
Spontaneous assembly of a class of small molecule prodrugs directed by SN38†
Zhenhai
Tang
a,
Wenning
Lan
a,
Kaiying
Wen
a,
Wenting
Li
a,
Tao
Wang
*b,
Dongdong
Zhou
 *a and
Hao
Su
*a
*a and
Hao
Su
*a
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: hsu@scu.edu.cn; zhoudd@scu.edu.cn
bDepartment of Pediatrics, West China Second University Hospital, Sichuan University, No. 20, 3rd Section, South Renmin Road, Chengdu 610041, China. E-mail: wangtao2001@scu.edu.cn
First published on 28th August 2024
Abstract
Small molecule self-assembling prodrugs (SAPDs) are an emerging class of amphiphilic monomers that can aggregate into supramolecular nanostructures with high drug loading identical to that of the individual prodrug. Despite great progress in creating nanodrugs via nanoprecipitation, the direct self-assembly of small molecule SAPDs in aqueous solution remains challenging, as the proper hydrophilic–hydrophobic balance and intermolecular interactions have to be rationally considered. We report a class of small molecule SAPDs by conjugating the anticancer drug SN38 as the structure-directing component with various hydrophilic auxiliaries (i.e., oligo ethylene glycol (OEG) of different lengths, amino, and carboxyl groups) via a self-immolative disulfanyl-ethyl carbonate linker. Driven by π–π interactions between SN38 units, these SAPDs spontaneously assembled into well-defined fibrous nanostructures. Variations in hydrophilic domains can robustly regulate the hydrophobicity of SAPDs, as well as the morphologies and surface features of supramolecular filaments, subsequently influencing cellular internalization behaviors. Furthermore, our study also reveals that the parent drug can be efficiently and controllably released in the presence of glutathione (GSH), exhibiting high in vitro toxicity against colorectal cancer cells. In this work, we present a delicate platform to design small molecule SAPDs that can spontaneously self-assemble into supramolecular filamentous assemblies directed by aromatic interaction of the parent drugs, providing a new strategy to optimize supramolecular drug delivery systems.
Introduction
Chemical modification of drugs into prodrugs has been a proven strategy to address their inherent properties, such as poor solubility, low permeability, chemical instability, undesirable pharmacokinetic characteristics, and serious adverse effects.1,2 For example, modifying a drug with ionized groups (e.g., piperidinyl, amino, and phosphate)3–5 or polar groups (e.g., hydroxyl)6 can significantly increase its solubility; masking a hydrophilic drug with a short hydrocarbon promoiety can improve its passive permeability across biological membranes;7,8 capping the unstable lactone ring in a drug can prevent its hydrolysis;9 conjugating a bioactive peptide epitope enables the active targeting of a drug to specific receptors.10 Although such a strategy enables optimized properties over the parent drugs, the resulting prodrugs still suffer from rapid renal clearance and premature degradation due to their small molecule nature.In some designs of prodrugs, chemically linking a hydrophilic auxiliary with a hydrophobic drug can endow the prodrug with amphiphilicity. Such self-assembling prodrugs (SAPDs) can associate into various nanostructures in aqueous solution via either nanoprecipitation or spontaneous assembly.11–15 As a result of their nano-size features, the resulting drug nanostructures may show reduced clearance, prolonged circulation, and enhanced accumulation at the disease site.16,17 In addition, the assembly–disassembly balance, drug release rate, surface decoration, and physicochemical properties of drug assemblies can be tuned via rational prodrug design.18–22 Especially, small molecule SAPDs created by modifying parent drugs with a low-molecular-weight promoiety have attracted increasing interest in recent years.19–22 The prodrug assemblies are essentially one-component nanomedicines with high drug loading identical to that of the individual prodrug and negligible promoiety-associated toxicity, enabling self-formulation and self-delivery. In addition, the small molecule feature enables mass production and simple purification by chromatography, which ensures the reproducibility of SAPDs. Various small molecule hydrophilic auxiliaries were used to construct the system, including oligoethylene glycol (OEG), short peptides, and targeting ligands such as folic acid and lactose.23–25 For example, Gu, Shen, and co-workers developed an amphiphilic prodrug composed of a hydrophobic drug SN38 and a hydrophilic OEG, which could self-assemble into nanocapsules in aqueous solution with a drug loading of 35%.26 Yan and colleagues constructed a DOX-lactose prodrug with a drug loading of 61.7%.27 This small molecule prodrug can form nanoparticles in water, showing increased cell uptake and enhanced efficacy. By coupling hydrophobic platinum(IV) and hydrophilic lactose, He et al. developed two amphiphiles that can self-assemble into vesicles or micelles with the increasing amount of lactose.28 Cui and co-workers reported a paclitaxel (PTX) drug amphiphile by coupling PTX with a β-sheet forming Tau peptide, which can self-assemble into well-defined nanofibers in water. This system significantly improved the solubility of PTX and achieved 41% drug loading.29 Despite recent advances, the rational design of small molecule SAPDs remains challenging. Most of the current strategies utilize the nanoprecipitation method to produce spherical aggregates of small molecule SAPDs,30–32 while examples of spontaneous self-assembly into various discrete nanostructures are often limited to peptide–drug conjugates,33–36 where hydrogen bonding interactions among peptides contribute greatly to the self-assembly process. The spontaneous self-assembly of small molecule SAPDs solely driven by the drug–drug interactions is rarely reported, since, unlike nanoprecipitation, a proper hydrophobic–hydrophilic balance and strong directional association in the hydrophobic domain need to be considered.
