
Synthesis of (−)-mesembrine using the quaternary carbon-constructing allylic substitution†‡
Takuri
Ozaki
and
Yuichi
Kobayashi
*
Department of Bioengineering, Tokyo Institute of Technology, Box B-52, Nagatsuta-cho 4259, Midori-ku, Yokohama 226-8501, Japan. E-mail: ykobayas@bio.titech.ac.jp
First published on 3rd February 2015
Abstract
Synthesis of (−)-mesembrine was studied by using the allylic substitution for construction of quaternary carbons. The required allylic picolinate 6 (R = TBS) with the three substituents on the olefin was synthesized by hydromagnesiation of propargylic alcohol (S)-13 followed by iodination, Pd-catalyzed coupling of the resulting vinyl iodide (S)-14 with CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)2MgBr and esterification with PyCO2H. The substitution proceeded stereo- and regioselectively to afford SN2′ product 9, which was transformed to keto aldehyde 29 by substitution with NsMeNH, Wacker oxidation and ozonolysis. Finally, aldol reaction and subsequent de-nosylation afforded the title compound. During the synthesis, the coupling of the vinyl iodide with alkylmetals was investigated under the Negishi conditions.
CH(CH2)2MgBr and esterification with PyCO2H. The substitution proceeded stereo- and regioselectively to afford SN2′ product 9, which was transformed to keto aldehyde 29 by substitution with NsMeNH, Wacker oxidation and ozonolysis. Finally, aldol reaction and subsequent de-nosylation afforded the title compound. During the synthesis, the coupling of the vinyl iodide with alkylmetals was investigated under the Negishi conditions.
Introduction
One significant challenge in modern synthetic organic chemistry is the stereoselective synthesis of all carbon quaternary stereogenic centers.1 Recently, we published an allylic substitution for construction of stereogenic centers as shown in Scheme 1, in which the use of Cu(acac)2 as a copper source and the picolinoxy leaving group are responsible for attaining anti SN2′ with high stereo- and regioselectivity.2 The method is suited to aryl copper reagents, which are less reactive than alkyl reagents. To demonstrate high efficiency and reliability of this method, we selected mesembrine (1)3 as a synthetic target. This