
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild cross-coupling of tertiary alkoxides with aryl chlorides enabled by a shelf-stable methylnaphthyl palladium NHC complex
Nikolaos V.
Tzouras
a,
Ruben
Fleischer
 a,
Pierpaolo
Satta
a,
Sourav
Manna
a,
Angelino
Doppiu
b and
Lukas J.
Gooßen
*a
a,
Pierpaolo
Satta
a,
Sourav
Manna
a,
Angelino
Doppiu
b and
Lukas J.
Gooßen
*a
aFaculty for Chemistry and Biochemistry, Ruhr Universität Bochum, Universitätsstr. 150, 44801, Bochum, Germany. E-mail: lukas.goossen@rub.de
bPrecious Metals Chemistry, Umicore AG & Co. KG, Rodenbacher Chaussee 4, 63457, Hanau–Wolfgang, Germany
First published on 6th November 2025
Abstract
The catalytic coupling of tertiary alkoxides with aryl halides has a high energetic barrier, which makes KOtBu an advantageous base in cross-couplings. A palladium methylnaphthyl (MeNAP) catalyst bearing the IPentCl ligand was found to promote this C–O coupling with high efficiency, enabling the synthesis of aryl t-alkyl ethers from abundant aryl chlorides.
The frequent occurrence of C(sp2)–O bonds in natural products, agrochemicals and pharmaceuticals has inspired intense efforts towards the development of efficient C–O bond-forming methodologies.1 The incorporation of tertiary alkyl ether groups is of specific interest in drug discovery as these groups increase lipophilicity and provide a defined steric bulk.2,3t-Butoxy groups can also serve as masked hydroxy functionalities. It can be synthetically advantageous to introduce phenolic hydroxyl groups in a protected form and liberate these sensitive functionalities at a late stage of the overall synthesis.4
The introduction of tertiary alkoxy substituents is traditionally achieved by metal-free methods via SNAr or aryne mechanisms starting from aryl fluorides or bromides,5–7 and multistep syntheses from arylammonium or aryliodonium salts.8,9 Cross-couplings of aryl halides with tertiary alcohols are promoted by Pd, Ni, and Cu catalysis, but in contrast to other C–O bond forming reactions,10 these couplings are still somewhat underdeveloped.11–13 Hartwig, Buchwald, Watanabe, and Maligres et al. found PtBu3,14,15 QPhos,16,17 and biphenylphosphine ligands18 to be most effective in palladium-catalyzed alkoxylations of aryl halides, whereas the JosiPhos-type ligand CyPF-tBu is particularly suited for heteroaryl chlorides (Scheme 1).19 Stradiotto and co-workers demonstrated that nickel/Dalphos systems promote alkoxylation of various (hetero)aryl electrophiles.20 However, all of these processes call for relatively high catalyst loadings and temperatures above 85 °C.
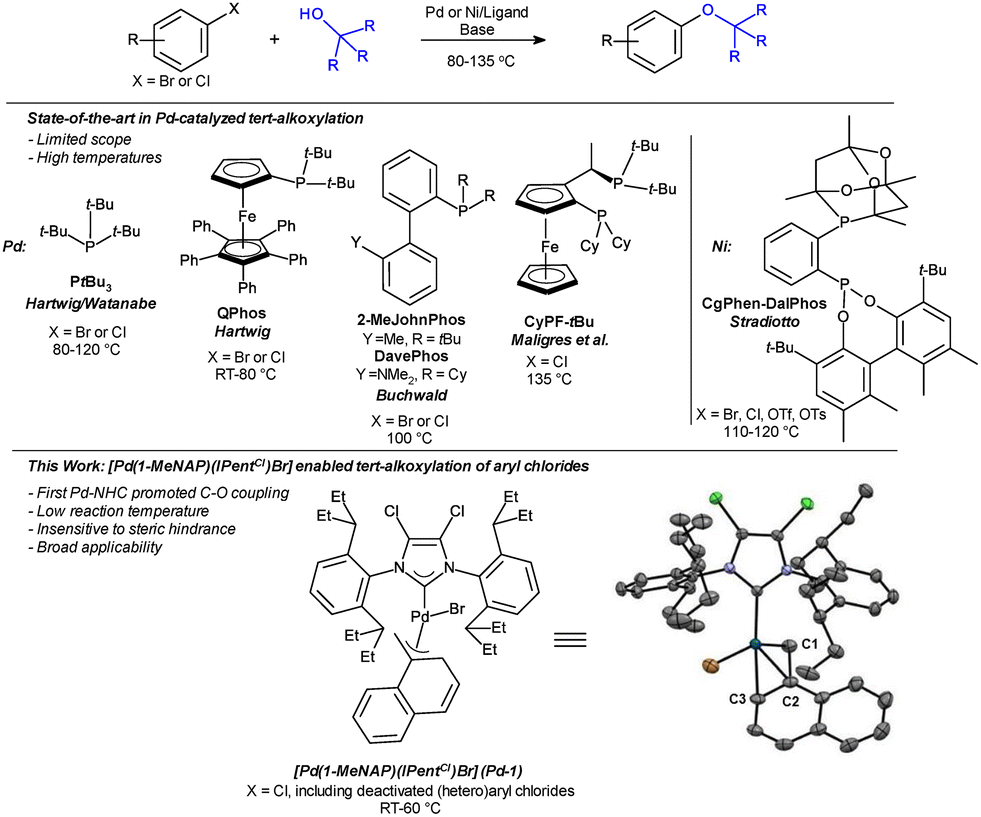 | ||