SN38, the parent drug of clinically used prodrug irinotecan (CPT-11), is a DNA topoisomerase I inhibitor, which exhibits poor solubility in water, but strong cytotoxic potency.3,37 Structurally, the strong hydrophobicity and aromaticity of SN-38 could provide directional driving forces for supramolecular assembly, making it a potential candidate for creating small molecule SAPDs. In our previous study, we reported the design of two SN38 prodrugs by conjugating pentaethylene glycol monomethyl ether (OEG5-OH) to the 10 and 20-site hydroxyl groups through a carbonate bond.38 Both SAPDs self-assembled into supramolecular nanostructures, indicating the feasibility of using SN-38 as a structure-directing component to create supramolecular drug assemblies. We also found that modifying the hydroxyl group at position 20 results in filamentous structures with a more compact and ordered internal molecular arrangement, leading to enhanced supramolecular stability. Taking on the same structural role as SN38, we report in this work a series of SN38 SAPDs by conjugating four different hydrophilic segments via a disulfanyl-ethyl carbonate (etcSS) linker. Our results revealed that these SAPDs can self-assemble into filamentous structures in aqueous solution with varying lengths and surface charges, subsequently influencing the cellular uptake of assemblies. Also, the incorporation of different small molecule hydrophilic promoieties affects the self-assembly capacity and molecular arrangements inside the assemblies. Furthermore, SAPDs enable efficient release of the parent drug SN38 through GSH-induced self-immolation of the etcSS linker and all of them exhibit strong in vitro cytotoxicity against colorectal cancer cells. Our results show the design of a class of prodrugs that can spontaneously self-assemble into supramolecular filamentous assemblies through strong directional associations among SN-38 units, providing strategies to create discrete well-defined drug nanostructures via small molecule prodrugs.
Results and discussion
Molecular design and synthesis of SN38 SAPDs
Fig. 1A illustrates the design of four SN38 SAPDs, comprising a hydrophobic drug SN38 with simple hydrophilic small molecules via an etcSS linker. This self-immolative etcSS linker enables efficient and controlled release of SN38 in the presence of glutathione (GSH), which is overexpressed in most cancer cells, at a rate much faster than that of the previously used carbonate bond.38 The introduction of the linker also provides a flexible region to separate the hydrophilic and hydrophobic segments, avoiding the disruption of π–π interactions via the hydration of hydrophilic moieties when two distinctive segments are too close. The strategy of constructing self-assembled nanostructures by introducing a simple hydrophilic group at the C20-hydroxyl group of SN38 has been demonstrated in our previous work.38 Here, in addition to the incorporation of the etcSS linker, we further expanded the library of small molecule SAPDs by conjugating various hydrophilic segments. Also, the morphologies and surface properties of the resulting supramolecular nanostructures can be adjusted by selecting hydrophilic small molecules with different functional groups (Fig. 1B), allowing for various application possibilities. The hydrophilic groups included here are oligoethylene glycol with different lengths (OEG7 and OEG11), positively charged amino group, and negatively charged carboxyl group, corresponding to drug loadings of 46%, 38%, 63%, and 58.5%, respectively. | ||
| Fig. 1 Schematic illustration of the design of SAPDs. The SN38 SAPDs were composed of three parts: a hydrophobic drug SN38, a small hydrophilic group, and an etcSS biodegradable linker (A). Cartoon representation of the assembled supramolecular nanofibers (B). HPLC traces of the library of SAPDs (C). | ||
To synthesize these small molecule SAPDs and achieve selective modification of the C20-hydroxyl group of SN38, the phenolic hydroxyl group was first protected with the tert-butyloxycarbonyl (Boc) group. The remaining C20-hydroxyl group was then capped via a carbonate bond with a linker (Pyr-SS-OH, Scheme S1, ESI†) containing pyridyl disulfide (PDS), which has been extensively used to produce disulfide bonds via thiol exchange reactions.39 After deprotection of the Boc group, SN38-SS-Pyr was obtained and characterized (Scheme S1 and Fig. S5, S6, ESI†). As shown in Fig. S6 (ESI†), the characteristic peaks located at 4.2–4.1 and 7–9 ppm could be attributed to the methylene group directly connected to the C20-hydroxyl group and the pyridine ring, indicating the successful synthesis of SN38-SS-Pyr. After coupling SN38-SS-Pyr with hydrophilic ligands possessing thiol groups through a thiol exchange reaction, the SN38 SAPDs were subsequently obtained (Scheme S2 and Section S1.2 in ESI†). All SAPDs were purified using preparative reversed-phase high-performance liquid chromatography (RP-HPLC). Their purities and chemical structures were thoroughly characterized by analytical HPLC and liquid chromatography–mass spectrometry (LC-MS). The analytical HPLC trace of each sample (Fig. 1C) exhibits a narrow and unimodal peak, confirming the high purity of the synthesized SAPDs. Meanwhile, significant shifts were observed with varying hydrophilic segments. SAPD3 with amino groups was eluted first, which may be attributed to the residual protonation in the acidic eluent. SAPD4 exhibited the longest elution time, since its carboxyl group cannot effectively ionize. Moreover, the peaks of OEGylated SAPD1 and SAPD2 were located between those of the samples with positive and negative charges. The trace of SAPD2 with a longer length of OEG shifts to a shorter elution time, revealing enhanced hydrophilicity. Furthermore, LC-MS further confirmed the molecular accuracy of SAPDs with a series of single peaks that precisely match with the calculated m/z values (Fig. S7–S10 in ESI†).
