Size-controlled PdO/graphene oxides and their reduction products with high catalytic activity†
Jingjing Lin,
Tao Mei,
Meijiao Lv,
Chang'an Zhang,
Zhenfeng Zhao and
Xianbao Wang*
Hubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry-of-Education Key laboratory for the Green Preparation and Application of Functional Materials, Faculty of Materials Science and Engineering Hubei University, Wuhan 430062, China. E-mail: wangxb68@aliyun.com; Tel: +86 27 8866 2132
First published on 23rd June 2014
Abstract
A simple two-step method for the preparation of Pd nanoparticles supported on reduced graphene oxide (Pd/RGO) is reported. Well-dispersed and size-controlled Pd/RGO was synthesized through reduction of PdO nanoparticles on graphene oxide (PdO/GO) with hydrazine hydrate and ammonia at 80 °C for 4 hours. The PdO/GO was successfully prepared by mixing GO and Pd(NO3)2 aqueous solution together without adding any additional chemicals. The sizes and morphologies of PdO nanoparticles can be controlled by changing the concentrations of Pd(NO3)2, reaction temperatures and times. The results suggest that a small scale increase of the loadings of Pd supported on RGO can significantly improve the catalytic activity of Pd/RGO, and the stability is much better than that of commercial Pd/C catalysts for methanol electrooxidation and the Suzuki reaction.
1. Introduction
Graphene, a two-dimensional sheet of sp2-hybrized carbon, has attracted plenty of interest for both theoretical studies and applications because of its unique structural, electronic, adsorption, mechanical and thermal properties.1–4 Graphene has been proposed for use in a wide range of areas including catalyst supports,5 supercapacitors,6 nanoelectronics, batteries, photovoltaics, solar cells, fuel cells, transparent conducting films, sensors7–9 and so on.Recently, Pd nanoparticles, which show greater electric catalytic properties10,11 than bulk catalysts because of their higher surface-to-volume ratio,12 have drawn a lot of attention. Pt-based catalysts have been widely studied for many years as the best catalysts for ethanol electro-oxidation in alkaline media. However, researches showed that the Pt based catalysts underwent poisoning by intermediate products like CO and losed their catalytic activity. Pd-based catalysts can replace Pt-based catalysts in polymer electrolyte membrane fuel cells because of their lower cost and greater resistance to intermediate products of CO.13
A high dispersion and size control are very needed to improve the catalytic activity, but these naked nanoparticles tend to aggregate and block the control of particle size. Catalyst support is a key point to promote the heterogeneous catalyst. The support has significant effect on the morphology, electronic state and catalytic activity of supported nanoparticles. Research based on first-principles calculations shows that Pd can interact with and bind more strongly to graphene because of more interaction states and transmission channels are generated between them, and Pd tends to grow into three-dimensional structures on graphene surfaces.14 This provides a hint that graphene and oxidized graphene sheets (GO) are promising catalyst supports for growing and anchoring Pd nanoparticles (Pd NPs) and graphene based Pd nanoparticles exhibit extraordinary eletrocatalytic properties for ethanol and methanol in alkaline media.
The Suzuki–Miyaura coupling reaction, a well-known reaction catalysed by Pd, is a very powerful and convenient synthetic method for generating biaryls, conducting polymers, and liquid crystals in organic chemistry.15–19 As an efficient catalyst in organic reactions, Pd loaded catalysts can offer the most favorable combination of activity and selectivity than homogeneous catalysts.20 They do not require working under an inert atmosphere and when dropped in recycling experiments, they are easy to recover.21
Here, we report a two-step way to prepare Pd-reduced graphene oxide (Pd/RGO). First, the synthesis of PdO/GO is performed by strongly anchoring the PdO NPs on a GO surface. The reaction is conducted at room temperature without adding any stabilizer and the reaction is a physical process, making it an environmentally friendly way. We also discuss the size of PdO on GO surface at different conditions (concentrations, temperatures, times). Second, by the reduction of hydrazine hydrate and ammonia, PdO/GO was successfully transformed into Pd/RGO. The Pd/RGO catalyst is used as an unusually higher activity for methanol oxidation in alkaline in comparison with commercial Pd/C catalyst, and an efficient semi-heterogeneous catalyst for the Suzuki–Miyaura cross-coupling reaction in aqueous solution without any ligand or surfactant under aerobic condition.
