dyes were at 690, 700 and 720 nm, respectively. JY13-based cell gave over 60% IPCE values from 350 to 610 nm with a maximum IPCE value of 86% at 500 nm, which ensured a good light-harvesting ability and a high photocurrent. The high IPCE value and broader absorption of JY13 explained the high Jsc value from the J–V measurement. The somewhat higher efficiency observed for JY13 was mainly due to the relatively larger Jsc. The best efficiency of JY13-based cell reached about 80% of the commercial N719-based standard cell that fabricated and measured under the same conditions, indicating the 3,3′-biindole skeleton is a promising candidate to construct effective double D–π–A branched organic dyes for DSSCs.
Electrochemical impedance spectroscopy
Electrochemical impedance spectroscopy (EIS) is a powerful technique of characterizing the important interfacial charge transfer and carrier transportation process in DSSCs.16 EIS Nyquist plots and Bode phase plots of DSSCs based on JY11, JY12 and JY13 measured in the dark under forward bias (−0.65 V) with frequency range of 100 kHz to 100 mHz are shown in Fig. 6. In the Nyquist plots, the larger semicircle in the lower frequency range represents the interfacial charge transfer resistances (Rct) at the TiO2–dyes–electrolyte interface.17 A larger radius of the major semicircle means a larger charge transfer resistance, indicating that the electron recombination in the devices are strongly reduced and therefore a smaller dark current and a larger value of photovoltage.18 The fitted Rct increased in the order of JY13 (44 Ω) < JY12 (77 Ω) < JY11 (120 Ω). This trend appeared to be consistent with the values of Voc of JY13 (663 mV) < JY12 (674 mV) < JY11 (701 mV). Besides, electron lifetime (τ) can be extracted from the chemical capacitance (Cμ) and Rct using τ = Rct × Cμ.19 In general, longer electron lifetime implies increased resistance between the injected electrons and the electrolyte, which consequently improves the Voc.20 The electron lifetime was in the order with the calculated values of JY13 (24.5 ms) < JY12 (34.4 ms) < JY11 (70.1 ms). So the higher Voc of JY11 could be further explained.
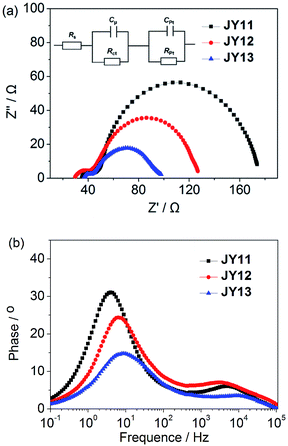 |
| | Fig. 6 (a) Nyquist plots and (b) Bode phase plots for DSSCs based on the three dyes measured in the dark under −0.65 V bias. | |
Conclusions
In conclusion, three novel double D–π–A branched organic dyes JY11, JY12 and JY13 composed of biindole donors, cyanoacrylic acid acceptors and aromatic π-bridges have been successfully designed, synthesized and applied as photosensitizers for DSSCs. Most importantly, the double D–π–A branched biindole dyes exhibit X-shaped structure, which is favorable for the suppression of the intermolecular interactions and finally results in the reduced charge recombination rates as well as good light-harvesting, large photocurrent and high PCE values in DSSCs. All three sensitizers have shown significant performance in DSSCs. The key difference of the photophysical characteristics among the three sensitizers locates at the absorption properties where a broader and more intense ICT band can be observed for sensitizer JY13 with bithiophene as the π-linker. The PCE value of the solar cell based on JY13 is up to 6.54%. This work emphasizes the potential of the application of double D–π–A branched biindole dyes for efficient DSSCs. Work focusing on the molecular modification of X-shape dye is ongoing in our lab.
Experimental
Materials and methods
All solvents were purified according to standard methods. Other chemicals (AR) obtained from commercial sources were used without any further purification. NMR solvents were used as received. 1H NMR and 13C NMR spectra were recorded on Bruker 400 MHz spectrometer using TMS as the internal standard. UV-vis spectra were obtained on a Varian Cary 300 Conc UV-visible spectrophotometer. HR-MS data was obtained on a Varian 7.0T FTMS. Cyclic voltammetry experiments and electrochemical impedance spectroscopy (EIS) were recorded on an electrochemical workstation (Zennium, Zahner Corporation). Cyclic voltammetry experiments were carried out using a conventional three-electrode system employing a glassy carbon electrode as the working electrode, a Ag/Ag+ electrode as the reference electrode, and a Pt wire as the counter electrode. The redox potentials were measured in dichloromethane, using 0.1 M n-Bu4NPF6 as the supporting electrolyte with a scan rate of 100 mV s−1.