Self-assembly of SAPDs
Given the structure-directing role of SN38, we envision that the supramolecular assembly process of SAPDs highly depends on the π–π interactions among SN38 molecules. With the fixed hydrophobic SN38 component, we next studied the self-assembling nature of SAPDs with various neutral OEGs, such as OEG4, OEG7, and OEG11, along with different functional groups such as amino and carboxyl residues. We intended to investigate the tolerance of SN-38 dominated self-assembly toward various hydrophilic moieties and the effect of hydrophilic–hydrophobic balance on supramolecular assembly. After dissolving SAPDs in water at a concentration of 1 mM and slowly cooling from 80 °C to room temperature, we used transmission electron microscopy (TEM) to examine their assembled morphologies. SAPD5 with OEG4 cannot fully dissolve at 1 mM (Fig. S11, ESI†), indicating that this prodrug molecule is too hydrophobic to achieve spontaneous assembly at this concentration. Fig. 2 reveals that all other four SAPDs self-assemble into well-defined fibrous nanostructures, indicating the critical role and strong tolerance of SN38 as the driving force. Taking a closer look, the length of OEG and the charge heads show certain effects on the self-assembly morphologies. Specifically, SAPD1 self-assembled into ordered nanofibers with a diameter of approximately 11.75 ± 1.51 nm (n > 50) and a length of several micrometers (Fig. 2A and Fig. S12, ESI†). When the OEG length increased to 11 repeat units, SAPD2 assembled into short fibers with a diameter of approximately 13.22 ± 1.68 nm (n > 50) and a length of 416 ± 96 nm (n > 50) (Fig. 2B and Fig. S13, ESI†). Interestingly, these fibers aggregated into spindle-shaped structures with a width of approximately 77.77 ± 10.65 nm (n > 50). Our previous work demonstrated that the length of supramolecular nanostructures is affected by attractive and repulsive interactions between monomers.40 Increasing the number of OEGs significantly reduces the length of nanofilaments. In our study, the change in the length of SAPD assemblies may be attributed to the intermolecular repulsion caused by the increasing length and volume of OEG chains. The aggregation of short fibers into spindle-shaped structures is likely attributed to the intermolecular interactions of OEG11 anchored on the surface of nanofibers. Furthermore, cationic SAPD3 and anionic SAPD4 were designed to validate the assembly protocol, and both formed well-defined nanofibers several micrometers in length. The diameters of SAPD3 and SAPD4 are 5 ± 1.1 nm (n > 50) and 4.8 ± 0.6 nm (n > 50), respectively (Fig. 2C, D and Fig. S14, S15, ESI†). The diameters of charged SAPDs are significantly shorter than those of OEGylated SAPDs, as a result of the longer chain length of OEG segments. These results suggest that the width of nanofibers can be adjusted by changing the size and length of the incorporated hydrophilic segments. The molecular packing in all these supramolecular filaments follows the core–shell cylindrical arrangement, with a diameter that is approximately twice the extended length of the motif (Fig. S16, ESI†). These nanofibers have a similar structure to those formed by most peptide drug conjugates. Additionally, the surface properties of these nanofibers were also characterized. As illustrated in Fig. S17 (ESI†), the ζ-potential values of the SAPDs at pH 7.4 are −23.4, −19.8, +30.3, and −50.9 mV, respectively. This further shows that our assembly strategy can modify the properties of the hydrophilic part, creating nanofibers with different surface characteristics suitable for various applications. | ||
| Fig. 2 TEM of supramolecular nanofiber of SAPD1 (A), SAPD2 (B), cationic SAPD3 (C), and anionic SAPD4 (D) at a concentration of 1 mM. | ||
To investigate the self-assembly propensity and the effect of hydrophilic segments on the self-assembly ability of SAPDs, we determined their critical micelle concentrations (CMCs) using dynamic light scattering (DLS).41 The count rate values remain low in the monomer solution. As the concentration increases, the signal shows a sudden rise, indicating the formation of nanostructures and the concentration at which is considered as the CMC value. As shown in Fig. 3, transitions were identified when the concentrations exceeded the CMC. The critical values at pH 7.4 were approximately 7.32, 37.11, 20.89, and 8.55 µM for SAPD1–4, respectively. As the OEG length increases, the SAPDs become more hydrophilic, resulting in higher solubility and increased CMC values. Additionally, it has been proven that amphiphilic molecules with carboxyl groups have stronger assembly ability than those with amino groups at pH 6.9.42 In our study, cationic SAPD3 also exhibits slightly higher CMC values than anionic SAPD4. Our results indicate that all SAPDs exhibited low CMC values, indicating a strong self-assembly capability of these prodrugs and our SN38 directed self-assembly of prodrugs could be a universal strategy for forming stable nanostructures.
