
Towards multicomponent MOFs via solvent-free synthesis under conventional oven and microwave assisted heating†
Mónica
Lanchas
,
Sandra
Arcediano
,
Garikoitz
Beobide
*,
Oscar
Castillo
*,
Antonio
Luque
and
Sonia
Pérez-Yáñez
Departamento de Química Inorgánica, Universidad del País Vasco, UPV/EHU, Apartado 644, E-48080, Spain. E-mail: oscar.castillo@ehu.es; Fax: +34-94601-3500; Tel: +34-94601-5991
First published on 11th February 2015
Abstract
Herein we prove the efficiency of the oven heating solvent-free synthesis, based on the acid–base reaction between a metal oxide/hydroxide, adenine (HAde) and monocarboxylic acids, to afford otherwise not accessible new MBioFs of formula [M2(μ3-Ade)2(μ2-OOC(CH2)xCH3)2]n expressed as 1_M@monocarboxylate [M(II): Ni or Zn; monocarboxylate: butanoato (But) and propanoato (Prop)]. Additionally, a microwave assisted solvent-free procedure has been carried out, leading to products with a somewhat lower adsorption performance but with the advantages of reducing the reaction times to the minute scale and incorporating randomly distributed additional meso/macropores generated during the release of the water vapour by-product. Both heating techniques provide the resulting products in a monolithic form. The N2 (77 K) and CO2 (273 K) adsorption isotherms indicate a great selectivity towards CO2 for the 1_Ni@But compound. On the other hand, a careful control over the solvent-free conditions provided good-quality single crystals of three new compounds based on the metal/nucleobase/carboxylate system: [Zn3(μ3-Ade)2(μ-OOCCH3)4]n·3H2O (2), [Zn(μ-Hypo)(μ-OOCCH3)]n (3) (Hypo: hypoxanthinate) and [Ni2(μ-HAde)2(μ-OOCH)2(OOCH)2(OH2)2]·2{(H2Ade)(HCOO)}·2HCOOH (4). Compounds 2 and 3 show lamellar structures without accessible voids. Compound 4 represents an intermediate stage between the initial reagent mixture and the final extended coordination polymers that depicts an insight into the reaction mechanism of the solvent-free approach. It also shows the difficulties in stabilizing the porous structure of 1 when short aliphatic monocarboxylic acids, such as acetic and formic acids, are employed.
Introduction
During the last decade, studies that focus on metal–organic frameworks (MOFs) have undergone an unremitting increase.1 MOFs constitute a rapidly progressing class of materials due to their permanent porosity, high surface area, large pore volume, and adjustable pore size, distribution, and shape.2 Moreover, these materials may exhibit additional properties such as catalytic activity,3 luminescence,4 magnetism,5 and/or ionic conductivity,6 since they benefit from their hybrid metal–organic nature.There are many reported synthetic routes to obtain MOFs. The most common one involves the reaction between a metal salt and a ligand under solvothermal or microwave assisted solvothermal conditions.7 In many cases, the employed solvents are expensive and/or toxic hindering the industrial scale production of MOFs. Thus, green solvents as water and alcohols have gained interest in solvothermal syntheses.8 Another option relies on mechano-chemical synthesis, where the reagents are subjected to a vigorous and continuous ball-mill grinding.9 The electrochemical synthesis of MOFs, in which metal ions are continuously supplied from a metal anode to the reaction media containing the ligand and a protic solvent, has attracted the interest of chemical companies.10 In 2012, it was shown that a solvent-free reaction of a metal oxide or a hydroxide with a diazole or triazole ligand yields zeolitic metal–azolate frameworks when the reaction mixture is heated in an oven for 24–48 hours.11 The key factor for its success lies in the melting of the diazole or triazole ligand under the reaction conditions to provide the necessary mobility for the reaction to progress. More recently, we have proven that this synthetic approach is also extrapolatable to carboxylate based MOFs employing low melting temperature metal salts, as the usually employed polycarboxylic acids do not melt before decomposing.12 In both cases the metal/ligand synthesis ratio fits the amounts required by the formula of the MOF, and as a result, heating of the reagent mixture promotes an acid–base reaction that leads to the desired MOF and to a stoichiometric minor amount of a by-product. The type of by-product (H2O, CH3COOH, HCl, or HNO3) depends on the metal source employed, but its volatility at the synthesis temperature favours its removal from the reagent mixture, fostering the reaction progress.
The lack of solvent makes this novel route more sustainable according to green chemistry principles and allows one to produce MOFs at lower prices. Moreover, its technical simplicity and ease of scaling make it highly appealing for massive MOF production. However, so far, only examples of two-component MOFs (1 metal + 1 ligand) have been reported. Therefore, in this work we assess the viability of the solvent-free synthesis in a more complex system based on two ligands (adenine/hypoxanthine and a monocarboxylato) and a divalent first-row transition cation. The cobalt(II) and copper(II) analogues were synthesized by Rosi and us using solvothermal syntheses and room condition aqueous synthesis, respectively.13 The structure of these compounds is composed of paddle-wheel shaped centrosymmetric dimeric units, [M2(μ3-Ade-κN3:κN7:κN9)2(μ2-OOC(CH2)xCH3-κO:κO′)2]n [MII: Cu and Co; x ranges from 0 (acetate) to 5 (heptanoate)], in which two metal(II) atoms are bridged by two adenine ligands coordinated by their N3 and N9 nitrogen atoms and two carboxylic ligands with a μ–κO:κO′ coordination mode. These units are cross-linked (Fig. 1) through the apical coordination of the imidazole N7 atom of the adeninato ligands in such a way that each paddle-wheel is linked to four adjacent entities to generate a 4-connected uninodal net with a lvt topology and a (42.84) point symbol, using as a node the dinuclear building unit. The net exhibits a three-dimensional system of intersecting cavities whose effective volume is directly related to the length of the aliphatic chain, which is pointing towards the inner portion of the channels, so that a longer chain implies less free volume.
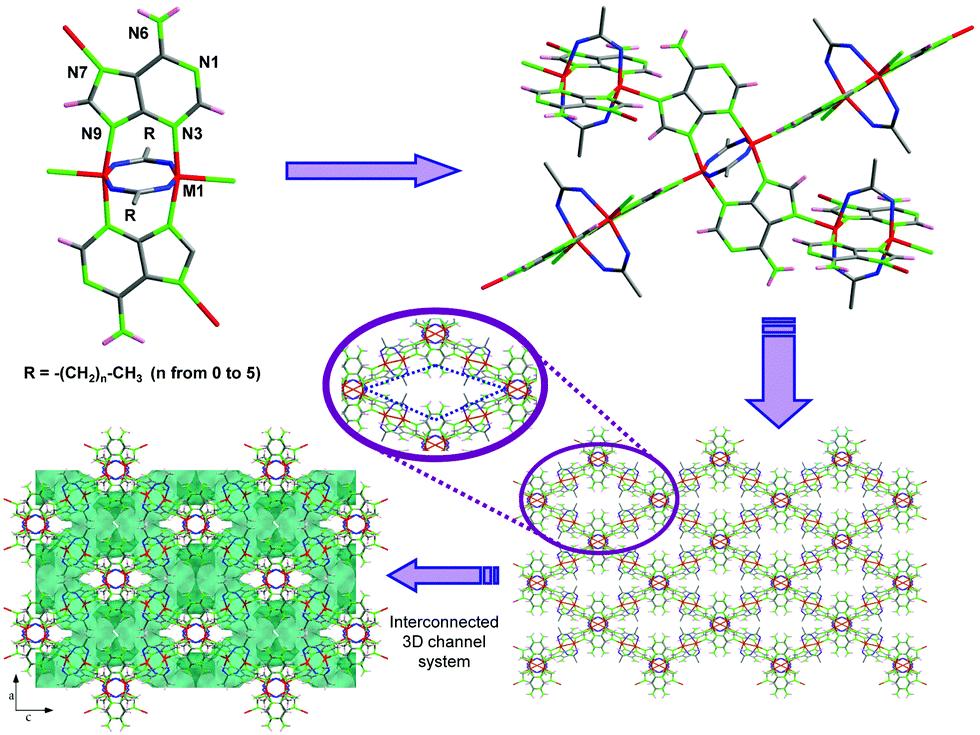 | ||
| Fig. 1 Porous crystal structure of [M(μ3-Ade)(μ2-carboxilato)]n compounds (M being Cu2+ or Co2+). | ||
Their crystal structure remains stable upon removal of the solvent molecules showing a relatively great adsorption selectivity towards CO2.14 In spite of the interest of this family of compounds, all the attempts to obtain analogous compounds using other transition metal centers by means of simple aqueous solvent synthesis or under solvothermal conditions were unsuccessful. Therefore, we focus here on the feasibility of the solvent-free route through the synthesis of otherwise not accessible Zn2+ and Ni2+ analogues. Additionally, many attempts are made employing different nucleobases to afford similar extended systems. Herein we report a first outcome of using hypoxanthine, a nucleobase closely related to adenine.