| Scheme 1 Catalytic couplings of aryl halides with tertiary alcohols. In the depicted X-ray of the [Pd(1-MeNAP)(IPentCl)Br] catalyst, displacement ellipsoids are shown at the 50% probability level, and hydrogens are omitted for clarity (CCDC = 2492026). Selected bond lengths (Å): CNHC–Pd = 2.049(3), Pd–C1 = 2.092(3), Pd–C2 = 2.242(3), Pd–C3 = 2.267(3). | ||
The use of N-heterocyclic carbenes (NHCs) has led to major advances in Pd-catalyzed cross-coupling with numerous efficient catalytic reactions being disclosed in the last decade.21 Backbone-modifications of NHCs have been shown to enhance their catalytic reactivity by adjusting steric and electronic properties.22–25 In this context, Organ et al. reported that the dichlorinated NHC ligand IPentCl promotes C–N,26 C–S,27 and C–C cross-coupling reactions more efficiently than its non-halogenated counterpart.28 The chloro-substituents are believed to induce a high but flexible steric bulk and increase the π-electron-accepting abilities of NHC ligands while maintaining their strong σ-donating character. This allows IPentCl systems to master both challenging oxidative addition and reductive elimination processes.29 IPentCl catalysts can be generated in situ from various palladium precursors, but stable, one-component IPentCl palladium pre-catalysts have so far only been synthesized as pyridine or cinnamyl complexes.30
During our studies on Pd-methylnaphthyl (MeNAP) complexes as bench-stable, rapidly activating palladium sources,31 we discovered that complexes generated from [Pd(MeNAP)X]2 and halogenated carbene ligands showed catalytic activity in challenging couplings of aryl chlorides with t-alkoxides. This raised our attention, since to the best of our knowledge, there is no literature precedence of an aryl ether synthesis promoted by a Pd-NHC system.
We systematically investigated the catalytic activity of various complexes generated from [Pd(1-MeNAP)Br]2 and phosphine or NHC ligands using the reaction of p-chloroanisol and potassium t-butoxide as a model system (Table 1). Comparative experiments revealed that state-of-the-art systems, which have been shown to promote this coupling at elevated temperatures, give only marginal conversion at 60 °C (entries 1 and 2). Even when switching to [Pd(1-MeNAP)Br]2 as the palladium source, none of the tested phosphine ligands promoted the target reaction, although many of them induced high catalytic activity in other Pd-mediated cross-couplings (entries 3–7). Non-halogenated NHC ligands were also ineffective (entries 8 and 9). In sharp contrast, the 4,5-dichlorinated NHC ligand IPentClHCl gave quantitative conversion and near quantitative yield of the desired aryl-t-butyl ether 3aa (Entry 11). The catalyst is very sensitive to variations of the chain length of the N-substituents (entries 10–12): For the smaller isopropyl substituted derivative IPrClHCl, a derivative of the widely used IPr NHC ligand, no conversion was