Fabrication and characterization of DSSCs
A TiO2 film (∼10 μm) for the transparent nanocrystalline layer was prepared according to the doctor-blade method by coating a commercial 20 nm TiO2 sol (China National Academy of Nanotechnology & Engineering) onto the treated FTO conductive glass (Nippon Sheet Glass, Japan, fluorine-doped SnO2 over layer, sheet resistance of 15 Ω sq−1, treated with TiCl4 (0.05 M) aqueous solution). The scattering layer (∼4 μm) was applied over the transparent layer by doctor-blade method, then gradually heated to 500 °C and sintered for 60 min. The resulting TiO2 electrodes were treated by TiCl4 (0.04 M) aqueous solution at 70 °C for 60 min and sintered again at 500 °C for 60 min. The Pt electrode was obtained by thermal deposition a platinum layer on the surface of FTO at 450 °C for 30 min. The TiO2 photoanodes were immersed in the commercial N719 dye solution (0.3 mM in ethanol) for 12 h. The adsorption of the biindole-based dyes on TiO2 was carried out with 0.3 mM dye solution in THF for 12 h. The electrolyte was composed of 0.3 M DMPII, 0.1 M LiI, 0.05 M I2 and 0.5 M 4-tert-butylpyridine in acetonitrile. The DSSCs were illuminated by a solar simulator (CHF-XM-500W, Trusttech Co. Ltd) under 100 mW cm−2 irradiation, which was calibrated by a standard silicon solar cell (91150V, Newport Corporation). The photocurrent intensity–voltage (J–V) characteristic curves of the DSSC under simulated sunlight were recorded using an electrochemical workstation (Zennium, Zahner Corporation). The measurement of the incident photon-to-current conversion efficiency (IPCE) was performed using a commercial setup (QTest Station 2000 IPCE Measurement System, CROWNTECH, USA).
Synthetic procedures
Synthesis of compound 2. Compound 2 was prepared according to literature procedure.12 To the solution of 2-[(2-aminophenyl)ethynyl]phenylamine (compound 1) (0.50 g, 2.4 mmol) and 4-octyloxybenzaldehyde (1.69 g, 7.2 mmol) in CH3CN (35 mL) was added concentrated hydrochloric acid (0.1 mL, 37% aqueous solution). The mixture was stirred at reflux and monitored by TLC. After 1 h, the solvent was evaporated and the residue was purified by silica-gel column chromatography (eluent: hexane–EtOAc = 95:5) to afford compound 2 as a pale yellow solid (yield: 82.0%). 1H NMR (400 MHz, acetone-d6) δ 10.58 (s, 2H), 7.51 (d, J = 8.6 Hz, 4H), 7.46 (d, J = 8.4 Hz, 2H), 7.13–7.03 (m, 4H), 6.87 (t, J = 7.5 Hz, 2H), 6.71 (d, J = 8.7 Hz, 4H), 3.87 (t, J = 6.5 Hz, 4H), 1.73–1.62 (m, 4H), 1.43–1.25 (m, 20H), 0.86 (t, J = 6.5 Hz, 6H). 13C NMR (101 MHz, acetone-d6) δ 158.34, 136.71, 135.17, 130.32, 127.86, 125.81, 121.54, 119.54, 119.11, 114.25, 110.92, 105.88, 67.50, 31.69, 29.14, 29.08, 28.43, 25.87, 22.44, 13.51.
Synthesis of compound 3. t-BuOK (0.97 g, 8.6 mmol) was added to a solution of compound 2 (1.54 g, 2.4 mmol) in dry THF (30 mL), and the resulting mixture was stirred at room temperature for 0.5 h. 1-Bromooctane (1.86 g, 9.6 mmol) was then added, and the mixture was heated at reflux for another 3 hours before evaporated. The crude product was purified by silica-gel column chromatography using CH2Cl2–petroleum ether (1:4) as the eluent to afford compound 3 as a colorless oil liquid (yield: 73.2%). 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 7.25 (s, 2H), 7.12 (t, J = 7.4 Hz, 2H), 6.67 (d, J = 8.3 Hz, 4H), 6.63 (d, J = 8.3 Hz, 4H), 4.03 (t, J = 7.1 Hz, 4H), 3.94 (t, J = 6.4 Hz, 4H), 1.87–1.75 (m, 4H), 1.56–1.47 (m, 4H), 1.41–1.11 (m, 40H), 0.95–0.85 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 158.05, 138.69, 136.54, 131.36, 129.56, 124.89, 120.96, 120.72, 118.91, 113.82, 109.62, 107.42, 67.96, 43.86, 31.88, 31.77, 29.80, 29.46, 29.33, 29.15, 29.06, 27.62, 26.74, 26.13, 22.72, 22.64, 14.16, 14.13. HR-MS (MALDI): m/z [M]+ calcd for C60H84N2O2, 864.6533, found, 864.6528.