We also tested the differences between conventional oven-heating and microwave assisted heating. Solvent based microwave-assisted techniques have been widely applied for rapid synthesis of nanoporous materials under hydro/solvothermal conditions.15 However, until very recently, they have not been applied to solvent-free MOF synthesis, showing a shortening of the reaction times to a few minutes and affording materials with porosity values slightly below those obtained by a conventional oven-heating solvent-free procedure and comparable to the results obtained for solvent based synthetic routes.12
Experimental section
Materials and methods
All the chemicals used were of analytical grade and were used without further purification.Synthesis
The synthesis of isostructural compounds [M2(μ3-Ade)2(μ2-OOC(CH2)xCH3)2]n with code 1_M@monocarboxylate [MII: Ni or Zn, monocarboxylate: butanoato (But) and propanoato (Prop)] was accomplished using 8 mmol of the corresponding metal oxide/hydroxide (ZnO and Ni(OH)2) and 8 mmol of adenine that were hand-ground in a mortar to provide a homogeneous reagent mixture, and transferred to a 45 mL Teflon-lined stainless steel autoclave. Later, an excess of propanoic or butanoic acid (27 mmol) was added to account for the amount of carboxylic acid that would go into the vapour phase during the heating process. Afterwards the reaction vessel was placed in a conventional oven with a heating rate of 1 °C h−1 up to 160 °C. Different metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :adenine:carboxylic acid ratios, heating rates and setting temperatures were evaluated but the best results correspond to the conditions described above. Similarly 1MW_M@monocarboxylate compounds were synthesized in a threaded PTFE reactor and microwave-heated on a household microwave oven (700 W) using reaction times between 6 and 60 minutes.
:adenine:carboxylic acid ratios, heating rates and setting temperatures were evaluated but the best results correspond to the conditions described above. Similarly 1MW_M@monocarboxylate compounds were synthesized in a threaded PTFE reactor and microwave-heated on a household microwave oven (700 W) using reaction times between 6 and 60 minutes.
Compounds [Zn3(μ3-Ade)2(μ-OOCCH3)4]n·3H2O (2) and [Zn(μ-Hypo)(μ-OOCCH3)]n (3) were obtained using again a solvent-free approach on a conventional oven with a heating rate of 1 °C h−1 up to 160 °C, but using an equimolar ratio of ZnO, adenine and acetic acid for 2 and a ZnO:hypoxanthine:acetic acid ratio of 1:1:5 for 3. Compound 2 was obtained in a relatively high yield (yield: 88.4% and sample purity: 95.9%), while compound 3 was mixed with some other unknown crystalline phases. Finally, single crystals of the compound [Ni2(μ-HAde)2(μ-OOCH)2(OOCH)2(OH2)2]·2{(H2Ade)(HCOO)}·2HCOOH (4), although mixed with some other non-identified compounds, were obtained on reacting Ni(OH)2, adenine and formic acid in a 1:1:3 ratio using a heating rate of 0.5 °C h−1 up to 120 °C. All the products were washed with ethanol to remove unreacted soluble reagents but a small amount of the unreacted Ni(OH)2 or ZnO is always present in the final product.
Characterization
Elemental analyses (C, H, and N) were performed on a Euro EA Elemental Analyzer, whereas the metal content was determined using an inductively coupled plasma atomic emission spectrometer (ICP-AES) from Horiba Yobin Yvon Activa. IR spectra (KBr pellets) were recorded on an FTIR 8400S Shimadzu spectrometer in the 4000–400 cm−1 spectral region. Thermal analyses (TG/DTA) were performed on a TA Instruments SDT 2960 thermal analyzer under a synthetic air atmosphere (79% N2/21% O2) with a heating rate of 5 °C min−1. Details of the characterization results are shown in the ESI.†Single-crystal X-ray diffraction data were collected at 100(2) K on an Agilent Technologies SuperNova diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) for 2 and 4 and with Cu Kα radiation (1.54185 Å) for 1_Ni@But and 3. Data reduction was done using the CrysAlis RED program.16 Structures of compounds 1_Ni@But and 2–4 were solved by direct methods using the SIR92 program17 and refined by full-matrix least-squares on F2 including all reflections (SHELXL97).18 The solvent molecules present in the channels of 1_Ni@But are highly disordered and their contribution to the diffraction pattern has been removed using the SQUEEZE subroutine as implemented in PLATON.19 All calculations for these structures were performed using the WINGX crystallographic software package.20 Relevant data acquisition and refinement parameters are shown in Table 1.
