DOI:
10.1039/C4RA09092F
(Paper)
RSC Adv., 2014,
4, 52307-52315
Direct arylation of heterocycles through C–H bond cleavage using metal–organic-framework Cu2(OBA)2(BPY) as an efficient heterogeneous catalyst†
Received
22nd August 2014
, Accepted 8th October 2014
First published on 8th October 2014
Abstract
A crystalline porous metal–organic-framework Cu2(OBA)2(BPY) was synthesized and characterized by X-ray powder diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), thermogravimetric analysis (TGA), Fourier transform infrared (FT-IR), atomic absorption spectrophotometry (AAS), and nitrogen physisorption measurements. The Cu-MOF was shown to be an efficient heterogeneous catalyst for direct C-arylation of a variety of heterocycles by iodoarenes. The optimal conditions employed tBuOLi in dioxane at elevated temperature. In addition, a leaching test was also conducted to investigate the heterogeneity. Gratifyingly, the MOF catalyst can be facilely recycled several times under identical conditions without remarkable loss in catalytic reactivity.
1. Introduction
The biaryl structural motif is often found in natural products as well as in many pharmaceutically relevant and biologically active compounds.1 As a consequence, for over a century, new and progressively more efficient aryl–aryl bond-forming methods have been developed. In general, transition metal catalysis is commonly used for the construction of aryl–aryl bonds.2 Traditional methods for biaryl preparation involve a reaction of aryl halide or pseudohalide with an organometallic reagent. However, the preparation of starting materials adds extra steps to the synthetic sequences. Catalytic direct arylation provides more efficient routes by using simple arenes in the coupling reactions. Since organic molecules usually display many non-equivalent C–H bonds with comparable dissociation energies, modern direct arylation methods that allow reactions to proceed with excellent selectivity are developed.3 A majority of the C–H bond arylation examples include palladium-, rhodium-, or ruthenium-catalyzed functionalization of five-membered-ring heterocycles and directing-group-containing arenes.4 Several recent reports have described copper-catalyzed deprotonative arylation of heterocycles and functionalization selectively occurred at the most acidic sp2 C–H bonds.5–7 It is worth to mention that, among heterocycles, azoles are frequently used in functionalization.8–10 Additionally, reported conditions for reactions of azoles are not active for other heterocycles such as thiazole, thiophens, or imidazoles.11,12 Therefore, functionalization of heterocycles other than azoles should be paid more attention since they are often structurally found in many valuable compounds. Recently, Zhang and co-workers demonstrated the first example of a heterogeneous C–H arylation reaction between benzoxazole, benzothiazole, and 1-methylbenzimidazole with aryl iodides in the presence of CuO nanoparticles as the catalyst.13 Under the view of green chemistry, organic transformations using heterogeneous catalysts would offer advantages in terms of the ease of handling, simple workup, and recyclability and reusability.14
Metal–organic frameworks (MOFs) are crystalline materials constructed from two components, metal ions or metallic clusters and polyfunctional organic linkers.15,16 Taking advantages of special physical properties from both organic and inorganic porous materials, these structures could offer potential applications in a variety of fields, including gas storage media, separations, chemical sensors, thin film devices, optics, drug carriers, biomedical application, and catalysis.17–24 The application of MOFs in catalysis has recently attracted significant attention as one of the newest developments of these porous materials.25–27 Indeed, many MOFs have been explored as catalysts or catalyst supports for various organic transformations, ranging from carbon–carbon28–31 to carbon-heteroatom forming reactions.32–39 Among numerous popular MOFs as catalysts, several copper-based structures could offer high activity for many organic reactions due to their unsaturated open copper metal sites.34,40–47 In this work, we present direct arylation of a variety of heterocycles through C–H activation using metal–organic framework Cu2(OBA)2(BPY) as an efficient and recyclable heterogeneous catalyst. To the best of our knowledge, the application of Cu2(OBA)2(BPY) as a heterogeneous catalyst for organic transformations was not previously mentioned in the literature.
2. Experimental section
2.1. Materials
All reagents and starting materials were obtained commercially from Sigma-Aldrich, Merck, and Acros Organics, and were used as received without any further purification unless otherwise noted. All reactions were conducted under inert (Argon) atmosphere.
2.2. Instrumentation
Nitrogen physisorption measurements were conducted using a Micromeritics 2020 volumetric adsorption analyzer system. Samples were pretreated by heating under vacuum at 150 °C for 3 h. A Netzsch Thermoanalyzer STA 409 was used for thermogravimetric analysis (TGA) with a heating rate of 10 °C min−1 under a nitrogen atmosphere. X-ray powder diffraction (XRD) patterns were recorded using a Cu Kα radiation source on a D8 Advance Bruker powder diffractometer. Scanning electron microscopy studies were conducted on a S4800 Scanning Electron Microscope (SEM). Transmission electron microscopy studies were performed using a JEOL JEM 1400 Transmission Electron Microscope (TEM) at 100 kV. The Cu2(OBA)2(BPY) sample was dispersed on holey carbon grids for TEM observation. Elemental analysis with inductive coupled plasma (ICP) was performed on an ICPE-9000 Shimadzu. Fourier transform infrared (FT-IR) spectra were obtained on a Nicolet 6700 instrument, with samples being dispersed on potassium bromide pallets.
Gas chromatographic (GC) analyses were performed using a Shimadzu GC 2010-Plus equipped with a flame ionization detector (FID) and an SPB-5 column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). The temperature program for GC analysis held samples at 100 °C for 1 min, heated samples from 100 to 180 °C at 40 °C min−1; heated them from 180 to 290 °C at 50 °C min−1 and held them at 290 °C for 2 min. Inlet and detector temperatures were set constant at 290 °C. n-Hexadecane was used as an internal standard to calculate reaction conversions. GC-MS analyses were performed using a Hewlett Packard GC-MS 5972 with a RTX-5MS column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.5 μm). The temperature program for GC-MS analysis heated samples from 60 to 280 °C at 10 °C min and held them at 280 °C for 2 min. Inlet temperature was set constant at 280 °C. MS spectra were compared with the spectra gathered in the NIST library. The 1H and 13C NMR were recorded on a Bruker AV 500 spectrometers using residual solvent peak as a reference.