observed, and IHeptClHCl bearing larger isoheptyl substituents gave a rather low yield. These results highlight the significant influence of the ligand's steric bulk, which primarily affects transmetallation and reductive elimination. We next probed the influence of the palladium source on the reaction outcome and found that the MeNAP complexes are clearly superior to established systems (entries 13–17). Neither the cinnamyl complex [Pd(cin)Cl]2 – the gold standard among Pd-allyl precursors,32 nor PEPPSI-type complexes or palladacycles of the Buchwald generation 3 type performed similarly well. This aligns with recent reports that MeNAP Pd-halide precursors decisively enhance the performance of cross-coupling catalysts both for phosphine and NHC systems.31,32b
|
|
|||
|---|---|---|---|
| Entry | Pd pre-catalyst | Ligand | 3aa (%) |
| a 0.5 mmol scale, 60 °C, 2 mL toluene, 16 hours, 2.5 mol% [Pd] and ligand (2.7 mol% of phosphine ligands), 2 eq. KOtBu, GC yield using tetradecane as an internal standard. b 1.2 eq. KOtBu. | |||
| 1 | Pd(dba)2 | QPhos | 2 |
| 2 | Pd(OAc)2 | 2-MeJohnPhos | 0 |
| 3 | [Pd(1-MeNAP)Br]2 | QPhos | 2 |
| 4 | [Pd(1-MeNAP)Br]2 | CyPF-tBu | 0 |
| 5 | [Pd(1-MeNAP)Br]2 | DavePhos | 2 |
| 6 | [Pd(1-MeNAP)Br]2 | PtBu3 | 0 |
| 7 | [Pd(1-MeNAP)Br]2 | PAd3 | 11 |
| 8 | [Pd(1-MeNAP)Br]2 | IPrHCl | 0 |
| 9 | [Pd(1-MeNAP)Br]2 | IPentHCl | 2 |
| 10 | [Pd(1-MeNAP)Br]2 | IPrClHCl | 0 |
| 11 | [Pd(1-MeNAP)Br]2 | IPentClHCl | 98 |
| 12 | [Pd(1-MeNAP)Br]2 | IHeptClHCl | 25 |
| 13 | [Pd(cin)Cl]2 | IPentClHCl | 76 |
| 14 | Pd G3 dimer | IPentClHCl | 20 |
| 15 | 3-Cl PEPPSI IPentCl | — | 23 |
| 16 | 2-Me PEPPSI IPentCl | — | 85 |
| 17 | [Pd(1-MeNAP)(IPentCl)Br] | — | >99b |
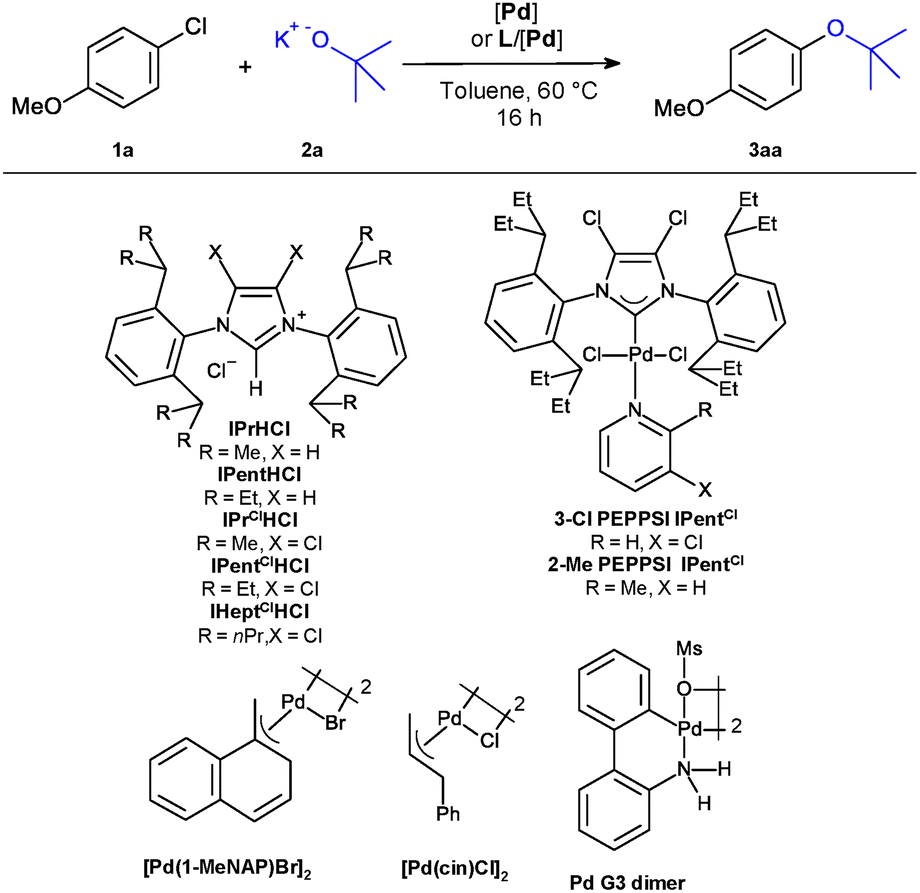
Comprehensive screening tables can be found in the SI. They show that the reaction is more effective with potassium than with sodium or lithium t-butoxide and that toluene and dioxane are the most effective solvents. NMR-studies revealed that the reaction of commercially available [Pd(1-MeNAP)Br]2, IPentClHCl and excess KOtBu proceeds quantitatively within minutes with the formation of the corresponding NHC complex. Under these conditions, activation takes place within 10 minutes, as judged by the disappearance of the characteristic MeNAP signals of Pd-1 in 1H-NMR. To obtain [Pd(1-MeNAP)(IPentCl)Br] (Pd-1) in a microanalytically pure form, we treated [Pd(1-MeNAP)Br]2 with the preformed carbene IPentCl. This way, Pd-1 was obtained in near quantitative yield as an air-stable, easy to administer complex. Its molecular structure was confirmed by single crystal X-ray diffraction analysis (see Scheme 1). The use of Pd-1 as a catalyst gives full conversion and near quantitative yields of 3aa, even when the base is used only in 1.2-fold excess (entry 17).