| 1 _Ni@But | 2 | 3 | 4 | |
|---|---|---|---|---|
| a S = [∑w(F02 − Fc2)2/(Nobs − Nparam)]1/2. b R 1 = ∑||F0| − |Fc||/∑|F0|. c wR2 = [∑w(F02 − Fc2)2/∑wF02]1/2; w = 1/[σ2(F02) + (aP)2 + b]. | ||||
| Empirical formula | C18H22N10Ni2O4 | C18H26N10O11Zn3 | C7H6N4O3Zn | C28H36N20Ni2O18 |
| Formula weight | 559.87 | 754.60 | 259.53 | 1058.19 |
| Crystal system | Tetragonal | Monoclinic | Monoclinic | Triclinic |
| Space group | I41/a | P21/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 15.1973(5) | 9.1709(4) | 4.7692(2) | 7.1579(4) |
| b (Å) | 15.1973(5) | 11.7306(4) | 10.4912(7) | 12.8195(7) |
| c (Å) | 22.1529(8) | 12.9958(4) | 17.9695(10) | 13.0602(7) |
| α (°) | 90.0 | 90.0 | 90.0 | 114.763(5) |
| β (°) | 90.0 | 96.400(3) | 104.874(5) | 100.349(5) |
| γ (°) | 90.0 | 90.0 | 90.0 | 100.859(5) |
| V (Å3) | 5116.4(4) | 1389.38(9) | 868.97(8) | 1022.83(10) |
| Z | 8 | 2 | 4 | 1 |
| GOFa | 1.064 | 1.041 | 1.095 | 1.039 |
| R int | 0.0668 | 0.0708 | 0.0509 | 0.0227 |
| Final R indices | ||||
| [I > 2σ(I)] R1b/wR2c |
0.1155/0.2708 | 0.0600/0.1265 | 0.0419/0.1131 | 0.0395/0.0948 |
| All data R1b/wR2c |
0.1204/0.2740 | 0.0918/0.1404 | 0.0474/0.1170 | 0.0543/0.1006 |
| Max/min residual (e− Å3) | 1.469/−0.731 | 1.401/−0.620 | 0.652/−0.659 | 0.559/−0.372 |
The powder X-ray diffraction (PXRD) patterns were collected on a Philips X'PERT powder diffractometer with Cu Kα radiation (λ = 1.5418 Å) over the range 5 < 2θ < 50° with a step size of 0.02° and an acquisition time of 2.5 s per step at 25 °C. Indexation of the diffraction profiles was made by means of the FULLPROF program (pattern matching analysis)21 on the basis of the space group and cell parameters found for the isostructural 1_Ni@But compound. The unit cell parameters obtained in the final refinement are listed in Table 2. The pattern matching analysis plots are available in the ESI.†
| 1 _Ni@Prop | 1 _Zn@Prop | |
|---|---|---|
| Empirical formula | C16H18N10Ni2O4 | C16H18N10O4Zn2 |
| Formula weight | 531.77 | 545.16 |
| Crystal system | Tetragonal | Tetragonal |
| Space group | I41/a | I41/a |
| a (Å) | 15.581(1) | 15.548(1) |
| c (Å) | 21.930(1) | 22.630(1) |
| V (Å3) | 5324(1) | 5470(1) |
The specific surface area was calculated from the adsorption branch in the relative pressure interval from 0.01 to 0.05 using the Brunauer–Emmett–Teller (BET) method,22 while the micropore volume was estimated by fitting the measured N2 isotherms with the t-plot method.23 The volumetric carbon dioxide physisorption data were recorded using a Micromeritics ASAP 2020 porosity analyser at 273 K.
Results and discussion
1 _M@monocarboxylate compounds show a similar unit cell volume to those of copper(II) and cobalt(II) analogues (5300–5400 Å3), except for 1_Ni@But that shows a significant shrinkage (5116 Å3). Coordination bond distances are listed in Table 3. We selected Ni(OH)2 and ZnO to react with the nucleobase and carboxylic acid because of the greenness of the by-product generated in the synthesis:| Ni(OH)2 + HAde + CH3(CH2)nCOOH → [Ni(μ3-Ade)(μ-OOC(CH2)nCH3)]n + 2H2O | (1) |
| ZnO + HAde + CH3(CH2)nCOOH → [Zn(μ3-Ade)(μ-OOC(CH2)nCH3)]n + H2O | (2) |
| a Symmetry codes for 1_Ni@But: (i) −x + 2, −y − 1, −z − 1; (ii) y + 3/4, −x + 3/4, z − 1/4; (iii) −y + 5/4, x − 3/4, −z + 1/4; (iv) y + 3/4, −x + 5/4,−z + 1/4; for 2: (i) −x + 1, y + 1/2, −z + 1/2; (ii) x, −y + 1/2, z + 1/2; for 3: (i) x − 1, y, z; (ii) −x + 1, y + 1/2, −z + 1/2; (iii) x − 1, y, z; for 4: (i) −x + 2, −y + 2, −z + 1. | |||
|---|---|---|---|
| Compound 1 _Ni@But | |||
| Ni1–O1 | 2.030(7) | Ni1–N17 | 2.028(6) |
| Ni1–O2i | 2.020(7) | Ni1–N19iii | 2.043(7) |
| Ni1–N13ii | 2.051(7) | ||
| Ni1⋯Ni1i | 2.854(3) | Ni1⋯Ni1iv | 5.951(2) |
| Compound 2 | |||
| Zn1–O1 | 2.146(4) | Zn2–O2 | 1.957(4) |
| Zn1–O3 | 2.196(4) | Zn2–O3 | 1.963(4) |
| Zn1–N9i | 2.052(5) | Zn2–N3ii | 2.046(5) |
| Zn2–N7 | 1.994(4) | ||
| Zn1⋯Zn2 | 3.3734(6) | Zn1⋯Zn2ii | 5.9415(7) |
| Compound 3 | |||
| Zn1–O1 | 1.991(2) | Zn1–N7 | 1.970(3) |
| Zn1–O2i | 1.977(2) | Zn1–N9ii | 1.988(2) |
| Zn1⋯Zn1ii | 5.8479(5) | Zn1⋯Zn1iii | 4.7692(2) |
| Compound 4 | |||
| Ni1–O1 | 2.072(2) | Ni1–O1w | 2.064(2) |
| Ni1–O1i | 2.066(2) | Ni1–N13 | 2.123(2) |
| Ni1–O3 | 2.025(2) | Ni1–N19i | 2.123(2) |
| Ni1⋯Ni1i | 3.0218(7) | ||
The reaction has been tested at different temperatures. Below 160 °C the resulting compounds show a lower crystallinity and the reaction yields also decrease. On the other hand, temperatures above 160 °C provide products with a brownish colour indicative of partial decomposition of the organic ligands. The heating rate also has a relevant effect on the crystallinity of the resulting sample and its particulate size, in such a way that both increase when a slower heating is employed. In fact, using a heating rate of 1 °C h−1, crystals of 1_Ni@But suitable for single-crystal X-ray diffraction characterization were obtained.
The same compounds were also synthesized applying a microwave assisted heating solvent-free procedure that allowed to reduce the synthesis time from days to just a few minutes. In general, the powder X-ray diffraction patterns of these samples show slightly less intense and broader peaks than those obtained from the conventional oven-heating procedure. Therefore, the crystallinity of the obtained samples is lower than that corresponding to the extended time oven-heating ones but the lowering of crystallinity is less than would be expected from such a great reduction in reaction time. For this experiment a household microwave oven has been used that does not provide control over the temperature but only over the microwave power output. The time required to complete the reaction depends on the microwave power changing from 6 to 60 min for ca. 4 g batches. The microwave power supply plays also a significant role in the crystallinity and adsorptive properties of the resulting samples as the crystallinity improves on using a lower microwave radiation output and, as a consequence, more extended reaction times. However, the use of very low microwave powers does not provide temperatures high enough to promote the reaction and negligible yields are achieved. The optimal conditions have been found to be around 50% of the maximum microwave output (reaction time: 12 min).
