 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst selective and regiodivergent O- to C- or N-carboxyl transfer of pyrazolyl carbonates: synthetic and computational studies†
Eoin
Gould
a,
Daniel M.
Walden
b,
Kevin
Kasten
a,
Ryne C.
Johnston
b,
Jiufeng
Wu
a,
Alexandra M. Z.
Slawin
a,
Thomas J. L.
Mustard
b,
Brittany
Johnston
b,
Tony
Davies
c,
Paul
Ha-Yeon Cheong
*b and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: ads10@st.andrews.ac.uk
bDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, Oregon 97333, USA
cMerck Sharp & Dohme Limited, Hertford Road, Hoddesdon, Hertfordshire EN11 9BU, UK
First published on 3rd July 2014
Abstract
The regiodivergent O- to C- or N-carboxyl transfer of pyrazolyl carbonates is described, with DMAP giving preferential N-carboxylation and triazolinylidenes promoting selective C-carboxylation (both with up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity). An enantioselective O- to C-carboxyl variant using NHC catalysis is demonstrated (up to 92% ee), while mechanistic and DFT studies outline the pathways operative in this system and provide insight into the reasons for the observed selectivity.
:1 regioselectivity). An enantioselective O- to C-carboxyl variant using NHC catalysis is demonstrated (up to 92% ee), while mechanistic and DFT studies outline the pathways operative in this system and provide insight into the reasons for the observed selectivity.
Introduction and background
The organocatalysed rearrangement of oxazolyl carbonates to the corresponding 4- or 2-carboxyazlactones was first described by Steglich and Höfle over forty years ago.1 This reaction process has since attracted considerable attention thanks to its potential to generate synthetically useful α,α-disubstituted α-amino acid derivatives and is often regarded as a benchmark for the evaluation of Lewis base-catalysed reaction processes. Initially promoted by the achiral Lewis bases DMAP or PPY, the groups of Fu,2 Vedejs,3 Richards4 and Gotor5 have shown that chiral DMAP or PPY derivatives can induce high enantiocontrol in this reaction process.6 Alternative chiral Lewis base catalysts that have been applied to this enantioselective rearrangement include chiral phosphines by Vedejs,3 asymmetric imidazoles by Zhang,7 and a variety of isothioureas by Gröger (acyl transfer),8 ourselves9 and Okamoto.10 A dual-catalytic approach that utilises DMAP and a chiral thiourea has been demonstrated by Seidel,11 while an ammonium betaine catalyst for this process has been utilised by Ooi.12 Within this area we have previously shown that N-heterocyclic carbenes (NHCs) are versatile catalysts for the racemic Steglich rearrangement of oxazolyl carbonates.13 Achiral triazolinylidenes promote this rearrangement process with low catalyst loadings and offer access to sterically hindered products, although chiral NHCs resulted in only modest enantiocontrol.14The potential of this strategy to access stereodefined products with a quaternary stereogenic centre has seen this process extended to incorporate the asymmetric rearrangement of furanyl, indolyl and benzofuranyl carbonates,15 alongside applications in total synthesis.16 Notably, Vedejs et al. have investigated the regio- and enantioselective O- to C-carboxyl transfer of 5-arylfuranyl carbonates using TADMAP 1,17 with the regioselectivity dependent upon the electronic nature of the 5-aryl substituent (Fig. 1). Electron-donating aryl substituents favour α-functionalisation (α:γ up to 92:8), while an electron-withdrawing substituent favours γ-functionalisation (α:γ up to 20:80). Building upon these precedents, we considered alternative molecular scaffolds upon which to investigate catalyst selective regio- and enantioselective O-to C-carboxyl transfer processes.18 While originally exploited in the dyeing and photographic industries, pyrazolinones and their derivatives have displayed a wide range of medicinal and pharmacological activities such as analgesic19 and antipyretic properties,20 anti-inflammatory,21 anti-tumor,22 anti-microbial,23 anti-retroviral24 as well as anti-ischemic effects25 and neuroprotective properties.26 These diverse applications have encouraged recent interest in novel synthetic methods to access enantioenriched pyrazolinones27,28 and served as inspiration for our studies concerning the regio- and enantioselective O- to C-carboxyl transfer of pyrazolyl carbonates. Notably, triazolinylidene NHCs promote the rearrangement to generate C(4)-α,α-disubstituted pyrazolinones with high regioselectivity (up to >99:1 C:N) and in up to 92% ee, while catalytic DMAP gives N(1)-carboxyl pyrazolinones with high regioselectivity (up to 1:99 C:N). A mechanistic rationale for this observed selectivity is provided by computational studies on a representative model system.