alkaloid possesses the 3a-(aryl)octahydroindole core unit, which is also found in other alkaloids such as crinine (2)4 and tazettine (3)5 (Fig. 1). Mesembrine (1), found in the plant species Sceletium tortuosum, is a potent inhibitor of 5-hydroxytryptamine (5-HT) uptake (IC50 27 nM)6 by binding to the 5-HT transporter.7 Previously, 1 was synthesized in optically active8 and racemic9 forms by using quaternary carbon-forming reactions, whereas our approach to 1 features allylic substitution.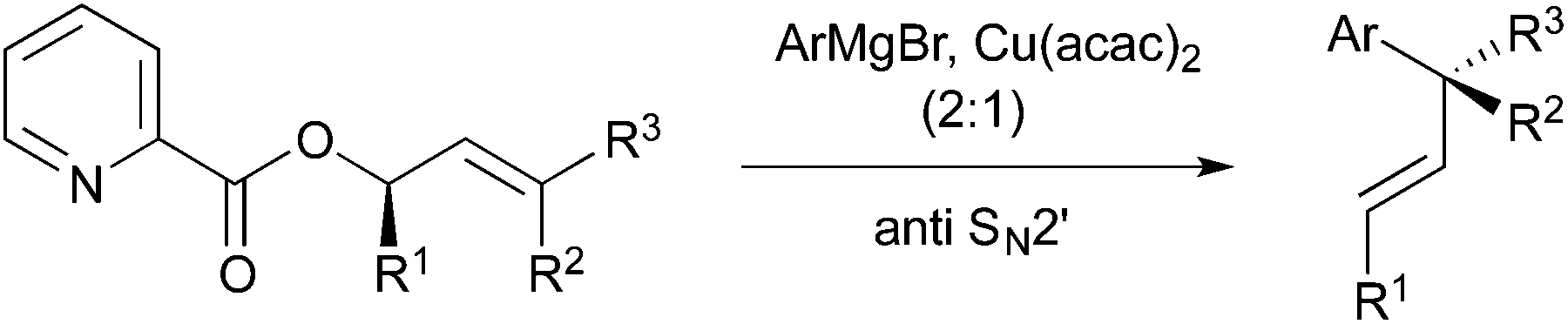 | ||
| Scheme 1 Allylic substitution developed recently. | ||
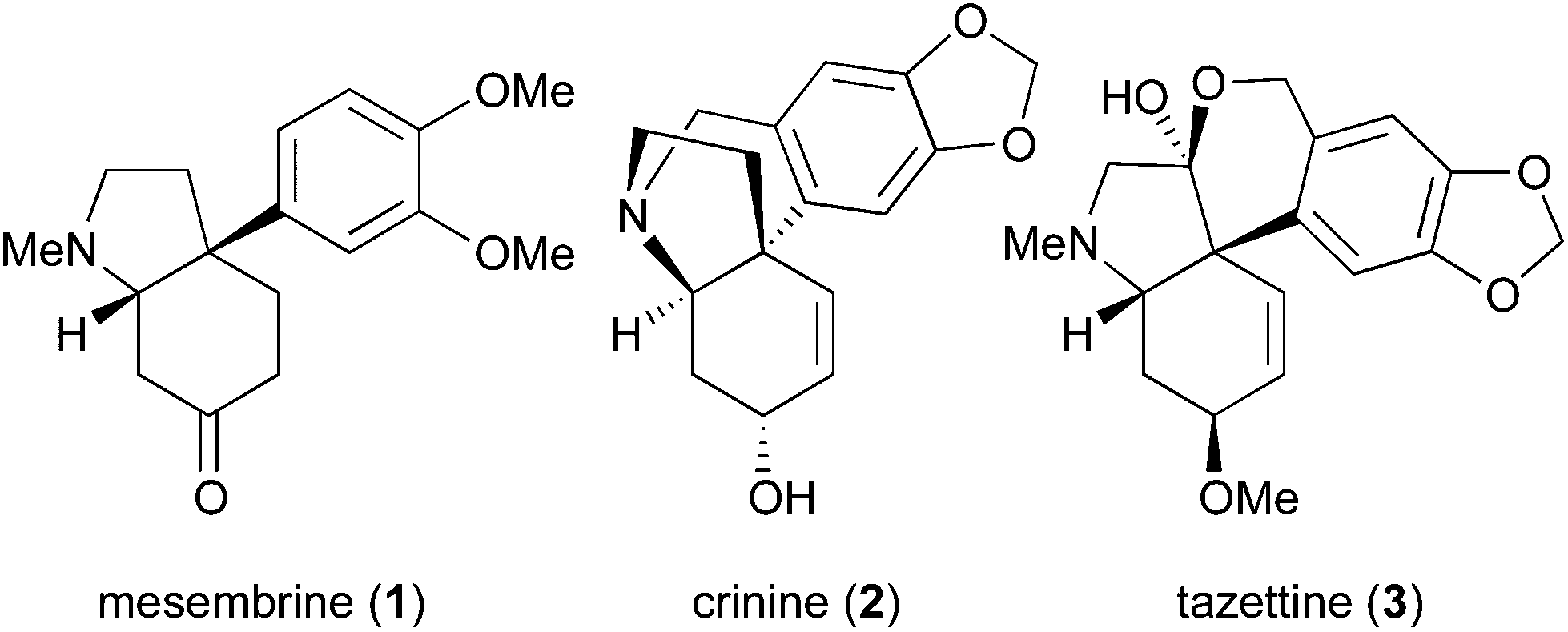 | ||
| Fig. 1 Alkaloids possessing the 3a-(aryl)octahydroindole substructure. | ||
Results and discussion
We envisaged that allylic substitution of picolinates 6 and/or 7 with copper reagent 8 would produce the key intermediate 9via the anti-SN2′ pathway and that the further transformation indicated in the structure of 9 would afford enone 10, which upon intramolecular addition of the nitrogen to the olefin should produce 1 (Scheme 2).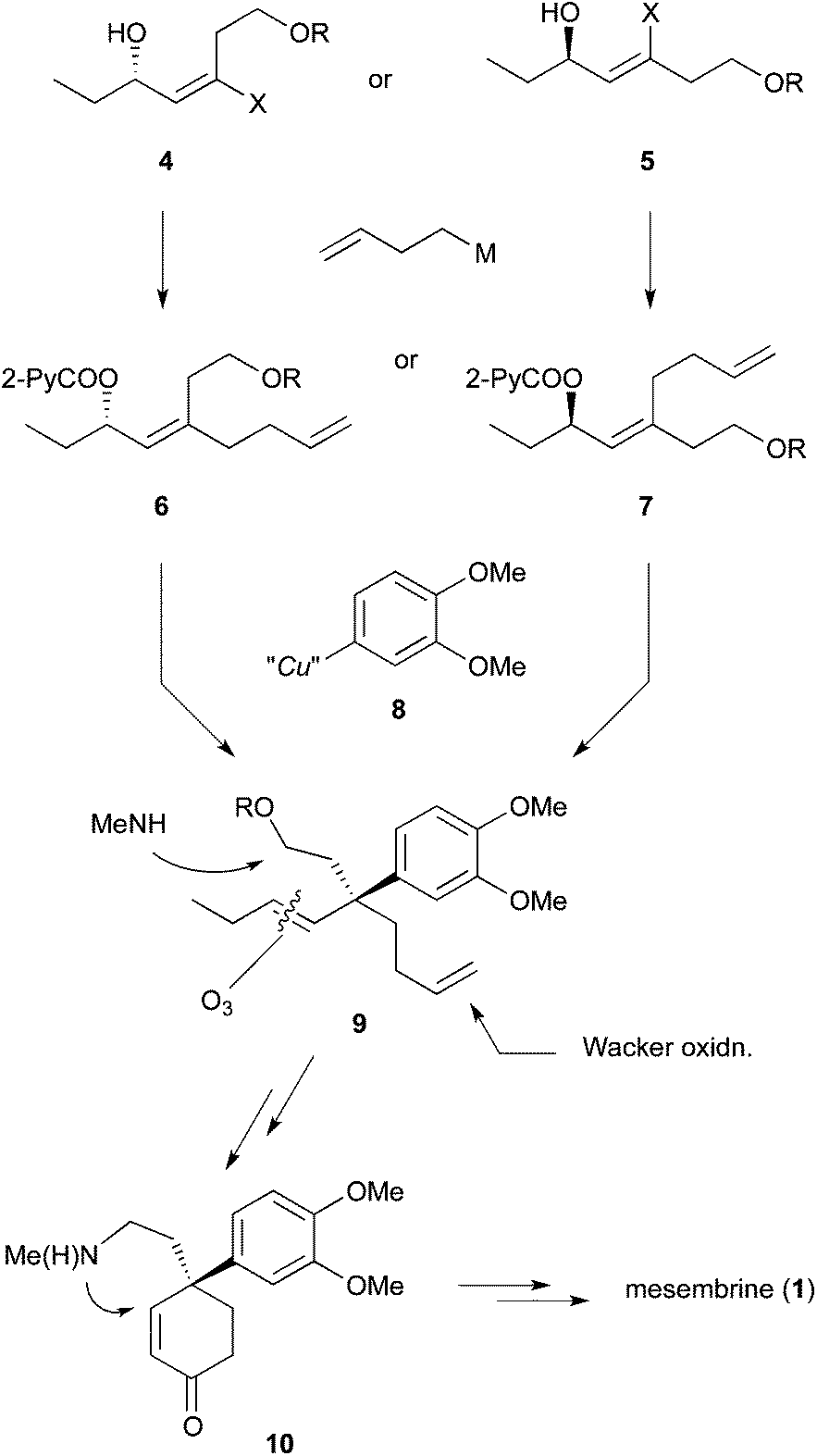 | ||
| Scheme 2 Synthetic plan of mesembrine (1). | ||
As an approach to 6 and 7, coupling of haloallylic alcohols 4, 5 or their derivatives with a 3-butenylmetal was conceived. Coupling of such secondary alcohol derivatives with alkylboranes is found in the literature,10 while that of sterically less hindered primary alcohol derivatives11 and simple alkenyl halides12,13 has been published. However, preparation of the required 3-butenylborane seemed difficult in our case. Consequently, (Z)- and (E)-iodoallylic alcohols and their derivatives 12a–c and 14, 16 were prepared by the methods14,15 delineated in Scheme 3 for a preliminary study of the coupling with model reagents of BuZnCl or BuMgBr (Scheme 4 and Table 1).
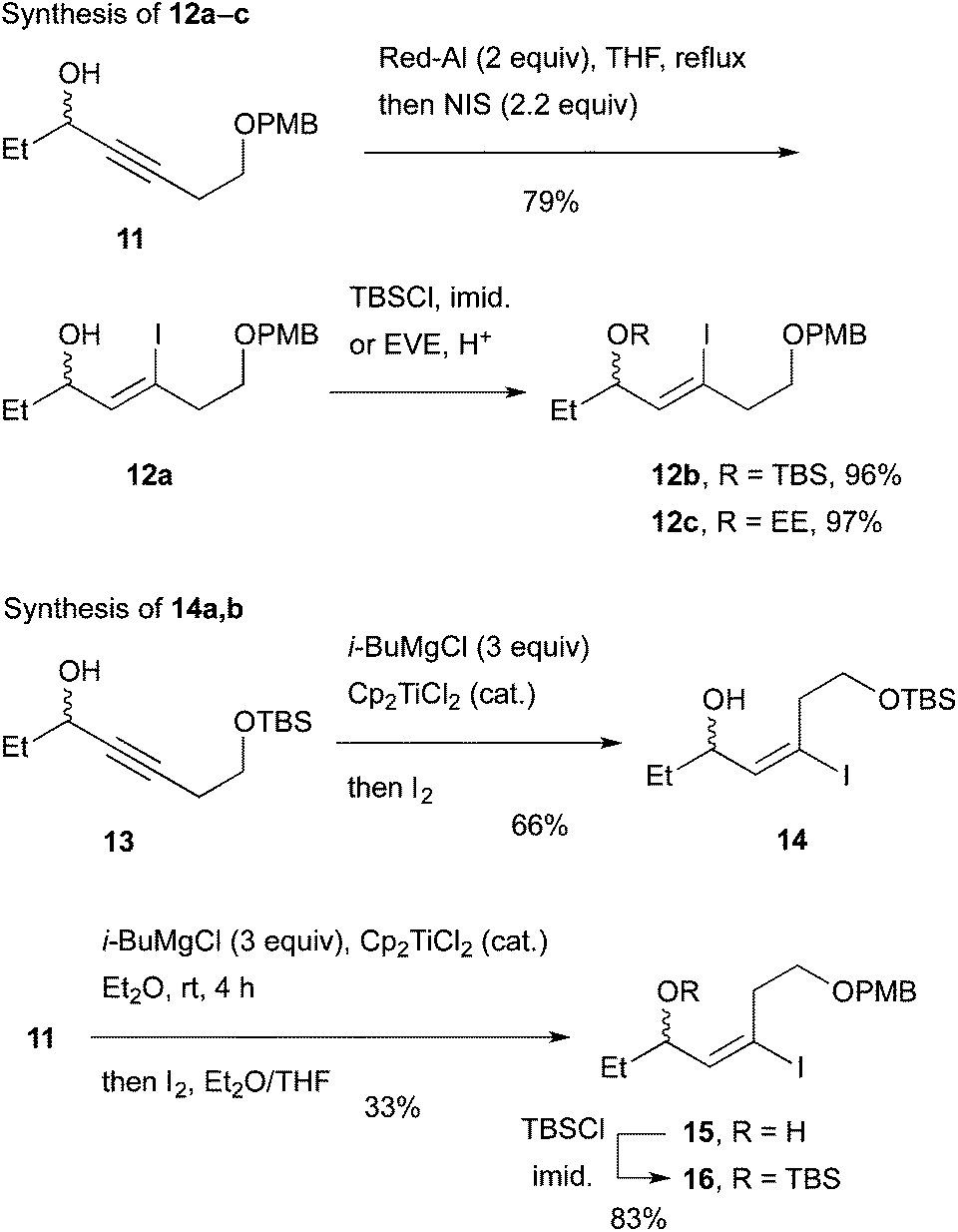 | ||
| Scheme 3 Synthesis of racemic iodides for a model study of coupling with Bu-M. | ||
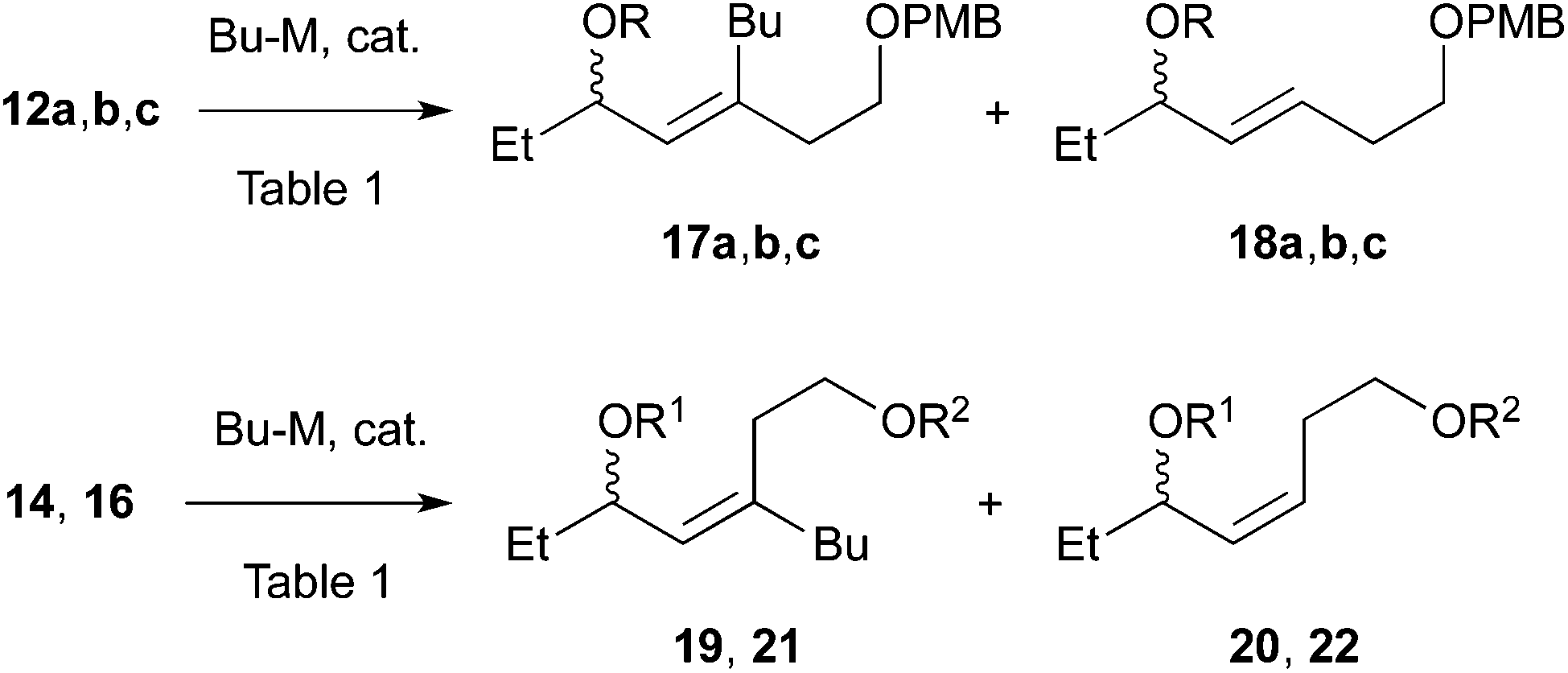 | ||
| Scheme 4 Coupling of (E)- and (Z)-iodides with Bu-M. R for 12, 17, and 18: a, H; b, TBS, c, EE. R1 and R2 for 14, 19, and 20: R1 = H, R2 = TBS. As above for 16, 21, and 22: R1 = TBS, R2 = PMB. | ||