2.3. Synthesis of the metal–organic framework Cu2(OBA)2(BPY)
In this work, the metal–organic framework Cu2(OBA)2(BPY) was synthesized by a solvothermal method (see ESI†).48 In a typical preparation, three solutions of copper(II) nitrate trihydrate (Cu(NO3)2·3H2O) (0.242 g, 1 mmol) in DMF (DMF = N,N′-dimethylformamide; 5 mL), 4,4′-oxybis(benzoic) acid (H2OBA) (0.258 g, 1 mmol) in DMF (3 mL), and 4,4′-bipyridine (BPY) (0.078 g, 0.5 mmol) in DMF (3 mL), respectively, were prepared. Distilled water was added dropwise into the DMF solution of Cu(NO3)2·3H2O (2 mL water) and the DMF solution of H2OBA (1 mL water), respectively, and the resulting solutions were magnetically stirred for 5 min. The solution of H2OBA and the solution of BPY were then added dropwise into the solution of Cu(NO3)2·3H2O, and the mixture was magnetically stirred until a clear solution was observed. The reaction solution was distributed into three 10 mL vials, and the vials were then heated at 85 °C in an isothermal oven for 48 h. After cooling the vials to room temperature, the solid product in each vial was removed by decanting with mother liquor and washed in DMF (3 × 20 mL) for 3 days. The material was then evacuated under vacuum at 150 °C for 6 h, yielding 0.284 g of Cu2(OBA)2(BPY) in the form of green light crystals (71% yield).
2.4. Catalytic activity
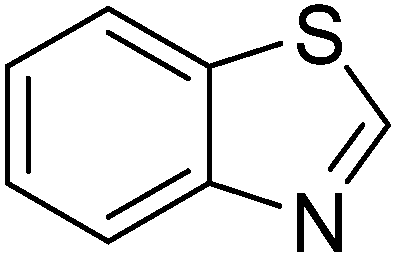
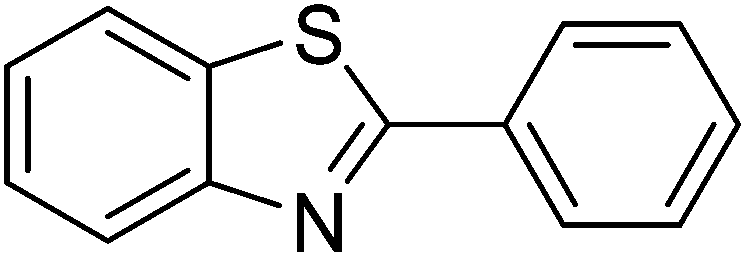
In reaction optimization, the Cu2(OBA)2(BPY) was used as a catalyst for direct arylation of benzothiazole with iodobenzene (Scheme 1). In a typical experiment, benzothiazole (0.11 mL, 1 mmol) were added to a vial with teflon cap containing 1,4-dioxane (4 mL) and n-hexadecane (0.1 mL) as internal standard. A mixture of iodobenzene (0.168 mL, 1.5 mmol), Cu2(OBA)2(BPY) (0.012 g, 3 mol%), and tBuOLi (0.192 g, 2 mmol) as a base were quickly added to the vial. The catalyst percentage was calculated based on the molar ratio of copper/benzothiazole. The reaction mixture was magnetically stirred at 120 °C for 6 h. The reaction conversion was monitored by withdrawing aliquots from the reaction mixture at different time intervals, quenching with an aqueous KOH solution (1%, 0.1 mL), diluting with diethyl ether, drying over anhydrous Na2SO4, analyzing by GC with reference to n-hexadecane. The product identity was further confirmed by GC-MS, 1H NMR, and 13C NMR. To investigate the recyclability of Cu2(OBA)2(BPY), the catalyst was filtered from the reaction mixture after the experiment, washed with copious amounts of DMF and water, dried at 150 °C under vacuum in 6 h. For the leaching test, a catalytic reaction was stopped after 1 h, analyzed by GC, and filtered to remove the solid catalyst. Solid tBuOLi was then added to the solution, and the reaction mixture was then stirred for a further 5 h. Reaction progress, if any, was monitored by GC as previously described.
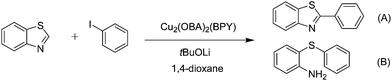 |
| | Scheme 1 Reaction of benzothiazole with iodobenzene using Cu2(OBA)2(BPY) catalyst. | |
3. Results and discussion
Synthesized catalyst characterization was carried out using several techniques, including XRD, SEM, TEM, TGA, FT-IR, ICP, and nitrogen physisorption measurements (Fig. S1–S7 in ESI†). The analysis results are in agreement with reported literature.36 Initial studies in optimization addressed the effect of temperature. Direct arylation reactions in 1,4-dioxane at 3 mol% Cu2(OBA)2(BPY) catalyst, with the benzothiazole–iodobenzene molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, in the presence of 2 equivalents of tBuOLi as the base, at 80 °C, 100 °C, 120 °C, and 140 °C were conducted. It was observed that the reaction carried out at 80 °C afforded 47% conversion after 6 h. As expected, increasing the reaction temperature led to a significant enhancement in the reaction rate, with 62% and 82% conversion being obtained after 6 h for the reaction carried out at 100 °C and 120 °C, respectively. Increasing the temperature to higher did not affect the conversion (Fig. 1). It should be noted that only 2-phenylbenzothiazole (product A, Scheme 1) was produced under these conditions. Daugulis and Do previously reported that the transformation could proceed readily at 140 °C.49 In the first example of the heterogeneous direct arylation of benzothiazole with iodobenzene using CuO nanoparticles catalyst, Wang and co-workers carried out the reaction at 162 °C for 8 h.13
:1.2, in the presence of 2 equivalents of tBuOLi as the base, at 80 °C, 100 °C, 120 °C, and 140 °C were conducted. It was observed that the reaction carried out at 80 °C afforded 47% conversion after 6 h. As expected, increasing the reaction temperature led to a significant enhancement in the reaction rate, with 62% and 82% conversion being obtained after 6 h for the reaction carried out at 100 °C and 120 °C, respectively. Increasing the temperature to higher did not affect the conversion (Fig. 1). It should be noted that only 2-phenylbenzothiazole (product A, Scheme 1) was produced under these conditions. Daugulis and Do previously reported that the transformation could proceed readily at 140 °C.49 In the first example of the heterogeneous direct arylation of benzothiazole with iodobenzene using CuO nanoparticles catalyst, Wang and co-workers carried out the reaction at 162 °C for 8 h.13
 |
| | Fig. 1 Effect of temperature on reaction conversion. | |
The effect of catalyst loading on the reaction conversion was next investigated. In the first example of the copper-catalyzed direct arylation of benzothiazole with iodobenzene, Daugulis and Do used 10 mol% CuI was employed for the transformation.49 Miura and co-workers employed up to 1 equivalent of CuI as catalyst for the same transformation, while 10 mol% CuI catalyst was required for the direct arylation of benzoxazole with iodobenzene.50 Huang and co-workers demonstrated that the copper catalyst concentration necessary for the transformation could be as low as 1 mol% when 0.25 mol% palladium as co-catalyst was employed.51 Wang and co-workers also carried out the direct arylation of benzothiazole with iodobenzene with 10 mol% CuO nanoparticles as the catalyst.13 The direct arylation reaction was then carried out in 1,4-dioxane at 120 °C, with the benzothiazole–iodobenzene molar ratio of 1:1.2, in the presence of 2 equivalents of tBuOLi as the base. It should be noted that no reaction occurred in the absence of the Cu2(OBA)2(BPY) catalyst. The reaction using 0.5 mol%, Cu2(OBA)2(BPY) catalyst proceeded to 53% conversion after 6 h, while 69% conversion was detected for the case of 1 mol% catalyst. Optimal conversion was obtained when 3% of catalyst was applied (Fig. 2). Furthermore, it was also observed that decreasing the benzothiazole–iodobenzene molar ratio resulted in a drop in the reaction rate. The reaction using 1.2 equivalent of iodobenzene afforded 75% conversion after 6 h, while only 65% conversion was detected after 6 h for that using 1 equivalent of iodobenzene (Fig. 3).