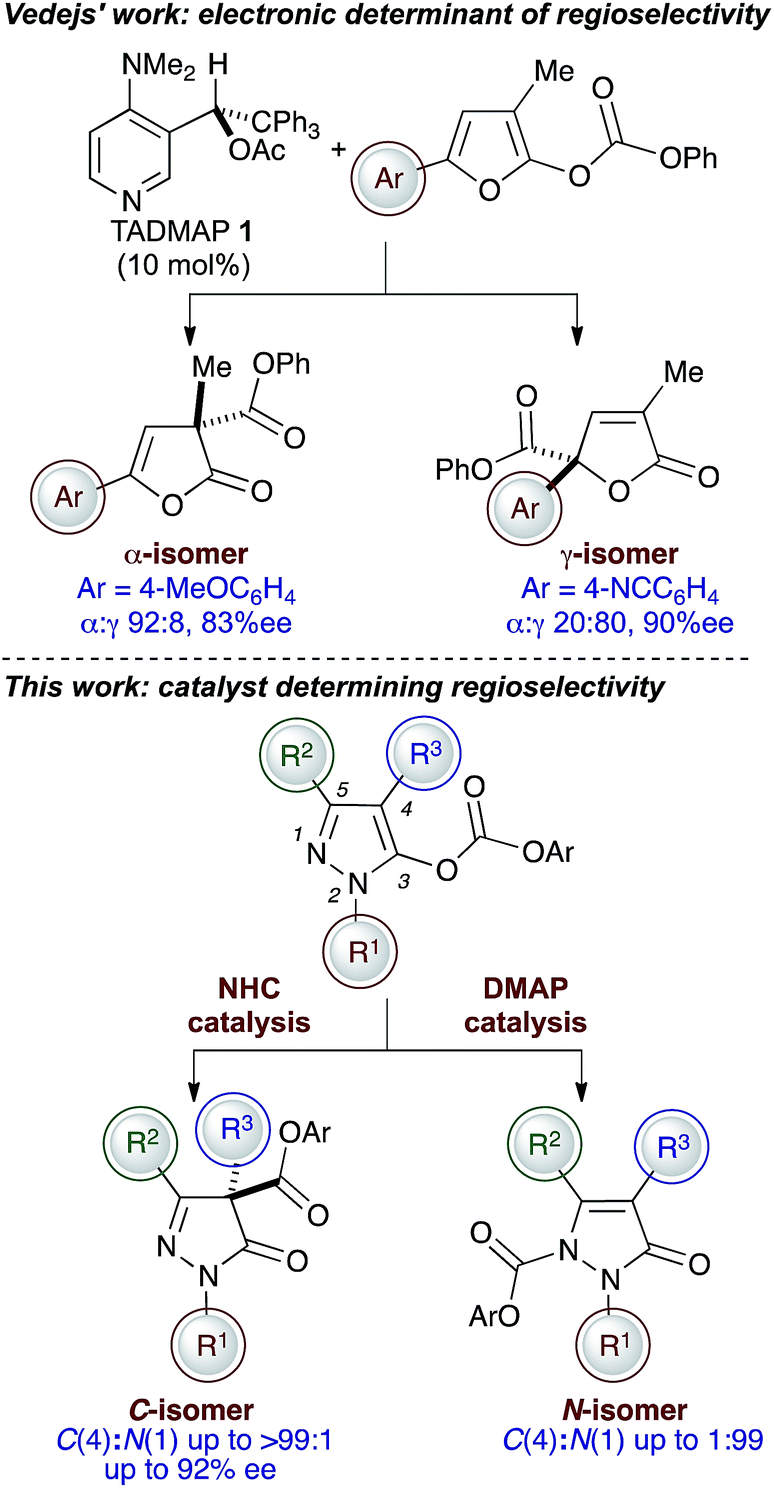 | ||
| Fig. 1 Lewis base-promoted regiodivergent and enantioselective O- to C- or N-carboxyl transfer reactions of pyrazolyl carbonates. | ||
Model studies: catalyst selective O- to C- or N-carboxyl transfer
Initial studies screened a range of Lewis base catalysts for their ability to promote the regioselective O- to C- or N-carboxyl transfer of a model N(2)-phenyl substituted pyrazolyl carbonate 2 that was readily prepared from commercially available materials (Table 1). In all cases, generation of the parent pyrazolinone as a side-product amounted to typically ∼5% of the crude reaction product mixture, so only a ratio of C- to N-regioisomeric products is given unless stated.29 Treatment of 2 with an NHC catalyst (generated in situ by deprotonation of the triazolium salt 5 with KHMDS) in THF gave the C-regioisomer 3 with high selectivity (>99:1 C:N), isolated in 44% yield (entry 1). Further optimisation showed that this NHC-promoted transformation could be performed using lower catalyst loadings in toluene (entries 2–4) while still giving 3 with excellent regioselectivity (>99:1 C:N). Remarkably, the use of DMAP in CH2Cl2 favoured N-carboxylation with high regioselectivity (7:93 C:N), giving 4 in 56% yield (entry 5). The regiochemistry of this carboxyl transfer was confirmed by X-ray crystal structure analysis of N-carboxylate 4.30,31 Rearrangement with isothiourea DHPB also favoured the N-carboxyl regioisomer but with lower reactivity compared to DMAP (entry 6). Further investigation of the DMAP-promoted reaction showed that THF and toluene proved poor solvents for this process, giving only ∼25% conversion to product with modest C:N ratios (entries 7 and 8). These results indicate that catalyst promoted regiodivergent selectivity is observed in this process under either NHC or DMAP catalysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Lewis base (mol%) | Solvent | Conv.a | C:N ratioa |
Yield (%) |
|
a Reaction conversion and C:N product ratio established by 1H NMR spectroscopic analysis of crude reaction mixture.
b NHC generated by deprotonation with KHMDS.
c 17% parent pyrazolinone generated.
|
|||||
| 1b | Precat 5 (20)b | THF | >95 | >99:1c |
44 (3) |
| 2 | Precat 5 (10) | Toluene | >95 | 99:1 |
49 (3) |
| 3 | Precat 5 (5) | Toluene | >95 | >99:1 |
— |
| 4 | Precat 5 (2) | Toluene | >95 | 99:1 |
— |
| 5 | DMAP (20) | CH2Cl2 | 73 | 7:93 |
56 (4) |
| 6 | DHPB (20) | CH2Cl2 | 66 | 9:91 |
— |
| 7 | DMAP (20) | Toluene | 25 | 18:82 |
— |
| 8 | DMAP (20) | THF | 25 | 23:77 |
— |

Scope and limitations
DMAP-catalysed selective O- to N-carboxyl transfer
The scope and limitations of these catalyst selective carboxyl transfer processes was next investigated through variation within the carbonate functionality and pyrazolyl scaffold at N(2)-, C(4)- and C(5)- (Table 2). Under DMAP catalysis in CH2Cl2, variation of the carbonate group gave the N-carboxyl products preferentially (≤16:84 C:N) that were isolated in good to moderate yield.31 Although benzyl carbonate 6 showed poor conversion even after extended reaction times, trichloroethyl and aryl N-carboxylate products containing both electron-withdrawing and electron-donating substituents were produced with good conversions. The effect of structural perturbation within the pyrazolyl skeleton was next investigated. With an N(2)-methyl substituent, C(5)-aryl substitution resulted in modest conversion but still preferential N-carboxylation to 10, while C(5)-methyl substitution gave preferential N-carboxyl product 11 with high selectivity and yield. Variation of the C(4) substituent also led to preferential N-carboxylation (products 12–16), irrespective of N(2)-Ph or -Me substitution.32
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | C:N ratioa |
N-Isomer | Yield (%) |
|
a C:N product ratios established by 1H NMR spectroscopic analysis of crude reaction mixture.