| Entry | Substrate | Bu-Ma (equiv.) | Catalystb (mol %) | Time (h) | Temp. | Result (ratio)c |
|---|---|---|---|---|---|---|
| a BuZnCl was prepared by mixing BuMgBr and ZnCl2. b Except Pd(PPh3)4 and PdCl2(MeCN)2 the other Pd catalysts were prepared in situ by mixing Pd2(dba)3·CHCl3 and the ligands. c The product ratios calculated by 1H NMR were given in parentheses. d Incomplete reactions at rt for 8 h (entry 5) and at rt for 17 h (entry 7). e (E)-7-(TBSO)-4-hepten-3-ol. | ||||||
| 1 | 12a | BuZnCl (4) | Pd(PPh3)4 (5) | 8 | rt | 18a + 12a |
| 2 | 12a | BuZnCl (4) | Pd(AsPh3)2 (5) | 14 | rt | 18a + 12a |
| 3 | 12a | BuZnCl (4) | PdCl2(MeCN)2 (5) | 20 | rt | No reaction |
| 4 | 12a | BuZnCl (4) | Pd(dppf) (5) | 14 | rt | No reaction |
| 5 | 12b | BuZnCl (2) | Pd(PPh3)4 (5) | 20d | rt |
17b + 18b (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25) :25) |
| 6 | 12b | BuMgBr (2) | Pd(PPh3)4 (5) | 9 | rt | 18b |
| 7 | 12c | BuZnCl (2) | Pd(PPh3)4 (10) | 4 | 50 °Cd |
17c + 18c (75:25) |
| 8 | 16 | BuZnCl (2) | Pd(PPh3)4 (5) | 40 | rt |
21 + 22 (90:10) |
| 9 | 16 | BuZnCl (2) | Pd(DavePhos)2 (5) | 40 | rt | 21 + 22 + 16 |
| 10 | 16 | BuZnCl (2) | Pd(IPr)2 (5) | 40 | rt | 21 + 22 + 16 |
| 11 | 14 | BuZnCl (4) | Pd(PPh3)4 (5) | 18 | 50 °C |
19 + 20 (89:11) |
| 12 | 14 | BuMgBr (5) | Pd(PPh3)4 (5) | 11 | 50 °C |
19 + 20 (87:13) |
| 13 | 14 | BuMgBr (5) | Ni(acac)2 (20) | 11 | 50 °C | trans-Olefine |
Since protection/deprotection manipulation adds two steps in synthesis of 1, coupling of alcohol 12a with BuZnCl and Pd(PPh3)4 was investigated first. The reaction under the Negishi conditions16 produced reduction product 18a (Table 1, entry 1), indicating that dehalogenation took place predominantly. The same olefin was produced with Ni catalysts such as Ni(acac)2 and NiCl2(dppp) as well (data not shown). The use of several other Pd catalysts shown in entries 2–4 resulted in almost no reaction or reduction to 18a. We then examined coupling of TBS ether 12b, which produced the desired product 17b and reduction product 18b in a 75:25 ratio by 1H NMR (entry 5). The reduction was predominant with BuMgBr to produce 18b (entry 6). Pd-catalyzed reaction of EE ether 12c with BuZnCl gave results similar to that of the TBS ether (entry 7).