We proceeded to investigate the scope of aryl-t-alkyl ether synthesis, varying both the aryl halide and the alkoxide component. As illustrated by the examples in Table 2, both electron-rich and electron-deficient aryl chlorides bearing substituents in the ortho-, meta- or para-position were successfully converted. 3aa was synthesized on a gram scale demonstrating the scalability of the process. Various functional groups were tolerated, e.g. thioethers, non-enolizable ketones, amides, and esters (3ca–3fa). Sulphur- and nitrogen-based heterocyclic scaffolds are also compatible (3ja, 3ka, 3pa, 3qa). Notably, electron-deficient aryl chlorides were found to be more reactive than electron-rich ones. For example, 1b was converted even with only 1% catalyst, and 1d gave high yields already at room temperature.

Still, even the extremely electron-rich aryl chloride 1o, bearing alkoxy substituents in the ortho and para positions, was successfully converted, albeit at slightly higher temperature. The catalyst is also remarkably tolerant of steric hindrance. Even aryl chlorides with two ortho methyl groups were cleanly converted. Compounds 3ma–3oa have never been accessed by catalytic alkoxylation reactions, which underlines the high efficiency of this protocol. The synthesis of a t-butoxylated derivative of the commercial drug cloperastine illustrates the synthetic utility of the transformation (3ta). Alkyne and alkene functionalities were left intact, without even traces of Heck-type products being observed (3ra, 3sa), whereas primary or secondary amino groups are incompatible, as they couple preferentially over the tertiary alkoxides. Acidic functionalities such as phenols, carboxylic acids, or enolizable carbonyl groups are not tolerated. The reaction scope with regard to the tertiary alcohols includes t-amyl and adamantyl derivatives (3ab, 3pb).33 The reaction protocol extends to tertiary alcohols, exclusively. Primary and secondary alcohols undergo redox reaction to form arenes and aldehydes, which is expected as the IPentCl ligand has an unsuitable steric profile to facilitate reductive elimination over β-hydride elimination.
A series of mechanistic experiments were conducted to shed some light on the reaction mechanism (Scheme 2a). Electron-rich aryl chloride 1a reacts more slowly than electron-poor 1b, both in parallel and one-pot competition reactions. This seems to point towards oxidative addition as the rate-limiting step. However, neither the starting concentration of the aryl chloride nor that of the alkoxide has any effect on the reaction rate (Scheme 2b), ruling out both the oxidative addition and transmetallation as rate-determining. Varying the catalyst loading revealed a first-order rate dependence on the catalyst (Fig. S1 in SI). Further experiments revealed that the reactivity increases with decreasing steric bulk in the series adamantanol, t-amyl alcohol and t-butanol (Scheme S2 in the SI). In contrast, Stradiotto et al. found that for Ni-based systems, bulkier alcohols react at higher rates.20
 | ||
| Scheme 2 (a) Competition experiments: 0.50 mmol aryl chloride, 1.00 mmol 2a. (b) Experiments conducted at 0.50 mmol scale, 60 °C, in 2 mL toluene, with 1.25 mol% [Pd(1-MeNAP)Br]2 and 2.5 mol% IPentClHCl: yield of 3aa vs time using increasing eq. of 1a (left) and conversion of 1avs. time using increasing eq. of 2b (right). GC yield/conversion using tetradecane as an internal standard. Each datapoint represents the average of two runs. | ||
The above findings align with mechanistic studies by Hartwig,11,34a as well as Wiedenhoefer and Buchwald.34b They found that reductive eliminations of aryl ethers from palladium complexes are accelerated by electron-withdrawing substituents. We can, thus, conclude that the reductive elimination of the aryl ether product is rate-determining for Pd-1. This explains why the electron-withdrawing chloro substituents on the bulky IPentCl enable mild, catalytic C–O bond formation.