b Overnight reaction.
|
|||||||
| 1b | Bn | Ph | Me | Me | >1:99 |
6 | 10 |
| 2 | CH2CCl3 | Ph | Me | Me | 16:84 |
7 | 64 |
| 3 | Ph | Ph | Me | Me | 7:93 |
4 | 56 |
| 4 | 4-FC6H4 | Ph | Me | Me | 6:94 |
8 | 66 |
| 5 | 4-OMeC6H4 | Ph | Me | Me | 7:93 |
9 | 32 |
| 6 | Ph | Me | Ph | Me | 6:94 |
10 | 14 |
| 7 | 4-FC6H4 | Me | Me | Me | >1:99 |
11 | 60 |
| 8 | 4-FC6H4 | Ph | Me | Et | 3:97 |
12 | 74 |
| 9 | 4-FC6H4 | Ph | Me | Bn | 3:97 |
13 | 80 |
| 10 | 4-FC6H4 | Me | Me | Et | >1:99 |
14 | 61 |
| 11 | 4-FC6H4 | Me | Me | Bn | 1:99 |
15 | 50 |
| 12 | 4-FC6H4 | Me | Me | i-Bu | 3:97 |
16 | 77 |

NHC-catalysed selective O- to C-carboxyl transfer
Having demonstrated the generality of the DMAP-promoted O- to N-carboxyl transfer process, the NHC-catalysed O- to C-carboxyl transfer process was explored with toluene chosen as the reaction solvent (Table 3). Using achiral NHC precursor 5, variation of the carbonate group (entries 1–5), as well as N(2)-, C(4)- and C(5)-substituents (entries 6–12) gave preferentially the C-carboxyl isomer with high selectivity (up to >99:1 C:N, up to 84% yield). The regioselectivity of the NHC-catalysed reaction appears essentially independent of the nature of the carbonate group and C(5)-substituent, however it is particularly sensitive to the steric constraint at C(4), with a C(4)-iso-butyl group giving reduced C:N selectivity (76:24 C:N, 27, entry 12) relative to less hindered methyl substitution (97:3 C:N, 22, entry 7). A more modest reduction in regioselectivity was observed on changing the N(2)-substituent from phenyl to methyl (for example, compare products 24 (99:1 C:N, entry 9) and 26 (86:14 C:N, entry 11)).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | C:N ratioa |
C-isomer | Yield (%) |
|
a C:N product ratios established by 1H NMR spectroscopic analysis of crude reaction mixture.29
b Overnight reaction.
c 20 mol% catalyst, 18 mol% KHMDS.
|
|||||||
| 1b | Bn | Ph | Me | Me | >99:1 |
17 | 12 |
| 2 | CH2CCl3 | Ph | Me | Me | >99:1 |
18 | 67 |
| 3 | Ph | Ph | Me | Me | >99:1 |
3 | 49 |
| 4 | 4-FC6H4 | Ph | Me | Me | >99:1 |
19 | 49 |
| 5 | 4-OMeC6H4 | Ph | Me | Me | 93:7 |
20 | 63 |
| 6c | Ph | Me | Ph | Me | >99:1 |
21 | 55 |
| 7 | 4-FC6H4 | Me | Me | Me | 97:3 |
22 | 71 |
| 8 | 4-FC6H4 | Ph | Me | Et | >99:1 |
23 | 84 |
| 9 | 4-FC6H4 | Ph | Me | Bn | 99:1 |
24 | 73 |
| 10 | 4-FC6H4 | Me | Me | Et | 97:3 |
25 | 68 |
| 11 | 4-FC6H4 | Me | Me | Bn | 86:14 |
26 | 31 |
| 12 | 4-FC6H4 | Me | Me | i-Bu | 76:24 |
27 | 45 |

Enantioselective NHC-catalysed selective O- to C-carboxyl transfer
With the achiral NHC derived from salt 5 established as a regioselective catalyst for the formation of C-carboxyl pyrazolinones, the expansion of this methodology to the synthesis of enantioenriched products using chiral NHCs was probed.14 A screen of chiral triazolium NHC catalysts for the asymmetric rearrangement of model substrate 2 into C-carboxyl 3 identified N-pentafluorophenyl precatalyst 28 as the optimal catalyst with regards to regio- and enantioselectivity (toluene was again preferred as solvent over THF as it gave superior product yields and enantioselectivity).33 The full scope and generality of this asymmetric process was then investigated using NHC precatalyst 28 (Table 4). Aryl carbonates containing both electron-withdrawing and electron-donating substituents were tolerated with moderate levels of enantioselectivity (up to 68% ee, entries 1–3). By contrast, trichloroethyl carbonate showed good reactivity but poor enantioselectivity (entry 4). With a common N-(2)-phenyl substituent, other C-(4)-alkyl substituents were tolerated with promising enantioselectivity (up to 69% ee, entries 5 and 6). With an N-(2)-methyl substituent, a mixture of N- and C-carboxyl products favouring the C-carboxyl products was observed, with good to excellent levels of enantioselectivity for the C-carboxyl product achieved with methyl, ethyl and iso-butyl C-(4)-substitution (88–92% ee, entries 7–10). The absolute configuration within C-regioisomer 29 (entry 8) was assigned by X-ray diffraction with all other configurations assigned by analogy.30|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | C:N ratioa |
C-isomer | Yield (%) | % eeb |
|
a C:N product ratios established by 1H NMR spectroscopic analysis of crude reaction mixture.
