 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The surface charge of anti-bacterial coatings alters motility and biofilm architecture†
Olena Rzhepishevskaa, Shoghik Hakobyana, Rohit Ruhala, Julien Gautrotb, David Barberoc and Madeleine Ramstedt*a
aDepartment of Chemistry, Umeå University, Umeå, SE-901 87, Sweden. E-mail: madeleine.ramstedt@chem.umu.se; Fax: +0046907867655; Tel: +0046907866328
bSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK
cDepartment of Physics, Umeå University, Umeå, SE-901 87, Sweden
First published on 4th March 2013
Abstract
Bacterial biofilms affect many areas of human activity including food processing, transportation, public infrastructure, and most importantly healthcare. This study addresses the prevention of biofilms and shows that the surface charge of an abiotic substrate influences bacterial motility as well as the morphology and physiology of the biofilm. Grafting-from polymerisation was used to create polymer brush surfaces with different characteristics, and the development of Pseudomonas aeruginosa biofilms was followed using confocal microscopy. Interestingly, two types of biofilms developed on these surfaces: mushroom structures with high levels of cyclic diguanylate (c-di-GMP) were found on negatively charged poly (3-sulphopropylmethacrylate) (SPM) and zwitterionic poly (2-(methacryloyloxy)ethyl)dimethyl-3-sulphoproyl) ammonium hydroxide) (MEDSAH), while flat biofilms developed on glass, positively charged poly (2-(methacryloyloxy)-ethyl trimethyl ammonium chloride) (METAC), protein-repellent poly oligo(ethylene glycol methyl ether methacrylate) (POEGMA) and hydrophobic polymethylmethacrylate (PMMA). The results show that of all the surfaces studied, overall the negatively charged polymer brushes were most efficient in reducing bacterial adhesion and biofilm formation. However, the increased level of regulatory c-di-GMP in mushroom structures suggests that bacteria are capable of a quick physiological response when exposed to surfaces with varying physicochemical characteristics enabling some bacterial colonization also on negatively charged surfaces.
Introduction
Bacterial biofilms are organized clusters of bacterial cells that are attached to a surface and produce extracellular substances, also called “matrix”, that mediate bacterial attachment and protect the cells.1 Pathogenic bacteria create highly resistant biofilms on medical devices and implants, resulting in chronic infections in humans.2 Biofilms are responsible for approximately 80% of all microbial infections, and cause 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually in the USA alone.3,4 Consequently, controlling biofilms is of high importance and several methods are being developed to inhibit their formation. One approach is to modify the properties of surfaces that bacteria attach to, for example, by coating the surface with a layer of polymer brushes. Polymer brushes are thin polymer films where the polymer chains are tethered to the surface of an underlying substrate. These films are convenient model substrates for attachment and biofilm studies since their chemistry can be tailored and controlled easily through monomer composition and polymerisation conditions.5 Surface charge and hydrophobicity are two properties of the substrate that are actively discussed in the literature.6–10 Positively charged polymer surfaces have been reported to be bactericidal supposedly because the positive charge can disrupt membrane potential of the cell or damage the membrane structure.7,8,11 Therefore, polycationic surfaces are often suggested to be efficient antibacterial coatings that bind (due to electrostatic interactions) and kill bacteria. Negatively charged surfaces on the other hand can be expected to repel bacterial adhesion due to electrostatic repulsion between the often negatively charged bacterial surface and the negatively charged polymer surface.9 Polymers and polymer brushes containing oligoethylene glycol subunits have been shown to be efficient in reducing bacterial attachment.5 Jiang and co-workers observed that zwitterionic brushes and brushes with oligoethylene glycol units both prevented biofilm formation and attachment of Pseudomonas aeruginosa PAO1 to a larger extent than monolayers of molecules carrying the same functionalities.10 It is, thus, clear that differences in substrate chemistry will influence bacterial attachment.
000 deaths annually in the USA alone.3,4 Consequently, controlling biofilms is of high importance and several methods are being developed to inhibit their formation. One approach is to modify the properties of surfaces that bacteria attach to, for example, by coating the surface with a layer of polymer brushes. Polymer brushes are thin polymer films where the polymer chains are tethered to the surface of an underlying substrate. These films are convenient model substrates for attachment and biofilm studies since their chemistry can be tailored and controlled easily through monomer composition and polymerisation conditions.5 Surface charge and hydrophobicity are two properties of the substrate that are actively discussed in the literature.6–10 Positively charged polymer surfaces have been reported to be bactericidal supposedly because the positive charge can disrupt membrane potential of the cell or damage the membrane structure.7,8,11 Therefore, polycationic surfaces are often suggested to be efficient antibacterial coatings that bind (due to electrostatic interactions) and kill bacteria. Negatively charged surfaces on the other hand can be expected to repel bacterial adhesion due to electrostatic repulsion between the often negatively charged bacterial surface and the negatively charged polymer surface.9 Polymers and polymer brushes containing oligoethylene glycol subunits have been shown to be efficient in reducing bacterial attachment.5 Jiang and co-workers observed that zwitterionic brushes and brushes with oligoethylene glycol units both prevented biofilm formation and attachment of Pseudomonas aeruginosa PAO1 to a larger extent than monolayers of molecules carrying the same functionalities.10 It is, thus, clear that differences in substrate chemistry will influence bacterial attachment.Bacterial attachment to surfaces is governed by both specific receptor–ligand interactions (adhesins) as well as more general physicochemical interactions. The physicochemical interactions play an important role allowing bacteria to come close enough to a surface to make adhesin binding possible.12,13 Gram-negative bacteria have the possibility to modify and alter the physicochemical properties of their outer membrane and thereby influence these physicochemical interactions, for example by changing the composition and structure of their lipopolysaccharide (LPS) layer.14 Altered composition of LPS has been reported for different strains of P. aeruginosa grown in biofilms compared to planktonic cells.15 Previous studies have shown that mutations in the pathways for LPS synthesis in Escherichia coli result in altered adhesion and biofilm formation on polystyrene. This adhesion appeared to be increased for strains with shorter LPS that were more hydrophobic.16 Similar observations have also been reported for LPS mutants of P. aeruginosa.17
P. aeruginosa is one of the most important Gram-negative bacteria causing biofilm-associated infections and at the same time it is a well-established model organism to study biofilm development. It has been shown that biofilm development is controlled by complex molecular mechanisms and influenced by environmental conditions, i.e. temperature, nutrients.2,18
Switching between sessile growth (biofilm) and free swimming in P. aeruginosa and other Gram-negative bacteria is regulated by the signaling molecule cyclic diguanylate (c-di-GMP).19 Low intracellular levels of c-di-GMP in P. aeruginosa increase motility and inhibit production of extracellular matrix components, while high levels of c-di-GMP inhibit motility and increase the production of adhesin CdrA20 and extracellular polysaccharide Pel.21 Though motility appears to be inhibited in biofilms, type IV pili-dependent (surface-associated twitching) motility is needed for the progress of biofilm development.18,22
Despite the growing research on antibacterial and anti-fouling surfaces, on the one hand, and biofilm biology, on the other hand, there is a gap in understanding the mechanisms behind decreased or increased biofilm formation on materials with different properties. One reason for this is the complexity of bacterial attachment and biofilm formation processes where many parameters contribute to the final result. This makes comparisons of the literature studies very difficult, if not impossible, and there is a need for studies where we can correctly judge whether one surface is superior to another, for example, in reducing biofilm formation. In order to accomplish this, well designed approaches are needed where several surfaces can be studied under the same conditions, for longer time periods and with well-defined bacterial strains.
In this work we have used several different approaches to carefully investigate the interaction between P. aeruginosa and a range of model surfaces, as well as to study how the surface physicochemical properties of the bacterium influence biofilm formation. We report on large differences in surface associated motility, attachment, biofilm formation and biofilm physiology following physicochemical changes in the model surface, as well as changes in biofilm properties due to differences in bacterial cell surface properties.