Another relevant aspect of this solvent-free approach, both for a conventional oven and microwave assisted heating, is the shaping of the resulting products, which appear in the form of robust monolithic blocks adopting the shape of the reaction vessel. Additionally, due to the nature of the reaction, a simple acid–base neutralization that involves the generation of water as a by-product, the resulting monolith contains interconnected meso- and macropores generated during the release of water vapour. This feature provides a final product in which we can distinguish pores in the micropore regime, due to the ordered structure of the MBioF, and meso/macropores randomly generated during the texturing of the sample under the synthesis conditions. This phenomenon is more notorious in the microwave assisted heating samples (Fig. 2). The robustness of the monolith can be inferred from the weight that it can stand, at least 200 times its mass. It is worth mentioning that nowadays there is a great interest in developing hierarchically ordered porous materials24 and also on developing techniques for shaping25 these materials. This synthetic route affords an easy and simple single-step solution to both issues.
 | ||
| Fig. 2 (a) Monolith of 1MW_Ni@Prop obtained using microwave assisted heating with an apparent density of 0.35 g cm−3. (b and c) SEM images of the same monolith showing the macroporous texture of the material. | ||
The N2 and CO2 adsorption isotherms of these compounds, measured at 77 and 273 K (Fig. 3 and Table 4) respectively, show that 1_Ni@But does not adsorb N2 but it does adsorb CO2, making this compound a very selective material for the capture of CO2. This selectivity towards CO2 was previously described for both Cu2+ and Co2+ butanoate analogues but it was not so great as to completely avoid the N2 adsorption at 77 K. The CO2 adsorption capacity of 1MW_Ni@But is strongly reduced but conclusions cannot be extrapolated from this fact because the purity of this sample is relatively low. The propanoate analogues adsorb both N2 and CO2 but it becomes clear that the performance of the oven-heating samples is better than that of the microwave assisted heating ones. Another interesting feature of the dinitrogen isotherms of the microwave assisted samples is the exponential increase taking place above P/P0 = 0.8–0.9, which correspond to capillary filling of the microstructural pores greater than 20–30 nm arising from the sample texturization. A similar but less pronounced effect is also found in the oven-heating samples, probably because the more prolonged reaction times promote a less abrupt release of the water vapour.
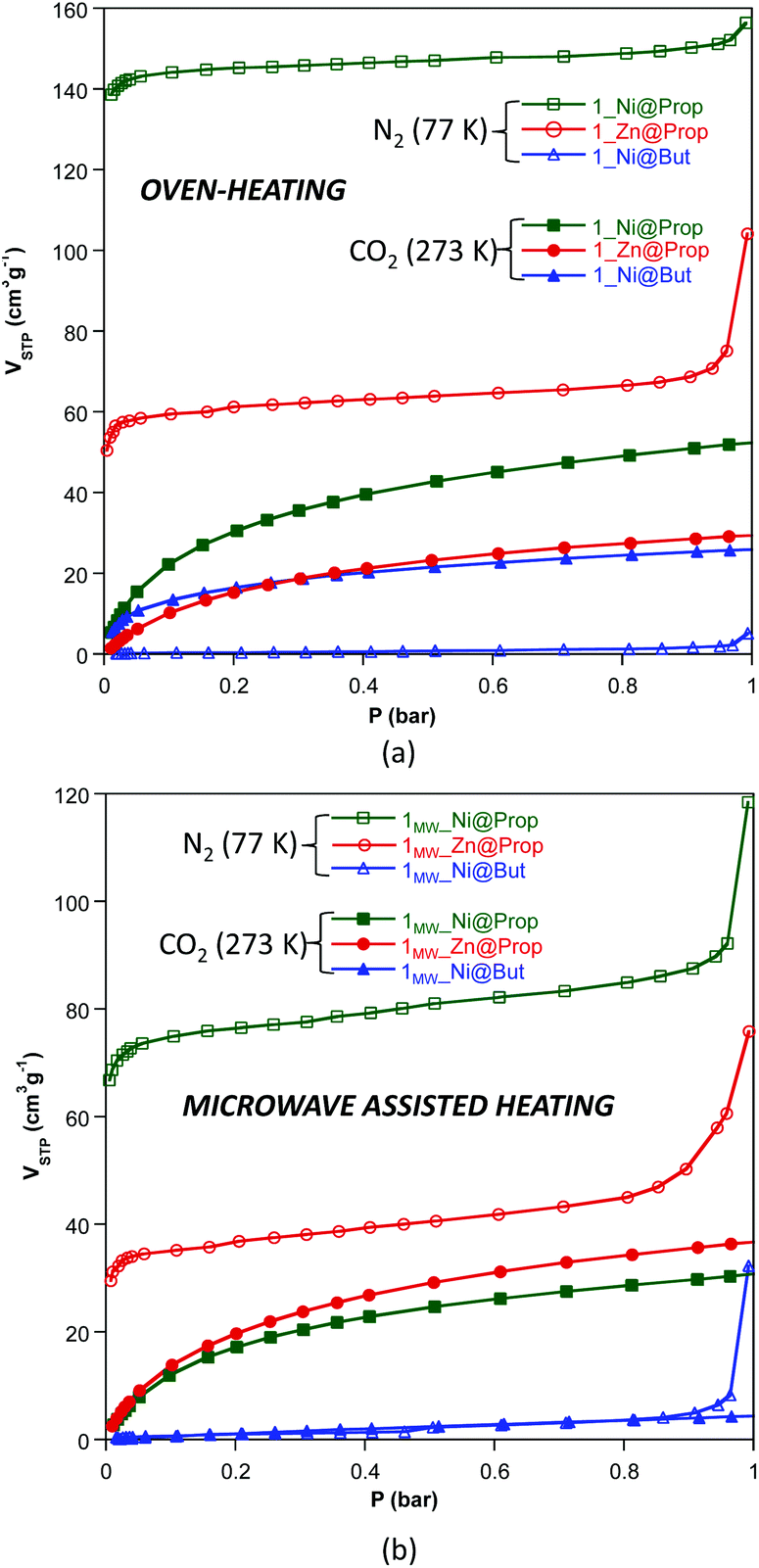 | ||
| Fig. 3 N2 (77 K) and CO2 (273 K) adsorption isotherms of compounds 1_M@monocarboxylate (a) and 1MW_M@monocarboxylate (b). | ||
| Yield (%) | Purity (%) | BETa (m2 g−1) | V STP (N2, P/P0: 0.15) (cm3 g−1) | V STP (CO2, 1 bar) (cm3 g−1) | V micro (cm3 g−1) | V meso/macro (cm3 g−1) | |
|---|---|---|---|---|---|---|---|
| a BET area estimated from the N2 adsorption isotherm at 77 K. | |||||||
| Oven heating (1 °C h −1 , 160 °C) | |||||||
| 1 _Ni@But | 98.2 | 99.4 | 1 | 0.4 | 26.1 | 0.000 | 0.007 |
| 1 _Zn@But | <10 | — | — | — | — | — | — |
| 1 _Ni@Prop | 97.3 | 99.0 | 601 | 144.7 | 52.6 | 0.218 | 0.242 |
| 1 _Zn@Prop | 96.5 | 98.9 | 232 | 60.0 | 29.6 | 0.086 | 0.161 |
| Microwave heating (power output: 50%, 12 min) | |||||||
| 1 MW _Ni@But | 58.8 | 88.2 | 3 | 0.86 | 4.6 | 0.000 | 0.008 |
| 1 MW _Zn@But | <10 | — | — | — | — | — | — |
| 1 MW _Ni@Prop | 94.0 | 97.8 | 307 | 75.9 | 31.9 | 0.103 | 0.183 |
| 1 MW _Zn@Prop | 99.2 | 99.8 | 146 | 35.8 | 36.9 | 0.044 | 0.117 |
As previously stated, the solvent-free synthetic procedure allowed us to obtain the propanoate and butanoate analogues but surprisingly these synthetic conditions did not provide the expected acetate analogue, but a non-porous new compound 2 with the [Zn3(μ3-Ade)2(μ-OOCCH3)4]n·3H2O formula is obtained at 160 °C. It presents a 2D coordination network composed of linear trimeric [Zn3(μ-Ade-κN3:κN9)2(μ-OOCCH3-κO:κO′)2(μ-OOCCH3-κO:κO)2] units (Fig. 4) that are further crosslinked among them through the adeninato ligands. Inside the trimeric entity there are two crystallographically independent metal centers. Zn1, placed in the middle of the trimer, presents a trans-N2O4 octahedral coordination environment and Zn2, located at both edges, shows a N2O2 tetrahedral coordination environment. Inside the trimeric entity, adjacent metal centers are triply bridged by an adeninato ligand with a μ–κN3:κN9 coordination mode and two acetate anions showing different coordination modes, μ–κO:κO′ and μ–κO:κO. The adeninate anion, apart from acting as a μ–κN3:κN9 bridging ligand inside the trimeric unit, also employs its N7 position to coordinate to a third zinc(II) atom placed at the edge of an adjacent trinuclear entity adopting therefore a μ3–κN3:κN7:κN9 bridging mode. Interestingly, this is the same bridging mode that it adopts in compounds 1_M@monocarboxylate. The connectivity array generated causes every trimeric unit to be bonded to four adjacent ones providing a herringbone 2D network that spreads in the (1 0 0) plane. Although the adeninate anion has its N3, N7 and N9 positions involved in the coordination network, its Watson–Crick face (N1/N6 positions) is exposed outwards from the 2D sheet being responsible for the supramolecular assembly of these sheets by means of complementary hydrogen bonding among the adeninates of adjacent sheets.