2. Experimental section
2.1 Synthesis of PdO/GO and Pd/RGO
GO was prepared according to the method of modified hummers based on our early report22 (see the ESI†). GO (50 mg) was dispersed in 100 mL 50 vol% methanol–water. After sonication for half an hour in an ultrasonic bath, the flask was placed on a magnetic stirrer. 20 mL of Pd(NO3)2 with different concentrations (2, 4, 8, 12 and 16 mmol L−1) was added drop-wise for ten minutes to produce PdO/GO NPs with different Pd loadings (8, 16, 32, 48 and 64 wt%). The mixture was stirred at room temperature for an hour and then separated by centrifugation. The sample was obtained by filtering and washing repeatedly with deionized water (Fig. S1†).The as-prepared PdO/GO (20 mg) was dispersed in 40 mL deionized water in an ultrasonic bath, then 20 μL hydrazine hydrate and 100 μL ammonia were added into with stirred at 80 °C for four hours. After cooling to room temperature, the graphene supported Pd catalyst was collected by centrifugation and washing several times with deionized.
2.2 Suzuki–Miyaura reaction catalyzed by Pd/RGO
In a typical experiment, potassium carbonate (276.5 mg, 2.0 mmol) was dissolved in a mixture of 20 mL water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Bromobenzene (157 mg, 1.0 mmol), phenylboronic acid (183 mg, 1.5 mmol), Pd/RGO catalyst (5 wt% Pd, 3 mg) were added into the mixture. For a comparison, commercial Pd/C (5 wt% Pd, 3 mg) was added in the same way to catalyze the reaction. The mixture was then stirred at 80 °C in oil bath for two hours and then extracted with chloroform (3 × 10 mL). The organic layers were combined, dried over anhydrous Na2SO4 and filtered, and the solvent was removed under a reduced pressure. The resulting product was analyzed by liquid chromatography (LC). The water layer was centrifuged and washed for several times by ethanol to separate Pd/RGO from the mixture. Then, the Pd/RGO was reused in the Suzuki reaction in the same manner for six times.
:1). Bromobenzene (157 mg, 1.0 mmol), phenylboronic acid (183 mg, 1.5 mmol), Pd/RGO catalyst (5 wt% Pd, 3 mg) were added into the mixture. For a comparison, commercial Pd/C (5 wt% Pd, 3 mg) was added in the same way to catalyze the reaction. The mixture was then stirred at 80 °C in oil bath for two hours and then extracted with chloroform (3 × 10 mL). The organic layers were combined, dried over anhydrous Na2SO4 and filtered, and the solvent was removed under a reduced pressure. The resulting product was analyzed by liquid chromatography (LC). The water layer was centrifuged and washed for several times by ethanol to separate Pd/RGO from the mixture. Then, the Pd/RGO was reused in the Suzuki reaction in the same manner for six times.