Overall, [Pd(1-MeNAP)(IPentCl)Br] is an easy-to-use, one-component catalyst precursor with unparalleled catalytic activity in the coupling of aryl halides with tertiary alkoxides. Our findings show how strongly the palladium precursor can impact a catalytic reaction. The unexpected discovery of a C–O coupling mediated by a Pd-NHC catalyst shows that there is still much left to explore in metal carbene catalysis.
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033–390677874 – RESOLV and the A. v. Humboldt Foundation (Fellowship to NVT). We thank Dr. Bert Mallick for help with the X-ray crystallography and M. Wüstefeld for HRMS measurements.
Conflicts of interest
UMICORE AG & Co. KG is commercializing many Pd complexes described in this work including [Pd(1-MeNAP)(IPentCl)Br].Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and copies of spectra. See DOI: https://doi.org/10.1039/d5cc05726d.CCDC 2492026 contains the supplementary crystallographic data for this paper.35
Notes and references
- A. Cook and S. G. Newman, Chem. Rev., 2024, 124, 6078 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516 CrossRef CAS.
- T. W. Johnson, R. A. Gallego and M. P. Edwards, J. Med. Chem., 2018, 61, 6401 CrossRef CAS PubMed.
- G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, E. Marcantoni, P. Melchiorre and L. Sambri, Adv. Synth. Catal., 2006, 348, 905 CrossRef CAS.
- T. F. Woiwode, C. Rose and T. J. Wandless, J. Org. Chem., 1998, 63, 9594 CrossRef CAS.
- Y. Dong, M. I. Lipschutz and T. D. Tilley, Org. Lett., 2016, 18, 1530 CrossRef CAS PubMed.
- M. Shigeno, K. Hayashi, O. Sasamoto, R. Hirasawa, T. Korenaga, S. Ishida, K. Nozawa-Kumada and Y. Kondo, J. Am. Chem. Soc., 2024, 146, 32452 CrossRef CAS PubMed.
- E. Lindstedt, E. Stridfeldt and B. Olofsson, Org. Lett., 2016, 18, 4234 CrossRef CAS.
- D. Wang, Z. Yang, C. Wang, A. Zhang and M. Uchiyama, Angew. Chem., Int. Ed., 2018, 57, 3641 CrossRef CAS PubMed.
- (a) H. Zhang, P. Ruiz-Castillo, A. W. Schuppe and S. L. Buchwald, Org. Lett., 2020, 22, 5369 CrossRef CAS PubMed; (b) Z. Chen, Y. Jiang, L. Zhang, Y. Guo and D. Ma, J. Am. Chem. Soc., 2019, 141, 3541 CrossRef CAS; (c) H. Zhang, P. Ruiz-Castillo and S. L. Buchwald, Org. Lett., 2018, 20, 1580 CrossRef CAS; (d) M. J. Strauss, M. E. Greaves, S. Kim, C. N. Teijaro, M. A. Schmidt, P. M. Scola and S. L. Buchwald, Angew. Chem., Int. Ed., 2024, 63, e202400333 CrossRef CAS; (e) P. M. MacQueen, J. P. Tassone, C. Diaz and M. Stradiotto, J. Am. Chem. Soc., 2018, 140, 5023 CrossRef CAS PubMed; (f) K. M. Morrison, E. C. Jackson, E. M. H. Elliott and M. Stradiotto, Chem. – Eur. J., 2025, 31, e202404352 CrossRef CAS.
- G. Mann and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 13109 CrossRef CAS.
- G. Mann and J. F. Hartwig, J. Org. Chem., 1997, 62, 5413 CrossRef CAS.
- M. Palucki, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 10333 CrossRef CAS.
- G. Mann, C. Incarvito, A. L. Rheingold and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 3224 CrossRef CAS.