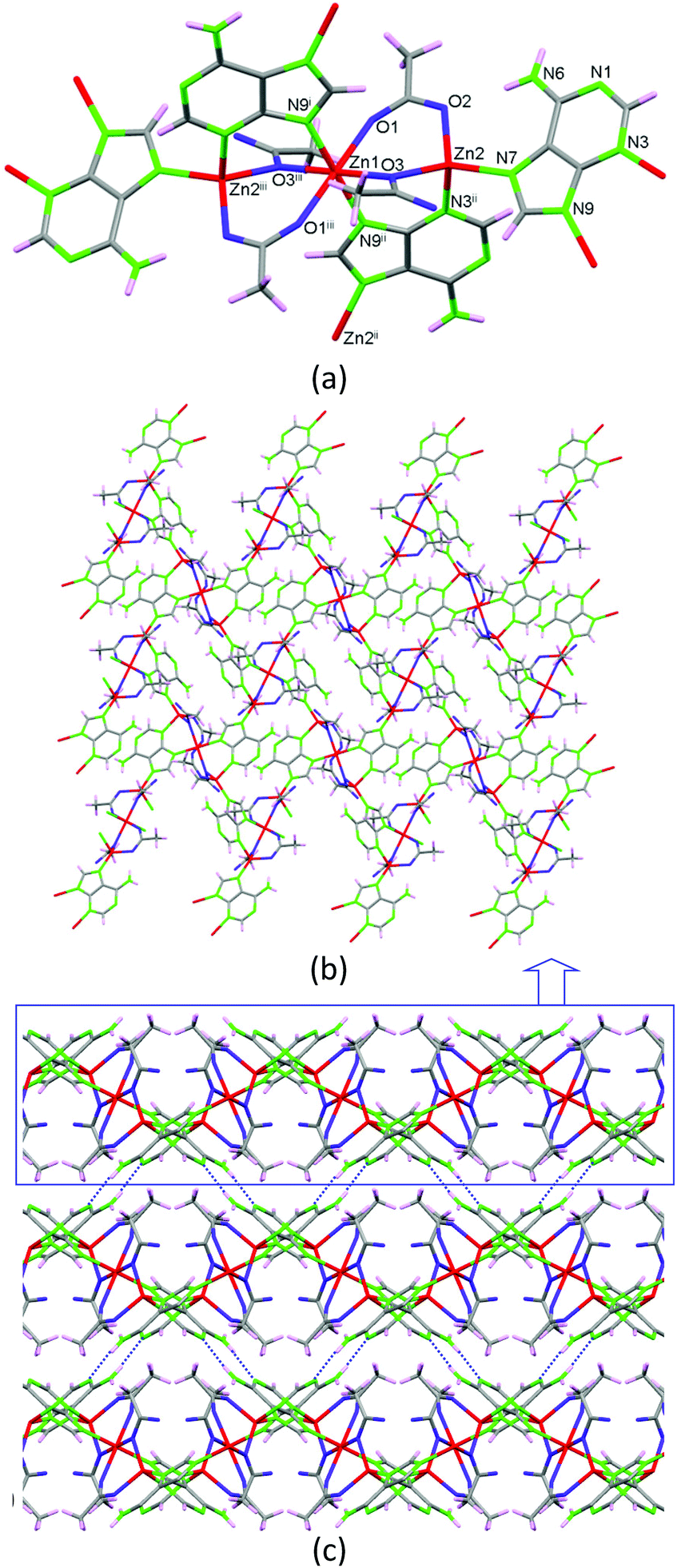 | ||
| Fig. 4 Trinuclear building units (a) crosslinked by means of μ3-adeninato ligands to afford 2D complex sheets (b) that are piled up by means of complementary hydrogen-bonding between the Watson–Crick faces of the adenine in compound 2 (c). | ||
However upon increasing the ratio of adenine with respect to the metal and the acetic acid, a mixture of compounds 1_Zn@Ace and 2 is achieved (Fig. 5). Unfortunately we have not found the synthetic conditions that could afford 1_Zn@Ace in a pure form but it provides some clues to the equilibrium between both compounds. Based on the equilibrium reaction between compounds 1_Zn@Ace and 2 (reaction (3)) it is easy to understand that an excess of adenine helps in displacing the equilibrium towards 1_Zn@Ace as it presents a higher Ade/Zn ratio in its formula. Apart from that, it becomes obvious that the aliphatic tails of the butanoate and propanoate bridging ligands have a templating effect, helping to stabilize the channel structure, avoiding in this way the growth of a compound analogous to 2. In fact during the synthesis of the copper(II) analogues in aqueous solution lower amounts of the monocarboxylic aliphatic acids were required as the aliphatic tail increases, supporting also the templating role of the aliphatic chain.