With the above results in mind, coupling of (E)-olefin 16 with the TBS group was examined with BuZnCl and Pd(PPh3)4 to afford 21 with better product selectivity over the reduction product 22 (entry 8). A sterically less congested reaction site (C–I) of 16 might be responsible for the higher selectivity. The Pd catalysts with the DavePhos and IPr ligands retarded the reaction (entries 9 and 10). Finally, coupling of alcohol 14 with BuZnCl afforded 19 with an almost similar product selectivity to that of 16 (entry 11, cf. entry 8). Surprisingly, the use of BuMgBr produced 19 as well (entry 12, cf. entry 6). In contrast, Ni(acac)2 produced the (E)-isomer of 20 (entry 13).
In total, (E)-iodoalkenyl isomers 14 and 16 gave 19 and 21 with 87–90% product selectivity. On the other hand, 15 (the precursor of 16) was synthesized only in 33% yield by the hydromagnesiation/iodination of alcohol 11, whereas 13 possessing the TBS group was transformed to 14 in good yield. On the basis of these results, allylic picolinate 6 (R = TBS) in Scheme 2 was selected as the key intermediate for the synthesis of 1.
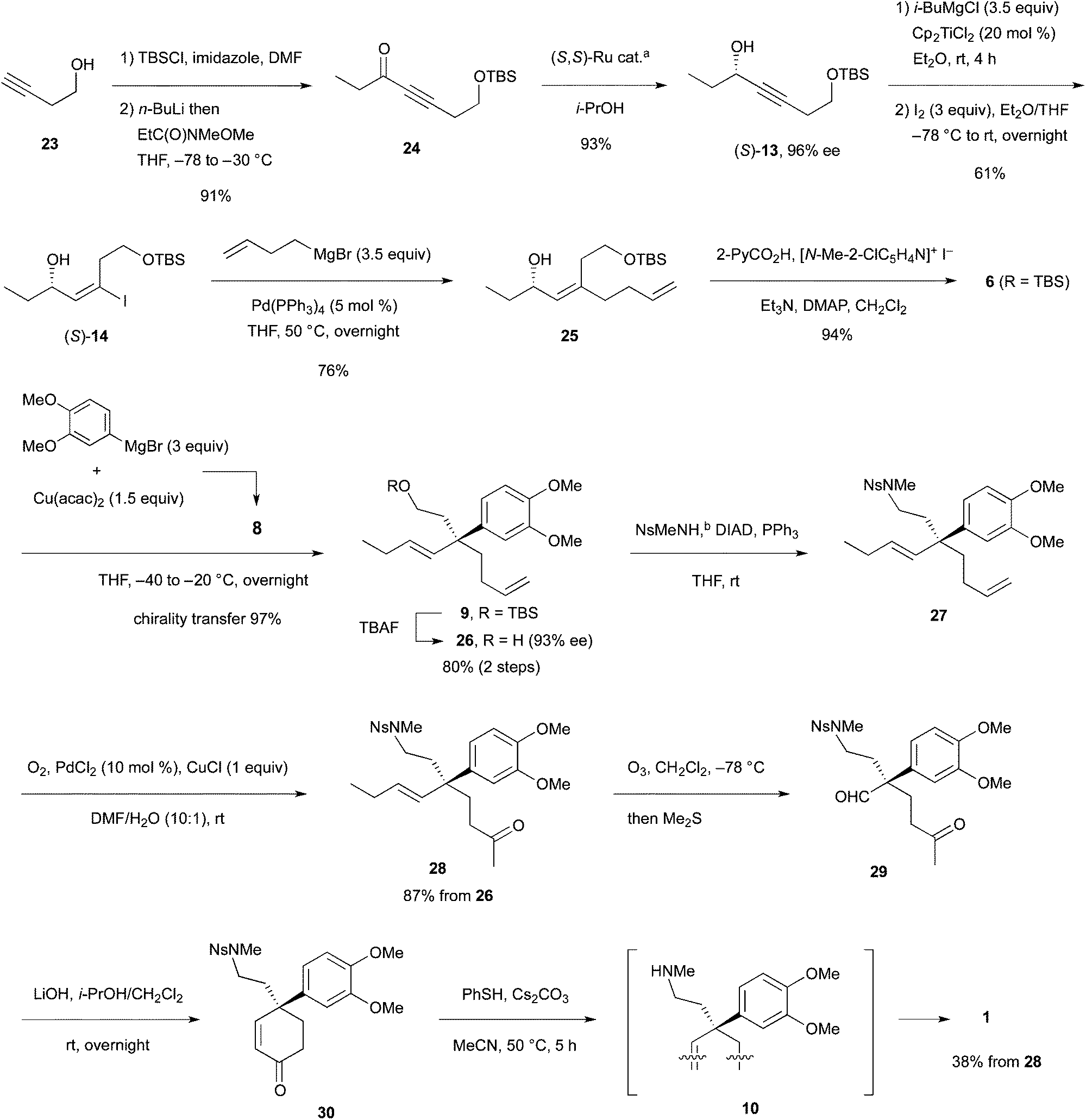
As delineated in Scheme 5, alcohol 23 was converted to ynone 2417 in two steps. Asymmetric hydrogen transfer18 of 24 afforded propargylic alcohol (S)-13 with 96% ee (chiral HPLC of the derived benzoate) in 93% yield. Hydromagnesiation15 of this alcohol with i-BuMgCl and Cp2TiCl2 in Et2O proceeded smoothly and the resulting vinylmagnesium species was subjected to iodination with I2 in THF to produce (S)-14 stereoselectively in 61% yield. Pd-catalyzed coupling of the iodide with 3-butenylmagnesium bromide under the conditions of Table 1, entry 12, proceeded smoothly to afford the desired product 25 in 76% yield after chromatography. Finally, esterification with 2-PyCO2H furnished the key intermediate 6 (R = TBS) in a high yield.
 | ||
| Scheme 5 Synthesis of (−)-mesembrine (1). aRu[(1S,2S)-TsDPEN](p-cymene). bNs: o-(NO2)C6H4SO2. | ||
Among the two types of reagents for the allylic substitution (ArMgBr/Cu(acac)2, 2:1; that is, 3:1 with ZnI2), the copper reagent 8 (1.5 equiv.) derived from 3,4-(MeO)2C6H3MgBr and Cu(acac)2 in a 2:1 ratio was investigated for the allylic substitution with 6. The substitution proceeded smoothly at −40 to −20 °C to afford 9, which upon deprotection with TBAF produced alcohol 26 in 80% yield over two steps. Absorbance corresponding to the regioisomer of 26 (structure not shown) was not detected by 1H NMR spectroscopy at the expected region (5.0–5.4 ppm) and 97% chirality transfer (CT) was determined by chiral HPLC analysis (93% ee of 26). Amination of alcohol 26 with NsMeNH19 (Ns: o-(NO2)C6H4SO2) under the Mitsunobu conditions afforded nosyl amine 27, which upon the Wacker oxidation20 produced methyl ketone 28 in 87% from alcohol 26 (two steps).