b Established by HPLC analysis on a chiral stationary phase.
c 3 h reaction.
d Overnight reaction.
|
|||||||
| 1 | Ph | Ph | Me | 99:1 |
3 | 77 | 62 |
| 2c | 4-FC6H4 | Ph | Me | >99:1 |
19 | 54 | 60 |
| 3d | 4-OMeC6H4 | Ph | Me | >99:1 |
20 | 57 | 68 |
| 4c | CH2CCl3 | Ph | Me | 99:1 |
18 | 75 | 10 |
| 5 | 4-FC6H4 | Ph | Et | >99:1 |
23 | 74 | 69 |
| 6 | 4-FC6H4 | Ph | Bn | 99:1 |
24 | 67 | 60 |
| 7 | 4-FC6H4 | Me | Me | 77:23 |
22 | 65 | 87 |
| 8d | Ph | Me | Me | 85:15 |
29 | 61 | 86 |
| 9c | 4-FC6H4 | Me | Et | 83:17 |
25 | 54 | 90 |
| 10d | 4-FC6H4 | Me | i-Bu | 55:45 |
27 | 23 | 92 |
Mechanistic investigations
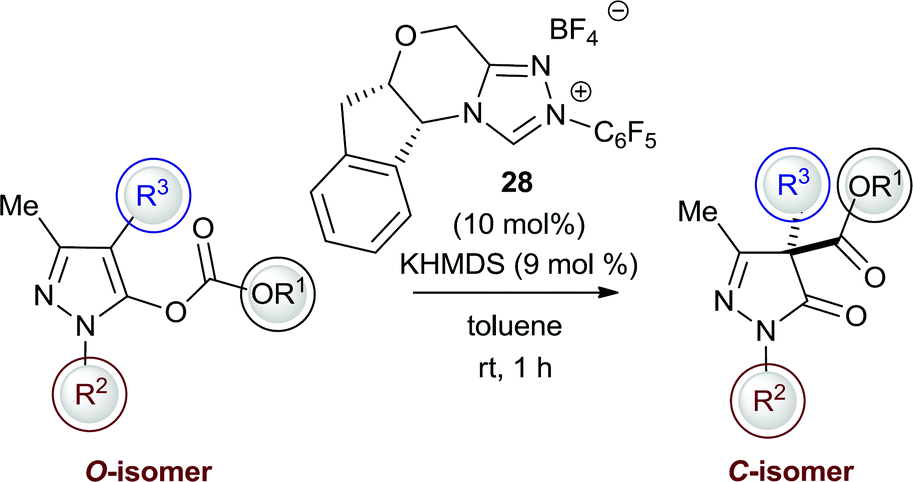
With this rearrangement reaction producing two regioisomeric products, the possibility of product interconversion due to the reversibility of the C–C and C–N bond-forming processes was investigated on model N(2)-Me substrates 11 and 22. First, N-carboxylate 11 (1:99 C:N) was resubmitted to both DMAP and NHC catalysis (Scheme 1). With DMAP, exclusively starting material was returned after overnight reaction, while treatment with an achiral NHC derived from 5 gave the C-regioisomer 22 (99:1 C:N). Furthermore, treatment of N-carboxylate 11 with the chiral NHC derived from precatalyst 28 gave 10% conversion to C-carboxylate 22 in 84% ee.
 | ||
| Scheme 1 Re-treatment of N-carboxyl 11 with DMAP and NHCs derived from 5 and 28. | ||
However, while treatment of enantioenriched C-carboxylate 22 (87% ee) with either DMAP or chiral NHC 28 returned 22 exclusively (87% ee), treatment with the achiral NHC derived from 5 gave C-carboxyl 22 in racemic form.34 Treatment of (±)-C-carboxylate 22 with chiral NHC 28 also returned (±)-22 (Scheme 2).
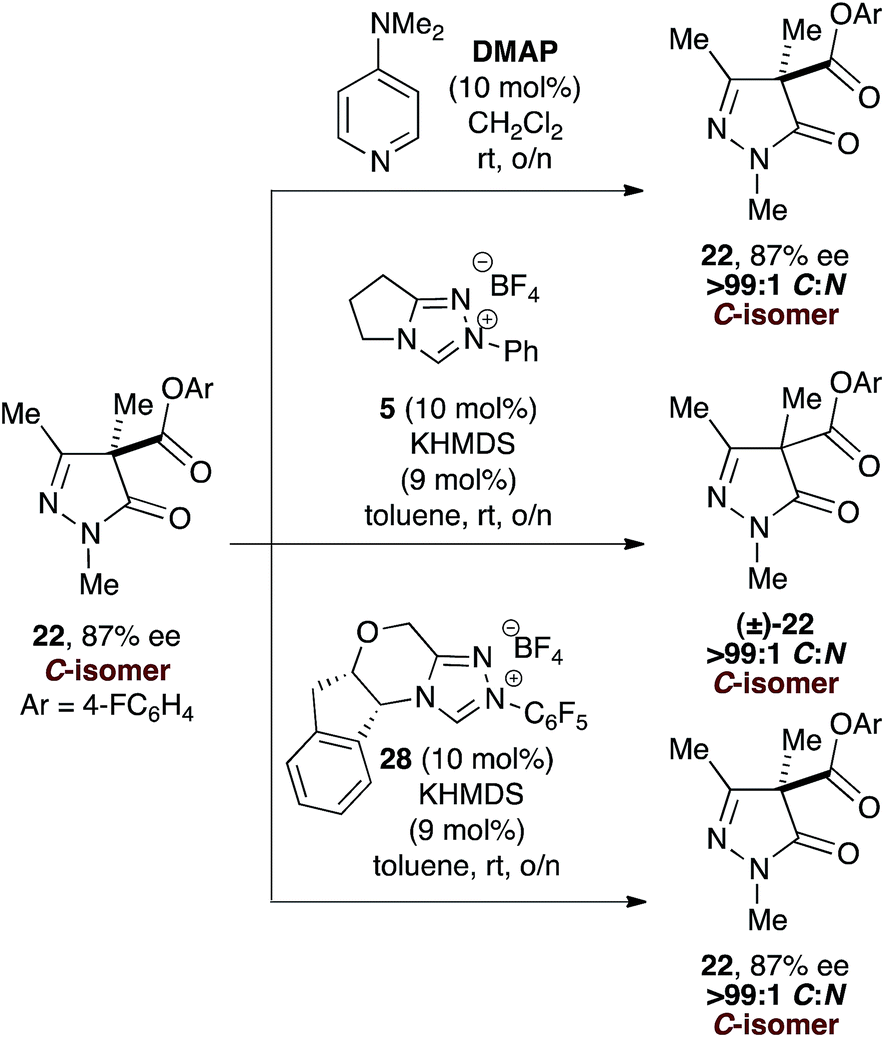 | ||
| Scheme 2 Re-treatment of C-carboxyl 22 with DMAP and NHCs derived from 5 and 28. | ||
These results, combined with a crossover experiment upon a mixture of N-carboxylate products,35 indicate that O- to C- or N-carboxyl transfer reactions with DMAP are irreversible in this model system, with N-carboxylation kinetically preferred; N- to C-carboxyl transfer is disfavoured with DMAP. With the achiral NHC derived from precatalyst 5, O- to C- or N-carboxyl transfer reactions are reversible, with the C-isomer thermodynamically preferred, while N- to C-carboxyl transfer is also favoured. However, with the chiral NHC, O- to C-carboxyl transfer is irreversible with high enantiocontrol, while N- to C-carboxyl transfer is also allowed with good enantiocontrol. To probe this latter hypothesis, the reaction conversion, C:N product ratio and ee of the O- to C-rearrangement of 30 to 22 using precatalyst 28 was monitored (Table 5). 1H NMR spectroscopic analysis of the reaction mixture showed increasing ratios of C:N products over time, further evidence of catalysed N- to C-carboxyl transfer.30 The ee of the C-carboxylate product 22 was however essentially independent of the reaction time and C:N ratio, consistent with our previous observations.