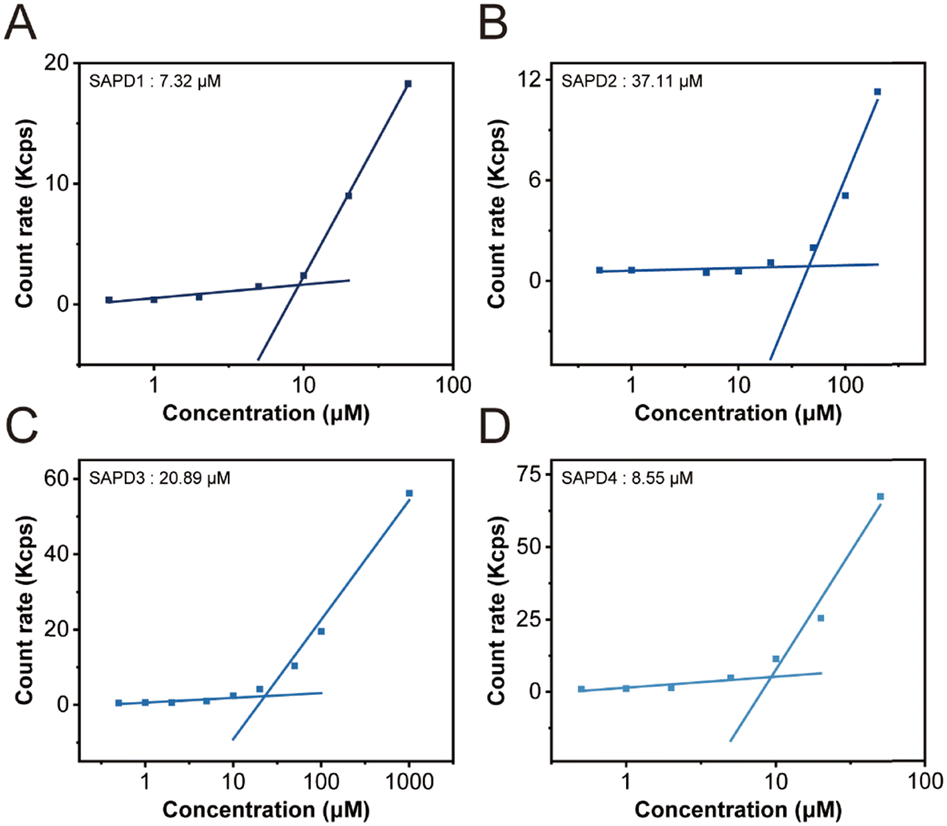 | ||
| Fig. 3 CMC transition curves of SAPD1 (A), SAPD2 (B), SAPD3 (C), and SAPD4 (D) at pH 7.4. | ||
To further illustrate the molecular arrangement within the assemblies, we recorded their circular dichroism (CD) spectra at a concentration of 200 µM. SAPD1 and SAPD2 show two negative peaks at 366 nm and 385 nm, along with a strong positive peak at 400 nm (Fig. 4A and B), indicating strong exciton coupling among neighboring SN38 rings, as reported previously.21 The exceptionally high intensity and bisignate features reflect the ordered aromatic packing and chiral orientation of SN38 units in the assemblies. The CD signal amplitude of SAPD2 in between 300–450 nm is significantly lower than that of SAPD1, indicating a lower degree of ordered packing in SAPD2. This observation is consistent with the TEM results, which show that SAPD2 formed shorter fibrous structures as the ordered molecular packing was disrupted by increased hydrophilic–hydrophobic balance and the steric repulsive force from the longer OEG chain. Additionally, SAPD2 exhibits a unique strong negative peak at 215 nm. We speculate that this might result from interactions among OEG11 units inside the fiber bundles.43 Interestingly, the strong positive peak at 400 nm is absent in SAPD3 and SAPD4 (Fig. 4C and D), suggesting looser molecular arrangements in these two assemblies compared to OEGylated ones. The loss of strong signals could be attributed to charge repulsion, resulting in relatively irregular packing of SN38 molecules. The observed signals at 250 nm (n–π*) and between 320 and 400 nm (π–π*) are still attributed to aromatic interactions among SN38 units. Despite differences in CD signals, all SAPD assemblies show strong associative interactions among SN38 molecules, indicating that this structure-directing component plays a primary role in the formation of fibrous morphology with different surface chemistries.
 | ||
| Fig. 4 CD spectra of SAPD1 (A), SAPD2 (B), SAPD3 (C), and SAPD4 (D) at a concentration of 200 µM. | ||
Cellular uptake
The physicochemical properties of supramolecular assemblies have attracted extensive attention due to their influence on both in vitro and in vivo behaviors. Next, we investigated the effect of nanostructure morphology and surface charge on cellular uptake by CT26 cancer cells. The pre-seeded cells were treated with cell media containing 100 µM SAPDs for 5 hours. After removing the cell culture medium containing SAPDs and washing with PBS, CT26 cells were treated with 200 µL of trypsin, centrifuged to remove the supernatant, and resuspended in PBS. After repeating the centrifugation and resuspension steps three times, the cells were ultimately resuspended in 300 µL of PBS. Then, 3 µL of propidium iodide (PI) dye solution (4 mg mL−1) was added to each sample to label dead cells. Flow cytometry (λex = 355 nm, λem = 450 nm) was then used to quantify the SN38 fluorescence within the cells. After gating sequentially for main cells, single cells, and live cells, the remaining cells were analyzed for SN38 fluorescence. Cell count and median fluorescence intensity were used as two detection indicators for assessing the degree of cellular uptake.In Fig. 5A, it is evident that the fluorescence signal of cells treated with SAPDs shifted compared to the control group, indicating that the SAPDs were internalized successfully to various extents. According to the results of SN38 fluorescence-positive cell counting, the cellular internalization rates of SAPDs 1–4 were 39.0%, 3.68%, 51.5%, and 15.3%, respectively (Fig. S18–S23 in ESI†). Moreover, median fluorescence intensity also showed significant differences in the uptake of SAPDs by cells based on their morphology and surface charge (Fig. 5B). Specifically, CT26 cells treated with long nanofibers SAPD1, SAPD3, and SAPD4 exhibited 2–4 times higher fluorescence compared to those treated with short nanofibers SAPD2. We speculate that the spindle-shaped structures formed by SAPDs are too large in all dimensions, which might hinder the cellular uptake.44 Furthermore, SAPD3 with positive surface charges, exhibits significantly higher cellular internalization compared to SAPD4 with negative surface charges. This observation is consistent with previous findings that cationic nanostructures often exhibit higher levels of cellular uptake than the anionic ones.45 The difference in cellular uptake may be explained by the electrostatic interaction between the positively charged cations on the nanostructure surface and the negatively charged anions on the cell membrane surface. Our results reveal that both the size and surface charge of SN38 prodrug assemblies affect cellular uptake, which can provide essential guidance for future prodrug design.