The last four-step conversion including ozonolysis of 28, aldol reaction, de-nosylation, and the addition of the free amino group to the enone was studied first using racemic olefin rac-28 prepared similarly (Scheme 6). Ozonolysis afforded keto aldehyde rac-29 in 67% yield. Subsequently, aldol reaction with LiOH in i-PrOH–CH2Cl2 (4:1) at room temperature overnight produced enone rac-30 in 63% isolated yield. The Ns group in rac-30 was removed with the standard reagent/solvent (PhSH, Cs2CO3, MeCN) at 50 °C for 3 h to afford racemic mesembrine (rac-1). Neither the free amine rac-10 nor the PhSH adduct to the enone (structure not shown) was detected by TLC and 1H NMR. Examined next was the one-pot synthesis of rac-1 from rac-29 by adding PhSH and MeCN into the i-PrOH–CH2Cl2 solution of the aldol mixture (containing rac-30 and LiOH). However, no de-nosylation took place and the enone intermediate rac-30 was isolated. Accordingly, the crude enone isolated was exposed to PhSH and LiOH in MeCN to furnish rac-1 in 50% yield over two steps from aldehyde rac-29. These results indicated that the de-nosylation was interfered with by i-PrOH. Indeed, aldol reaction with rac-29 conducted in MeCN with Cs2CO3 was followed by reaction with PhSH to afford rac-1 in 36% yield in one pot.
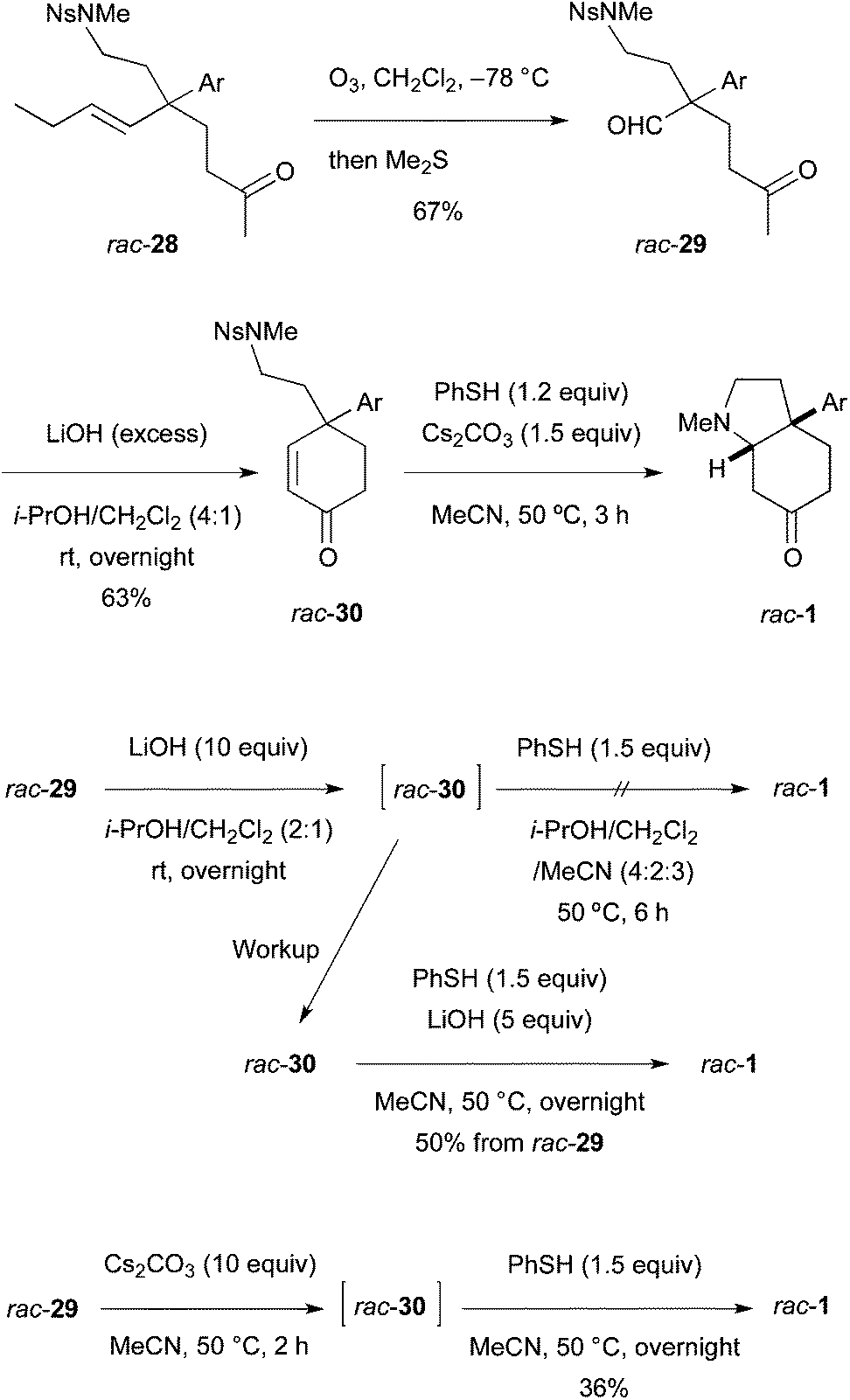 | ||
| Scheme 6 Study of the last four steps furnishing racemic mesembrine. Ar: 3,4-(MeO)2C6H3. | ||
Among the two-step and one-pot methods for the synthesis of rac-1 from keto aldehyde rac-29, the high yielding former method was applied to synthesis of optically active mesembrine from keto olefin 28 (Scheme 5). Thus, ozonolysis of 28 afforded crude keto aldehyde 29, which upon aldol reaction with LiOH gave enone 30. Finally, enone 30 was exposed to PhSH in MeCN at 50 °C to afford (−)-mesembrine (1) in 38% yield from 28. The yield including ozonolysis was almost comparable to that of the synthesis of racemic mesembrine (rac-1).
Conclusions
The allylic substitution for construction of quaternary carbons was applied to the synthesis of (−)-mesembrine. The substitution proceeded with high stereo- and regioselectivity. Furthermore, the required allylic picolinate with three substituents on the olefin was synthesized stereoselectively from the propargylic alcohol through Pd-catalyzed coupling of the vinyl iodide intermediate with CH2CH(CH2)2MgBr. The method would be applicable to synthesis of alkaloids with 3a-(aryl)octahydroindole.
Experimental section
General information
The 1H (300 MHz) and 13C NMR (75 MHz) spectroscopic data were recorded in CDCl3 using Me4Si (δ = 0 ppm) and the centerline of the triplet (δ = 77.1 ppm), respectively, as internal standards. Signal patterns are indicated as br s (broad singlet), s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are given in hertz (Hz). Chemical shifts of carbons are accompanied by minus (for C and CH2) and plus (for CH and CH3) signs of the attached proton test (APT) experiments. The solvents that were distilled prior to use are THF (from Na/benzophenone), Et2O (from Na/benzophenone), and CH2Cl2 (from CaH2).:1). 1H NMR (300 MHz, CDCl3) δ 0.07 (s, 6H), 0.90 (s, 9H), 1.00 (t, J = 7.5 Hz, 3H), 1.61–1.77 (m, 3H), 2.43 (dt, J = 1.8, 7.2 Hz, 2H), 3.72 (t, J = 7.2 Hz, 2H), 4.30 (tq, J = 1.8, 5.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ −5.3 (CH3, +), 9.5 (CH3, +), 18.3 (C, −), 23.1 (CH2, −), 25.9 (CH3, +), 31.1 (CH2, −), 61.9 (CH2, −), 63.9 (CH, +), 82.27 (C, −), 82.29 (C, −). HRMS (FAB) calcd for C13H27O2Si [(M + H)+] 243.1780, found 243.1780.