Results and discussion
Characterization of polymer brushes
The different model surfaces (Fig. 1A) had dry thicknesses around 100 nm and their chemical composition was confirmed using XPS (data not shown). In the hydrated state, the hydrophilic brushes have been reported to have up to double thickness depending on conditions such as ionic strength and brush charge.23–25 The surfaces used in this study spanned a wide range of hydrophilicity, as measured by the contact angle formed by a sessile drop of water placed on the various surfaces (Fig. 1C). The contact angles were <10° for METAC and SPM, 39° ± 1 for MEDSAH, 49° ± 1 for POEGMA and 76° ± 2 for PMMA. The charge density was 0.30 ± 0.16 μmol cm−2 and 0.09 ± 0.01 μmol cm−2 for METAC and SPM brushes, respectively.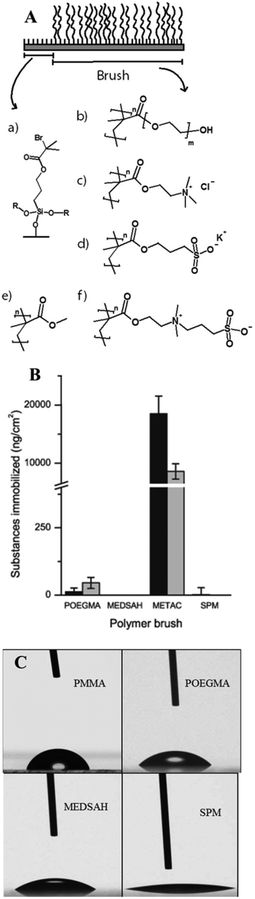 | ||
| Fig. 1 Polymer brush surfaces used in the study and their characteristics; A – chemical structure; polymerisation was initiated from self-assembled monolayers of initiator molecules (a) on cleaned glass slides. R represents crosslinking to other initiator molecules or the glass surface. The polymerisation resulted in polymer brush films of (b) POEGMA, (c) METAC, (d) SPM, (e) PMMA, and (f) MEDSAH. B – adsorption from medium components to different brushes (the black bar represents tryptic soy broth and the grey bar isosensitest medium); C – contact angles for polymer brushes; drops on METAC spread in the same way as on SPM (not shown). | ||
Exposure of the model surfaces to pure bacterial growth media with differences in medium composition (Fig. 1B) was performed to investigate whether medium components produce a conditioning film that could enhance bacterial attachment. Such conditioning films have been shown to form on most types of medical devices and serve as attachment sites for colonising bacteria.26 Consequently, if reduction of bacterial attachment is desired, it is important to use surfaces that are subject to low or, ideally, no surface conditioning from surrounding fluids. SPR experiments showed that of the hydrophilic brushes only METAC adsorbed elevated amounts of substances from the growth media tested especially from the rich tryptic soy broth (TSB). POEGMA brushes adsorbed very low levels, whereas SPM and MEDSAH did not adsorb substances from the media. Consequently, the METAC and to some extent POEGMA surfaces can be expected to have displayed an altered surface chemistry due to surface conditioning from the medium in our bacterial experiments. The adsorption of media components onto the hydrophilic brushes can be explained by the electrostatic interactions between medium components and the polymer brush, and be predicted by the zeta potential of the brush. (The zeta potential of a charged particle will be influenced by the surface charge as well as ionic strength of the surrounding medium.) The zeta potential for METAC brushes was positive whereas the zeta potential for SPM was negative (approximately +27 mV and −35 mV, respectively, in PBS), correlating to the charge of the polymer brushes. Both MEDSAH and POEGMA displayed a slightly negative zeta potential (approximately −16 mV and −2 mV, respectively, in PBS) which should result in repulsion of negatively charged proteins and substances from the growth medium, although more so in the case of MEDSAH.
Attachment and biofilm formation on different surfaces
Attachment of P. aeruginosa PAO1 to different polymer brushes was first studied under shear force condition in a flow chamber. POEGMA and MEDSAH were previously shown to have low bacteria binding properties10 and this was also observed in our experimental conditions as less than 10 cells/4.03 × 105 μm2 were bound to these surfaces (Fig. 2A). In contrast, many more bacteria (9100 ± 3000 cells/4.03 × 105 μm2) were bound to untreated glass, and binding to positively charged METAC was the most extensive (Fig. 2). Despite low cell binding to POEGMA and PMMA, flat biofilms covering the whole surface with occasional mushroom structures (type I) developed on these polymers after 72 hours (Fig. 3A). Another type of biofilm (type II) developed on SPM and MEDSAH, where the biofilm consisted of occasional mushroom structures with a regular shape, but without any trace of a flat biofilm (Fig. 3A and B). These two distinct types of biofilm architecture (flat and mushroom) could already be observed 18 h post inoculation and were maintained up to 96 hours, i.e. the longest experimental period in our study (Fig. 4). However, the amount of biomass was found to be larger for type I biofilms in all experiments (Fig. 3C). Interestingly, flat biofilms displayed approximately the same thickness (about 20 μm) in all experiments, while mushroom structures ranged in size with the largest being about 60 μm in diameter and the smallest consisting of only a group of several cells (Fig. 3A and 7B). This range of sizes probably reflects the “seeding process” when single cells from mushroom structures can be released, manage to attach to the surface and start a new colony. These results agree with a recently published study where biofilms of another organism, E. coli, formed sparse, heterogeneous and mushroom-like colonies on a negatively charged surface in a flow chamber6 indicating that this behaviour might be a common trait for Gram-negative bacteria. However, the long term biofilm result, presented here, contradict previously published data for POEGMA and MEDSAH,10 which showed low deposition of biofilm for both surfaces in flow chamber experiments. This could be explained by differences in experimental conditions such as the culture medium and flow speed between our study and previous work.11 SPR analysis on POEGMA surfaces (Fig. 1B) indicated that a low level of surface conditioning occurred from iso-sensitest medium, which perhaps facilitated biofilm formation in the present study. However, neither changes nor additional substances could be detected using XPS on POEGMA surfaces immersed for three days in isosensitest medium (data not shown). This could indicate that if there are changes occurring in the surface composition of the POEGMA, they are below the detection limit of XPS or they require the presence of bacteria to occur.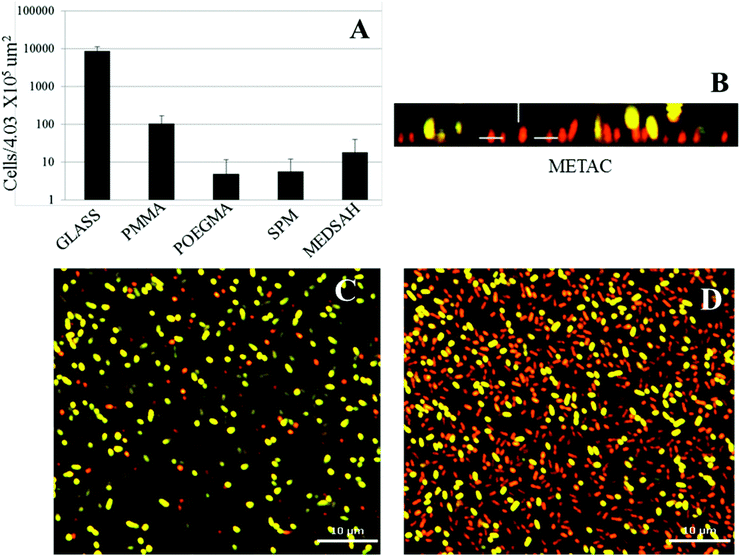 | ||
| Fig. 2 Attachment of P. aeruginosa PAO1 to different surfaces; A – cell counts of bacteria attached to five surfaces in the flow chamber 1 h post inoculation; B, C, D – P. aeruginosa cells attached to the METAC surface, stained by Live–Dead staining and measured by confocal microscopy 1 h post inoculation. It was difficult to perform accurate counting of cells attached to METAC due to their high number density. Representative images are shown for attachment to METAC (B, C, D). B – side view of a confocal image (the METAC surface is at the bottom of image B); C and D – slice images of the same group of attached cells as B at different distances from the surface. C shows the layer of cells most distant from the METAC surface where most of the cells are stained green and hence viable. D shows a layer of cells closest to the METAC surface where most cells are stained red and hence non-viable. Scale bar represents 10 μm in C and D. | ||
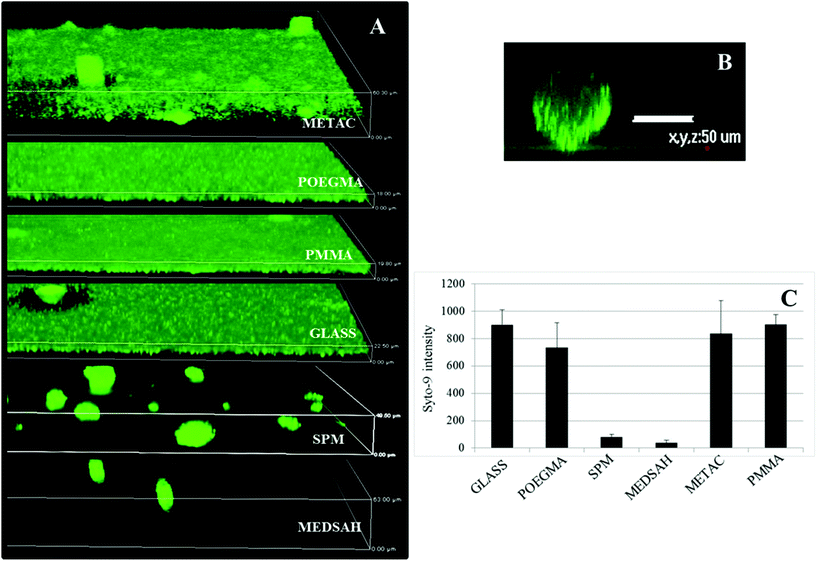 | ||