 | ||
| Fig. 5 Effect of charge and shape of SAPDs nanostructures on the cellular uptake efficiency by CT26 cancer cells characterized using flow cytometry. Flow cytometry spectra comparing fluorescence intensity (A), and fluorescence intensity measurement (subtract control) (B). | ||
Drug release
To exert a therapeutic effect, the parent drug SN38 needs to be released from the SAPDs. We next explored the release behavior of our SN38 prodrugs. Given the similar chemical structure, but different supramolecular stability, OEGylated SAPD1 and SAPD2 were selected as model drugs for the study. The release properties of the SAPDs were assessed by incubating 200 µM solutions in 10 mM PBS buffer at 37 °C with or without 10 mM GSH, respectively. Both SAPDs remained stable and exhibited negligible free drug release in PBS without GSH in 24 h, suggesting that the hydrolysis of carbonate bonds is relatively slow and the protection of the degradable linker in the hydrophobic core after assembly also retards the release (Fig. 6A, C and Fig. S24, ESI†). Moreover, SAPDs were rapidly converted to the bioactive form in the presence of GSH, indicating the responsiveness of the etcSS linker to the reducing agent and demonstrating their ability for controlled release. Fig. 6B and D exhibit the representative analytical RP-HPLC traces of drug release in the GSH environment at different time points. The emergence of a single and sharp peak located at 10.2 min corresponds to the free SN38 drug, suggesting the effective release via the self-immolative etcSS linker. However, there is a difference in the degradation rate of the two SAPDs. SAPD2 released approximately 40% of free SN38 within 1 h, whereas SAPD1 took 12 h to achieve the same release ratio. Besides, SAPD2 accounted for around 10% of the total SN38-containing components in the solution after 24 hours, while SAPD1 still comprised 43%. The divergence in drug release can be attributed to the different supramolecular stability as reflected by CMC results. SAPD1, with higher supramolecular stability, has fewer free monomers in solution, reducing the contact with GSH and thus lowering the drug release rate. Our results reveal that SAPDs with the etcSS linker can be effectively released via GSH-induced degradation, and the assembly nature influences the release rate to a certain extent. | ||
| Fig. 6 Drug release of SAPDs at 200 µM in PBS with or without GSH. Drug release plots of SAPDs: SAPD1 (A) and SAPD2 (C). Comparison of HPLC traces at different time points with GSH: SAPD1 (B) and SAPD2 (D). | ||
Cytotoxicity
Given the verified effective drug release, we then assessed the in vitro cytotoxicity of SAPDs against mouse (CT26) and human (HT29) colorectal cancer cells, using the parent drug SN38 and the clinically used prodrug irinotecan as controls, at various concentrations. The SAPDs were incubated with tumor cells for 48 hours, and their efficacies were evaluated using CCK-8 assay. Fig. 7 illustrates that the IC50 values of irinotecan were approximately 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 062 nM for CT26 cancer cells and 15725 nM for HT29 cancer cells. All SAPDs exhibit significantly lower IC50 values, roughly two orders of magnitude lower than the prodrug irinotecan. This observation indicates that the fast release rate of the etcSS linker in our SN38 prodrugs significantly enhances the drug's efficacy upon contact with intracellular GSH. In addition, the IC50 values of the SAPDs were similar to those of the parent drug SN38 because of the ultra-fast self-immolative release into SN38 in the cellular environment. It should be noted that SAPD1 and SAPD2 exhibited similar cytotoxicities, even though they showed different in vitro drug release rates. The difference may be attributed to the experimental concentrations. The drug release rates were partially controlled by the supramolecular stabilities when the concentrations exceeded CMC values. In the cytotoxicity study, the concentrations of SAPDs were below their CMCs; therefore the toxicity highly depended on the degradation of prodrugs into the parent drug SN38. There were no significant differences in cytotoxicities among the SAPDs, leading to the conclusion that all SAPDs show excellent in vitro cytotoxicity, highlighting their substantial potential for anticancer treatment.
062 nM for CT26 cancer cells and 15725 nM for HT29 cancer cells. All SAPDs exhibit significantly lower IC50 values, roughly two orders of magnitude lower than the prodrug irinotecan. This observation indicates that the fast release rate of the etcSS linker in our SN38 prodrugs significantly enhances the drug's efficacy upon contact with intracellular GSH. In addition, the IC50 values of the SAPDs were similar to those of the parent drug SN38 because of the ultra-fast self-immolative release into SN38 in the cellular environment. It should be noted that SAPD1 and SAPD2 exhibited similar cytotoxicities, even though they showed different in vitro drug release rates. The difference may be attributed to the experimental concentrations. The drug release rates were partially controlled by the supramolecular stabilities when the concentrations exceeded CMC values. In the cytotoxicity study, the concentrations of SAPDs were below their CMCs; therefore the toxicity highly depended on the degradation of prodrugs into the parent drug SN38. There were no significant differences in cytotoxicities among the SAPDs, leading to the conclusion that all SAPDs show excellent in vitro cytotoxicity, highlighting their substantial potential for anticancer treatment.