:1). 1H NMR (300 MHz, CDCl3) δ 0.115 (s, 3H), 0.118 (s, 3H), 0.918 (s, 9H), 0.922 (t, J = 7.5 Hz, 3H), 1.43–1.70 (m, 2H), 2.55 (dt, J = 14.4, 3.8 Hz, 1H), 2.94–3.06 (m, 2H), 3.67–3.83 (m, 2H), 4.21 (tq, J = 1.7, 6.3 Hz, 1H), 6.42 (d, J = 7.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ −5.4 (CH3, +), −5.3 (CH3, +), 9.7 (CH3, +), 18.7 (C, −), 26.1 (CH3, +), 29.1 (CH2, −), 42.8 (CH2, −), 60.8 (CH2, −), 70.2 (CH, +), 104.1 (C, −), 147.1 (CH, +). HRMS (FAB) calcd for C13H28O2SiI [(M + H)+] 371.0903, found 371.0906.
To a solution of the above product (320 mg, 1.07 mmol) in CH2Cl2 (10 mL) were added picolinic acid (197 mg, 1.60 mmol), Et3N (0.450 mL, 3.23 mmol), DMAP (131 mg, 1.07 mmol), and 2-chloro-1-methylpyridinium iodide (410 mg, 1.60 mmol). The mixture was stirred at rt overnight, and diluted with brine with vigorous stirring. The layers were separated and the aqueous layer was extracted with CH2Cl2 three times. The combined extracts were washed with brine, dried over MgSO4, and concentrated to give a residue, which was purified by chromatography on silica gel (hexane–EtOAc) to afford 6 (407 mg, 94%) as a pale yellow oil: Rf = 0.36 (hexane–EtOAc, 3:1). 1H NMR (300 MHz, CDCl3) δ 0.05 (s, 6H), 0.88 (s, 9H), 0.96 (t, J = 7.4 Hz, 3H), 1.64–1.79 (m, 1H), 1.82–1.98 (m, 1H), 2.08–2.24 (m, 4H), 2.29–2.42 (m, 1H), 2.55–2.66 (m, 1H), 3.60–3.78 (m, 2H), 4.92 (d, J = 10.2 Hz, 1H), 4.99 (d, J = 17.1 Hz, 1H), 5.37 (d, J = 9.3 Hz, 1H), 5.70–5.85 (m, 2H), 7.45 (ddd, J = 7.8, 4.8, 1.2 Hz, 1H), 7.83 (dt, J = 7.8, 1.2 Hz, 1H), 8.12 (dt, J = 7.8, 1.2 Hz, 1H), 8.77 (ddd, J = 4.8, 1.2, 1.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ −5.3 (CH3, +), 9.9 (CH3, +), 18.4 (C, −), 26.0 (CH3, +), 28.2 (CH2, −), 32.2 (CH2, −), 34.6 (CH2, −), 36.7 (CH2, −), 62.3 (CH2, −), 74.2 (CH, +), 114.7 (CH2, −), 124.9 (CH, +), 125.1 (CH, +), 126.6 (CH, +), 136.9 (CH, +), 138.1 (CH, +), 141.8 (C, −), 148.7 (C, −), 149.9 (CH, +), 164.6 (CO, −). HRMS (FAB) calcd for C23H38NO3Si [(M + H)+] 404.2621, found 404.2630.
To a solution of the above product in THF (15 mL) was added TBAF (1.0 M in THF, 2.50 mL, 2.50 mmol). The mixture was stirred at rt overnight, and diluted with saturated NH4Cl and EtOAc with vigorous stirring. The layers were separated and the aqueous layer was extracted with EtOAc three times. The combined extracts were washed with brine, dried over MgSO4, and concentrated to give a residue, which was purified by chromatography on silica gel (hexane–EtOAc) to afford 26 (379 mg, 80% from 6, 93% ee, 97% CT) as a colorless oil. The enantiomeric information was determined by HPLC analysis: Chiralcel OD-H; hexane–i-PrOH = 95/5, 1 mL min−1, 30 °C; tR (min) = 18.9 (S), 21.8 (R): Rf = 0.11 (hexane–EtOAc, 3:1). 1H NMR (300 MHz, CDCl3) δ 1.03 (t, J = 7.5 Hz, 3H), 1.12 (t, J = 5.4 Hz, 1H), 1.73–1.94 (m, 4H), 1.97–2.17 (m, 4H), 3.51–3.62 (m, 2H), 3.86 (s, 3H), 3.87 (s, 3H), 4.88–5.02 (m, 2H), 5.52 (d, J = 15.8 Hz, 1H), 5.58 (dt, J = 15.8, 5.7 Hz, 1H), 5.70–5.85 (m, 1H), 6.74–6.87 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 14.2 (CH3, +), 26.0 (CH2, −), 28.6 (CH2, −), 37.9 (CH3, −), 40.7 (CH3, −), 45.1 (C, −), 55.77 (CH3, +), 55.81 (CH3, +), 59.7 (CH2, −), 110.6 (CH, +), 110.8 (CH, +), 114.1 (CH2, −), 119.0 (CH, +), 130.5 (CH, +), 136.0 (CH, +), 138.7 (C, −), 139.0 (CH, +), 147.2 (C, −), 148.5 (C, −). HRMS (FAB) calcd for C19H28O3 [(M)+] 304.2038, found 304.2040.
To a solution of the above mixture in aqueous DMF (DMF–H2O = 10:1, 2.75 mL) were added PdCl2 (4.8 mg, 0.027 mmol) and CuCl (25.8 mg, 0.261 mmol). The mixture was stirred at rt under an atmosphere of O2 (balloon used) overnight, and diluted with saturated NH4Cl and EtOAc with vigorous stirring. The layers were separated and the aqueous layer was extracted with EtOAc three times. The combined extracts were washed with saturated NH4Cl and brine, dried over MgSO4, and concentrated to give a residue, which was purified by chromatography on silica gel (hexane–EtOAc) to afford 28 (116 mg, 87% from 26) as a pale yellow oil: Rf = 0.40 (hexane–EtOAc, 1:1). 1H NMR (300 MHz, CDCl3) δ 1.03 (t, J = 7.4 Hz, 3H), 1.95–2.18 (m, 6H), 2.10 (s, 3H), 2.25–2.33 (m, 2H), 2.85 (s, 3H), 2.95–3.03 (m, 2H), 3.86 (s, 3H), 3.88 (s, 3H), 5.44 (d, J = 15.9 Hz, 1H), 5.61 (dt, J = 15.9, 6.3 Hz, 1H), 6.77–6.82 (m, 3H), 7.52–7.74 (m, 3H), 7.83 (dd, J = 7.5, 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.2 (CH3, +), 26.0 (CH2, −), 30.2 (CH3, +), 31.7 (CH3, −), 34.9 (CH3, +), 36.4 (CH3, −), 38.6 (CH3, −), 44.6 (C, −), 46.6 (CH2, −), 55.8 (CH3, +), 55.9 (CH3, +), 110.5 (CH, +), 110.6 (CH, +), 119.0 (CH, +), 124.1 (CH, +), 130.6 (CH, +), 131.5 (CH, +), 131.6 (CH, +), 132.3 (C, −), 133.5 (CH, +), 134.5 (CH, +), 137.3 (C, −), 147.4 (C, −), 148.1 (C, −), 148.7 (C, −), 208.9 (CO, −). HRMS (FAB) calcd for C26H34N2O7S [(M)+] 518.2087, found 518.2079.