| Fig. 3 Biofilm of P. aeruginosa PAO1 on different polymer surfaces formed after 72 h in the flow cell. A – confocal microscopy images; although flat biofilms are dominating on glass, POEGMA, PMMA and METAC, occasional mushroom structures are seen; B – zoomed side view of a typical mushroom structure formed on SPM and MEDSAH; C – bacterial biomass of P. aeruginosa PAO1 attached to the surface; the height of the 3D-box in panel A represents 60 μm for METAC, 18 μm for POEGMA, 20 μm for PMMA, 23 μm for glass, 50 μm for SPM and 63 μm for MEDSAH. | ||
 | ||
| Fig. 4 P. aeruginosa PAO1 biofilm development on glass (1) and MEDSAH (2); A – after 1 h, B – 18 h, C – 72 h, and D – 96 h. It is evident that flat biofilms and mushroom structures are present from the beginning of the experiment on glass and MEDSAH; the height of the 3D-boxes in the figure represents 1B – 20 μm, 1C – 36 μm, 1D – 49 μm, 2B – 49 μm, 2C – 63 μm and 2D – 48 μm; scale bar in A represents 10 μm. Note that the images for different time points represent different fields of view. | ||
Positively charged surfaces have been suggested to be antibacterial and it has previously been shown that polycationic surfaces like poly-lysine11 and poly(2-(dimethylamino)ethyl methacrylate (pDMAEMA) modified with alkyl bromides7 are bactericidal and disrupt the proton motive force of bacteria.11 Hence, it was interesting that polycationic METAC surfaces in our study were covered with biofilm similarly to untreated glass. Live–dead staining of cells attached 1 h post inoculation showed that most of the cells in direct contact with the surface were, in fact, non-viable while the majority of the cells at the top of the non-viable cell layer were undamaged and viable (Fig. 2). Moreover, 5 hours after the beginning of the experiment, clusters of actively dividing cells were detected on the METAC surface (data not shown), which suggests that the cells surviving during the attachment form a biofilm on top of the layer of dead attached cells. This implies that polycationic surfaces may not form long-term bactericidal surfaces unless they are constantly cleaned or refreshed.
Surface associated motility
A biofilm pattern similar to the one observed for SPM and MEDSAH (mushroom structures) has previously been described for P. aeruginosa associated with surfaces coated with mucin,27 a negatively charged glycoprotein,28 while flat biofilm developed on glass.27 Mucin was shown to affect surface-associated motility of bacteria through the proposed mucin-FilD specific interaction,27 thereby promoting the formation of a biofilm with mushroom structures. In our study, bacterial motility was measured as spreading of bacteria between agar and a glass slide with or without polymer. P. aeruginosa is classified as an aerobic bacterium29 and, hence, the rationale of the motility test was that the bacteria would move from a relatively oxygen poor environment under the glass slide, towards the edge of the slide where oxygen was abundant. Indeed, we saw that bacteria covered with a glass slide on an agar plate moved along the surface of the glass towards the edge and rich growth was seen around the slide (Fig. 5). Bacteria behaved similarly in the case of POEGMA or PMMA-coated surfaces. In contrast, for METAC, SPM and MEDSAH, growth only occurred in a small circular area where a drop of the bacterial suspension was initially placed indicating that bacterial motility was inhibited by these surfaces. An additional explanation for the absence of motility on METAC is that the cells were killed by the dense positive charge at the surface and that progression of surviving bacteria was considerably slowed down or restricted by the toxicity of the brush surrounding the initial colony.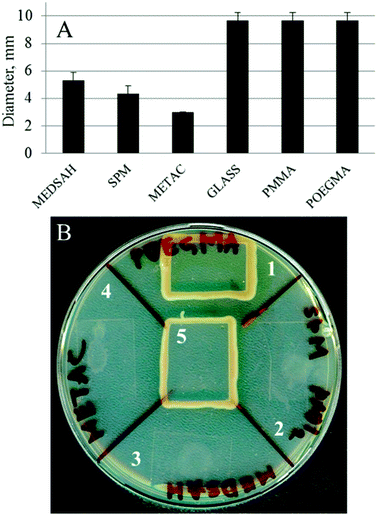 | ||
| Fig. 5 Motility of P. aeruginosa PAO1 associated with polymer surfaces; a drop of bacterial culture was applied onto an agar plate, dried and covered with a polymer-coated glass slip. Untreated glass was used as a control. Panel A shows the diameter of the ring of bacteria spreading under the coated glass slip after 18 h. Panel B shows a similar experiment after two days. In the case of untreated glass, POEGMA and PMMA, the bacteria placed in the center of the surface reached the edge of the glass slip and started growing around it. In the case of SPM, METAC and MEDSAH, the bacteria only spread in a small circle under the surface. 1 – POEGMA, 2 – SPM, 3 – MEDSAH, 4 – METAC, and 5 – untreated glass. | ||
To better understand the attachment, the dynamics of P. aeruginosa interactions with the polymer surfaces was studied using live-cell microscopy in the absence of shear force. The micrographs (Fig. 6) and the movies (ESI†) show that bacteria attach transiently to the glass, under these conditions, and are constantly moving. Fewer cells seemed to be in contact with the glass under these conditions compared to the flow-chamber (Fig. 6A). This corresponds to the previous observations showing increased bacterial attachment to glass surfaces under flow conditions compared to static conditions.30 In the case of METAC and SPM, bacteria displayed a more stable contact with the surface in static conditions. The whole bacterial cell appeared to be in contact with the METAC surface for the majority of cells (Fig. 6C). For SPM, on the other hand, bacteria were mainly seen as “dots” i.e. oriented perpendicular to the surface (Fig. 6B). These data illustrate that positively charged METAC surfaces attract negatively charged bacterial cells while negatively charged SPM surfaces repel cells that have the same charge and make them change their spatial orientation to minimize the exposure to the polymer. The cells on SPM also appeared to be retained at the surface during static conditions and did not exhibit the same dynamic movement as bacteria on untreated glass (ESI†). In other words, motility of P. aeruginosa appears to be disrupted when associated with highly charged surfaces under static conditions, as was also seen in the motility assay on agar plates. The reason for this immobilization could be electrostatics or, in the case of negatively charge polymers, presence of mechanosensitive bonds on some part of the bacterium that are disrupted by the force from the flow in the flow chamber experiments.31
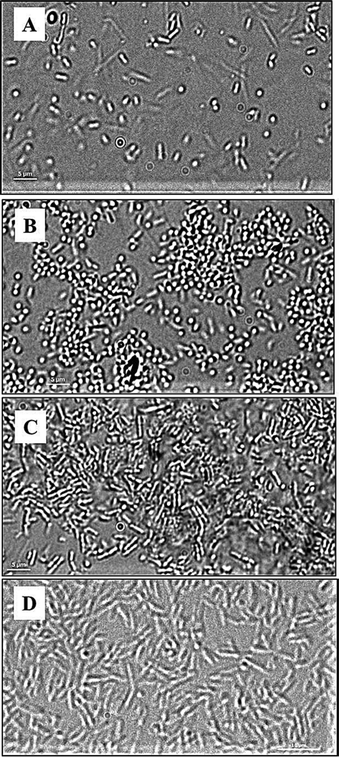 | ||
| Fig. 6 DIC images of P. aeruginosa PAO1 (A, B, C) and P. aeruginosa PAO1 ΔpilAΔfliC double mutant (D) at the surface of glass (A), SPM (B and D), and METAC (C); P. aeruginosa PAO1 bacteria associated with SPM (B) are seen as “dots” as they are perpendicularly oriented with respect to the SPM brush surface. This is not seen in P. aeruginosa PAO1 associated with METAC or in the P. aeruginosa PAO1 ΔpilAΔfliC double mutant on SPM. Note that cells of the double mutant have a larger size than the wild type, are non-motile, and did not come in contact with the SPM surface (making focusing difficult and giving rise to the less sharp image in D). | ||
Previous reports have shown that flagella of P. aeruginosa can bind to negatively but not positively charged substances,32 and that the FliD protein of P. aeruginosa, located at the distal end of a flagellum, plays a key role in binding to the negatively charged glycoprotein mucin.27 Pili have also been shown to enable bacteria to orient vertically33 and enhance bacterial attachment to charged surfaces.34 Moreover, it has been shown that curli of E. coli can overcome repulsion forces between bacterial cells and negatively charged particles allowing cells to associate with these particles.35 Hence, it is possible that cell appendices could loosely anchor P. aeruginosa to the SPM surface.