 | ||
| Fig. 7 In vitro cell cytotoxicity of SAPDs against CT-26 mouse colorectal cancer cells (A) and HT-29 human colorectal cancer cells (B), with both free SN38 and irinotecan as controls (48-h incubation). | ||
Conclusions
In this study, we designed and synthesized a series of SAPDs by coupling SN38 and diverse hydrophilic segments via a self-immolative etcSS linker, and investigated the self-assembly behaviors and biological functions. Due to the aromatic interactions of SN38, these SAPDs assembled into well-defined fibrous nanostructures with strong assembly capability, exhibiting different morphologies and surface properties due to the varying hydrophilic segments. SAPD1 with OEG7 self-assembled into ordered nanofibers several micrometers in length with a regular molecular arrangement, due to the directional aromatic interaction of SN38. On further increasing the length of OEG, the assemblies of SAPD2 transformed to short fibers, which may be attributed to the loss of hydrophobic–hydrophilic balance, as well as the increasing molecular size that disrupts the regular molecular packing of SN38. Both cationic SAPD3 and anionic SAPD4 also formed supramolecular filaments with oppositely charged surfaces. The morphology and surface feature of SAPDs can further affect their uptake by cancer cells. Due to its larger diameters, SAPD2 was harder to internalize than others, while SAPD3 with positive charges exhibited the highest level of cellular internalization. Moreover, all SAPDs could achieve efficient and controlled release via GSH-induced degradation and show high in vitro toxicity in colorectal cancer cells, significantly exceeding that of the clinical drug irinotecan. Overall, our results demonstrated that concise SN38 prodrugs can utilize aromatic interactions as directional intermolecular forces to create supramolecular drug assemblies, and exhibit strong tolerance towards different hydrophilic moieties. In addition, the chemical structures of hydrophilic segments also influence their self-assembly behaviors, cellular uptake, and controlled release. We hope that this work can serve as a new strategy to optimize supramolecular drug delivery systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. T. and H. S. designed research; Z. T., K. W., W. L., W. L., and T. W. performed research; Z. T., D. Z., and H. S. analysed data; and Z. T., D. Z., and H. S. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52273136 to H. S.), the Sichuan Science and Technology Program (2023YFH0071 to H. S., 2023NSFSC0306 to D. Z.) and the Opening Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (grant no. sklpme2022-3-14 to H. S.). This work was also financially supported by the Med-X Innovation Program of Med-X Center for Materials, Sichuan University (MCM202304 to H. S.) and the Fundamental Research Funds for the Central Universities (YJ202210 to H. S.). We are also grateful to Minghua Zhang from the College of Polymer Science and Engineering at Sichuan University for his help in TEM analysis. We acknowledge Dandan Yuan from the College of Polymer Science and Engineering at Sichuan University for providing assistance with characterization experiments.Notes and references
- Z. Fralish, A. Chen, S. Khan, P. Zhou and D. Reker, The Landscape of Small-Molecule Prodrugs, Nat. Rev. Drug Discovery, 2024, 23(5), 365–380 CrossRef PubMed.
- H.-H. Han, H.-M. Wang, P. Jangili, M. Li, L. Wu, Y. Zang, A. C. Sedgwick, J. Li, X.-P. He, T. D. James and J. Seung Kim, The Design of Small-Molecule Prodrugs and Activatable Phototherapeutics for Cancer Therapy, Chem. Soc. Rev., 2023, 52(3), 879–920 RSC.
- C. Bailly, Irinotecan: 25 Years of Cancer Treatment, Pharmacol. Res., 2019, 148, 104398 CrossRef CAS PubMed.
- A. Bentley, M. Butters, S. P. Green, W. J. Learmonth, J. A. MacRae, M. C. Morland, G. O’Conno and J. Skuse, The Discovery and Process Development of a Commercial Route to the Water Soluble Prodrug, Fosfluconazole, Org. Process Res. Dev., 2002, 6(2), 109–112 CrossRef CAS.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, The Expanding Role of Prodrugs in Contemporary Drug Design and Development, Nat. Rev. Drug Discovery, 2018, 17(8), 559–587 CrossRef CAS PubMed.
- I. Tranoy-Opalinski, T. Legigan, R. Barat, J. Clarhaut, M. Thomas, B. Renoux and S. Papot, β-Glucuronidase-Responsive Prodrugs for Selective Cancer Chemotherapy: An Update, Eur. J. Med. Chem., 2014, 74, 302–313 CrossRef CAS PubMed.
- J. Stangier, K. Rathgen, H. Stähle, D. Gansser and W. Roth, The Pharmacokinetics, Pharmacodynamics and Tolerability of Dabigatran Etexilate, a New Oral Direct Thrombin Inhibitor, in Healthy Male Subjects, Br. J. Clin. Pharmacol., 2007, 64(3), 292–303 CrossRef CAS PubMed.
- Q. Liu, Q. Yang, W. Sun, P. Vogel, W. Heydorn, X.-Q. Yu, Z. Hu, W. Yu, B. Jonas, R. Pineda, V. Calderon-Gay, M. Germann, E. O’Neill, R. Brommage, E. Cullinan, K. Platt, A. Wilson, D. Powell, A. Sands, B. Zambrowicz and Z. Shi, Discovery and Characterization of Novel Tryptophan Hydroxylase Inhibitors That Selectively Inhibit Serotonin Synthesis in the Gastrointestinal Tract, J. Pharmacol. Exp. Ther., 2008, 325(1), 47–55 CrossRef CAS PubMed.
- E. L. Cyphert, J. D. Wallat, J. K. Pokorski and H. A. Von Recum, Erythromycin Modification That Improves Its Acidic Stability While Optimizing It for Local Drug Delivery, Antibiotics, 2017, 6(2), 11 CrossRef PubMed.