| 3[Zn(μ3-Ade)(μ-OOCCH3)]n + CH3COOH ↔ [Zn3(μ3-Ade)2(μ-OOCCH3)4] + HAde | (3) |
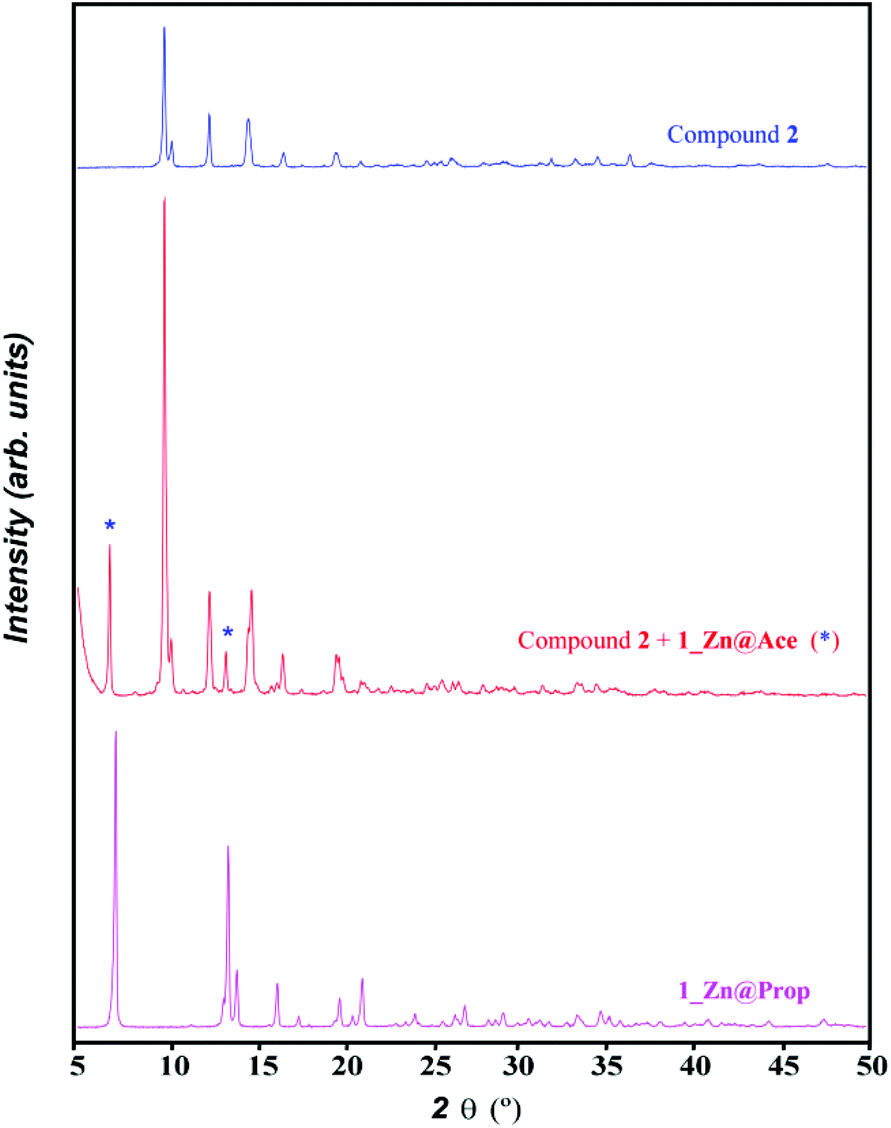 | ||
| Fig. 5 XRPD pattern of the product resulting from the reaction of ZnO, adenine and acetic acid in a 1:2:1 ratio by slowly heating up to 160 °C (1 °C h−1) and that of compounds 1_Zn@Prop and 2. | ||
Performing the same synthesis as that for 2 but using hypoxanthine instead of adenine, compound 3 with formula [Zn(μ-Hypo)(μ-OOCCH3)]n (3) (Hypo: hypoxanthinate) is achieved. The crystal structure is composed of tetrahedral Zn(II) metal centers that are bridged by two hypoxanthinate ligands using the imidazolic N7 and N9 positions and by two acetate anions that adopt a syn–anti bridging mode (Fig. 6). Although hypoxanthine behaves mostly as a monodentate ligand, it can also act as a bidentate bridging ligand (usually as μ-N7,N9 or μ-N3,N9) or even as a tetradentate ligand. The μ-N7,N9 coordination mode, which appears in compound 3, is more frequent than the μ-N3,N9 mode that commonly appears in paddle-wheel shaped entities.26 This connectivity among the metal centers provides a corrugated 2D structure at which the hypoxanthinate, in its ketonic tautomeric form with the hydrogen atom attached to N1, exposes the O6/N1 face outwards the sheets. Such an arrangement allows the piling up of the sheets by means of hydrogen bonding interactions between the hypoxanthinate moieties. As in compound 2, this new crystal structure does not provide any accessible void.
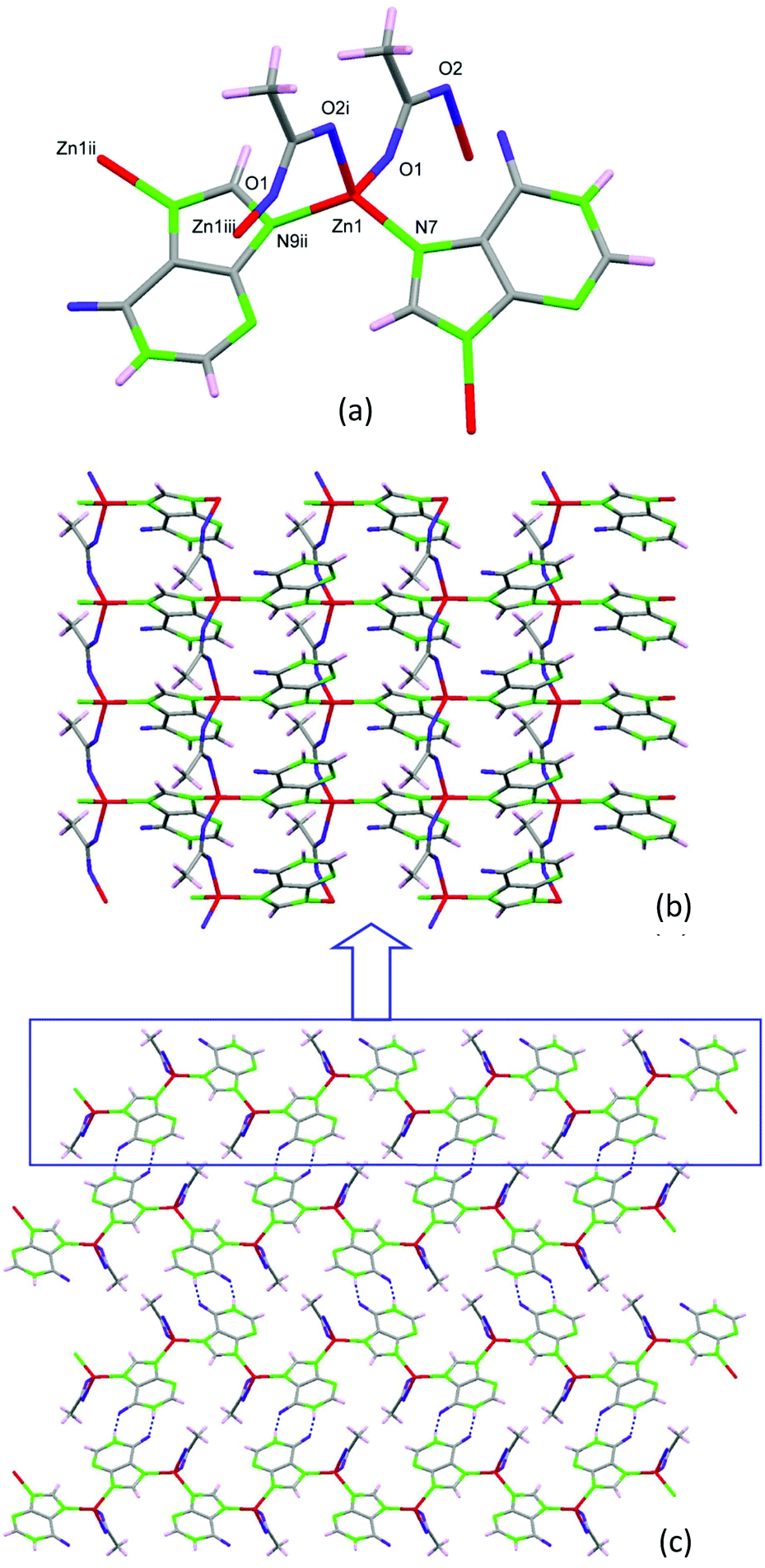 | ||
| Fig. 6 Coordination environment (a) and 2D complex sheets (b) piled up by means of complementary hydrogen-bonding between the hypoxanthinate ligands (c) in compound 3. | ||
Crystals of compound 4, [Ni2(μ-HAde)2(μ-OOCH)2(OOCH)2(OH2)2]·2{(H2Ade)(HCOO)}·2HCOOH, were obtained at significantly lower temperature (120 °C) and its crystal structure can be considered as a frozen intermediate structure on the path towards more extended structures in which the acid–base neutralization reaction has been completed. It is composed of centrosymmetric isolated Ni(II) dimers in which two neutral adenines (μ–κN3:κN9) and two formate anions (μ–κO:κO) act as bridging ligands to afford an overall paddle-wheel shaped entity (Fig. 7). An additional formate and a water molecule complete the octahedral coordination environment around the Ni(II) metal centers. The Watson–Crick faces of the coordinated adenines belonging to adjacent dimeric entities self-recognize to generate supramolecular chains. Non-coordinated formate anions, non-deprotonated formic acid molecules and protonated adeninium cations also cocrystallize with the dimeric entities establishing a complex network of hydrogen bonding interactions. The presence of neutral and protonated adenines altogether with formate and formic acid clearly indicates that this compound is generated at the beginning of the acid–base reaction among Ni(OH)2, adenine and formic acid. The relatively low temperatures employed in the synthesis have allowed its isolation. Another fact that reinforces this interpretation is that only a few crystals of this compound were obtained in a mixture of different polycrystalline and amorphous phases, as evidenced from the powder X-ray diffraction data.