Bacterial cell polarity has also been suggested to orient bacteria with respect to a surface. Jones et al.35 showed that cells of E. coli devoid of both flagella and pili could attach to negatively charged polystyrene particles in an oriented manner.
To test whether cell appendices were involved in vertical orientation of P. aeruginosa on SPM surfaces, we performed live-cell microscopy with a ΔfliCΔpilA double mutant of P. aeruginosa PAO1.36 Mutants at the surface of SPM did not behave like wild type bacteria; instead most of the cells were seen as rods e.g. cells were oriented parallel to the surface. This suggests that cell appendices such as flagella and pili may be involved in the vertical orientation of the cells with respect to the SPM surface rather than cell polarity. However, anchoring through flagella or pili does not appear to be sufficient under flow conditions as few cells were found attaching to SPM in the flow chamber experiments.
Results from the motility experiments provide a clue why mushroom structures were found on SPM and MEDSAH instead of flat biofilms. It has previously been shown that flat and mushroom-like biofilm phenotypes of P. aeruginosa in flow cells develop under distinct conditions (such as presence/absence of particular nutrients or mucin).18,27 Here we observe that cells attached to SPM and MEDSAH display poor surface motility, and probably cannot move over the surface to create a flat biofilm but instead develop mushroom structures.
Flat and mushroom biofilms represent two physiologically different phenotypes of P. aeruginosa
Since the bacterial motility and biofilm architecture on SPM/MEDSAH and glass/POEGMA/PMMA/METAC were different, we tested whether these effects could be mediated through c-di-GMP signaling. Production of c-di-GMP was detected in mushroom-like biofilm (Fig. 7) but not in the flat biofilms. This indicates that biofilms formed on SPM and MEDSAH are different from flat biofilms not only in their appearance and amount of biomass, but also in their physiology. It is well established that high c-di-GMP levels in bacteria are responsible for extensive secretion of exopolymers.20,21,37 Consequently, it is possible that secretion of these exopolymers allows bacteria to alter their surface properties and attach also to negatively charged surfaces. | ||
| Fig. 7 Production of c-di-GMP in biofilms formed on glass (A) and SPM (B). To assess the levels of c-di-GMP, a reporter was used, where GFP was expressed under the control of a c-di-GMP sensitive promoter; GFP (green) is expressed mainly in mushroom structures demonstrating that c-di-GMP levels are higher in this type of biofilm. Biofilm was counterstained with propidium iodide (red in panel A). SPM polymer also binds propidium iodide and this accounts for the red background color in panel B; the height of the 3D-box in the image represents 23 μm for A and 46 μm for B. | ||
Biofilm formation by P. aeruginosa LPS mutants
LPS-mutants (with and without o-antigen) exhibit altered surface physicochemical properties, which have been shown to influence biofilm formation.16 Wild type P. aeruginosa PAO1 produces both A-band and B-band o-antigen, while the ΔwbpA mutant produces only A-band. PAO1ΔrmlC mutant does not produce A-band but instead produces B-band (Fig. 8E and F). However, due to the truncated LPS core, the o-antigen of PAO1ΔrmlC is not attached to the LPS core38 and is not expected to be exposed at the surface of the bacterium. Measurements of zeta potential and hydrophobicity of these strains show that PAO1ΔrmlC had more negative zeta potential than the wild type but a similar cell hydrophobicity. In contrast, the zeta potential of PAO1ΔwbpA was similar to wild type while cell hydrophobicity for this mutant was higher (Fig. 8A and B) probably due to the presence of the relatively hydrophobic A-band. The differences in zeta potential and hydrophobicity in the wild type and mutants were mainly due to distinct LPS phenotypes as the outer membrane proteins of these strains did not differ significantly apart from a small intensity variation in a protein band identified as outer membrane lipoprotein OprI precursor around 10 kDa (Fig. 8D).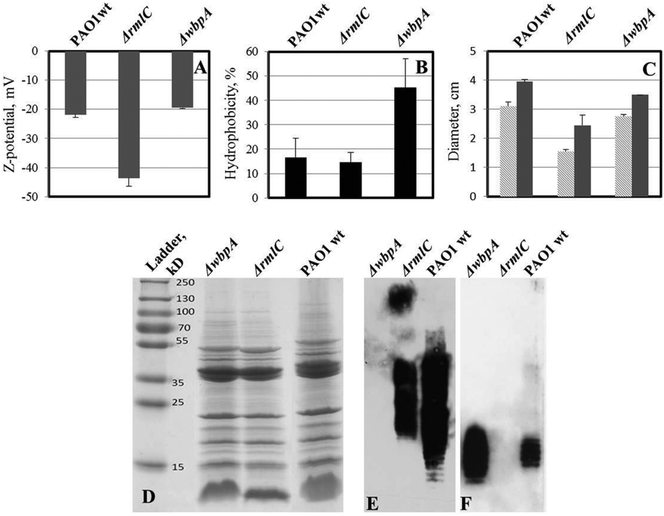 | ||
| Fig. 8 Properties of LPS mutants of P. aeruginosa PAO1 (compared to wild type). A – zeta potential of bacterial cells; two-tailed P ≤ 0.035 for zeta potential values of ΔrmlC mutant while zeta potential of wbpA is not significantly different from wild type PAO1. B – cell hydrophobicity; two-tailed P ≤ 0.0016 for ΔwbpA compared to the wild type, while the difference is not significant between ΔrmlC and wild type. C – swimming (light grey) and twitching (dark grey) motility of bacteria in polystyrene Petri dishes; both swimming and twitching motility were significantly inhibited in the ΔrmlC mutant (P < 0.001); D – outer membrane protein (OMP) profile of P. aeruginosa PAO1 and its LPS mutants; E – LPS Western blot with B-band specific, MF15-4 antibody; F – LPS Western blot with A-band specific, N1F10 antibody. No major difference in outer membrane proteins was found, which indicates that differences in hydrophobicity and surface charge were due to differences in LPS structure. | ||
Biofilms formed on glass by PAO1ΔwbpA and PAO1ΔrmlC mutants differed from the wild type. The PAO1ΔwbpA mutant formed flat but grainy biofilms and PAO1ΔrmlC formed thick biofilms (about 50 μm thick) resembling merged mushroom structures with uneven edges (Fig. 9). Such a mushroom biofilm of PAO1ΔrmlC may be due to less efficient swimming and twitching motility (Fig. 8C), properties that previously have been shown to influence biofilm phenotype in P. aeruginosa.18,27 When grown on SPM, PAO1ΔrmlC produced scarce biofilm while PAO1ΔwbpA developed multiple mushroom-like structures (Fig. 9). This scarce biofilm can be explained by the physicochemical properties of the bacterial cell. The PAO1ΔrmlC mutant exhibits high zeta potentials and can, consequently, be more strongly repelled by a negatively charged polymer brush surface in comparison to the wild type or the PAO1ΔwbpA mutant. The increase in the number of mushroom colonies for the PAO1ΔwbpA mutant could be a result of the higher hydrophobicity and relatively low zeta potential of the PAO1ΔwbpA mutant. This would result in lower repulsive forces between the bacterium and the negatively charged polymer brush, and perhaps some increased interactions with the polymer backbone of the surface, enabling the bacterium to better attach to the film or at pin-hole defects or scratches in the surface film. However, the number of colonies attaching in the different experiments was most probably not only a function of the amount of defects in the polymer film, since identical SPM films were used in the experiments with different strains. The amount of pin-holes also seems to be low. XPS data from the brush surfaces showed no or less than 0.1 atom% of Si at the surface. The exact mechanism for the increased interactions with the negative surface in the case of the PAO1ΔwbpA mutant, as well as the influence of extracellular substances, is a subject for further studies.