- M. Watanabe, M. Nishiyama and Y. Koie, Tetrahedron Lett., 1999, 40, 8837 CrossRef CAS.
- Q. Shelby, N. Kataoka, G. Mann and J. Hartwig, J. Am. Chem. Soc., 2000, 122, 10718 CrossRef CAS.
- N. Kataoka, Q. Shelby, J. P. Stambuli and J. F. Hartwig, J. Org. Chem., 2002, 67, 5553 CrossRef CAS PubMed.
- C. A. Parrish and S. L. Buchwald, J. Org. Chem., 2001, 66, 2498 CrossRef CAS.
- P. E. Maligres, J. Li, S. W. Krska, J. D. Schreier and I. T. Raheem, Angew. Chem., Int. Ed., 2012, 51, 9071 CrossRef CAS.
- K. M. Morrison, R. T. McGuire, M. J. Ferguson and M. Stradiotto, ACS Catal., 2021, 11, 10878 CrossRef CAS.
- S. S. Bera, G. Utecht-Jarzyńska, S. Yang, S. P. Nolan and M. Szostak, Chem. Rev., 2025, 125, 5349 CrossRef CAS.
- Y. Zhang, G. Lavigne, N. Lugan and V. César, Chem. – Eur. J., 2017, 23, 13792 CrossRef.
- F. Izquierdo, C. Zinser, Y. Minenkov, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, F. Nahra, C. S. J. Cazin and S. P. Nolan, ChemCatChem, 2018, 10, 601 CrossRef.
- A. Bugarin, S. A. Patil, R. Q. Tran and K. O. Marichev, Inorg. Chim. Acta, 2024, 572, 122263 CrossRef PubMed.
- M. Ohashi, K. Ando, S. Murakami, K. Michigami and S. Ogoshi, J. Am. Chem. Soc., 2023, 145, 23098 CrossRef PubMed.
- (a) S. Sharif, R. P. Rucker, N. Chandrasoma, D. Mitchell, M. J. Rodriguez, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2015, 54, 9507 CrossRef PubMed; (b) M. Pompeo, J. L. Farmer, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2014, 53, 3223 CrossRef PubMed; (c) A. Khadra, S. Mayer and M. G. Organ, Chem. – Eur. J., 2017, 23, 3206 CrossRef PubMed; (d) S. Sharif, D. Mitchell, M. J. Rodriguez, J. L. Farmer and M. G. Organ, Chem. – Eur. J., 2016, 22, 14860 CrossRef PubMed.
- (a) M. Sayah, A. J. Lough and M. G. Organ, Chem. – Eur. J., 2013, 19, 2749 CrossRef PubMed; (b) M. Sayah and M. G. Organ, Chem. – Eur. J., 2013, 19, 16196 CrossRef PubMed.
- (a) M. Pompeo, R. D. J. Froese, N. Hadei and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 11354 CrossRef PubMed; (b) N. Sinha, P. A. Champagne, M. J. Rodriguez, Y. Lu, M. E. Kopach, D. Mitchell and M. G. Organ, Chem. – Eur. J., 2019, 25, 6508 CrossRef PubMed; (c) N. Sinha, D. Heijnen, B. L. Feringa and M. G. Organ, Chem. – Eur. J., 2019, 25, 9180 CrossRef PubMed.
- (a) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650 RSC; (b) F. Nahra, D. J. Nelson and S. P. Nolan, Trends Chem., 2020, 2, 1096 CrossRef.
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244 CrossRef CAS PubMed.
- (a) N. Sivendran, N. Pirkl, Z. Hu, A. Doppiu and L. J. Gooßen, Angew. Chem., Int. Ed., 2021, 60, 25151 CrossRef CAS; (b) B. Xue, F. Papp, M. Yang, J. Shen, A. Doppiu and L. J. Gooßen, ACS Catal., 2024, 14, 11172 CrossRef CAS.
- (a) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed; (b) G. Carì, R. Saito, L. P. Zorba, T. Ruggiero, K. Van Hecke, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2025, e01967 CrossRef.
- K. L. Mears, C. R. Stennett, J. C. Fettinger, P. Vasko and P. P. Power, Angew. Chem., Int. Ed., 2022, 61, e202201318 CrossRef CAS PubMed.
- (a) J. P. Stambuli, Z. Weng, C. D. Incarvito and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7674 CrossRef CAS; (b) R. A. Widenhoefer and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6504 CrossRef CAS.
- CCDC 2492026: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pn4yg.
| This journal is © The Royal Society of Chemistry 2025 |