 |
| | Fig. 2 Effect of catalyst concentration on reaction conversion. | |
 |
| | Fig. 3 Effect of benzothiazole–iodobenzene molar ratio on reaction conversion. | |
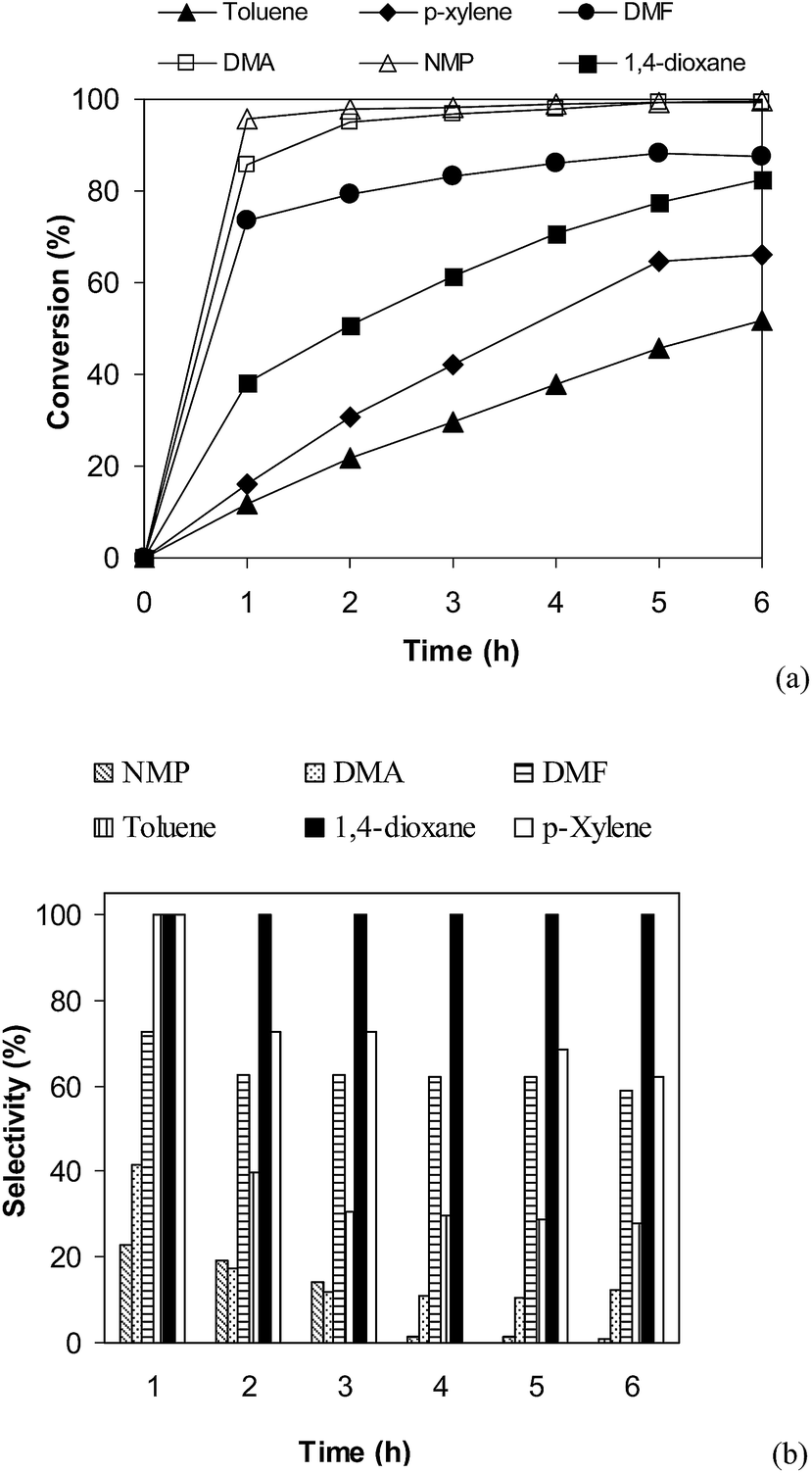
Effect of different solvents on cross-coupling reaction of benzothiazole with iodobenzene was also examined.52,53 It was observed that the direct arylation reaction could proceed to 95% conversion after 1 h in N-methylpyrrolidone (NMP). However, experimental results indicated that 2-(phenylthio)benzenamine (product B, Scheme 1) was mainly formed in reaction mixture. The identity of 2-(phenylthio)benzenamine was confirmed by GC-MS, 1H NMR, and 13C NMR (see ESI†). Similarly, poor selectivity was observed in DMA, DMF, p-xylene, and toluene (Fig. 4). As mentioned, good conversion and undetectable by-products was achieved in 1,4-dioxane solvent. In the first example of the heterogeneous direct arylation of benzothiazole with iodobenzene using CuO nanoparticles as the catalyst, Wang and co-workers carried out the reaction in diglyme at reflux temperature (160 °C), and only trace amount of 2-phenylbenzothiazole was detected in 1,4-dioxane at reflux temperature.13
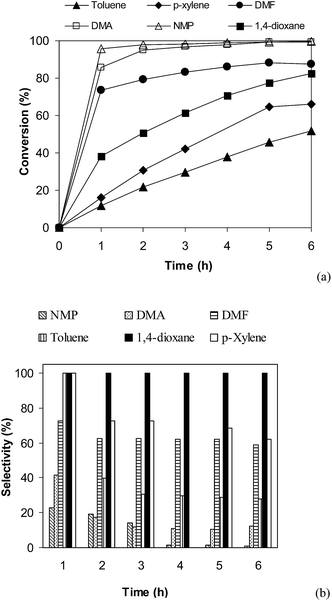 |