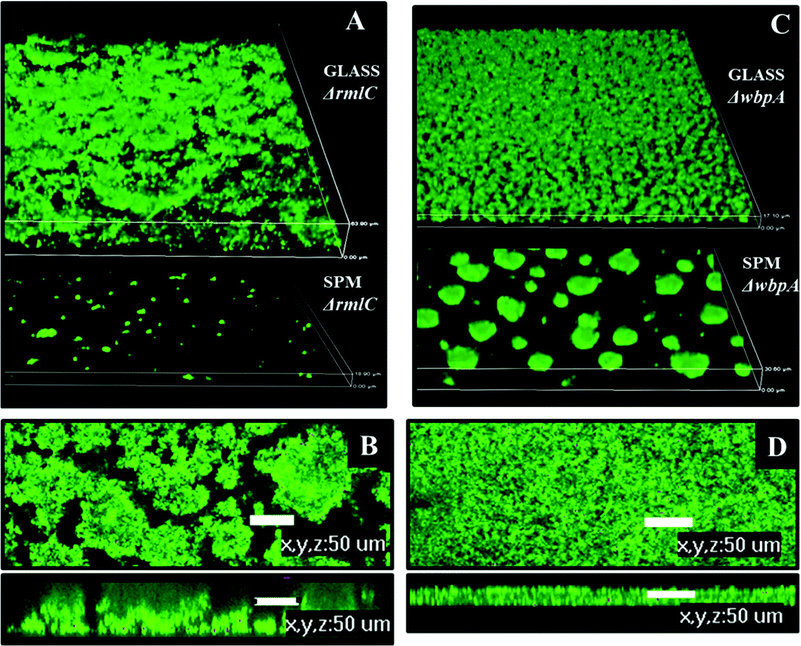 | ||
| Fig. 9 Biofilm architecture of P. aeruginosa PAO1 LPS mutants on SPM and on glass, 72 hours post inoculation. A – biofilm of PAO1 ΔrmlC on glass and SPM, 3D view, height of the 3D-box represents 63 μm for glass and 18 μm for SPM; B – biofilm of PAO1 ΔrmlC on glass, top and side view, scale bar represents 50 μm; C – biofilm of PAO1 ΔwbpA on glass and SPM, 3D view, height of the 3D-box represents 17 μm for glass and 30 μm for SPM; D – PAO1 (wild type) biofilm on glass, top and side view, scale bar represents 50 μm. | ||
Experimental
Strains and growth conditions
Strains and plasmids used in this study are described in Table 1. Bacteria were cultured either on blood agar plates or in Isosensitest broth (Oxoid LTD, Hampshire, England) supplemented with antibiotics when needed. pCdrA::gfpS reporter plasmid was introduced into P. aeruginosa PAO1 (J. Lam group) by electroporation. P. aeruginosa PAO1 expressing pCdrA::gfpS was cultured in the same medium as the wild type but with the addition of carbenicilin (100 μg mL−1).| Strain/plasmid | Relevant characteristics | Source |
|---|---|---|
| P. aeruginosa PAO1 | Wild type | Joseph Lam lab14 |
| PAO1ΔwbpA | Deficient in B-band LPS biosynthesis | Joseph Lam lab14 |
| PAO1ΔrmlC | Truncated LPS core | Joseph Lam lab14 |
| PAO1 ΔfliCΔpilA | Deficient in flagella and pili | Alain Filloux lab36 |
| pJBA129 | pME6030 PA1/04/03-gfp-T0–T1, Tcr; constitutive GFP expression | Michael Givskov lab1 |
| pCdrA::gfpS | pUCP22Not-PcdrARBS-CDS-RNaseIIIgfp(Mut3)-T0–T1, Ampr | Matthew Parsek lab49 |
Composition of growth media; Iso sensitest: hydrolysed casein 11 g L−1, peptones 3 g L−1, glucose 2 g L−1, NaCl 3 g L−1, starch 1 g L−1, Na2HPO4 2 g L−1, Na acetate 1 g L−1, Mg glycerophosphate 0.2 g L−1, Ca gluconate 0.1 g L−1, CoSO4 0.001 g L−1, CuSO4 0.001 g L−1, ZnSO4 0.001 g L−1, FeSO4 0.001 g L−1, MnCl2 0.002 g L−1, menadione 0.001 g L−1, cyanocobalamine 0.001 g L−1, L-cystein hypochloride 0.02 g L−1, L-tryptofan 0.02 g L−1, pyridoxine 0.003 g L−1, pantothenate 0.003 g L−1, nicotineamide 0.003 g L−1, biotin 0.0003 g L−1, thiamine 0.00004 g L−1, adenine 0.01 g L−1, guanine 0.01 g L−1, xanthine 0.01 g L−1, uracil 0.01 g L−1; TSB: casein peptone (pancreatic) 17 g L−1, K2HPO4 2.5 g L−1, glucose 2.5 g L−1, NaCl 5 g L−1 and soya peptone (papain digest) 3 g L−1.
Materials
All chemicals were of analytical reagent grade and were used as received from the manufacturer unless stated otherwise. (3-Trimethoxylsilyl)propyl 2-bromo-2-methylpropionate was purchased from Fluorochem Lt, UK. 3-Sulfopropylmethacrylate (98%) in the form of potassium salt, oligo (ethylene glycol)methacrylates [∼360 g mol−1 (OEGMA6)], [2-methacryloyloxy)ethyl] trimethylammonium chloride (80 wt% in water), methyl methacrylate (MMA) and [2-(Methacryloyloxy)ethyl]dimethyl-3-sulfopropyl)ammonium hydroxide, 2,2′-bipyridyl(99+%) (BiPy), copper(II)chloride (97%) (Cu(II)Cl2 and copper(I)chloride (99995+%) (Cu(I)Cl) were obtained from Sigma-Aldrich (stored under vacuum until needed). Solvents (ethanol, methanol, sulfuric acid, hydrochloric acid, and hexane (dried using molecular sieves)) were obtained from Fisher. Deionized water with a resistance of 18.2 MΩ cm was prepared with a Millipore Milli-Q Plus 185 system. Coverslips N2, 60 × 24 mm, were purchased from VWR, USA (VWR micro cover glasses).Polymer synthesis on glass slides
The coverslips were cleaned as follows: first they were sonicated in a (1:1) solution of ethanol and deionized water for 10 min. Further, the coverslips were immersed in piranha solution [H2SO4:H2O2 (7:3)] for 30 min at 100–150 °C and then in a mixture of ammonia (25%), H2O2 (30%) and water (1:1:5) for 15 min at 70 °C followed by a solution of hydrochloric acid for 15 min at room temperature (HCl:H2O, 1:6).39 The slides were rinsed between every step and at the end with deionized water. Finally, they were dried under a stream of N2. A monolayer of initiator molecules was vapor deposited onto the glass slides as described by Brown et al.40 The polymerisations were carried out using aqueous ATRP with conditions adapted from previously published procedures.23,25,41,42 Typical polymerisations occurred as follows:SPM polymerisation: 51.89 g of monomer was dissolved by stirring in 60 mL of methanol and 31.5 mL of water at room temperature. To this solution, 1.967 g of BiPy and 0.0454 g of Cu(II)Cl2 were added. The mixture was stirred and degassed by N2(g) for 30 min before 0.50256 g of Cu(I)Cl was added. The mixture was left for 15 min under N2. Initiator-coated glass slides were sealed in a Schlenk tube and degassed (4 × high vacuum pump/N2 refill cycles). The reaction mixture was injected with a syringe into the tube to cover the glass slides completely, and the mixture was left for 3 h under a stream of N2 (g). After that, the glass slides were removed and thoroughly rinsed with deionized water and dried.