2.3 Preparation of the modified GCE
Pd/RGO was dissolved in 1 mL N,N-dimethylformamide (DMF) and sonicated for 1 hour to get a homogeneous solution. Then 20 μL Nafion (NA: perfluorosulfonic acid–PTFE copolymer, 5%, w/w solution, purchased from Alfa Aesar) was added into the above solution. Then the mixture was placed in an ultrasonic bath for 30 min to get an about 1 mg mL−1 Pd/RGO-Nafion suspension. Prior to the surface modification, the bare GCE (Φ = 3 mm) was polished with 0.05 μm alumina power and rinsed thoroughly with doubly distilled water. Then it was cleaned successively with anhydrous ethanol and doubly distilled water in an ultrasonic bath and dried under nitrogen. 10 μL Pd/RGO-Nafion suspension was coated on an electrode and dried in air to obtain a Pd/RGO-Nafion-GC. For comparison, Pd/RGO (16 wt% Pd, 32 wt% Pd, 48 w% Pd, 64 wt% Pd)-Nafion-GC, Pd/RGO (5 wt% Pd), Pd/C (5 wt% Pd) were prepared in the same manner.2.4 Characterization
The structural and morphological characterization of PdO/GO and Pd/RGO were performed with a D8A25 X-ray diffractometer and transmission electron microscopy (TEM, Tecnai F20) and high resolution transmission electron microscopy (HRTEM, JEM-2100F STEM/EDS). The structure characterizations of biphenyl were performed with Fourier transformed infrared (FTIR, NTCOLET iS10) and 1H NMR and 13C NMR (WIPM 400). All electrochemical measurements were performed with an IM6 electrochemical workstation in a standard three-electrode system. A bare or modified GCE (Φ = 3 mm) served as a working electrode; a platinum electrode and a saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode. The catalytic activity of Pd/RGO was measured by LC, which was performed on a Uitimate 3000 system.2.5 Materials
All reagents used in this work were of analytical grade and employed without further purification. Natural graphite powder was purchased from Duratight Sealing Product Co., Ltd. Qingdao, China. Methyl alcohol, ethanol, hydrazine hydrate, potassium carbonate and palladium nitrate were all obtained from Sinopharm Chemical Reagent Co. Ammonium hydroxide was purchased from Wuhan Zhongtian Chemical Co., Ltd. Phenylboronic acid and bromobenzene were obtained from Aladdin Chemistry Co., Ltd. Unless otherwise stated, all solutions were prepared with double-distilled water.3. Results and discussion
Well-dispersed PdO NPs decorated on GO surfaces could be obtained by simple mixing Pd(NO3)2 and GO under constant stirring. The as-prepared PdO/GO can be easily transformed into Pd/RGO by reduction of hydrazine hydrate and ammonia at 80 °C. The complete transformation of the crystal phase was detected in the XRD patterns. Fig. 1a shows that PdO/GO has a typical XRD pattern with the diffraction peaks at the 2θ angles of 33.6° (002), 42.0° (110), 54.9° (112), 60.3° (103) and 71.7° (211). After reduction, PdO transformed into Pd (Fig. 1b) with its diffraction peaks at 39.8°, 46.2°, 67.6°, and 81.4°, which are attributes to the (111), (200), (220) and (311) planes of face-centered cubic