| | Fig. 4 Effect of solvent on reaction conversion (a) and selectivity (b). | |
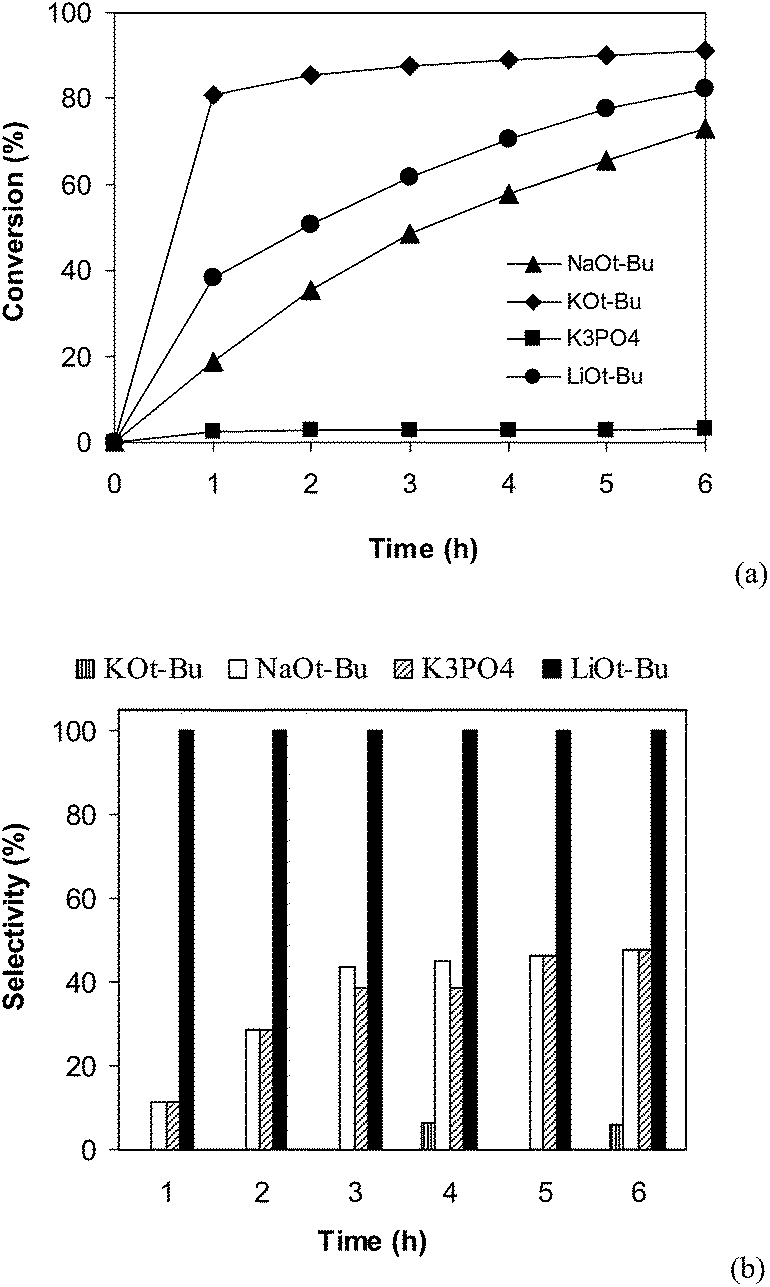
Different bases including tBuOK, tBuONa, and K3PO4 were then employed. The experimental results indicated that K3PO4 was not unsuitable with less than 5% conversion being detected after 6 h. This observation was different from results of Miura and co-workers, where the CuI-catalyzed direct arylation reaction between benzothiazole and iodobezene could efficiently produce 2-phenylbenzothiaazole with K3PO4 base.50 The direct arylation reaction using tBuOK and tBuONa as the base could proceed to 92% and 73% conversion, respectively, after 6 h. However, 2-(phenylthio)benzenamine but not 2-phenylbenzothiaazole was obtained as the main product (Fig. 5). It is well-known that benzothiozole can undergo ring-opening under basic conditions.54–57 Thus, formation of by-products can be explained by reaction sequence in Scheme 2.58 Moreover, it was observed that using more than 2 equivalents of tBuOLi was not necessary as the reaction conversion did not increase any further, and using less than 2 equivalents of tBuOLi resulted in a drop in the reaction rate (Fig. 6).