METAC polymerisation: The polymerisation solution was prepared by dissolving 39.63 mL of monomer in 60 mL of methanol and 30 mL of water. 1.9658 g of BiPy and 0.03483 g of Cu(II)Cl2 were added and the mixture was degassed for 30 min. Then, 0.50274 g of Cu(I)Cl was added and the solution was degassed for 15 more minutes. Glass slides were removed after 7 hours.
Polymerisation of OEGMA: 0.7499 g of BiPy and 0.04275 g of Cu(II)Cl2 were added to the solution of monomer (32 mL) in water (50 mL). After stirring for 30 min and degassing by N2(g), 0.1944 g of Cu(I)Cl was added and the mixture was left again for 15 min. The polymerisation time was 2 hours.
Polymerisation of MMA: A quantity of 0.8333 g of BiPy and 0.108 g of Cu(II)Cl2 was added into the solution of monomer (35 mL) in 10 mL of water and 40 mL of methanol. After degassing and stirring for 30 min, 0.216 g of CuCl was added and the mixture was kept under a stream of N2 (g) for 15 more min. The polymerisation reaction was stopped after 7.5 hours.
MEDSAH polymerisation: 37.5 g of monomer was completely dissolved by stirring in a solution of methanol (60 mL) and water (15 mL) while degassing with N2 (g). In a separate vessel a mixture of BiPy (1.05 g), CuCl2 (0.075 g) and CuCl (0.278 g) was degassed and left under nitrogen for 5 min. A volume of 8 mL of methanol and 2 mL of water were added to the catalyst mixture and the solution was stirred under N2(g) for 15 min. Thereafter the catalyst mixture was added to the monomer solution and left under N2(g) for 20 min. The polymerisation was carried out for 6.5 hours and the glass slides were washed thoroughly with warm water (65 °C).
Thickness of polymer brushes
The thickness of polymer brushes was measured on a J.A. Woollam alfa-SE spectroscopic ellipsometer in the dry state using translucent adhesive tape applied to the back surface of the slide to suppress backside reflection.X-ray photoelectron spectroscopy
The composition of the brush surfaces was analysed using X-ray Photoelectron Spectroscopy (XPS) on a Kratos Axis Ultra DLD electron spectrometer using a monochromatic AlKα source operated at 150 W. An analyzer pass energy of 160 eV was used for survey spectra and 20 eV for individual photoelectron lines. The spectrometer charge neutralising system was used to compensate for sample charging during measurement and the binding energy scale was referenced to the C 1s aliphatic carbon peak at 285.0 eV.Charge density of charged polymer brushes
The surface density of cationic (METAC) and anionic (SPM) polymer brushes was estimated by colorimetric methods using UV-Vis spectrophotometry adapted to our system.43,44 The procedure assumes that counter ions from the charged brush get replaced by charged dye molecules to give a 1:1 complex between one charged dye molecule and one charged functional group. Orange II dye was used for cationic brushes and Toluidine Blue O (TB) dye for anionic brushes. At pH 3 Orange II has an absorption peak at 485 nm, with an extinction coefficient of 19476 L mol−1 cm−1, while TB has an absorption peak at 633 nm and an extinction coefficient of 50000 L mol−1 cm−1.METAC: A volume of 50 mL of 0.5 mM aqueous Orange II solution was prepared and adjusted to pH 3 with a 1 mM solution of HCl (solution 1). Samples were placed in the solution and left overnight at 30 °C. Thereafter each sample was rinsed with water and immersed in 100 mL of 1 mM NaOH solution under stirring, to remove physically adsorbed dye from the brush (solution 2). After 24 h the pH of solution 2 was adjusted to pH 3 with 100 mM HCl and colorimetric analyses were carried out on both solutions. The amount of dye bound to the brush is deduced from the difference between the two solutions. SPM: A volume of 50 mL of 0.5 mM aqueous TB dye solution was prepared and adjusted to pH 10 using a buffer (Na2CO3/NaHCO3). Samples were placed in this solution and kept at 30 °C for 6 h. After washing with NaOH, 0.5 mM, each sample was placed in a 50% aqueous solution of acetic acid for 24 h to dissociate the dye from the brush. By analyzing the latter solution with UV-Vis the amount of absorbed dye was obtained.
SPR measurements
In order to monitor the conditioning of substrates during incubation in culture media, adsorption was followed via surface plasmon resonance (SPR), using a Biacore 3000. SPR sensor chips (Ssens), on which polymer brushes had been grown, were mounted onto a substrate holder and docked in the instrument before priming twice with a buffer (PBS). The substrates were allowed to equilibrate for 30 min at 20 μL min−1 in PBS, until a stable baseline was obtained. To monitor adsorption from culture media, the substrates were washed and equilibrated for 5 min whilst recording the baseline signal detected. The surfaces were then exposed to culture media for 5 min before washing with PBS for 30 min. The reading of the amount of material deposited on the surface was carried out after 30 min equilibration. The flow rate was 20 μL min−1 and measurements were carried out in triplicate.Contact angle measurement
Sessile drop contact angles were measured on the substrates with an optical tensiometer (KSV Instruments). A drop of 3–4 μL of deionized water was deposited on the surface of the substrate from an automated syringe and pictures of the water drop on the surface were taken during and after deposition at 250 ms intervals. The contact angle of the drop at each time was measured to assess the evolution of the drop shape and a possible change in the surface properties of the substrate over time. The Young Laplace method (Attension software) was used to fit the shape of the drops and to measure the contact angles. The values reported are averages of 3 to 4 drops deposited at different locations on one substrate. All samples with contact angles above ≈15° had stable drop shapes over time (without taking into account slow evaporation of the water droplet). The error associated with each value was low and indicates that the surfaces were homogeneous and the contact angles were reproducible on each substrate.Flow chamber biofilm and confocal laser scanning microscopy (CLSM)
Bacteria were cultured in Isosensitest broth diluted 10 times with MQ water. Flow chamber BST FC270, glass flow break FB50, and bubble trap FC34, were all purchased from Biosurface Technology Corporation, USA. These parts were connected with silicon tubing, 2 mm bore diameter; Marprene tubing, 0.8 mm bore diameter (Alitea, Sweden), was used in the place of contact with a pump head. After the flow chamber was autoclaved, functionalized glass slides sterilized in 70% ethanol were inserted in sterile conditions. For inoculation, bacteria from the late exponential growth phase were pelleted and re-suspended in NaCl 0.9% to the concentration of 2 × 109 cell mL−1. 2 mL of this bacterial suspension was injected to each channel of the flow chamber with a syringe. The flow in the chamber was stopped for 30 min to allow bacterial attachment. All through the experiment the system was operated at 1.2 mL min−1 using a 405U/L2 double-channel pump (Watson Marlow, Alitea, Sweden). Bacteria were visualized either through expression of green fluorescent protein (GFP) or by staining with Syto-9 fluorescent dye (Invitrogen, Molecular Probes, USA). When Syto-9 was used, 1 mL of media containing 5 nM Syto-9 was injected into each channel and the flow was stopped for 10 min to allow staining. Flow chamber experiments were performed at least in duplicate and images were captured at several positions on each slide.Confocal (3-D) biofilm images were captured with a Nikon Eclipse90i fluorescent microscope equipped with a Nikon D-eclipse C1+ laser system (Nikon Corporation, Japan). Images were acquired and the intensity of the signal from the biofilm was measured at 510–530 nm wavelength using EZ-C1 ver.3.80 and NIS-Elements Advanced Research ver.3.2 software (Nikon Corporation). Biomass was measured through the intensity of syto-9 in confocal 3D images; the same settings were used during image acquisition. Attached bacteria were counted using free ImageJ software (rsb.info.nih.gov/ij/) and presented as the number of cells in a field of view, which was 4.03 × 105 μm2.