structure of Pd (JCPDS no. 46-1043), respectively. Fig. 2a and b show the HRTEM images of PdO/GO and Pd/RGO, respectively. The inter-planar spacing of the particle lattices are 0.26 nm and 0.21 nm (Fig. 2a), which are consistent with the (002) and (110) lattice spacing of PdO. The inter-planar spacing of the particle lattice of Pd is 0.22 nm (Fig. 2b), which is consistent with the (111) lattice spacing of face-centered cubic Pd. RGO is clearly visible in the HRTEM images of Pd/RGO (as shown with a white arrow). The reaction mechanism is proposed as follows:| Pd(NO3)2 + H2O + GO → PdO/GO + 2HNO3 |
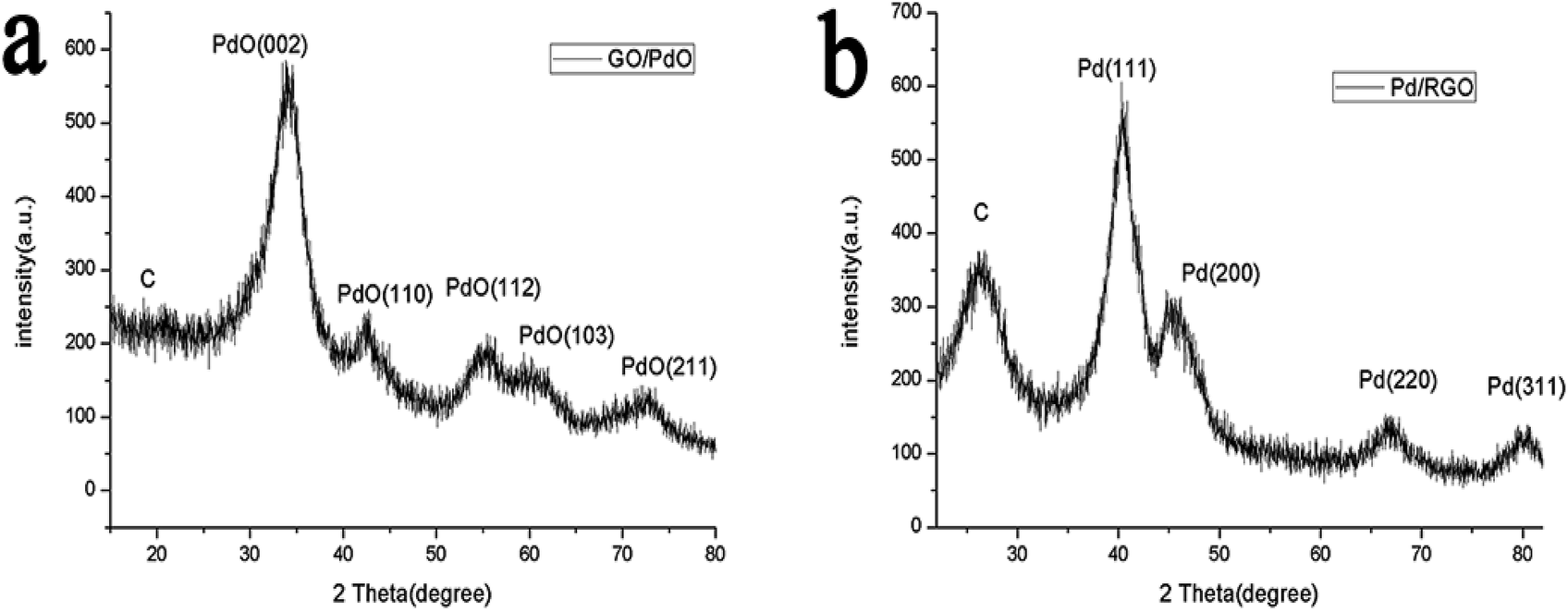 | ||
| Fig. 1 XRD patterns of (a) PdO/GO and (b) Pd/RGO. | ||
 | ||
| Fig. 2 HRTEM images of (a) PdO/GO and (b) Pd/RGO. | ||
In order to get a clear vision of the effect of the concentrations of Pd(NO3)2, TEM images of the PdO/GO prepared in 50% methanol–water with different concentrations of Pd(NO3)2 were displayed in Fig. 3. It is clear that GO has been decorated with a large amount of well-dispersed PdO NPs. PdO/GO (Fig. 3a) prepared with the lowest concentration has the smallest PdO NPs with an average size of 1.57 nm. When Pd(NO3)2 amounts increased from 2 mmol L−1 to 4, 8, 12 and 16 mmol L−1, the size of PdO NPs increased from 1.57 nm to 1.82, 2.20, 2.40 and 4.17 nm, respectively. Such observations indicate that the loading amounts and sizes of PdO NPs on GO could be controlled by the concentrations of Pd(NO3)2. The growth of the PdO NPs on GO is related to the oxygen functional groups on the surface of GO. There is a strong interaction between the PdO atoms and the functional groups, which permits only the adjacent Pd atoms to combine together. Therefore the size of PdO NPs is related to the loading amounts of PdO, which mainly depends on the concentration of Pd(NO3)2.
 | ||
| Fig. 3 TEM images and particle size distributions of PdO/GO with different concentrations of Pd(NO3)2 (A: 2 mmol L−1; B: 4 mmol L−1; C: 8 mmol L−1; D: 12 mmol L−1; E: 16 mmol L−1). Reaction time: 1 hour, reaction temperature: 25 °C. The scale bars of all the images are 20 nm. | ||
Fig. 4 shows the TEM images of PdO/GO prepared at the temperatures of 0, 25 and 50 °C. When the materials reacted in an ice bath (0 °C), as shown in Fig. 4a, PdO NPs is very small and tend to aggregate with each other. When the temperature increased from 0 to 50 °C, the size of PdO NPs increased. There is a strong anchoring effect between the PdO nuclei and the GO surface. When the temperature increased, on the one hand, the hydrolysis of Pd(NO3)2 is an endothermic reaction, and a higher temperature can accelerate its hydrolysis. On the other hand, Ostwald ripening obviously occurred, resulting in a decrease in the number of PdO nuclei and an increase in the particles size. Such a phenomenon indicates that an increase in the mobility between PdO nuclei and GO surface due to weakening of the anchoring ability at higher temperature.23 These results suggest that temperature can affect the size of PdO on GO.