A mixture of the above aldehyde and LiOH (34.6 mg, 1.44 mmol) in i-PrOH–CH2Cl2 (4:1, 15 mL) was stirred at rt overnight, and diluted with saturated NH4Cl and CH2Cl2 with vigorous stirring. The layers were separated and the aqueous layer was extracted with CH2Cl2 three times. The combined extracts were washed with brine, dried over MgSO4, and concentrated to give enone 30, which was used for the next reaction without further purification: 1H NMR (300 MHz, CDCl3) δ 2.06–2.45 (m, 6H), 2.87 (s, 3H), 3.00–3.23 (m, 2H), 3.880 (s, 3H), 3.884 (s, 3H), 6.19 (d, J = 10.5 Hz, 1H), 6.76–6.85 (m, 3H), 7.05 (d, J = 10.5 Hz, 1H), 7.56–7.72 (m, 3H), 7.86 (dm, J = 7.2 Hz, 1H).
To a solution of the above enone in MeCN (12 mL) were added Cs2CO3 (142 mg, 0.437 mmol) and a solution of PhSH (0.030 mL, 0.292 mmol) in MeCN (3 mL) dropwise. The mixture was stirred at 50 °C for 5 h and diluted with brine and EtOAc with vigorous stirring. The layers were separated and the aqueous layer was extracted with EtOAc three times. The solution was concentrated to afford a residue, which was purified by preparative TLC (EtOAc) to afford (−)-mesembrine (1) (16.1 mg, 38% from 28) as a yellow oil: Rf = 0.17 (EtOAc). 1H NMR (300 MHz, CDCl3) δ 2.02–2.29 (m, 5H), 2.32 (s, 3H), 2.34–2.52 (m, 4H), 2.61 (d, J = 3.9 Hz, 2H), 2.95 (t, J = 3.5 Hz, 1H), 3.10–3.19 (m, 1H), 3.89 (s, 3H), 3.91 (s. 3H), 6.85 (d, J = 8.4 Hz, 1H), 6.90 (d, J = 2.1 Hz, 1H), 6.93 (dd, J = 8.4, 2.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 35.3 (CH2, −), 36.3 (CH2, −), 38.9 (CH2, −), 40.2 (CH3, +), 40.6 (CH2, −), 47.5 (C, −), 54.9 (CH2, −), 55.9 (CH3, +), 56.0 (CH3, +), 70.4 (CH, +), 109.9 (CH, +), 110.9 (CH, +), 117.9 (CH, +), 140.2 (C, −), 147.5 (C, −), 149.0 (C, −), 211.6 (CO, −). The 1H and 13C NMR spectra were consistent with those reported.8m,x [α]20D −43.6 (c 0.76, MeOH); lit.8m [α]30D −53.0 (c 0.24, MeOH); lit.8x [α]20D +43.0 (c 0.8, MeOH) for the (+)-isomer.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture, Japan.Notes and references
- (a) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890–1932 CrossRef CAS PubMed; (b) M. Fañanás-Mastral, M. Pérez, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 1922–1925 CrossRef PubMed; (c) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC; (d) P. P. Das, I. L. Lysenko and J. K. Cha, Angew. Chem., Int. Ed., 2011, 50, 9459–9461 CrossRef CAS PubMed; (e) M. Shimazu, Angew. Chem., Int. Ed., 2011, 50, 5998–6000 CrossRef PubMed; (f) J. A. Dabrowski, F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 4778–4781 CrossRef CAS PubMed; (g) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC; (h) C. Uyeda, A. R. Rötheli and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 9753–9756 CrossRef CAS PubMed; (i) F. Gao, Y. Lee, K. Mandai and A. H. Hoveyda, Angew. Chem., Int. Ed., 2010, 49, 8370–8374 CrossRef CAS PubMed; (j) I. Marek and G. Sklute, Chem. Commun., 2007, 1683–1691 RSC; (k) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS PubMed; (l) H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 4435–4439 CrossRef CAS PubMed; (m) A. H. Hoveyda, A. W. Hird and M. A. Kacprzynski, Chem. Commun., 2004, 1779–1785 RSC; (n) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688–1690 CrossRef CAS PubMed.
- C. Feng and Y. Kobayashi, J. Org. Chem., 2013, 78, 3755–3766 CrossRef CAS PubMed.
- (a) M. T. Smith, N. R. Crouch, N. Gericke and M. Hirst, J. Ethnopharmacol., 1996, 50, 119–130 CrossRef CAS; (b) N. Gericke and A. M. Viljoen, J. Ethnopharmacol., 2008, 119, 653–663 CrossRef CAS PubMed.
- W. E. Campbell, J. J. Nair, D. W. Gammon, C. Codina, J. Bastida, F. Viladomat, P. J. Smith and C. F. Albrecht, Phytochemistry, 2000, 53, 587–591 CrossRef CAS.
- (a) K. Likhitwitayawuid, C. K. Angerhofer, H. Chai, J. M. Pezzuto, G. A. Cordell and N. Ruangrungsi, J. Nat. Prod., 1993, 56, 1331–1338 CrossRef CAS; (b) J. R. Lewis, Nat. Prod. Rep., 1998, 15, 107–110 RSC; (c) Z. Jin, Nat. Prod. Rep., 2005, 22, 111–126 RSC.
- N. P. Gericke and W. B.-E. Van, PCT Int. Appl, WO9746234, 1997 Search PubMed.
- A. L. Harveya, L. C. Younga, A. M. Viljoenb and N. P. Gerickec, J. Ethnopharmacol., 2011, 137, 1124–1129 CrossRef PubMed.