Live cell microscopy
To monitor more precisely the interaction between polymer surfaces and bacteria, live cell microscopy was performed as follows: Petri dishes 3.5 cm in diameter were completely filled with tryptic soy agar (TSA) medium and after solidification 1 μL of bacterial suspension (8 × 1010 cell mL−1) in NaCl 0.9% was applied on the top of the agar. Bacteria were covered with a glass slide with or without polymer coating and directly observed using a Nikon Eclipse Ti-E inverted microscope equipped with ×100 objective. Differential interference contrast (DIC) images were captured using an Andor iXon+ EMCCD camera. Sequences of images taken with 5 s intervals were assembled in a movie (130 ms interval) using NIS-Elements 3.2 Software.Surface associated motility
25 mL of TSA-agar (BD, BLL™) medium was poured into a Petri dish with an inner diameter of 88 mm. After solidification plates were dried at 37 °C for 20 min. Bacterial suspension (2 μL, 1 × 109 cell mL−1) was dropped on the surface of agar and allowed to dry. Glass slides coated with polymer were sterilized in 70% ethanol for 10 min, rinsed in MQ water and dried. The glass slides were placed on the top of a dried drop of bacterial suspension. After incubation at 37 °C the growth associated with glass slides was observed.Isolation and analysis of outer membrane proteins
Outer membrane proteins (OMPs) were isolated as described previously with some modifications.45 In brief, 25 mL of overnight grown cultures were centrifuged at 10000 rpm for 15 minutes at 4 °C, re-suspended in 25 mL of milliQ water and sonicated on ice. Cell debris and intact cells were removed by centrifugation at 7000 rpm for 10 minutes at 4 °C. The supernatant fraction was treated with 2% N-lauryl sarcosyl at room temperature. This solution was ultra-centrifuged twice at 29000 rpm for 1.5 hours in a 45Ti Beckmann rotor and the pellet containing OMPs was resuspended in milliQ water. OMPs were separated by 15% polyacrylamide gel electrophoresis.Cell hydrophobicity
Hydrophobicity was measured as previously described.16 Briefly: an overnight culture was centrifuged at 10000 rpm for 15 min and re-suspended in phosphate buffered saline (PBS) to give an OD595 of approximately 1(A0). Then, hexadecane was added to the suspension in the ratio of 4:1 (bacterial suspension:hexadecane). The optical density of the aqueous phase was measured at 595 nm (A). The hydrophobicity was calculated according to the equation: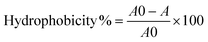
Zeta potential
Zeta potential was measured by dynamic light scattering using Malvern Nano ZS zetasiser and clear disposable zeta cells (Malvern). Before measuring, bacteria were cultured overnight, washed and re-suspended in phosphate buffer with an ionic strength of 20 mM (1 × 109 cell mL−1).Swimming and twitching motility
The swimming and twitching of P. aeruginosa PAO1 and its LPS mutants were evaluated in the Isosensitest medium complemented with 0.3% agar as described elsewhere.46 Briefly: 12 mL of the medium was poured into 53 mm diameter Petri dishes and 5 μL of bacteria suspension with ABS600 = 1 was stabbed into the agar in the middle of the plate. Swimming was represented by a cloudy ring within the agar, while twitching was represented by a ring of thin film between the agar and the plastic bottom of the Petri dish. The diameter of both kinds of motility rings was measured 15 h after the inoculation.Polyacrylamide gel electrophoresis (PAGE) and Western immunoblotting
Cultures of P. aeruginosa PAO1 and respective mutants were grown to ABS600 = 1. Cells from 1 mL of the culture were collected by centrifugation and LPS preparation and staining was done as previously described.47 LPS was resolved on gradient 4 to 12% gel. LPS differences in mutants were confirmed by staining with an Emerald Green LPS Kit (Invitrogen, Molecular Probes, USA) and Western blot analysis with N1F10 (A-band specific) and MF15-4 (B-band specific) monoclonal antibodies.Live–dead staining
Live–dead staining (a combination of syto-9 and propidium iodide staining) was performed to discriminate between viable and non-viable cells attached to the glass/brush in the flow cell. Syto-9 (Invitrogen, Molecular Probes, USA) was used according to manufacturer's instructions. Viable cells were stained green with Syto-9 dye and cells with compromised membrane were stained red.Conclusions
Using P. aeruginosa as a model bacterium, this work illustrates some of the complexity of bacterial attachment and biofilm formation and how it is dependent on many interlocking parameters of both the abiotic and the bacterial surface. Expected antifouling and antibacterial surfaces such as brushes with ethylene glycol subunits or cationic brushes were found to become covered with biofilm in the same way as glass reference surfaces. However, on negatively charged surfaces the biofilm formation was strongly reduced. Both attachment and motility were found to be inhibited for bacterial cells associated with negative SPM and zwitterionic MEDSAH surfaces. Additionally, bacteria that were attached to SPM and MEDSAH showed high levels of c-di-GMP, which indicates increased production of biofilm matrix components (exopolysaccharides). This suggests that Gram-negative bacteria, such as P. aeruginosa, can modify their cell surfaces when in contact with negatively charged substrates and this, in turn, influences the development and architecture of the biofilm. This work also shows that LPS structures that confer high zeta potential on the bacterial cell decrease biofilm formation on negatively charged surfaces. On the other hand, LPS structures that increase cell hydrophobicity could facilitate the formation of biofilm on polymer surfaces with negative charge. Biofilms on SPM and MEDSAH consisted of characteristic mushroom structures, but the quantity of biofilm was much lower than on glass, POEGMA and PMMA. Similar mushroom structures were previously reported by other authors to be more resistant to antibiotics than flat biofilms,27,48 which should be investigated further and taken into consideration in the development of antifouling surfaces. Taken together, this study shows that to successfully design antifouling surfaces, emphasis needs to be placed on understanding the dynamics of the bacterial cell surface in relation to the abiotic material in question.Abbreviations
| SPM | poly (3-sulphopropylmethacrylate) |
| MEDSAH | poly (2-(methacryloyloxy)ethyl)dimethyl-3-sulphoproyl) ammonium hydroxide) |
| METAC | poly (2-(methacryloyloxy)-ethyl trimethyl ammonium chloride) |
| POEGMA | poly oligo(ethylene glycol methyl ether methacrylate) |
| PMMA | polymethylmethacrylate |
| c-di-GMP | cyclic diguanylate |