Synthesis of compound 4a. A solution of N-bromosuccinimide (NBS) (166 mg, 0.93 mmol) in CH2Cl2 (15 mL) was added dropwise to a solution of compound 3 (402 mg, 0.46 mmol) in CH2Cl2 (40 mL) at 0 °C. The mixture was then slowly warmed to room temperature and stirred for 1 h before it was poured into water. The organic phase was separated and dried over anhydrous Na2SO4. After the solvent was evaporated, the crude product was transferred to a two-neck round-bottomed flask. Then, 5-formylfuran-2-boronic acid (167 mg, 1.20 mmol), Pd(dppf)Cl2 (38 mg, 0.046 mmol), K2CO3 (730 mg, 2.30 mmol), toluene (15 mL) and methanol (5 mL) were added. The flask was charged with N2. The mixture was reflux for 5 h before it was poured into water. The organic phase was separated and dried over anhydrous Na2SO4. After the solvent was removed under vacuum, the crude product was purified by silica-gel column chromatography using CH2Cl2 as the eluent to afford compound 4a as an orange thickness liquid (yield: 76.4%). 1H NMR (400 MHz, CDCl3) δ 9.64 (s, 2H), 7.95 (s, 2H), 7.54–7.45 (m, 4H), 7.38 (s, 2H), 6.88 (s, 2H), 6.72 (d, J = 8.1 Hz, 4H), 6.66 (d, J = 7.9 Hz, 4H), 4.13 (t, J = 7.0 Hz, 4H), 3.94 (t, J = 6.3 Hz, 4H), 1.85–1.76 (m, 4H), 1.54–1.45 (m, 4H), 1.39–1.07 (m, 40H), 0.92 (t, J = 5.8 Hz, 6H), 0.86 (t, J = 7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 176.63, 161.97, 158.50, 151.56, 141.46, 136.60, 131.14, 130.66, 124.03, 121.96, 120.69, 117.26, 114.05, 107.61, 107.06, 106.72, 68.00, 43.94, 31.85, 31.73, 29.87, 29.72, 29.43, 29.29, 29.09, 28.98, 26.58, 26.10, 22.69, 22.61, 14.13, 14.10. HR-MS (MALDI): m/z [M]+ calcd for C70H88N2O6, 1052.6642, found, 1052.6662.
Synthesis of compound 4b. The synthesis method resembles that of compound 4a and the compound was purified by silica-gel column chromatography using CH2Cl2 as the eluent to afford compound 4b as an orange thickness liquid (yield: 80.2%). 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 2H), 7.73 (d, J = 17.9 Hz, 4H), 7.51 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.4 Hz, 4H), 6.69 (d, J = 8.4 Hz, 4H), 6.65 (d, J = 8.1 Hz, 4H), 4.07 (t, J = 7.0 Hz, 4H), 3.93 (t, J = 5.9 Hz, 4H), 1.84–1.76 (m, 4H), 1.53–1.45 (m, 4H), 1.39–1.11 (m, 40H), 0.90 (t, J = 6.4 Hz, 6H), 0.85 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 182.66, 158.50, 157.04, 141.25, 141.16, 137.90, 136.70, 131.14, 130.53, 126.26, 123.96, 123.12, 120.88, 118.22, 114.06, 108.08, 107.47, 68.01, 44.00, 31.87, 31.75, 29.85, 29.74, 29.44, 29.31, 29.12, 29.00, 26.68, 26.12, 22.71, 22.64, 14.16, 14.14. HR-MS (MALDI): m/z [M]+ calcd for C70H88N2O4S2, 1084.6186, found, 1084.6210.