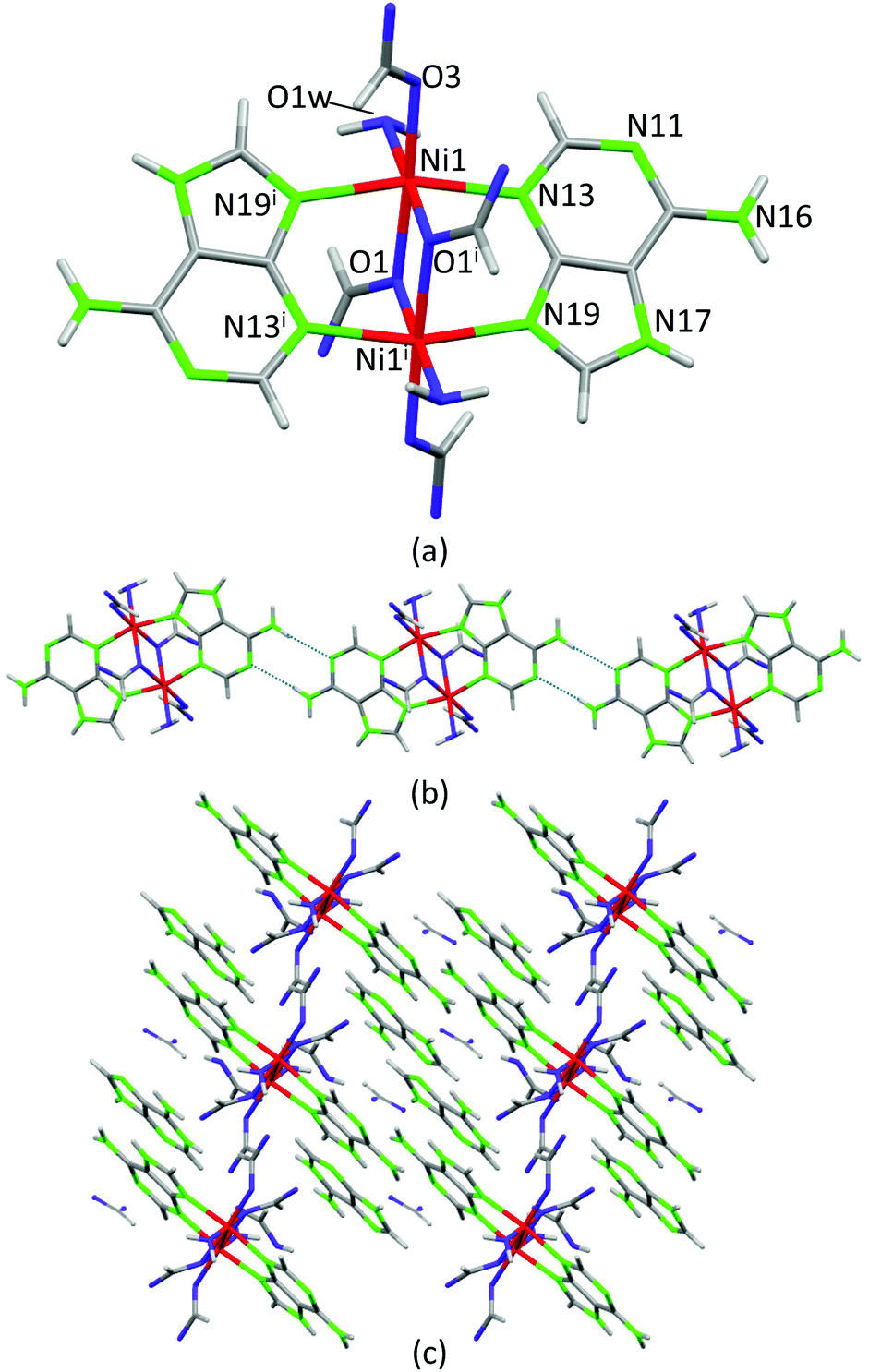 | ||
| Fig. 7 Dimeric entity (a) and supramolecular dimeric chains (b) assembled together by an extensive hydrogen-bonding network involving the cocrystallizing adeninium, formate and formic acid molecules (c) in compound 4. | ||
Conclusions
In this contribution we assess, for the first time, the viability of the solvent-free route by conventional oven heating and by microwave assisted heating to obtain three-component MOFs. There have been synthesized, based on the acid–base reaction between a metal oxide/hydroxide, adenine, and a carboxylic acid, new [M2(μ3-Ade-κN3:κN7:κN9)2(μ2-OOC(CH2)xCH3-κO:κO′)2]n MBioFs [where MII: Ni or Zn, x: 1 or 2]. These compounds are not accessible by simple aqueous solvent synthesis or under solvothermal conditions as the previously reported ones for CuII and CoII analogues. The solvent-free reaction takes place upon heating the reagent mixture by conventional oven-heating but also when using microwave heating. Both heating procedures provide the resulting products in a monolithic form but the microwave heating procedure, despite giving a lower surface area value, incorporates randomly distributed additional meso/macropores generated during the release of the water vapour by-product. The adsorptive performance of the resulting product is related to the crystallinity/purity degree of the samples and obviously to the length of the aliphatic chain of the carboxylate ligand that partially occupies the space within the voids. In fact, we have observed that the longer the aliphatic chain the more selective the adsorption towards CO2, as it occurs in Cu(II) and Co(II) analogues.13,14Moreover, three new compounds based on a metal/nucleobase/carboxylate system have been characterized by means of single-crystal X-ray diffraction, providing also evidence of the feasibility of the synthetic procedure to achieve good quality single-crystals. The crystal structure of compounds 2 and 3 corresponds to lamellar structures without any accessible void and reflects the difficulties in stabilizing the porous structure of 1 when short aliphatic monocarboxylic acids are employed. On the other hand, compound 4, containing coordinated formate and adenines together with non-coordinated formate anions, non-deprotonated formic acid molecules and protonated adeninium cations, represents an intermediate stage between the reagent mixture and the extended final coordination polymers. Thus, it gives an insight into the reaction mechanism of the solvent-free approach.
Finally, we would like to emphasize the greenness of this procedure for MOF synthesis, which is atomically efficient, environmentally friendly and apparently easy to scale up. Moreover, although previous studies have analyzed its applications in bicomponent systems, herein we have proven its viability for more complex multi-component systems.