Acknowledgements
P. aeruginosa PAO1 strain expressing the pJBA129 plasmid was kindly provided by Jens Bo Andersen and Michael Givskov, University of Copenhagen. We are grateful to Joseph Lam, University of Guelph and Mathew Parsek, University of Washington for generously providing us with P. aeruginosa LPS mutants, anti-LPS antibodies and c-di-GMP reporter plasmid, respectively. Alain Filloux, Imperial College London is acknowledged for the gift of P. aeruginosa PAO1 ΔfliCΔpilA double mutant. We thank Joseph D. Mougous, University of Washington and Helena Lindgren, Umeå University for fruitful discussions. The Curth Nilsson Foundation for Strategic Research, the Carl Trygger Foundation for Scientific Research, the Swedish Research Council and the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) are acknowledged for funding.Notes and references
- A.P. Stapper, G. Narasimhan, D. E. Ohman, J. Barakat, M. Hentzer, S. Molin, A. Kharazmi, N. Høiby and K. Mathee, J. Med. Microbiol., 2004, 53, 679–690 CrossRef CAS
.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS
- D. Davies, Nat. Rev. Drug Discov., 2003, 2, 114–122 CrossRef CAS
- R. M. Klevens, J. R. Edwards, C. L. Richards Jr., T. C. Horan, R. P. Gaynes, D. A. Pollock and D. M. Cardo, Public Health Rep., 2007, 122, 160–166 Search PubMed
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC
- A. Terada, K. Okuyama, M. Nishikawa, S. Tsuneda and M. Hosomi, Biotechnol. Bioeng., 2012, 109, 1745–1754 CrossRef CAS
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS
- B. Zdyrko, V. Klep, X. W. Li, Q. Kang, S. Minko, X. J. Wen and I. Luzinov, Mater. Sci. Eng., C, 2009, 29, 680–684 CrossRef CAS
- D. Montag, M. Frant, H. Horn and K. Liefeith, Biofouling, 2012, 28, 315–327 CrossRef CAS
- G. Cheng, Z. Zhang, S. F. Chen, J. D. Bryers and S. Y. Jiang, Biomaterials, 2007, 28, 4192–4199 CrossRef CAS
- H. Strahl and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12281–12286 CrossRef CAS
- A. Clements, F. Gaboriaud, J. F. Duval, J. L. Farn, A. W. Jenney, T. Lithgow, O. L. Wijburg, E. L. Hartland and R. A. Strugnell, PLoS One, 2008, 3, e3817 Search PubMed
- F. Gosselin, J. F. L. Duval, J. Simonet, C. Ginevra, F. Gaboriaud, S. Jarraud and L. Mathieu, Colloids Surf., B, 2011, 82, 283–290 CrossRef CAS
- J. S. Lam, V. L. Taylor, S. T. Islam, Y. Hao and D. Kocincova, Front. Microbiol., 2011, 2, 118 CrossRef CAS
- C. D. Ciornei, A. Novikov, C. Beloin, C. Fitting, M. Caroff, J. M. Ghigo, J. M. Cavaillon and M. Adib-Conquy, Innate. Immun., 2010, 16, 288–301 CrossRef CAS
- R. Nakao, M. Ramstedt, S. N. Wai and B. E. Uhlin, PLoS One, 2012, 7, e51241 CAS
- C. A. Flemming, R. J. Palmer, A. A. Arrage, H. C. Van der Mei and D. C. White, Biofouling, 1999, 13, 213–231 CrossRef
- M. Klausen, A. Heydorn, P. Ragas, L. Lambertsen, A. Aaes-Jorgensen, S. Molin and T. Tolker-Nielsen, Mol. Microbiol., 2003, 48, 1511–1524 CrossRef CAS
- R. Simm, M. Morr, A. Kader, M. Nimtz and U. Romling, Mol. Microbiol., 2004, 53, 1123–1134 CrossRef CAS
- B. R. Borlee, A. D. Goldman, K. Murakami, R. Samudrala, D. J. Wozniak and M. R. Parsek, Mol. Microbiol., 2010, 75, 827–842 CrossRef CAS
- V. T. Lee, J. M. Matewish, J. L. Kessler, M. Hyodo, Y. Hayakawa and S. Lory, Mol. Microbiol., 2007, 65, 1474–1484 CrossRef CAS
- G. A. O'Toole and R. Kolter, Mol. Microbiol., 1998, 30, 295–304 CrossRef CAS
- O. Azzaroni, S. Moya, T. Farhan, A. A. Brown and W. T. S. Huck, Macromolecules, 2005, 38, 10192–10199 CrossRef CAS
- O. Azzaroni, A. A. Brown and W. T. S. Huck, Angew. Chem., Int. Ed., 2006, 45, 1770–1774 CrossRef CAS
- J. E. Gautrot, W. T. S. Huck, M. Welch and M. Ramstedt, ACS Appl. Mater. Inter., 2010, 2, 193–202 CrossRef CAS
- L. Ploux, A. Ponche and K. Anselme, J. Adhes. Sci. Technol., 2010, 24, 2165–2201 CrossRef CAS
- R. M. Landry, D. An, J. T. Hupp, P. K. Singh and M. R. Parsek, Mol. Microbiol., 2006, 59, 142–151 CrossRef CAS
- R. Bansil and B. S. Turner, Curr. Opin. Colloid Interface, 2006, 11, 164–170 CrossRef CAS
- Medical Microbiology, ed. P. Murray, K. Rosenthal and M. Pfaller, Elsevier MOSBY, Philadelphia, 5th edn, 2005, p. 357 Search PubMed
- H. H. Rijnaarts, W. Norde, E. J. Bouwer, J. Lyklema and A. J. Zehnder, Appl. Environ. Microbiol., 1993, 59, 3255–3265 CAS
- M. Chabria, S. Hertig, M. L. Smith and V. Vogel, Nat. Commun., 2010, 1 CAS
- L. D. Hazlett and X. L. Rudner, Ophthalmic Res., 1994, 26, 375–379 CrossRef CAS
- J. C. Conrad, M. L. Gibiansky, F. Jin, V. D. Gordon, D. A. Motto, M. A. Mathewson, W. G. Stopka, D. C. Zelasko, J. D. Shrout and G. C. L. Wong, Biophys. J., 2011, 100, 1608–1616 CrossRef CAS
- B. Pidhatika, J. Moller, E. M. Benetti, R. Konradi, E. Rakhmatullina, A. Muhlebach, R. Zimmermann, C. Werner, V. Vogel and M. Textor, Biomaterials, 2010, 31, 9462–9472 CrossRef CAS
- J. F. Jones, J. D. Feick, D. Imoudu, N. Chukwumah, M. Vigeant and D. Velegol, Appl. Environ. Microbiol., 2003, 69, 6515–6519 CrossRef CAS
- S. de Bentzmann, M. Aurouze, G. Ball and A. Filloux, J. Bacteriol., 2006, 188, 4851–4860 CrossRef CAS
- E. Karatan and P. Watnick, Microbiol. Mol. Biol. Rev., 2009, 73, 310–347 CrossRef CAS
- R. Rahim, L. L. Burrows, M. A. Monteiro, M. B. Perry and J. S. Lam, Microbiology, 2000, 146, 2803–2814 CAS
- S. Tugulu, A. Arnold, I. Sielaff, K. Johnsson and H. A. Klok, Biomacromolecules, 2005, 6, 1602–1607 CrossRef CAS
- A. A. Brown, N. S. Khan, L. Steinbock and W. T. S. Huck, Eur. Polym. J., 2005, 41, 1757–1765 CrossRef CAS
- M. Ramstedt, N. Cheng, O. Azzaroni, D. Mossialos, H. J. Mathieu and W. T. S. Huck, Langmuir, 2007, 23, 3314–3321 CrossRef CAS
- N. Cheng, A. A. Brown, O. Azzaroni and W. T. S. Huck, Macromolecules, 2008, 41, 6317–6321 CrossRef CAS
- E. Uchida, Y. Uyama and Y. Ikada, Langmuir, 1993, 9, 1121–1124 CrossRef CAS
- M. Ciobanu, A. Siove, V. Gueguen, L. J. Gamble, D. G. Castner and V. Migonney, Biomacromolecules, 2006, 7, 755–760 CrossRef CAS
- T. Song, F. Mika, B. Lindmark, Z. Liu, S. Schild, A. Bishop, J. Zhu, A. Camilli, J. Johansson, J. Vogel and S. Wai, Mol. Microbiol., 2008, 70, 100–111 CrossRef CAS
- M. H. Rashid and A. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4885–4890 CrossRef CAS
- X. H. Lai, R. L. Shirley, L. Crosa, D. Kanistanon, R. Tempel, R. K. Ernst, L. A. Gallagher, C. Manoil and F. Heffron, PLoS One, 2010, 5, e11857 Search PubMed
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877–888 CrossRef CAS
- M. T. Rybtke, B. R. Borlee, K. Murakami, Y. Irie, M. Hentzer, T. E. Nielsen, M. Givskov and M. R. Parsek, Appl Environ Microbiol, 2012, 78, 5060–5069 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Three films related to Fig. 6a–c are available free of charge. See DOI: 10.1039/c3bm00197k |
| This journal is © The Royal Society of Chemistry 2013 |