Synthesis of compound 4c. The synthesis method resembles that of compound 4a and the compound was purified by silica-gel column chromatography using CH2Cl2 as the eluent to afford compound 4c as a red thickness liquid (yield: 70.5%). 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 2H), 7.67 (d, J = 3.8 Hz, 2H), 7.61 (s, 2H), 7.50 (d, J = 8.2 Hz, 2H), 7.38 (dd, J = 4.9, 2.5 Hz, 4H), 7.31 (d, J = 3.8 Hz, 2H), 7.27 (d, J = 3.9 Hz, 2H), 6.67 (d, J = 8.7 Hz, 4H), 6.63 (d, J = 8.8 Hz, 4H), 4.05 (t, J = 6.8 Hz, 4H), 3.91 (t, J = 6.5 Hz, 4H), 1.85–1.73 (m, 4H), 1.51–1.43 (m, 4H), 1.40–1.10 (m, 40H), 0.89 (t, J = 6.7 Hz, 6H), 0.84 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 182.43, 158.37, 148.70, 147.79, 141.06, 140.53, 137.57, 136.79, 133.75, 131.19, 129.73, 127.31, 126.77, 124.22, 123.57, 123.24, 120.89, 117.93, 114.00, 107.49, 107.26, 68.01, 43.91, 31.86, 31.77, 29.80, 29.73, 29.44, 29.40, 29.31, 29.12, 28.99, 26.67, 26.11, 22.70, 22.65, 14.14. HR-MS (MALDI): m/z [M]+ calcd for C78H92N2O4S4, 1248.5940, found, 1248.5938.
Synthesis of JY11. A mixture of compound 4a (80 mg, 0.15 mmol), cyanoacetic acid (38 mg, 0.45 mmol), piperidine (0.10 mL), CHCl3 (10 mL) and CH3CN (10 mL) was heated at reflux for 12 h and then acidified with 2 M aqueous hydrochloric acid (10 mL). The crude product was extracted into CHCl3, and the organic layer was washed with water and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the crude product was purified by silica-gel column chromatography using CH2Cl2–CH3OH (8:1) as the eluent to afford JY11 as a red solid (yield: 80.5%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 2H), 8.04 (s, 2H), 7.54 (d, J = 3.4 Hz, 2H), 7.50 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 3.3 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.2 Hz, 4H), 6.73 (d, J = 8.2 Hz, 4H), 4.23–4.04 (m, 4H), 3.87 (t, J = 5.3 Hz, 4H), 1.72–1.61 (m, 4H), 1.59–1.49 (m, 4H), 1.39–1.32 (m, 4H), 1.29–1.19 (m, 16H), 1.18–1.11 (m, 4H), 1.09–0.95 (m, 16H), 0.83 (t, J = 6.4 Hz, 6H), 0.77 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 164.69, 161.34, 158.60, 147.31, 141.72, 139.64, 137.70, 136.76, 131.27, 130.01, 125.36, 123.83, 121.99, 120.32, 117.56, 117.26, 114.46, 109.22, 107.54, 96.16, 67.68, 34.84, 31.70, 31.59, 30.86, 29.19, 29.16, 29.01, 28.94, 28.62, 26.12, 25.96, 22.55, 22.50, 14.36, 14.32. HR-MS (MALDI): m/z [M]+ calcd for C76H90N4O8, 1186.6759, found, 1186.6755.
Synthesis of JY12. JY12 was synthesized according to the procedure described for JY11 and the compound was purified by silica-gel column chromatography using CH2Cl2–CH3OH (8:1) as the eluent to afford JY12 as a red solid (yield: 82.1%). 1H NMR (400 MHz, DMSO-d6) δ 8.47 (s, 2H), 8.08–7.95 (m, 4H), 7.80 (d, J = 3.9 Hz, 2H), 7.32 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 8.3 Hz, 2H), 6.88 (d, J = 8.4 Hz, 4H), 6.73 (d, J = 8.6 Hz, 4H), 4.25–4.07 (m, 4H), 3.89 (t, J = 6.2 Hz, 4H), 1.72–1.62 (m, 4H), 1.55–1.46 (m, 4H), 1.41–1.33 (m, 4H), 1.29–1.21 (m, 16H), 1.17–1.10 (m, 4H), 1.06–0.92 (m, 16H), 0.84 (t, J = 6.6 Hz, 6H), 0.77 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 164.30, 158.62, 155.94, 146.82, 141.99, 141.75, 136.92, 133.90, 131.27, 130.20, 126.04, 124.59, 123.90, 120.54, 118.62, 117.28, 114.53, 108.79, 107.45, 97.78, 67.75, 43.48, 31.70, 31.57, 29.43, 29.19, 29.15, 29.02, 28.92, 28.63, 26.12, 25.96, 22.55, 22.49, 14.39, 14.35. HR-MS (MALDI): m/z [M]+ calcd for C76H90N4O6S2, 1218.6302, found, 1218.6297.