Computational insight
Computations were next performed on a simplified model substrate to elucidate the mechanism and origins of the observed regioselectivity. We employed M06-2X36/6-31+G**37/PCM38//M06-2X/6-31G*/PCM in toluene for NHC catalysis and dichloromethane for DMAP, as implemented in Gaussian09.39 Manual, exhaustive conformational searches were performed to ensure all relevant intermediates and transition state structures (TSs) were located. Intrinsic reaction coordinate (IRC) computations were performed on all TSs to verify reaction pathways. C- and N-carboxylations share the same general mechanism shown in Fig. 2. Initial O-carboxylate attack by catalyst (TS-II) and subsequent tetrahedral intermediate collapse (TS-IV) leads to common intermediates, enolate 30 and carboxylated catalyst (CO2Me-Cat). Regiodivergence occurs by recapture of carboxyl group by enolate 30 at either C(4)- or N(1)- (TS-VI). The dissociation of the catalyst from the resulting tetrahedral intermediate (TS-VIII) releases the final products. | ||
| Fig. 2 Detailed proposed mechanism for C- and N-carboxylation. | ||
DMAP catalysis
The DMAP-mediated carboxyl transfer preferentially results in N-carboxylation. Shown in Fig. 3, initial O-carboxylate attack (TS-II) by DMAP affords tetrahedral intermediate III, collapse of which (TS-IV, 20.4 kcal mol−1) affords ion pair intermediates V (CO2Me-DMAP and enolate 30). The C- vs. N-regiocontrol is established when the substrate enolate 30 attacks the carboxylated DMAP either via C(4)- or N(1). Consistent with the experimental results, the N-carboxylation process is favoured computationally by ∼5 kcal mol−1 (DMAP-TS-VI-N, ΔG‡ = 21.9 kcal mol−1vs.DMAP-TS-VI-C, ΔG‡ = 26.6 kcal mol−1). Interestingly, the N-carboxylation is stepwise addition of enolate and extrusion of catalyst, whereas the C-carboxylation process (black, Fig. 3) proceeds via a concerted, asynchronous carboxylation.40 This difference in concerted/stepwise behaviour reflects the lack of electrostatic stabilizing effects in DMAP-TS-VI-Cvs.DMAP-TS-VI-N. In DMAP-TS-VI-N, there is a substantial spatial overlap between the positively charged carboxylated DMAP and the attacking enolate 30, as the enolate oxygen and π bond is in closer proximity to the carboxylated DMAP. This is in contrast to DMAP-TS-VI-C, where there is a relatively poor spatial overlap, with only the enolate oxygen in proximity to the positively charged DMAP ring.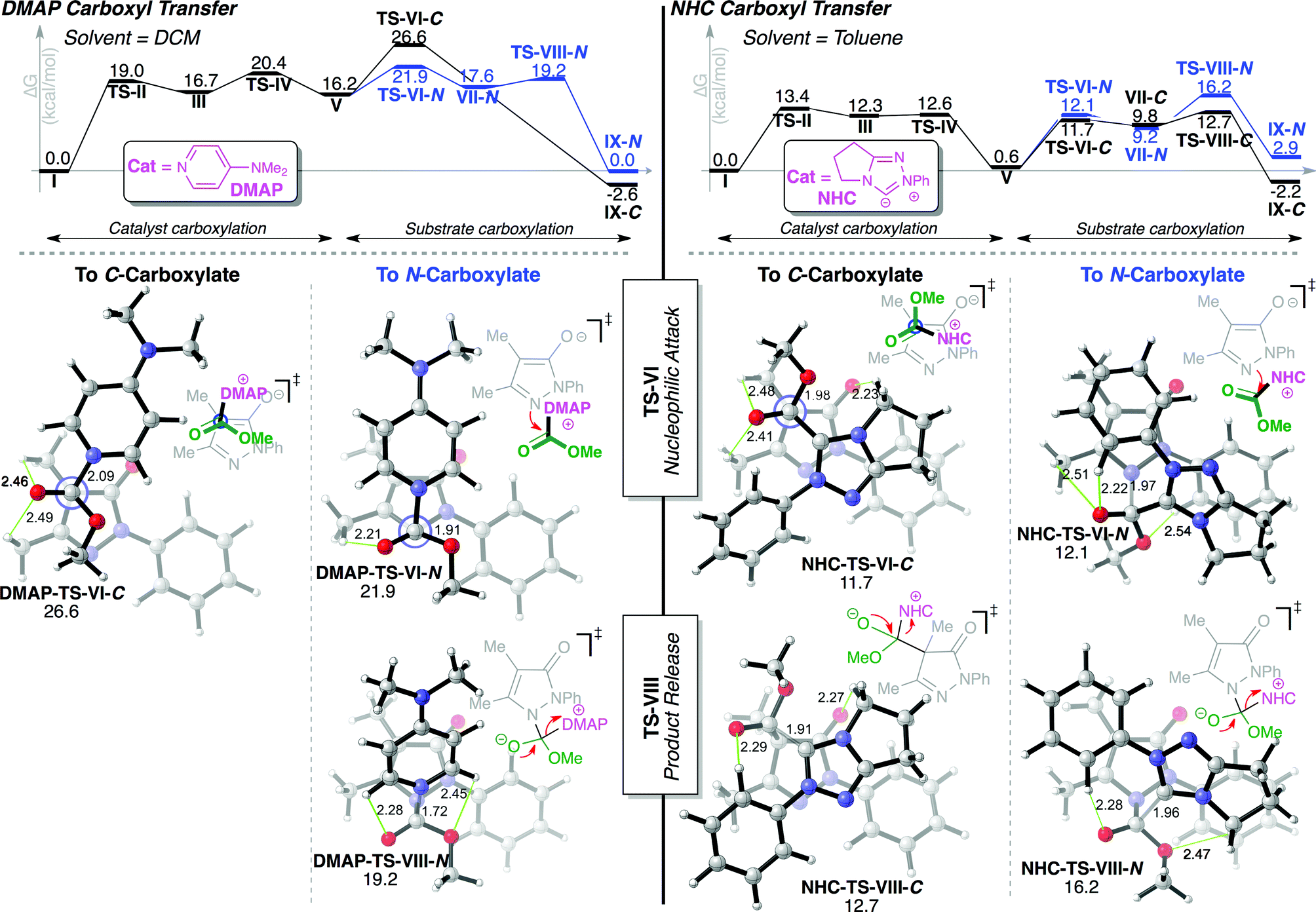 | ||
| Fig. 3 Reaction coordinate diagrams (top) and regioisomeric TSs (bottom) for DMAP-catalysed (left) and NHC-promoted (right) carboxyl transfer for major C-carboxylate (black) and minor N-carboxylate (blue). Green lines indicate stabilizing C–H⋯O hydrogen bonds, grey lines & Newman projections forming/breaking bond. | ||
NHC catalysis