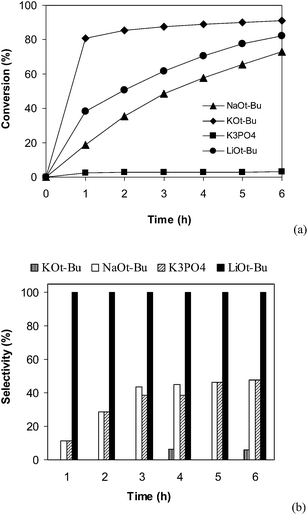 |
| | Fig. 5 Effect of base on reaction conversion (a) and selectivity (b). | |
 |
| | Scheme 2 Plausible reaction pathway for the formation of 2-(phenylthio)benzenamine by-product. | |
 |
| | Fig. 6 Effect of base concentration on reaction conversion. | |
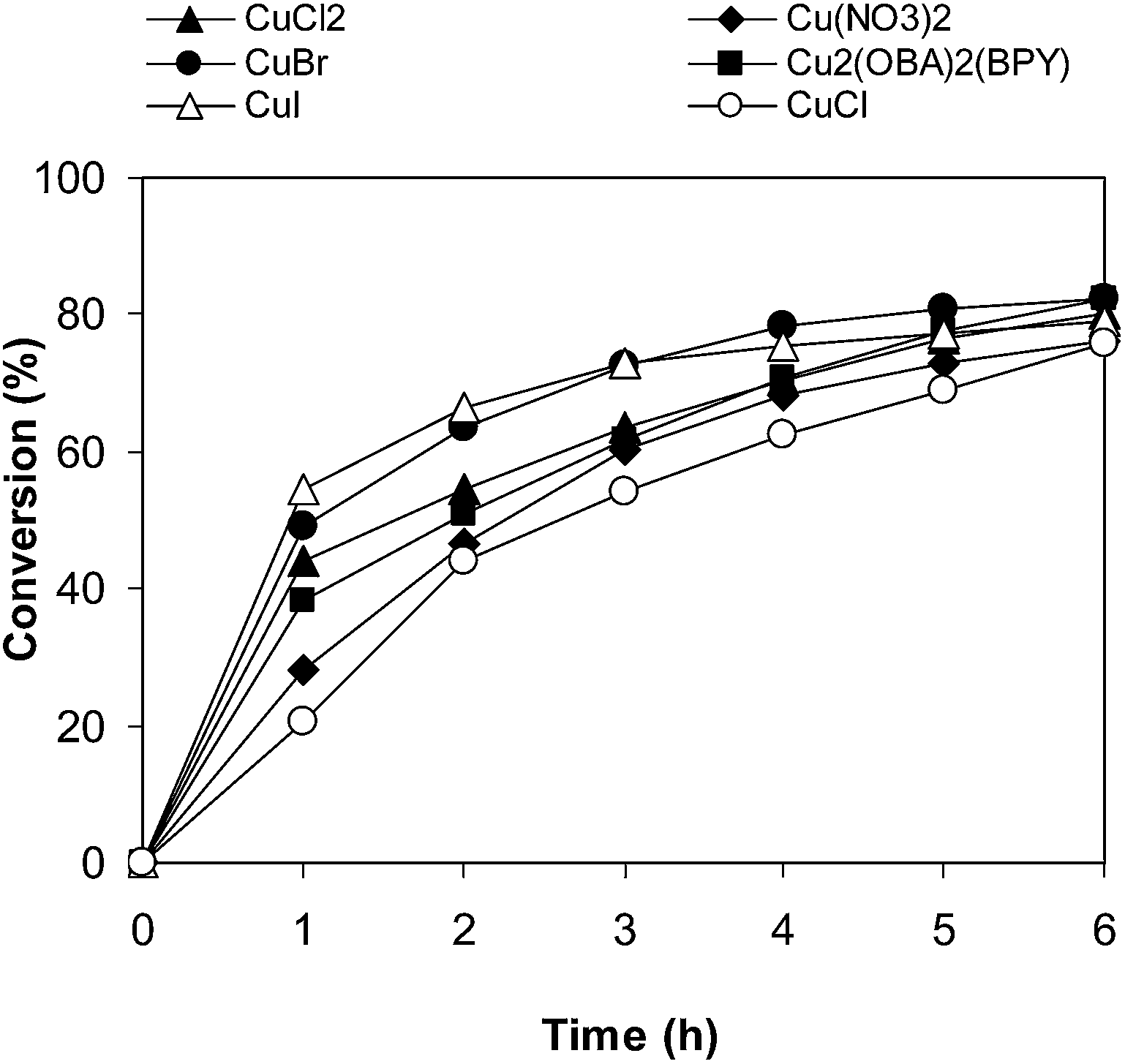
To highlight the advantages of the Cu2(OBA)2(BPY), other Cu-MOFs including Cu3(BTC)2, Cu(BDC), Cu2(BDC)2DABCO, Cu2(BPDC)2DABCO, and Cu(DBA)(BPY) were used as catalysts under optimal conditions. Although the well-known Cu3(BTC)2 offered high activity in several previous organic reactions,59–66 less than 5% conversion was detected. Reactions catalyzed by Cu(DBA)(BPY) and Cu2(BPDC)2DABCO afforded lower conversions. Similar results, 76% conversion, were obtained when Cu(BDC) and Cu2(BDC)2DABCO were employed. Compared to other tested Cu-MOFs catalysts, Cu2(OBA)2(BPY) exhibited the best activity, with 82% conversion being achieved after 6 h (Fig. 7). The 70% isolated yield of desired product indicated the correlation between conversion and selectivity of reactions. Furthermore, comparable reaction conversions were observed when other common copper salts such as Cu(NO3)2, CuCl2, CuCl, CuBr, and CuI were used under identical conditions (Fig. 8). However, it should be noted that these copper salts suffer disadvantages in terms of catalyst separation and recycling, and a heterogeneous catalyst should be favored for the transformation.
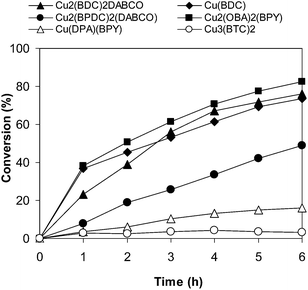 |
| | Fig. 7 Effect of different Cu-MOFs as catalysts on reaction conversion. | |
 |
| | Fig. 8 Effect of different copper salts as catalysts on reaction conversion. | |
Leaching test was next studied to explore whether active copper species dissolved from the solid Cu2(OBA)2(BPY) catalyst could contribute to the total conversion. A control experiment using optimized conditions was carried out using a simple filtration during the course of the reaction. The solid Cu-MOF catalyst was separated from the reaction mixture after 1 h reaction time at 37% conversion by simple filtration. The liquid phase was then transferred to a new vial, two equivalents of tBuOLi were added, and the resulting mixture was then stirred for an additional 5 h min at 120 °C with aliquots being sampled at different time intervals, and analyzed by GC. It was found that no further conversion was detected after the solid Cu2(OBA)2(BPY) catalyst was separated from the reaction mixture. In a second experiment, the solid Cu-MOF was removed from the reaction mixture by filtration after 6 h. The liquid phase was transferred to a new reactor vessel, and fresh reagents were then added to the solution under the same condition to the first run. The resulting mixture was stirred for 6 h at 120 °C with aliquots being sampled at different time intervals, and analyzed by GC. It was also observed that the solution phase from the first run could not catalyze the transformation (Fig. 9). These observations indicated that there should be no contribution from catalytically active copper species in the liquid phase.