Synthesis of JY13. JY13 was synthesized according to the procedure described for JY11 and the compound was purified by silica-gel column chromatography using CH2Cl2–CH3OH (6:1) as the eluent to afford JY13 as a deep red solid (yield: 85.2%). 1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 2H), 7.95 (d, J = 3.9 Hz, 2H), 7.87 (s, 2H), 7.60 (d, J = 3.4 Hz, 2H), 7.56 (d, J = 3.5 Hz, 2H), 7.54 (d, J = 3.7 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 6.82 (d, J = 8.1 Hz, 4H), 6.68 (d, J = 8.1 Hz, 4H), 4.39–4.03 (m, 4H), 3.96–3.73 (m, 4H), 1.75–1.55 (m, 4H), 1.58–1.40 (m, 4H), 1.37–0.93 (m, 40H), 0.81 (t, J = 6.8 Hz, 6H), 0.75 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 164.13, 158.51, 148.24, 146.61, 146.54, 141.98, 140.89, 137.01, 133.97, 133.17, 131.28, 129.47, 128.78, 126.57, 124.83, 124.70, 124.13, 120.36, 118.04, 117.16, 114.45, 107.93, 107.41, 98.30, 67.75, 43.40, 31.71, 31.60, 29.45, 29.21, 29.16, 29.05, 28.93, 28.66, 26.14, 25.98, 22.55, 22.50, 14.37, 14.34. HR-MS (MALDI): m/z [M]+ calcd for C84H94N4O6S4, 1382.6056, found, 1382.6050.
Acknowledgements
We are grateful to the 973 Program (2011CB932502), and the National Natural Science Foundation of China (nos: 21172126 and 21272123) for their generous financial support.
Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
-
(a) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed;
(b) J. Mao, N. He, Z. Ning, Q. Zhang, F. Guo, L. Chen, W. Wu, J. Hua and H. Tian, Angew. Chem., Int. Ed., 2012, 51, 9873–9876 CrossRef CAS PubMed.
-
(a) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–633 CrossRef CAS PubMed;
(b) A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977 CrossRef CAS PubMed;
(c) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
-
(a) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed;
(b) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC;
(c) N. Cai, J. Zhang, M. Xu, M. Zhang and P. Wang, Adv. Funct. Mater., 2013, 23, 3539–3547 CrossRef CAS;
(d) K. Ladomenou, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2014, 4, 21379–21404 RSC;
(e) E. Gabrielsson, H. Ellis, S. Feldt, H. Tian, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Energy Mater., 2013, 3, 1647–1656 CrossRef CAS.
-
(a) Z. Wang, M. Liang, Y. Hao, Y. Zhang, L. Wang, Z. Sun and S. Xue, J. Mater. Chem. A, 2013, 1, 11809–11819 RSC;
(b) J. Zhao, X. Yang, M. Cheng, S. Li, X. Wang and L. Sun, J. Mater. Chem. A, 2013, 1, 2441–2446 RSC;
(c) Y. Tan, M. Liang, Z. Lu, Y. Zheng, X. Tong, Z. Sun and S. Xue, Org. Lett., 2014, 16, 3978–3981 CrossRef CAS PubMed;
(d) Z. Yao, L. Yang, Y. Cai, C. Yan, M. Zhang, N. Cai, X. Dong and P. Wang, J. Phys. Chem. C, 2014, 118, 2977–2986 CrossRef CAS;
(e) X. Guo, H. N. Tsao, P. Gao, D. Xia, C. An, M. K. Nazeeruddin, M. Baumgarten, M. Grätzel and K. Müllen, RSC Adv., 2014, 4, 54130–54133 RSC.
- A. Baheti, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, J. Mater. Chem. A, 2014, 2, 5766–5779 CAS.
-
(a) X. Ren, S. Jiang, M. Cha, G. Zhou and Z.-S. Wang, Chem. Mater., 2012, 24, 3493–3499 CrossRef CAS;
(b) M. Cai, X. Pan, W. Liu, J. Sheng, X. Fang, C. Zhang, Z. Huo, H. Tian, S. Xiao and S. Dai, J. Mater. Chem. A, 2013, 1, 4885–4892 RSC.