NHC catalysis leads preferentially to C-carboxypyrazolinone product. NHC attack of the O-carboxylate substrate (TS-II, 13.4 kcal mol−1) and subsequent collapse (TS-IV, 12.6 kcal mol−1) of the tetrahedral intermediate (III, 12.3 kcal mol−1) leads to ion pair intermediates V (CO2Me–NHC and enolate 30). The NHC favours the C-carboxylation pathway by 3.5 kcal mol−1 (>99:1 C:N, NHC-TS-VIII-C and NHC-TS-VIII-N), in agreement with experiments. This selectivity arises due to the large relative instability of NHC-TS-VIII-N, where the close proximity of the enolate oxygen and the relatively negatively charged areas of the carboxylated NHC results in a destabilizing repulsive interaction. This is in contrast to NHC-TS-VIII-C, where this repulsive interaction is replaced by stabilizing C–H⋯O hydrogen bonds41 between the NHC and the enolate 30. The computed reaction coordinates corroborate the experimentally observed reversibility of the NHC-catalysed process. NHC addition to the N-carboxylated product (NHC-TS-VIII-N) is energetically accessible, with a reverse barrier of 13.3 kcal mol−1 (from NHC-IX-N). The forward process leading to the C-carboxylation is favoured by 2.6 kcal mol−1 over the forward process for the N-carboxylation (Fig. 3), resulting in exclusive production of C-carboxylated product upon retreatment of N-carboxylated product with achiral NHC (as observed in Scheme 1).
Structural comparison of enolate π vs. σ reactivity
The remarkable switch in regioselectivity observed between DMAP and NHC catalysis in this system is a result of the markedly different reactivity patterns of the intermediate carboxylated DMAP or NHC and their interaction with the pyrazoline enolate as illustrated in Fig. 3. This is most striking in DMAP-TS-VI-N, where favoured nucleophilic attack from the substrate does not originate from the N(1)-lone pair of the substrate enolate in the σ-plane, but rather the π-system of the extended enolate. This is in contrast to the analogous (disfavoured) NHC-TS-VI-N, where nucleophilic attack is predicted to occur from the N(1)-lone pair of the substrate enolate in the σ-plane. As yet, the exact origins of this π vs. σ reactivity are unknown. Our working hypothesis is that the relatively sterically unencumbered conjugated DMAP promotes π-π electrostatic interactions,42 allowing the π-system of the extended enolate to be an energetically more competent nucleophile compared with the σ-N(1)-lone pair.Conclusion
In conclusion, the regiodivergent O- to C- or N-carboxyl transfer of pyrazolyl carbonates has been investigated, with DMAP giving preferential N-carboxylation and triazolinylidenes promoting selective C-carboxylation (both with up to 99:1 regioselectivity). An enantioselective O- to C-carboxyl variant using NHC catalysis is demonstrated (up to 92% ee), while mechanistic and DFT studies outline the pathways operative in this system and delineate insight into the structural reasons for the observed selectivity. Current investigations from within our groups are focused upon the demonstration of further Lewis base-mediated organocatalytic transformations and developing further computational insight into these transformations.