 |
| | Fig. 9 Leaching test indicated no contribution from homogeneous catalysis of active species leaching into reaction solution. | |
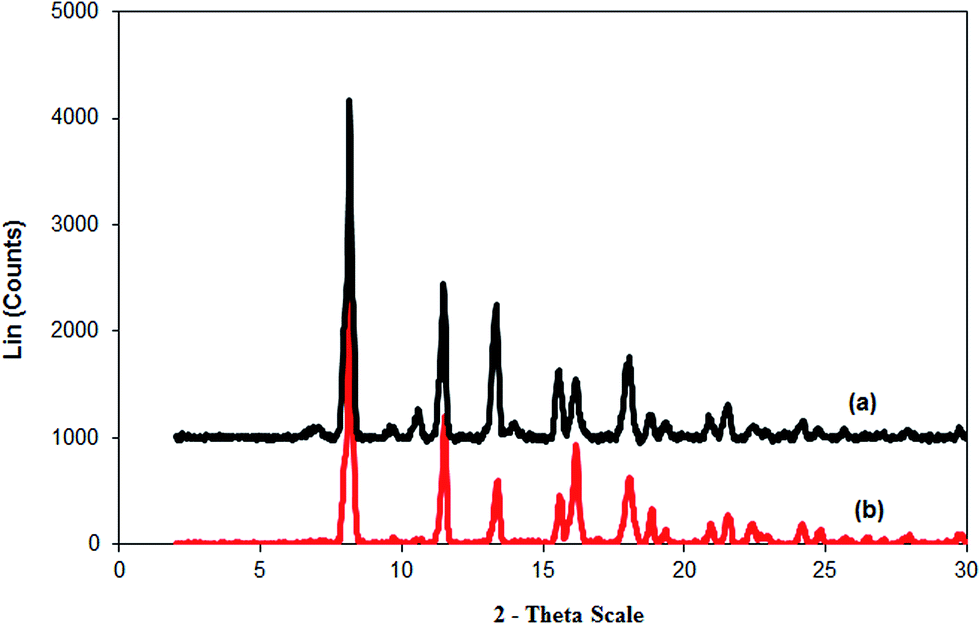
In this work, the recoverability and reusability of the Cu2(OBA)2(BPY) catalyst was then investigated over seven successive runs, by repeatedly separating the Cu-MOF catalyst from the reaction mixture, washing it and then reusing it. The direct arylation reaction was carried out at 120 °C in 1,4-dioxane, with the benzothiazole–iodobenzene molar ratio of 1:1.2, in the presence of 2 equivalents of tBuOLi as the base, at 3 mol% Cu2(OBA)2(BPY) catalyst. After each run, the Cu-MOF catalyst was removed from the reaction mixture by filtration, washed with copious amounts of DMF and water, dried at 150 °C under vacuum in 6 h. The recovered Cu2(OBA)2(BPY) was then reused in further direct arylation reaction under identical conditions to those of the first run. It was found that the Cu2(OBA)2(BPY) catalyst could be recovered and reused several times without a significant degradation in catalytic activity. Indeed, a conversion of 73% was still obtained in the 7th run (Fig. 10). In addition, the XRD result of the recovered Cu2(OBA)2(BPY) catalyst showed a slight difference in the diffractogram, as compared to that of the fresh catalyst. However, it was apparent that the recovered Cu2(OBA)2(BPY) was still highly crystalline (Fig. 11). Moreover, the FT-IR spectra of the recovered Cu-MOF revealed a similar absorption as compared to that of the fresh catalyst (Fig. 12).
 |
| | Fig. 10 Catalyst recycling studies. | |
 |
| | Fig. 11 X-ray powder diffractograms of the fresh (a) and reused (b) Cu2(OBA)2(BPY) catalyst. | |
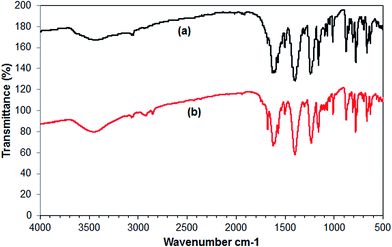 |
| | Fig. 12 FT-IR spectra of the fresh (a) and reused (b) Cu2(OBA)2(BPY) catalyst. | |
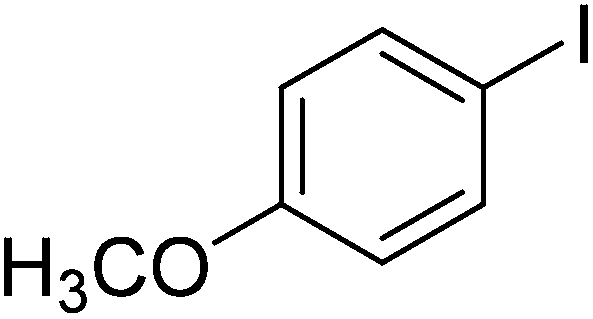
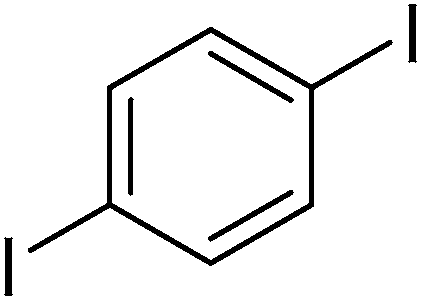
The optimized conditions were then employed for the arylation of benzothiazole using various different aryl iodides (Table 1). Direct arylation using electron-rich iodobenzene, 4-iodoanisole and 4-iodotoluen, was possible and products were obtained in excellent conversions (entries 1 and 2). Iodobenzene with electron-withdrawing groups was also active and arylated products were achieved in good conversion (entry 3). Interestingly, reactive amino functionality was tolerated and arylation by 4-iodoaniline afforded product in 78% conversion (entry 5). Trace amount of product was observed when 4-iodophenol was employed (entry 4). Selective mono-arylation was feasible with 1,4-diiodobenzene coupling partners (entry 6).