 | ||
| Fig. 4 TEM images of PdO/GO prepared at different temperatures (a: 0 °C; b: 25 °C; c: 50 °C). Reaction time: 1 hour. Pd(NO3)2: (16 mmol). | ||
TEM images and size distributions of the PdO/GO after reaction for 1, 3, 6, and 9 h are shown in Fig. 5. Fig. 5A showed that PdO nuclei with an average size of 6.69 nm were formed. When the reaction time increased from 1 to 3 h, there was an increase in the density of PdO particles while the sizes of PdO particles almost remain constant, indicating a continuous hydrolysis process of Pd(NO3)2. When the reaction time was over 3 h, some particles started to combine together to form a bigger nuclei, meaning that the hydrolysis reaction was already finished, and the average size of PdO NPs increased to 8.51 nm. In Fig. 5D, there was already no small particle, and some particles grew to several tens of nanometers with 18.07 nm of the average size of PdO NPs. This phenomenon suggests that the reaction time can affect the morphology of particles.
 | ||
| Fig. 5 TEM images and particle size distributions of PdO/GO prepared at different reaction times (A: 1 hour; B: 3 hours; C: 6 hours; D: 9 hours). Reaction temperature: 25 °C. Pd(NO3)2: (16 mmol). The scale bars of all the images are 100 nm. | ||
When the concentration of Pd(NO3)2 is increased, after reduction, the loading amounts and sizes of Pd NPs on reduced GO are increased too. We investigated the performance of Pd/RGO with different loadings of Pd NPs for methanol electrooxidation in alkaline media. Fig. 6a shows the performance of Pd/RGO with different loadings of Pd NPs (Pd1/RGO, Pd2/RGO, Pd3/RGO, Pd4/RGO, Pd5/RGO) for methanol oxidation. The CVs were obtained in the potential ranges of −0.8 to 0.3 V with the scan rate of 50 mV s−1. There are two peaks of methanol oxidation under anodic condition. The oxidation peak in the forward scan was corresponding to the oxidation of freshly chemisorbed species coming from methanol adsorption, and the reverse oxidation peak was primarily associated with removal of carbonaceous species not completely oxidized in the forward scan. The oxidation peak in the forward scan indicated the electrocatalytic activity of the electrocatalysts. It is obvious that the Pd/RGO electrocatalyst with a higher loading of Pd NPs exhibits a higher current density, indicating that the loadings of metals can affect the catalytic property. Although the loading of Pd stay the same, the smaller size of Pd NPs can provide a higher surface area to exhibit a higher activity for methanol oxidation.24 In a small scope the loading increase of Pd NPs leads to an increase of PdNP size, which also can exhibit a higher activity. When the loading of Pd is very little and has a good dispersion, and a small scale increase of Pd NPs can provide more active sites to have a higher activity. When the loadings of Pd NPs increase over a certain level, the nanoparticles tend to aggregate to form a bigger one, causing the decrease of active surface area and catalytic activity. The stabilities of the catalytic performance recorded at −0.1 V for 900 s are displayed in Fig. 6b. The rapid decrease in the current density could be attributed to the poisoning of the electrocatalysts, due to the formation of intermediate and some poisoning species during the methanol oxidation.25 It is found that the current decay on the Pd5/RGO is significantly slower than the others. This demonstrated that Pd5/RGO enhanced the electrochemical stability for methanol electrooxidation in alkaline media.