- (a) S. Yamada and G. Otani, Tetrahedron Lett., 1971, 16, 1133–1136 CrossRef; (b) G. Otani and S. Yamada, Chem. Pharm. Bull., 1973, 21, 2130–2135 CrossRef CAS; (c) S. Takano, C. Murakata, Y. Imamura, N. Tamura and K. Ogasawara, Heterocycles, 1981, 16, 1291–1294 CrossRef CAS; (d) S. Takano, Y. Imamura and K. Ogasawara, Tetrahedron Lett., 1981, 22, 4479–4482 CrossRef CAS; (e) A. I. Meyers, R. Hanreich and K. T. Wanner, J. Am. Chem. Soc., 1985, 107, 7776–7778 CrossRef CAS; (f) S. Takano, K. Samizu and K. Ogasawara, Chem. Lett., 1990, 19, 1239–1242 CrossRef; (g) T. Yokomatsu, H. Iwasawa and S. Shibuya, Tetrahedron Lett., 1992, 33, 6999–7002 CrossRef CAS; (h) H. Kosugi, Y. Miura, H. Kanna and H. Uda, Tetrahedron: Asymmetry, 1993, 4, 1409–1412 CrossRef CAS; (i) H. Nemoto, T. Tanabe and K. Fukumoto, Tetrahedron Lett., 1994, 35, 6499–6502 CrossRef CAS; (j) H. Nemoto, T. Tanabe and K. Fukumoto, J. Org. Chem., 1995, 60, 6785–6790 CrossRef CAS; (k) T. Yoshimitsu and K. Ogasawara, Heterocycles, 1996, 42, 135–139 CrossRef CAS; (l) S. E. Denmark and L. R. Marcin, J. Org. Chem., 1997, 62, 1675–1686 CrossRef CAS; (m) M. Mori, S. Kuroda, C.-S. Zhang and Y. Sato, J. Org. Chem., 1997, 62, 3263–3270 CrossRef CAS PubMed; (n) T. Kamikubo and K. Ogasawara, Chem. Commun., 1998, 783–784 RSC; (o) O. Yamada and K. Ogasawara, Tetrahedron Lett., 1998, 39, 7747–7750 CrossRef CAS; (p) P. I. Dalko, V. Brun and Y. Langlois, Tetrahedron Lett., 1998, 39, 8979–8982 CrossRef CAS; (q) D. F. Taber and T. D. Neubert, J. Org. Chem., 2001, 66, 143–147 CrossRef CAS; (r) D. F. Taber and Y. He, J. Org. Chem., 2005, 70, 7711–7714 CrossRef CAS PubMed; (s) T. Paul, W. P. Malachowski and J. Lee, Org. Lett., 2006, 8, 4007–4010 CrossRef CAS PubMed; (t) S. Arns, M.-E. Lebrun, C. M. Grise, I. Denissova and L. Barriault, J. Org. Chem., 2007, 72, 9314–9322 CrossRef CAS PubMed; (u) E. A. Ilardi, M. J. Isaacman, Y. Qin, S. A. Shelly and A. Zakarian, Tetrahedron, 2009, 65, 3261–3269 CrossRef CAS PubMed; (v) Q. Gu and S.-L. You, Chem. Sci., 2011, 2, 1519–1522 RSC; (w) Q.-Q. Zhang, J.-H. Xie, X.-H. Yang, J.-B. Xie and Q.-L. Zhou, Org. Lett., 2012, 14, 6158–6161 CrossRef CAS PubMed; (x) K. Geoghegan and P. Evans, J. Org. Chem., 2013, 78, 3410–3415 CrossRef CAS PubMed.
- Recent syntheses: (a) J. H. Rigby and W. Dong, Org. Lett., 2000, 2, 1673–1675 CrossRef CAS PubMed; (b) M. G. Kulkarni, R. M. Rasne, S. I. Davawala and A. K. Doke, Tetrahedron Lett., 2002, 43, 2297–2298 CrossRef CAS; (c) Z. L. Song, B. M. Wang, Y. Q. Tu, C. A. Fan and S. Y. Zhang, Org. Lett., 2003, 5, 2319–2321 CrossRef CAS PubMed; (d) S. P. Chavan, D. A. Khobragade, A. B. Pathak and U. R. Kalkote, Tetrahedron Lett., 2004, 45, 5263–5265 CrossRef CAS PubMed; (e) N. T. Tam and C.-G. Cho, Org. Lett., 2007, 9, 3391–3392 CrossRef CAS PubMed; (f) C. Roe, E. J. Sandoe, G. R. Stephenson and C. E. Anson, Tetrahedron Lett., 2008, 49, 650–653 CrossRef CAS PubMed; (g) T. Ishikawa, K. Kudo, K. Kuroyabu, S. Uchida, T. Kudoh and S. Saito, J. Org. Chem., 2008, 73, 7498–7508 CrossRef CAS PubMed; (h) Y. Zhao, Y. Zhou, L. Liang, X. Yang, F. Du, L. Li and H. Zhang, Org. Lett., 2009, 11, 555–558 CrossRef CAS PubMed; (i) K. C. Guerard, C. Sabot, L. Racicot and S. Canesi, J. Org. Chem., 2009, 74, 2039–2045 CrossRef CAS PubMed; (j) K. C. Guerard, C. Sabot, M.-A. Beaulieu, M.-A. Giroux and S. Canesi, Tetrahedron, 2010, 66, 5893–5901 CrossRef CAS PubMed; (k) T. Honda, H. Arai, N. Yamamoto and K. Takahashi, Heterocycles, 2012, 84, 327–331 CrossRef CAS PubMed.
- E. Roulland, C. Monneret, J.-C. Florent, C. Bennejean, P. Renard and S. Léonce, J. Org. Chem., 2002, 67, 4399–4406 CrossRef CAS PubMed.
- (a) E. Negishi, Y. Zhang, F. E. Cederbaum and M. B. Webb, J. Org. Chem., 1986, 51, 4080–4082 CrossRef CAS; (b) L. Anastasia, Y. R. Dumond and E. Negishi, Eur. J. Org. Chem., 2001, 3039–3043 CrossRef CAS; (c) D. B. Berkowitz, B. Wu and H. Li, Org. Lett., 2006, 8, 971–974 CrossRef CAS PubMed.
- E. Negishi, L. F. Valente and M. Kobayashi, J. Am. Chem. Soc., 1980, 102, 3298–3299 CrossRef CAS.
- (a) T. Hayashi, M. Konishi and M. Kumada, Tetrahedron Lett., 1979, 1871–1874 CrossRef CAS; (b) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158–163 CrossRef CAS; (c) A. B. Smith III, T. J. Beauchamp, M. J. LaMarche, M. D. Kaufman, Y. Qiu, H. Arimoto, D. R. Jones and K. Kobayashi, J. Am. Chem. Soc., 2000, 122, 8654–8664 CrossRef.
- (a) N. Kawai, J.-M. Lagrange and J. Uenishi, Eur. J. Org. Chem., 2007, 2808–2814 CrossRef CAS; (b) Y. Sato, Y. Aso and M. Shindo, Tetrahedron Lett., 2009, 50, 4164–4166 CrossRef CAS PubMed.
- (a) F. Sato, H. Ishikawa and M. Sato, Tetrahedron Lett., 1981, 22, 85–88 CrossRef CAS; (b) F. Sato, H. Ishikawa, H. Watanabe, T. Miyake and M. Sato, J. Chem. Soc., Chem. Commun., 1981, 718–720 RSC; (c) F. Sato, J. Organomet. Chem., 1985, 285, 53–64 CrossRef CAS; (d) F. Sato and Y. Kobayashi, Org. Synth., Coll., 1993, 8, 507 Search PubMed.
- (a) E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (c) E. Negisi, Bull. Chem. Soc. Jpn., 2007, 80, 233–257 CrossRef.
- (a) T. Jensen, H. Pedersen, B. Bang-Andersen, R. Madsen and M. Jørgensen, Angew. Chem., Int. Ed., 2008, 47, 888–890 CrossRef CAS PubMed; (b) C. Z. Rotsides, C. Hu and K. A. Woerpel, Angew. Chem., Int. Ed., 2013, 52, 13033–13036 CrossRef CAS PubMed; (c) T. E. Nielsen, M. A. C. de Dios and D. Tanner, J. Org. Chem., 2002, 67, 7309–7313 CrossRef CAS PubMed.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
- (a) T. Kan and T. Fukuyama, Chem. Commun., 2004, 353–359 RSC; (b) G. Pan and R. M. Williams, J. Org. Chem., 2012, 77, 4801–4811 CrossRef CAS PubMed; (c) M. Gans and F. Bracher, Tetrahedron, 2014, 70, 1084–1090 CrossRef CAS PubMed.
- (a) J. Tsuji, I. Shimizu and K. Yamamoto, Tetrahedron Lett., 1976, 34, 2975–2976 CrossRef; (b) J. Tsuji, H. Nagashima and H. Nemoto, Org. Synth., Coll., 1990, 7, 137 Search PubMed; (c) J. Tsuji, Synthesis, 1984, 369–384 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00353e |
| This journal is © the Partner Organisations 2015 |