Table 1 Arylation scope with respect to aryl halidesa
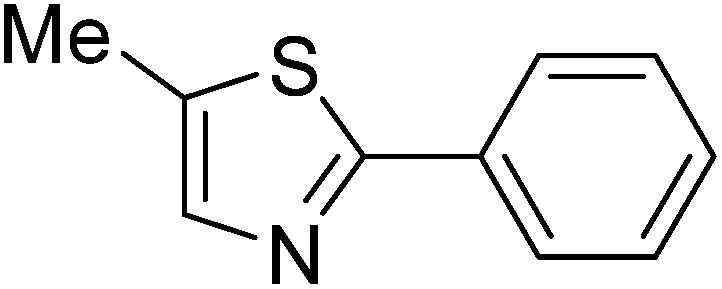
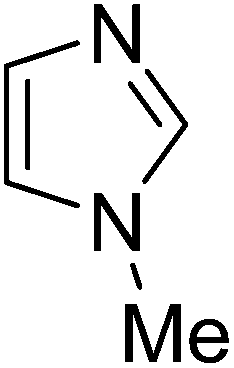
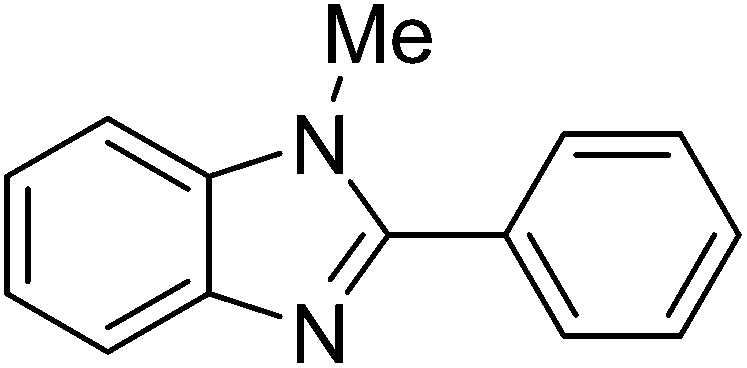
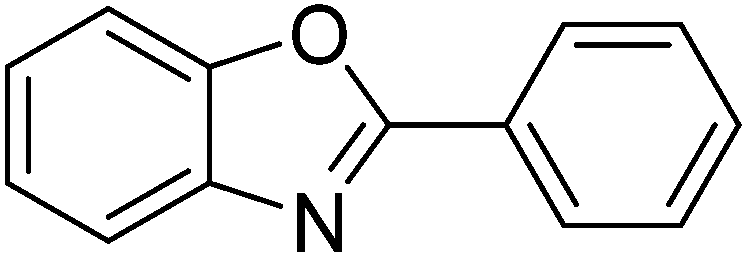
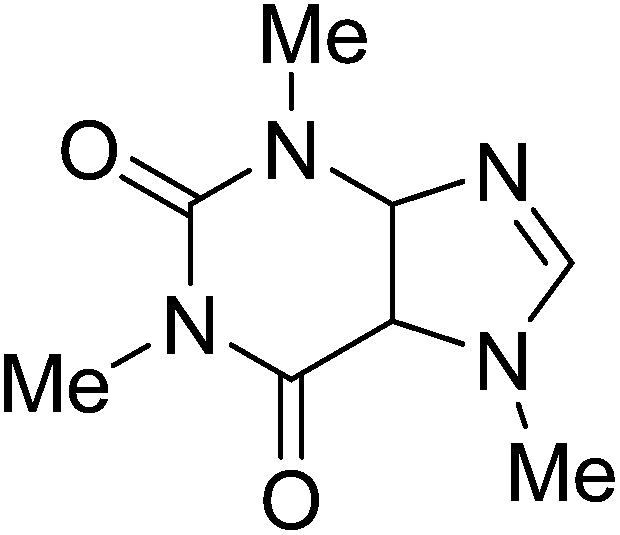
Reaction scope with respect to a variety of heterocycles is shown in Table 2. It was concluded that arylation using iodobenzene occurred at most acidic C–H bonds. Thiazole derivatives including benzothiazole and 4-methylthiazole were phenylated in good conversions (entries 1, 2). Arylation of benzimidazole resulted in the desired product in 76% conversion (entry 4). 1-Methylimidazole was also active and phenylated product was obtained in reasonable conversion (entry 3). Excellent conversion was achieved when benzoxazole was employed (entry 5). Gratifyingly, arylation of caffeine was also possible and moderate conversion of product was obtained (entry 6).
Table 2 Arylation scope with respect to heterocycles
4. Conclusions
In summary, the metal–organic framework Cu2(OBA)2(BPY) was synthesized and characterized by XRD, SEM, TEM, FT-IR, TGA, AAS, and nitrogen physisorption measurements. The Cu2(OBA)2(BPY) could be used as an efficient heterogeneous catalyst for direct arylation reaction of heterocyles. The optimal conditions employed tBuOLi base in dioxane solvent at 120 °C. A variety of heterocycles and iodoarenes were showed to be active under our conditions. Good conversion as well as functional group tolerance has been observed. Furthermore, the Cu-MOF catalyst could be facilely separated from the reaction mixture and reused without significant degradation in catalytic activity. The Cu2(OBA)2(BPY) could offer higher catalytic activity than other Cu-MOFs such as Cu3(BTC)2, Cu(BDC), Cu2(BDC)2DABCO, Cu2(BPDC)2DABCO, and Cu(DBA)(BPY). Our results here would propose the feasibility of using the Cu2(OBA)2(BPY) as an efficient heterogeneous catalyst for several copper-catalyzed organic transformations.
Acknowledgements
The Viet Nam National Foundation for Science and Technology Development NAFOSTED is acknowledged for financial support through project code 104.99-2012.25.
References
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- H.-Q. Do, R. M. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185 CrossRef CAS PubMed.
- S. Yotphan, R. G. Bergman and J. A. Ellman, Org. Lett., 2009, 11, 1511 CrossRef CAS PubMed.
- H. T. N. Le, T. T. Nguyen, P. H. L. Vu, T. Truong and N. T. S. Phan, J. Mol. Catal. A: Chem., 2014, 391, 74 CrossRef CAS PubMed.
- Y. K. Lingkui Meng, K. Muto, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2013, 52, 10048–10051 CrossRef PubMed.
- D. K. H. Mayuko Nishino, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 6993–6997 CrossRef PubMed.
- D. K. H. Tomoyuki Yao, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775–779 CrossRef PubMed.
- K. H. Hitoshi Hachiya, T. Satoh and M. Miura, Org. Lett., 2009, 11, 1737–1740 CrossRef PubMed.
- F. B. a. S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553–9556 CrossRef PubMed.
- W. Zhang, Q. Zeng, X. Zhang, Y. Tian, Y. Yue, Y. Guo and Z. Wang, J. Org. Chem., 2011, 76, 4741–4745 CrossRef CAS PubMed.
- M. L. Kantam, S. Laha, J. Yadav and S. Jha, Tetrahedron Lett., 2009, 50, 4467–4469 CrossRef CAS PubMed.