- L. Battistini, K. Bugatti, A. Sartori, C. Curti and F. Zanardi, RGD Peptide-Drug Conjugates as Effective Dual Targeting Platforms: Recent Advances, Eur. J. Org. Chem., 2021, 2506–2528 CrossRef CAS.
- H. Su, J. M. Koo and H. Cui, One-Component Nanomedicine, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Self-Assembling Prodrugs, Chem. Soc. Rev., 2017, 46(21), 6638–6663 RSC.
- C. Zhang, S. Jin, X. Xue, T. Zhang, Y. Jiang, P. C. Wang and X.-J. Liang, Tunable Self-Assembly of Irinotecan-Fatty Acid Prodrugs with Increased Cytotoxicity to Cancer Cells, J. Mater. Chem. B, 2016, 4(19), 3286–3291 RSC.
- Y. Zheng, X. Ying, Y. Su, X. Jin, Q. Xu and Y. Li, Kinetically-Stable Small-Molecule Prodrug Nanoassemblies for Cancer Chemotherapy, Int. J. Pharm., 2021, 597, 120369 CrossRef CAS PubMed.
- X. Wen, R. Zhang, Y. Hu, L. Wu, H. Bai, D. Song, Y. Wang, R. An, J. Weng, S. Zhang, R. Wang, L. Qiu, J. Lin, G. Gao, H. Liu, Z. Guo and D. Ye, Controlled Sequential in Situ Self-Assembly and Disassembly of a Fluorogenic Cisplatin Prodrug for Cancer Theranostics, Nat. Commun., 2023, 14(1), 800 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Shape Effects of Filaments versus Spherical Particles in Flow and Drug Delivery, Nat. Nanotechnol., 2007, 2(4), 249–255 CrossRef CAS PubMed.
- J. Andrew MacKay, M. Chen, J. R. McDaniel, W. Liu, A. J. Simnick and A. Chilkoti, Self-Assembling Chimeric Polypeptide–Doxorubicin Conjugate Nanoparticles That Abolish Tumours after a Single Injection, Nat. Mater., 2009, 8(12), 993–999 CrossRef PubMed.
- H. Su, W. Zhang, H. Wang, F. Wang and H. Cui, Paclitaxel-Promoted Supramolecular Polymerization of Peptide Conjugates, J. Am. Chem. Soc., 2019, 141(30), 11997–12004 CrossRef PubMed.
- H. Su, F. Wang, Y. Wang, A. G. Cheetham and H. Cui, Macrocyclization of a Class of Camptothecin Analogues into Tubular Supramolecular Polymers, J. Am. Chem. Soc., 2019, 141(43), 17107–17111 CrossRef PubMed.
- A. G. Cheetham, Y.-C. Ou, P. Zhang and H. Cui, Linker-Determined Drug Release Mechanism of Free Camptothecin from Self-Assembling Drug Amphiphiles, Chem. Commun., 2014, 50(45), 6039–6042 RSC.
- A. G. Cheetham, P. Zhang, Y. Lin, L. L. Lock and H. Cui, Supramolecular Nanostructures Formed by Anticancer Drug Assembly, J. Am. Chem. Soc., 2013, 135(8), 2907–2910 CrossRef PubMed.
- S. Fu, A. Zheng, L. Wang, J. Chen, B. Zhao, X. Zhang, V. A. McKenzie, Z. Yang, M. R. Leblanc, R. Prabhakar and F. Zhang, Tuneable Redox-Responsive Albumin-Hitchhiking Drug Delivery to Tumours for Cancer Treatment, J. Mater. Chem. B, 2024, 12(27), 6563–6569 RSC.
- Y. Yuan, J. Zhang, X. Qi, S. Li, G. Liu, S. Siddhanta, I. Barman, X. Song, M. T. McMahon and J. W. M. Bulte, Furin-Mediated Intracellular Self-Assembly of Olsalazine Nanoparticles for Enhanced Magnetic Resonance Imaging and Tumour Therapy, Nat. Mater., 2019, 18(12), 1376–1383 CrossRef CAS PubMed.
- X. Han, K. Cheng, Y. Xu, Y. Wang, H. Min, Y. Zhang, X. Zhao, R. Zhao, G. J. Anderson, L. Ren, G. Nie and Y. Li, Modularly Designed Peptide Nanoprodrug Augments Antitumor Immunity of PD-L1 Checkpoint Blockade by Targeting Indoleamine 2,3-Dioxygenase, J. Am. Chem. Soc., 2020, 142(5), 2490–2496 CrossRef CAS PubMed.
- Z. Xu, M. Hou, X. Shi, Y.-E. Gao, P. Xue, S. Liu and Y. Kang, Rapidly Cell-Penetrating and Reductive Milieu-Responsive Nanoaggregates Assembled from an Amphiphilic Folate-Camptothecin Prodrug for Enhanced Drug Delivery and Controlled Release, Biomater. Sci., 2017, 5(3), 444–454 RSC.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Tumor Redox Heterogeneity-Responsive Prodrug Nanocapsules for Cancer Chemotherapy, Adv. Mater., 2013, 25(27), 3670–3676 CrossRef CAS PubMed.
- Q. Mou, Y. Ma, X. Zhu and D. Yan, A Small Molecule Nanodrug Consisting of Amphiphilic Targeting Ligand–Chemotherapy Drug Conjugate for Targeted Cancer Therapy, J. Controlled Release, 2016, 230, 34–44 CrossRef CAS PubMed.