 | ||
| Fig. 6 (a) CVs of methanol oxidation on Pd1/RGO, Pd2/RGO, Pd3/RGO, Pd4/RGO, Pd5/RGO in N2 saturated 1M KOH + 1M CH3OH solution at a scan rate of 50 mV s−1; (b) Chronoamperometry of Pd1/RGO, Pd2/RGO, Pd3/RGO, Pd4/RGO, Pd5/RGO in 1 M KOH + 1 M CH3OH solution at an operation potential of −0.1 V; (c) CVs of the Pd/RGO and Pd/C in N2 saturated 1 M KOH + 1 M CH3OH solution at a scan rate of 50 mV s−1; (d) Chronoamperometric tests of Pd/RGO and Pd/C at −0.1 V in the same solution as above. | ||
The electrocatalytic activity of the Pd/RGO (5 wt% Pd) and commercial Pd/C (5 wt% Pd) for methanol oxidation are displayed in Fig. 6c. It is obvious that the Pd/RGO electrocatalyst gave a much higher mass activity than Pd/C at the same conditions. One main reason for the higher mass activity of the Pd/RGO is due to the higher electrochemical active surface area (ECSA). The ECSA value for Pd/RGO is much higher than that of the Pd/C electrode.26–29 These probably enhance the active sites for the electrooxidation reaction of methanol. Fig. 6d shows the stability of the Pd/RGO and Pd/C catalytic performance. The current decay on the Pd/RGO was slower than that of the Pd/C, demonstrating that Pd/RGO enhanced the electrochemical stability. This result suggests that the reduced graphene plays a critical role in promoting the methanol oxidation, which could be attributed to the good electrical conductivity of reduced graphene and good dispersion of Pd NPs on RGO.
The catalytic activity of the Pd/RGO in the formation of biaryl carbon–carbon bonds was investigated using the Suzuki reaction of bromobenzene with phenylboronic acid (Scheme 1). This reaction was conducted in a mixture of ethanol and water (1:1) containing K2CO3 under aerobic conditions (Fig. S2†). The results from FTIR, 1H NMR and 13C NMR (Fig. S3–S5†) showed that biphenyl can be synthesized by the Pd/RGO catalyst through the Suzuki–Miyaura cross-coupling reaction. In this study, we used commercial Pd/C as a reference and investigated the recyclability of the Pd/RGO catalysts.
 | ||
| Scheme 1 The Suzuki–Miyaura cross-coupling reaction. | ||
Table 1 summarizes the performance of the Pd/RGO and Pd/C catalysts with the same Pd content (5%) under the same reaction conditions. The results confirm that Pd/RGO is more catalytically active than that of Pd/C, which are probably due to the structure of the support RGO. The inert behavior of the support C may lead to the poor performance of commercial Pd/C. The circulating experiments of Pd/RGO catalysts were successfully carried out for the Suzuki–Miyaura cross-coupling reaction. We investigated the activity of the Pd/RGO catalyst after reused in six circles. In the first run, the yield was 93.96%. The activity dramatically dropped in the next three runs, as in the sixth run, showing just 4.14% conversion. Fig. 7 shows the TEM images of Pd/RGO before the reaction and recovered after the sixth circle. These pictures showed after the sixth run, the Pd particles tend to agglomerate. Many correlated aspects have to be taken into account when commenting on the reasons for the reused catalysts. The shape and size of the nanoparticles and their size distribution are the most easily understandable aspect. The dispersion is supposed to be even higher during the reaction than suggested by the final particle size measured after complete reaction.18 This result indicates that the mechanism of deactivation is likely to involve the formation and dispersion of agglomerated Pd nanoparticles. Reuse of the Pd catalysts can be achieved, albeit with loss in activity depending on the recycling procedure. However, recovery of the Nobel metal is possible because of very low leaching.
| Catalysts | Pd/C | Pd/RGO 1st | Pd/RGO 2nd | Pd/RGO 3rd | Pd/RGO 4th | Pd/RGO 5th | Pd/RGO 6th |
|---|---|---|---|---|---|---|---|
| Yield (%) | 88.48 | 93.96 | 72.27 | 40.08 | 17.62 | 13.68 | 4.14 |
 | ||
| Fig. 7 Micrographs of Pd/RGO: (a) TEM image of Pd/RGO before the reaction and (b) TEM image of Pd/RGO recovered after the sixth circle. | ||
4. Conclusions
In summary, we have successfully synthesized the Pd/RGO catalyst through a two-step way. Well-dispersed and size-controlled PdO supported on GO was fabricated by mixing GO and Pd(NO3)2 with continuous stirring. After reduction, the Pd/RGO exhibit significantly enhanced catalytic activity and stability than that of commercial Pd/C catalysts towards methanol electrocatalysts for DMFCs. These results suggest that Pd/RGO could be considered a good electrocatalyst material for use in alkaline fuel cell cathode materials. The Pd/RGO can be used as an excellent heterogeneous catalyst for the Suzuki–Miyaura cross-coupling reaction under aerobic conditions.Acknowledgements
This work was supported by National Natural Science Foundation (51272071), Ministry of Education (20114208110005), Hubei Provincial Department of Education (D20111002), and Wuhan Science and Technology Bureau (201271130447), China.References
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed
.