Acknowledgements
We thank the Royal Society for a University Research Fellowship (ADS) and the EPSRC and Merck Sharp & Dohme Ltd (CASE award to EG) for funding, and the EPSRC National Mass Spectrometry Service Centre (Swansea). The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) / ERC grant agreement n° 279850.Notes and references
- (a) W. Steglich and G. Höfle, Angew. Chem., 1968, 80, 78 CrossRef; (b) W. Steglich and G. Höfle, Tetrahedron Lett., 1970, 11, 4727–4730 CrossRef . For an early review of this area, see: ; (c) G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed., 1978, 17, 569–583 CrossRef.
- J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532–11533 CrossRef CAS.
- S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368–13369 CrossRef CAS PubMed.
- H. V. Nguyen, D. C. D. Butler and C. J. Richards, Org. Lett., 2006, 8, 769–772 CrossRef CAS PubMed.
- E. Busto, V. Gotor-Fernández and V. Gotor, Adv. Synth. Catal., 2006, 348, 2626–2632 CrossRef CAS.
- For selected reviews on azlactone rearrangements and related reactions, see: (a) J. S. Fisk, R. A. Mosey and J. J. Tepe, Chem. Soc. Rev., 2007, 36, 1432–1440 RSC; (b) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC; (c) R. A. Mosey, J. S. Fisk and J. J. Tepe, Tetrahedron: Asymmetry, 2008, 19, 2755–2762 CrossRef CAS PubMed; (d) A. Moyano, N. El-Hamdouni and A. Atlamsani, Chem.–Eur. J., 2010, 16, 5260–5273 CrossRef CAS PubMed; (e) A.-N. R. Alba and R. Rios, Chem.–Asian J., 2011, 6, 720–734 CrossRef CAS PubMed.
- Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef CAS PubMed.
- (a) F. R. Dietz and H. Gröger, Synlett, 2008, 663–666 CAS; (b) H. Gröger, Synthesis, 2009, 4208–4218 Search PubMed.
- C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed.
- B. Viswambharan, T. Okimura, S. Suzuki and S. Okamoto, J. Org. Chem., 2011, 76, 6678–6685 CrossRef CAS PubMed.
- C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed.
- D. Uraguchi, K. Koshimoto, S. Miyake and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 5567–5569 CrossRef CAS PubMed.
- (a) J. E. Thomson, K. Rix and A. D. Smith, Org. Lett., 2006, 8, 3785–3788 CrossRef CAS PubMed; (b) J. E. Thomson, C. D. Campbell, C. Concellón, N. Duguet, K. Rix, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2008, 73, 2784–2791 CrossRef CAS PubMed; (c) C. D. Campbell, N. Duguet, K. A. Gallagher, J. E. Thomson, A. G. Lindsay, A. O'Donoghue and A. D. Smith, Chem. Commun., 2008, 3528–3530 RSC; (d) J. E. Thomson, A. F. Kyle, C. Concellón, K. A. Gallagher, P. Lenden, L. C. Morrill, A. J. Miller, C. Joannesse, A. M. Z. Slawin and A. D. Smith, Synthesis, 2008, 2805–2818 CAS; (e) J. E. Thomson, A. F. Kyle, K. B. Ling, S. R. Smith, A. M. Z. Slawin and A. D. Smith, Tetrahedron, 2010, 66, 3801–3813 CrossRef CAS PubMed.
- (a) C. D. Campbell, C. Concellón and A. D. Smith, Tetrahedron: Asymmetry, 2011, 22, 797–811 CrossRef CAS PubMed; (b) Y. P. Rey and R. Gilmour, Beilstein J. Org. Chem., 2013, 9, 2812–2820 CrossRef CAS PubMed.
- For selected examples of these rearrangements processes see: (a) A. H. Mermerian and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 4050–4051 CrossRef CAS PubMed; (b) I. D. Hills and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 3921–3924 CrossRef CAS PubMed; (c) T. A. Duffey, S. A. Shaw and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14–15 CrossRef CAS PubMed; (d) M. Ismail, H. V. Nguyen, G. Ilyashenko, M. Motevalli and C. J. Richards, Tetrahedron Lett., 2009, 50, 6332–6334 CrossRef CAS PubMed; (e) C. Joannesse, L. C. Morrill, C. D. Campbell, A. M. Z. Slawin and A. D. Smith, Synthesis, 2011, 1865–1879 CAS.
- J. E. DeLorbe, S. Y. Jabri, S. M. Mennen, L. E. Overman and F.-L. Zhang, J. Am. Chem. Soc., 2011, 133, 6549–6552 CrossRef CAS PubMed.
- S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va and E. Vedejs, J. Am. Chem. Soc., 2006, 128, 925–934 CrossRef CAS PubMed.
- For an excellent review that details the power of catalyst selective synthesis see: J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954–10990 CrossRef CAS PubMed.
- T. Nishiyama and M. Ogawa, Acta Anaesthesiol. Scand., 2005, 49, 147–151 CrossRef CAS PubMed.
- G. G. Graham and K. F. Scott, Inflammopharmacology, 2003, 11, 401–413 CrossRef CAS PubMed.
- (a) C. Calvet, R. Cuberes, C. Pérez-Maseda and J. Frigola, Electrophoresis, 2002, 23, 1702–1708 CrossRef CAS; (b) S. M. Sondhi, M. Dinodia, J. Sinigh and R. Rani, Curr. Bioact. Compd., 2007, 3, 91–108 CrossRef CAS.