-
(a) A. Venkateswararao, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 2524–2535 CrossRef PubMed;
(b) S. Jiang, S. Fan, X. Lu, G. Zhou and Z.-S. Wang, J. Mater. Chem. A, 2014, 2, 1–11 Search PubMed.
-
(a) N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org. Chem., 2014, 32, 7069–7086 CrossRef;
(b) X. Lu, X. Jia, Z.-S. Wang and G. Zhou, J. Mater. Chem. A, 2013, 1, 9697–9706 RSC.
-
(a) A. Abbotto, V. Leandri, N. Manfredi, F. De Angelis, M. Pastore, J. H. Yum, M. K. Nazeeruddin and M. Grätzel, Eur. J. Org. Chem., 2011, 31, 6195–6205 CrossRef;
(b) V. Leandri, R. Ruffo, V. Trifiletti and A. Abbotto, Eur. J. Org. Chem., 2013, 30, 6793–6801 CrossRef;
(c) R. Y.-Y. Lin, F.-L. Wu, C.-H. Chang, H.-H. Chou, T.-M. Chuang, T.-C. Chu, C.-Y. Hsu, P.-W. Chen, K.-C. Ho, Y.-H. Lo and J. T. Lin, J. Mater. Chem. A, 2014, 2, 3092–3101 RSC.
- X. Qian, Y.-Z. Zhu, J. Song, X.-P. Gao and J.-Y. Zheng, Org. Lett., 2013, 15, 6034–6037 CrossRef CAS PubMed.
- A. Arcadi, M. Chiarini, G. D'Anniballe, F. Marinelli and E. Pietropaolo, Org. Lett., 2014, 16, 1736–1739 CrossRef CAS PubMed.
- Y.-Y. Lin, H.-W. Lin, Y.-S. Yen, C.-H. Chang, H.-H. Chou, P.-W. Chen, C.-Y. Hsu, Y.-C. Chen, J. T. Lin and K.-C. Ho, Energy Environ. Sci., 2013, 6, 2477–2486 Search PubMed.
- Y. Liang, B. Peng, J. Liang, Z. Tao and J. Chen, Org. Lett., 2010, 12, 1204–1207 CrossRef CAS PubMed.
- P. Gao, H. N. Tsao, M. Grätzel and M. K. Nazeeruddin, Org. Lett., 2012, 14, 4330–4333 CrossRef CAS PubMed.
-
(a) D. Kumar, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, J. Org. Chem., 2014, 79, 3159–3172 CrossRef CAS PubMed;
(b) W.-Q. Liu, Z.-G. Liang, D.-X. Kou, L.-H. Hu and S.-Y. Dai, Electrochim. Acta, 2013, 88, 395–403 CrossRef CAS PubMed.
-
(a) L.-Y. Chang, C.-P. Lee, R. Vittal, J.-J. Lin and K.-C. Ho, J. Mater. Chem. A, 2013, 1, 3055–3060 RSC;
(b) H. Choi, S. Paek, K. Lim, C. Kim, M.-S. Kang, K. Song and J. Ko, J. Mater. Chem. A, 2013, 1, 8226–8233 RSC;
(c) J. Tian, X. Yang, J. Zhao, L. Wang, W. Wang, J. Li and L. Sun, RSC Adv., 2014, 4, 34644–34648 RSC;
(d) X. Yang, J. Zhao, L. Wang, J. Tian and L. Sun, RSC Adv., 2014, 4, 24377–24383 RSC.
-
(a) J. Liu, X. Yang, A. Islam, Y. Numata, S. Zhang, N. T. Salim, H. Chen and L. Han, J. Mater. Chem. A, 2013, 1, 10889–10897 RSC;
(b) S. Zhu, Z. An, X. Chen, P. Chen and Q. Liu, RSC Adv., 2014, 4, 42252–42259 RSC.
-
(a) L.-Y. Lin, M.-H. Yeh, C.-P. Lee, J. Chang, A. Baheti, R. Vittal, K. R. J. Thomas and K.-C. Ho, J. Power Sources, 2014, 247, 906–914 CrossRef CAS PubMed;
(b) Z. Wan, C. Jia, Y. Duan, X. Chen, Z. Li and Y. Lin, RSC Adv., 2014, 4, 34896–34903 RSC.
- G. E. Zervaki, M. S. Roy, M. K. Panda, P. A. Angaridis, E. Chrissos, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2013, 52, 9813–9825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all new compounds. See DOI: 10.1039/c4ra12888e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.