- G. G. Wildgoose, C. E. Bands and R. G. Compton, Small, 2006, 2, 182–193 CrossRef CAS PubMed
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
- H. K. He and C. Gao, Molecules, 2010, 15, 4670–4694 CrossRef PubMed
- H. Tang, J. H. Chen, Z. P. Huang, D. Z. Wang, Z. F. Ren, L. H. Nie, Y. F. Kuang and S. Z. Yao, Carbon, 2004, 42, 191–197 CrossRef CAS PubMed
- C. M. Yang, Y. J. Kim, M. Endo, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 20–21 CrossRef CAS PubMed
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed
- J. Wu, W. Pisula and K. Mullen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS
- A. K. Manocchi, N. E. Horelik, B. Lee and H. Yi, Langmuir, 2010, 26, 3670–3677 CrossRef CAS PubMed
- C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed
- S. Guo, S. Dong and E. Wang, Energy Environ. Sci., 2010, 3, 1307–1310 CAS
- P. A. Khomyakov, G. Giovannetti, P. C. Rusu, G. Brocks, J. van den Brink and P. J. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195425 CrossRef
- N. Z. Shang, C. Feng, H. Y. Zhang, S. T. Gao, R. X. Tang, C. Wang and Z. Wang, Catal. Commun., 2013, 40, 111–115 CrossRef CAS PubMed
- J. F. Hu, Y. P. Wang, M. Han, Y. M. Zhou, X. Q. Jiang and P. P. Sun, Catal. Sci. Technol, 2012, 2, 2332–2340 CAS
- J. Lemo, K. Heuze and D. Astruc, Chem. Commun., 2007, 4351–4353 RSC
- G. M. Scheuermann, L. G. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed
- A. Corma, H. Garcia and A. Leyva, Tetrahedron, 2005, 61, 9848–9854 CrossRef CAS PubMed
- H. U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3–18 CrossRef CAS
- H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67, 2721–2722 CrossRef CAS PubMed
- M. J. Lv, X. B. Wang, J. Li, X. Y. Yang, C. A. Zhang, J. Yang and H. Hu, Electrochim. Acta, 2013, 108, 412–420 CrossRef CAS PubMed
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, Z. X. Xie and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed
- Y. W. Zhang, G. H. Chang, S. Liu, J. Q. Tian, L. Wang, W. B. Lu, X. Y. Qin and X. P. Sun, Catal. Sci. Technol., 2011, 1, 1636–1640 CAS
- Y. C. Zhao, L. Zhan, J. N. Tian, S. L. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS PubMed
- D. S. Yuan, C. W. Xu, Y. L. Liu, S. Z. Tan, X. Wang, Z. D. Wei and P. K. Shen, Electrochem. Commun., 2007, 9, 2473–2478 CrossRef CAS PubMed
- A. Safavi, H. Kazemi and S. H. Kazemi, J. Power Sources, 2014, 256, 354–360 CrossRef CAS PubMed
- S. S. Li, J. J. Lv, Y. Y. Hu, J. N. Zheng, J. R. Chen, A. J. Wang and J. J. Feng, J. Power Sources, 2014, 247, 213–218 CrossRef CAS PubMed
- L. Y. Chen, N. Chen, Y. Hou, Z. C. Wang, S. H. Lv, T. Fujita, J. H. Jiang, A. Hirata and M. W. Chen, ASC Catal., 2013, 3, 1220–1230 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02855d |
| This journal is © The Royal Society of Chemistry 2014 |