Acknowledgements
This work has been funded by the Ministerio de Economía y Competitividad (MAT2013-46502-C2-1-P), Eusko Jaurlaritza/Gobierno Vasco (grant IT477-10 and S-PE13UN016) and UPV/EHU (UFI 11/53, EHUA14/09, postdoctoral fellowship for S.P.Y.). Technical and human support provided by SGIKer (UPV/EHU, MINECO, GV/EJ, ERDF, and ESF) is gratefully acknowledged.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 975 CrossRef PubMed.
- (a) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed; (b) D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2009, 38, 1257 RSC.
- (a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC; (b) C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef; (c) A. Dhakshinamoorthy and H. García, Chem. Soc. Rev., 2014, 43, 5750 RSC; (d) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
- (a) M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem. – Eur. J., 2011, 17, 11372 CrossRef CAS PubMed; (b) M. Alvaro, E. Carbonell, B. Ferrer, F. X. Labrés i Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106 CrossRef CAS PubMed.
- (a) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (b) E. Coronado and G. Mínguez-Espallargas, Chem. Soc. Rev., 2013, 42, 1525 RSC.
- (a) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed; (b) K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 234 CrossRef PubMed; (c) S. V. Elisseva and J. C. G. Bunzli, New J. Chem., 2011, 35, 1165 RSC.
- (a) Y.-R. Lee, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2013, 30, 1667 CrossRef CAS; (b) A. Czaja, E. Leung, N. Trukhan and U. Müller, in Metal-Organic Frameworks - Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, ch. 14, pp. 337–352 Search PubMed.
- (a) A. García-Márquez, A. Demessence, A. E. Platero-Prats, D. Heurtaux, P. Horcajada, C. Serre, J.-S. Chang, G. Férey, V. A. de la Peña-O'Shea, C. Boissière, D. Grosso and C. Sanchez, Eur. J. Inorg. Chem., 2012, 5165 CrossRef; (b) A. F. Gross, E. Sherman and J. J. Vajo, Dalton Trans., 2012, 41, 5458 RSC; (c) F.-K. Shieh, S.-C. Wang, S.-Y. Leo and K. C.-W. Wu, Chem. – Eur. J., 2013, 19, 11139 CrossRef CAS PubMed; (d) A. C. Kathalikkattil, D.-W. Kim, J. Tharun, H.-G. Soek, R. Roshan and D.-W. Park, Green Chem., 2014, 16, 1607 RSC; (e) B. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS; (f) Y. Xiao, Y. Cui, Q. Zheng, S. Xiang, G. Qian and B. Chen, Chem. Commun., 2010, 46, 5503 RSC.
- (a) H. Sakamoto, R. Matsuda and S. Kitagawa, Dalton Trans., 2012, 41, 3956 RSC; (b) T. Frišcic and L. Fábián, CrystEngComm, 2009, 11, 743 RSC; (c) S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS PubMed; (d) M. Schlesinger, S. Schulze, M. Hietschold and M. Mehring, Microporous Mesoporous Mater., 2010, 132, 121 CrossRef CAS PubMed; (e) A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211 RSC; (f) M. Klimakow, P. Klobes, A. F. Thünemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216 CrossRef CAS.
- (a) A. M. Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489 CrossRef; (b) U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626 RSC; (c) I. Richter, M. Schubert and U. Müller, PCT Patent, 2007, 131, 955 Search PubMed.
- (a) M. Lanchas, D. Vallejo-Sánchez, G. Beobide, O. Castillo, A. T. Aguayo, A. Luque and P. Román, Chem. Commun., 2012, 48, 9930 RSC; (b) J.-B. Lin, R.-B. Lin, X.-N. Cheng, J.-P. Zhang and X.-M. Chen, Chem. Commun., 2011, 47, 9185 RSC.
- (a) M. Lanchas, S. Arcediano, A. T. Aguayo, G. Beobide, O. Castillo, J. Cepeda, D. Vallejo-Sánchez and A. Luque, RSC Adv., 2014, 4, 60409 RSC.
- (a) J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38 CrossRef CAS PubMed; (b) S. Pérez-Yáñez, G. Beobide, O. Castillo, J. Cepeda, A. Luque, A. T. Aguayo and P. Román, Inorg. Chem., 2011, 50, 5330 CrossRef PubMed; (c) T. Li, D.-L. Chen, J. E. Sullivan, M. T. Kozlowski, J. K. Johnson and N. L. Rosi, Chem. Sci., 2013, 4, 1746 RSC.
- S. Pérez-Yáñez, G. Beobide, O. Castillo, M. Fisher, F. Hoffmann, M. Fröba, J. Cepeda and A. Luque, Eur. J. Inorg. Chem., 2012, 5921 CrossRef.
- (a) S. H. Jhung, J. H. Lee, J. W. Yoon, C. Serre, G. Ferey and J. S. Chang, Adv. Mater., 2007, 19, 121 CrossRef CAS; (b) Y. S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592 CrossRef CAS PubMed; (c) N. A. Khan, E. Haque and S. H. Jhung, Phys. Chem. Chem. Phys., 2010, 12, 2625 RSC; (d) Z. X. Zhao, X. M. Li, S. S. Huang, Q. B. Xia and Z. Li, Ind. Eng. Chem. Res., 2011, 50, 2254 CrossRef CAS; (e) E. Haque and S. H. Jhung, Chem. Eng. J., 2011, 173, 866 CrossRef CAS PubMed; (f) J. Klinowski, F. A. Almeida Paz, P. Silva and J. Rocha, Dalton Trans., 2011, 40, 321 RSC.
- CrysAlis RED, version 1.171.33.55, Oxford Diffraction, Wroclaw, Poland, 2010 Search PubMed.
- A. Altomare, M. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- (a) J. Rodríguez-Carvajal, FULLPROF, a Program for Rietveld Refinement and Pattern Matching Analysis. Abstracts of the Satellite Meeting on Powder Diffraction of the XVth Congress of the IUCr, Toulouse, France, 1990, p. 127 Search PubMed; (b) J. Rodríguez-Carvajal, FULLPROF 2000, version 2.5d, Laboratoire Léon Brillouin (CEA-CNRS), Centre d’Études de Saclay, Gif sur Yvette Cedex, France, 2003 Search PubMed.
- S. Brunauer, P. H. Emmet and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- J. H. de Boer, B. C. Lippens, B. G. Linsen, J. C. P. Broekhoff, A. van den Heeuvel and T. V. Osinga, J. Colloid Interface Sci., 1966, 21, 405 CrossRef CAS.
- (a) S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700 RSC; (b) L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C.-Y. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed.
- (a) S. Aguado, J. Canivet and D. Farrusseng, Chem. Commun., 2010, 46, 7999 RSC; (b) Y.-R. Lee, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2013, 30, 1667 CrossRef CAS; (c) B. Böhringer, R. Fischer, M. R. Lohe, M. Rose, S. Kaskel and P. Küsgens, in Metal-Organic Frameworks – MOF shaping and inmovilization, ed. D. Farrusseng, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, ch. 15, pp. 353–380 Search PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed (v. 5.35 – May 2014).
Footnote |
| † Electronic supplementary information (ESI) available: Details of the syntheses, elemental analyses, IR bands, X-ray powder diffraction analyses, TG curves, and CIF files. CCDC 1035438–1035441. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00208c |
| This journal is © the Partner Organisations 2015 |