- X. Zhang, F. X. L. i. Xamena and A. Corma, J. Catal., 2009, 265, 155–160 CrossRef CAS PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
-
(a) R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed;
(b) F.-K. Shieh, S.-C. Wang, S.-Y. Leo and K. C.-W. Wu, Chem.–Eur. J., 2013, 19, 11139–11142 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
-
(a) J.-R. Li, J. Sculley and H.-C. Zho, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed;
(b) R. C Huxford, J. D. Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef PubMed.
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed;
(b) N. L. Torad, Y. Li, S. Ishihara, K. Ariga, Y. Kamachi, H.-Y. Lian, H. Hamoudi, Y. Sakka, W. Chaikittisilp, K. C.-W. Wu and Y. Yamauchi, Chem. Lett., 2014, 43, 717–719 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
-
(a) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed;
(b) Y.-C. Sue, J.-W. Wu, S.-E. Chung, C.-H. Kang, K.-L. Tung, K. C.-W. Wu and F.-K. Shieh, ACS Appl. Mater. Interfaces, 2014, 6, 5192–5198 CrossRef CAS PubMed.
-
(a) A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef PubMed;
(b) G. M. Shi, T. Yang and T. S. Chung, J. Membr. Sci., 2012, 415–416, 577–586 CrossRef CAS PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- Z.-Y. Gu, J. Park, A. Raiff, Z. Wei and H.-C. Zhou, ChemCatChem, 2014, 6, 67–75 CrossRef CAS.
- P. Valvekens, F. Vermoortelea and D. D. Vos, Catal. Sci. Technol., 2013, 3, 1435–1445 CAS.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2540 CAS.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- A. S. Roy, J. Mondal, B. Banerjee, P. Mondal, A. Bhaumik and S. M. Islam, Appl. Catal., A, 2014, 469, 320–327 CrossRef CAS PubMed.
- N. T. S. Phan, T. T. Nguyen, P. Ho and K. D. Nguyen, ChemCatChem, 2013, 5, 1822–1831 CrossRef CAS.
- N. T. S. Phan, C. K. Nguyen, T. T. Nguyen and T. Truong, Catal. Sci. Technol., 2014, 4, 369–377 CAS.
- M. Savonnet, S. Aguado, U. Ravon, D. Bazer-Bachi, V. Lecocq, N. Bats, C. Pinel and D. Farrusseng, Green Chem., 2009, 11, 1729–1732 RSC.
- E. Pérez-Mayoral, Z. Musilová, B. Gil, B. Marszalek, M. Položij, P. Nachtigall and J. Čejka, Dalton Trans., 2012, 41, 4036–4044 RSC.
- M. Opanasenko, M. Shamzhy and J. Čejka, ChemCatChem, 2013, 5, 1024–1031 CrossRef CAS.
- N. T. S. Phan, P. H. L. Vu and T. T. Nguyen, J. Catal., 2013, 306, 38–46 CrossRef CAS PubMed.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS PubMed.
- Y. Luan, N. Zheng, Y. Qi, J. Tang and G. Wang, Catal. Sci. Technol., 2014, 4, 925 CAS.
- F. G. Cirujano, A. Leyva-Pérez, A. Corma and F. X. L. i. Xamena, ChemCatChem, 2013, 3, 538–549 CrossRef.
- M. Pintado-Sierra, A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2013, 299, 137–145 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2009, 351, 2271–2276 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 711–717 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 3022–3030 CrossRef CAS.
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS PubMed.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2010, 276, 134–140 CrossRef CAS PubMed.
- N. T. S. Phan, T. T. Nguyen, K. D. Nguyen and A. X. T. Vo, Appl. Catal., A, 2013, 464–465, 128–135 CrossRef CAS PubMed.
- N. T. S. Phan, T. T. Nguyen and P. H. L. Vu, ChemCatChem, 2013, 5, 3068–3077 CrossRef CAS.
- L. Tang, D. Li, F. Fu, Y. Wu, Y. Wang, H. Hu and E. Wang, J. Mol. Struct., 2008, 888, 344–353 CrossRef CAS PubMed.
- H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404–12405 CrossRef CAS PubMed.
- T. Yoshizumi, H. Tsurugi, T. Satoh and M. Miura, Tetrahedron Lett., 2008, 49, 1598–1600 CrossRef CAS PubMed.
- J. Huang, J. Chan, Y. Chen, C. J. Borths, K. D. Baucom, R. D. Larsen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 3674–3675 CrossRef CAS PubMed.
- G. Langhendries, D. E. D. Vos, G. V. Baron and P. A. Jacobs, J. Catal., 1997, 187, 453–463 CrossRef.
- N. T. S. Phan and C. W. Jones, J. Mol. Catal. A: Chemical, 2006, 253, 123–131 CrossRef CAS PubMed.
- M. F. Giuseppe Bartoli, F. Ciminale, E. Paolo and J. C. S. Todesco, Chem. Commun., 1974, 732 RSC.
- G. B. F. Babudri, F. Ciminale, S. Florio and G. Ingrosso, Tetrahedron Lett., 1984, 25, 2047–2050 CrossRef.
- V. T.-K. Livio Racané, Z. Mihalić, G. Pavlović and G. Karminski-Zamola, Tetrahedron, 2008, 64, 11594–11602 CrossRef PubMed.
- W. W. Daqian Xu, C. Miao, Q. Zhang, C. Xia and W. Sun, Green Chem., 2013, 15, 2975–2980 RSC.
- R. K. G. C. G. Bates and D. Venkataraman, Org. Lett., 2002, 4, 2803 CrossRef PubMed.
- L. H. Wee, S. R. Bajpe, N. Janssens, I. Hermans, K. Houthoofd, C. E. A. Kirschhock and J. A. Martens, Chem. Commun., 2010, 46, 8186–8188 RSC.
- L. T. L. Nguyen, T. T. Nguyen, K. D. Nguyen and N. T. S. Phan, Appl. Catal., A, 2012, 425–426, 44–52 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, J. Catal., 2009, 267, 1–4 CrossRef CAS PubMed.
- D. Jiang, T. Mallat, F. Krumeich and A. Baiker, Catal. Commun., 2011, 12, 602–605 CrossRef CAS PubMed.
- L. Alaerts, E. Seguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. D. Vos, Chem.–Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- I. Luz, F. X. L. i. Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS PubMed.
- E. Pérez-Mayoral, Z. Musilová, B. Gil, B. Marszalek, M. Položij, P. Nachtigall and J. Čejka, Dalton Trans., 2012, 41, 4036–4044 RSC.
- E. Pérez-Mayoral and J. Cejka, ChemCatChem, 2011, 3, 157–159 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09092f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.