- S. He, C. Li, Q. Zhang, J. Ding, X.-J. Liang, X. Chen, H. Xiao, X. Chen, D. Zhou and Y. Huang, Tailoring Platinum(IV) Amphiphiles for Self-Targeting All-in-One Assemblies as Precise Multimodal Theranostic Nanomedicine, ACS Nano, 2018, 12(7), 7272–7281 CrossRef CAS PubMed.
- R. Lin, A. G. Cheetham, P. Zhang, Y. Lin and H. Cui, Supramolecular Filaments Containing a Fixed 41% Paclitaxel Loading, Chem. Commun., 2013, 49(43), 4968–4970 RSC.
- Z. Xu, X. Shi, M. Hou, P. Xue, Y.-E. Gao, S. Liu and Y. Kang, Disassembly of Amphiphilic Small Molecular Prodrug with Fluorescence Switch Induced by pH and Folic Acid Receptors for Targeted Delivery and Controlled Release, Colloids Surf., B, 2017, 150, 50–58 CrossRef CAS PubMed.
- L. Lu, B. Li, C. Lin, K. Li, G. Liu, Z. Xia, Z. Luo and K. Cai, Redox-Responsive Amphiphilic Camptothecin Prodrug Nanoparticles for Targeted Liver Tumor Therapy, J. Mater. Chem. B, 2020, 8(17), 3918–3928 RSC.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, Prodrugs Forming High Drug Loading Multifunctional Nanocapsules for Intracellular Cancer Drug Delivery, J. Am. Chem. Soc., 2010, 132(12), 4259–4265 CrossRef CAS PubMed.
- J. Wang, S. Hu, W. Mao, J. Xiang, Z. Zhou, X. Liu, J. Tang and Y. Shen, Assemblies of Peptide-Cytotoxin Conjugates for Tumor-Homing Chemotherapy, Adv. Funct. Mater., 2019, 29(7), 1807446 CrossRef.
- Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, Supramolecular “Trojan Horse” for Nuclear Delivery of Dual Anticancer Drugs, J. Am. Chem. Soc., 2017, 139(8), 2876–2879 CrossRef CAS PubMed.
- Y. Cai, B. Zhu, X. Shan, L. Zhou, X. Sun, A. Xia, B. Wu, Y. Yu, H. H. Zhu, P. Zhang and Y. Li, Inhibiting Endothelial Cell-Mediated T Lymphocyte Apoptosis with Integrin-Targeting Peptide-Drug Conjugate Filaments for Chemoimmunotherapy of Triple-Negative Breast Cancer, Adv. Mater., 2024, 36(3), 2306676 CrossRef CAS PubMed.
- H. Wang, H. Su, T. Xu and H. Cui, Utilizing the Hofmeister Effect to Induce Hydrogelation of Nonionic Supramolecular Polymers into a Therapeutic Depot, Angew. Chem., Int. Ed., 2023, 62(43), e202306652 CrossRef CAS PubMed.
- R. H. J. Mathijssen, R. J. van Alphen, J. Verweij, W. J. Loos, K. Nooter, G. Stoter and A. Sparreboom, Clinical Pharmacokinetics and Metabolism of Irinotecan (CPT-11), Clin. Cancer Res., 2001, 7(8), 2182–2194 CAS.
- Z. Tang, J. Zhang, W. Li, K. Wen, Z. Gu, D. Zhou and H. Su, Supramolecular Assembly of Isomeric SN-38 Prodrugs Regulated by Conjugation Sites, J. Mater. Chem. B, 2024, 12, 6146–6154 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, Self-Cross-Linked Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform, J. Am. Chem. Soc., 2010, 132(48), 17227–17235 CrossRef CAS PubMed.
- H. Su, F. Wang, H. Wang, W. Zhang, C. F. Anderson and H. Cui, Propagation-Instigated Self-Limiting Polymerization of Multiarmed Amphiphiles into Finite Supramolecular Polymers, J. Am. Chem. Soc., 2021, 143(44), 18446–18453 CrossRef CAS PubMed.
- L. Degrand, R. Garcia, K. Crouvisier Urion and W. Guiga, Dynamic Light Scattering for the Determination of Linoleic Acid Critical Micelle Concentration. Effect of pH, Ionic Strength, and Ethanol, J. Mol. Liq., 2023, 388, 122670 CrossRef CAS.
- J. R. Wester, J. A. Lewis, R. Freeman, H. Sai, L. C. Palmer, S. E. Henrich and S. I. Stupp, Supramolecular Exchange among Assemblies of Opposite Charge Leads to Hierarchical Structures, J. Am. Chem. Soc., 2020, 142(28), 12216–12225 CrossRef CAS PubMed.
- A. C. French, A. L. Thompson and B. G. Davis, High-Purity Discrete PEG-Oligomer Crystals Allow Structural Insight, Angew. Chem., Int. Ed., 2009, 48(7), 1248–1252 CrossRef CAS PubMed.
- M. Ueda, S. Seo, B. G. Nair, S. Müller, E. Takahashi, T. Arai, T. Iyoda, S. Fujii, S. Tsuneda and Y. Ito, End-Sealed High Aspect Ratio Hollow Nanotubes Encapsulating an Anticancer Drug: Torpedo-Shaped Peptidic Nanocapsules, ACS Nano, 2019, 13(1), 305–312 CrossRef CAS PubMed.
- L. Chen, J. M. Mccrate, J. C.-M. Lee and H. Li, The Role of Surface Charge on the Uptake and Biocompatibility of Hydroxyapatite Nanoparticles with Osteoblast Cells, Nanotechnology, 2011, 22(10), 105708 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01429d |
| This journal is © The Royal Society of Chemistry 2024 |