- M. Johnson, B. Younglove, L. Lee, R. LeBlanc, H. Holt Jr, P. Hills, H. Mackay, T. Brown, S. L. Mooberry and M. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 5897–5901 CrossRef CAS PubMed.
- (a) T. Rosu, S. Pasculescu, V. Lazar, C. Chifiriuc and R. Cernat, Molecules, 2006, 11, 904–914 CrossRef CAS; (b) M. J. Seo, J. K. Kim, B. S. Son, B. G. Song, Z. No, H. G. Cheon, K.-R. Kim, Y. S. Sohn and H. R. Kim, Bull. Korean Chem. Soc., 2004, 25, 1121–1123 CrossRef CAS; (c) S. Bondock, R. Rabie, H. A. Etman and A. A. Fadda, Eur. J. Med. Chem., 2008, 43, 2122–2129 CrossRef CAS PubMed.
- V. Hadi, Y.-H. Koh, T. W. Sanchez, D. Barrios, N. Neamati and K. W. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 6854–6857 CrossRef CAS PubMed.
- (a) R. Oishi, Y. Itoh, M. Nishibori, T. Watanabe, H. Nishi and K. Saeki, Stroke, 1989, 20, 1557–1564 CrossRef CAS; (b) T. Yamaguchi, K. Oishi, M. Uchida and H. Echizen, Biol. Pharm. Bull., 2003, 26, 1706–1710 CrossRef CAS.
- (a) D. J. Hlasta, F. B. Casey, E. W. Ferguson, S. J. Gangell, M. R. Heimann, E. P. Jaeger, R. K. Kullnig and R. J. Gordon, J. Med. Chem., 1991, 34, 1560–1570 CrossRef CAS; (b) L. Savini, P. Massarelli, C. Nencini, C. Pellerano, G. Biggio, A. Maciocco, G. Tuligi, A. Carrieri, N. Cinone and A. Carotti, Bioorg. Med. Chem., 1998, 6, 389–399 CrossRef CAS; (c) M. G. Ferlin, G. Chiarelotto, S. Dall'Acqua, E. Maciocco, M. P. Mascia, M. G. Pisu and G. Biggio, Bioorg. Med. Chem., 2005, 13, 3531–3541 CrossRef CAS PubMed; (d) A. Kimata, H. Nakagawa, R. Ohyama, T. Fukuuchi, S. Ohta, T. Suzuki and N. Miyata, J. Med. Chem., 2007, 50, 5053–5056 CrossRef CAS PubMed; (e) W. J. Yuan, T. Yasuhara, T. Shingo, K. Muraoka, T. Agari, M. Kameda, T. Uozumi, N. Tajiri, T. Morimoto, M. Jing, T. Baba, F. Wang, H. Leung, T. Matsui, Y. Miyosh and I. Date, BMC Neurosci., 2008, 9, 75 CrossRef PubMed; (f) G. Mariappan, B. P. Saha, L. Sutharson, Ankit, S. Garg, L. Pandey and D. Kumar, J. Pharma Res., 2010, 3, 2856–2859 CAS.
- (a) E. Gould, T. Lebl, A. M. Z. Slawin, M. Reid and A. D. Smith, Tetrahedron, 2010, 66, 8992–9008 CrossRef CAS PubMed; (b) M. Reid, T. Davies and A. D. Smith, Org. Biomol. Chem., 2013, 11, 7877–7892 RSC.
- The enantioselective conjugate addition of pyrazolinone enolates to electrophiles has been reported: (a) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928–4932 CrossRef CAS PubMed; (b) Y.-H. Liao, W.-B. Chen, Z.-J. Wu, X.-L. Du, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Adv. Synth. Catal., 2010, 352, 827–832 CrossRef CAS.
- For full details of the observed product ratios, see the ESI.†.
- See ESI for further details.†.
- For substrates where N(2) = Ph, DMAP gave predominantly the C-carboxylate product at extended reaction times (18 h). See ESI for further details.†.
- In all cases, phenyl carboxyl substrates were also screened with very similar results. See ESI for details.†.
- See ESI for details of enantioselective catalyst screening.†.
- This is in contrast to analogous reactions with oxazolyl carbonates in which the ee remained unchanged after treatment with an achiral NHC: C. D. Campbell, C. J. Collett, J. E. Thomson, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2011, 9, 4205–4218 CAS.
- A crossover experiment performed with a mixture of N-carboxylates in the presence of DMAP displayed no mixing of the carbonate groups despite extended reaction time, further evidence for the irreversibility of the DMAP catalysed process in this system. See ESI for further details.†.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem.–Eur. J., 2005, 11, 4751–4757 CrossRef CAS PubMed.
- (a) R. C. Johnston and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057–5064 RSC; (b) R. C. Johnston, D. T. Cohen, C. C. Eichman, K. A. Scheidt and P. H.-Y. Cheong, Chem. Sci., 2014, 5, 1974–1982 RSC; (c) M. D. Pierce, R. C. Johnston, S. Mahapatra, H. Yang, R. G. Carter and P. H.-Y. Cheong, J. Am. Chem. Soc., 2012, 134, 13624–13631 CrossRef CAS PubMed; (d) O. Pattawong, T. J. L. Mustard, R. C. Johnston and P. H.-Y. Cheong, Angew. Chem., Int. Ed., 2013, 52, 1420–1423 CrossRef CAS PubMed; (e) I. V. Alabugin, M. Manoharan, S. Peabody and F. Weinhold, J. Am. Chem. Soc., 2003, 125, 5973–5987 CrossRef CAS PubMed.
- A. Schmidt, A. Lindner, M. Nieger, M. Ruiz-Delgado and F. J. Ramirez, Org. Biomol. Chem., 2006, 4, 3056–3066 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic details for all novel compounds. CCDC 987861–987864. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00879k |
| This journal is © The Royal Society of Chemistry 2014 |