
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring co-crystallisation as a technique for taste-masking of nevirapine†
Yichun
Shen
 a,
Marique
Aucamp
b,
Hend E.
Abdelhakim
c,
Xiunan
Li
d,
Yusuf
Ghazali
b and
Katharina
Edkins
*ae
a,
Marique
Aucamp
b,
Hend E.
Abdelhakim
c,
Xiunan
Li
d,
Yusuf
Ghazali
b and
Katharina
Edkins
*ae
aSchool of Health Sciences, University of Manchester, Stopford Building, Oxford Road, Manchester M13 9PT, UK
bSchool of Pharmacy, Faculty of Natural Sciences, University of the Western Cape, Bellville 7535, South Africa
cGlobal Business School for Health, University College London (UCL), 7 Sidings Street, London, E20 2AE, UK
dDepartment of Pharmaceutics, UCL School of Pharmacy, 29-39 Brunswick Square, London WC1N 1AX, UK
eInstitute of Pharmacy and Biomedical Sciences, Strathclyde University, 161 Cathedral Street, Glasgow G4 0RE, UK. E-mail: katharina.edkins@strath.ac.uk
First published on 26th February 2024
Abstract
The bitter taste of pharmaceuticals majorly impacts patient adherence. Co-crystallisation has been proposed as a novel way for taste masking using sweetener-based co-formers, while other co-formers also have a positive effect. We hypothesise that the sweetness of the co-formers is not the key factor but rather the molecular aggregation between the drug and co-former in solution, i.e., the stronger the interaction, the better the taste masking effect. Here, we explore the solution aggregation between the bitter-tasting drug nevirapine and five co-formers by 1H NMR spectroscopy. The co-formers benzoic acid, salicylic acid and maleic acid show strong interaction with nevirapine, while glutaric acid and saccharin have weak and no interaction, respectively. The taste of the resulting co-crystal, as assessed by the electrical taste sensing system e-tongue, reveals that the bitterness of nevirapine has been covered with the co-crystal benzoic acid, maleic acid and glutaric acid but not saccharin or salicylic acid. From the taste results we deduct that both solution aggregation and the taste of the pure co-former play an important role in taste masking. It is likely that a large variety of co-formers can be used to cover bitter drugs and we show that the investigation of molecular aggregation in solution can help screen the co-formers before any in vitro or in vivo taste test.
1. Introduction
Taste plays a significant role in the development of pharmaceutical formulations, especially for paediatric and geriatric patients, as it directly affects patient compliance and adherence.1 Humans perceive five primary tastes, namely sour, sweet, umami, bitter and salty, among which bitterness is the most problematic for patient adherence2 and hence pharmaceutical formulation.3 The poor adherence may cause drug resistance, deliberate forgetfulness and treatment failure.4 The bitter taste is detected by receptors on the taste buds that have evolved to recognise a wide range of structurally diverse compounds, a necessity in order to identify numerous potentially harmful substances.5 Therefore, it can be challenging to convince children to take bitter drugs. It is thus important to improve bitter-tasting drugs using innovative techniques to increase the palatability of dosage forms.Various taste masking techniques have been used to cover the bitter taste of drugs. The conventional method involves adding sweeteners and flavouring agents, however, they are not necessarily effective at masking strong bitterness and an unpleasant aftertaste could persist due to the short duration of the sweetener taste.6 The barrier forming strategy is another common approach, encompassing coating, emulsion formation7 and molecular encapsulation with cyclodextrins,3 to prevent the drug's contact with the taste buds and hence reduce the unpleasant taste. A novel and promising way for taste masking is co-crystallisation, in which a co-former interacts with the drug molecules forming single phase crystalline structures through intermolecular interactions.8 Co-crystallisation has been used to improve many drug properties such as solubility and dissolution rate,9 and the use of sugar-based co-formers is considered an effective approach for taste masking10 together with the improvement of other physiochemical properties. Maeno et al.11 reported a new co-crystal of paracetamol with trimethylglycine (TMG) with an improved compressibility, dissolution rate and a sweeter taste compared with pure paracetamol. Similarly, saccharin was used to increase the sweetness of theophylline by forming a co-crystal, improving stability and ensuring sustained drug release.12 However, the researchers merely tested the sweetness of the co-crystal, while an increase in sweetness does not necessarily indicate effective coverage of bitterness but rather that the taste is a mix of sweet and bitter, which might taste even worse. Besides, Ogata et al. found that some acids, which are not sweeteners, can also have a positive effect on taste masking of the bitter drug propiverine.13 Hence, we hypothesise a new mechanism of taste-masking through co-crystallisation: the molecular aggregation of drug and co-former dissolved in saliva is different to that of pure drugs, and human taste receptors fail to detect the original taste of the bitter pure drugs in the co-crystal systems. Therefore, some co-formers, despite lacking sweetness, can effectively mask the bitter taste of drugs based on their ability to form small transient clusters with drug molecules upon dissolution in the mouth. On the other hand, not all sweet or umami co-formers will improve the taste of bitter drugs due to the lack of this clustering in solution. Thus, it is important to investigate the interaction and assembly of the drug and co-formers in solution state and build a systematic way to guide the subsequent taste masking tests.
The main approaches to the investigation of solution aggregation include various spectroscopic methods such as Fourier transform infrared (FTIR),14 ultraviolet-visible (UV-vis)15 and nuclear magnetic resonance (NMR) spectroscopy.16–18 Beside these, neutron total scattering19–22 and molecular dynamics simulations23 are also applied. NMR spectroscopy is one of the most widely used quantitative analytical methods. The changes in chemical shift of one compound (e.g. the drug) caused by the change in its concentration or by addition of a second compound (e.g. the co-former) can be used to study the aggregation of complexes in the solution state.24 Since in pharmaceutical compounds hydrogen bonds are of particular interest, 1H is the most important nucleus to study.
The taste masking effect can be assessed through two main methods: in vivo and in vitro analysis. The in vivo techniques include human taste panels and animal models. Human perception is considered the most widely used and reliable method for taste assessment.25 However, the limitation of the method is due to the difference in tasting between individuals and the translatability of adult volunteers’ assessment to children's application. Animal models such as the rodent Brief-Access Taste Aversion (BATA) is currently developed as a useful tool,26 while further validation work is still needed.3 The in vitro methods including dissolution rate test and the electrical taste sensing system (e-tongue) are easier, cheaper and more convenient than in vivo techniques. The dissolution rate testing shows the amount of drug released into solution with time as a crude indication of the amount of bitter taste exposure to the receptors.27 The e-tongue consists of sensors with various lipid membranes to measure the electric potential difference, representing the taste quality.28,29 The system, despite its inability to perfectly duplicate the mouth environment, possesses the capability to provide an unbiased and secure approach for evaluating taste-masking effects and is still continuously undergoing improvements.25
The antiretroviral drug nevirapine (NVP, Scheme 1), used for the treatment of HIV-1 infection and AIDS is used in this study due to the high HIV infection rate in babies and children especially in sub-Saharan Africa and the strongly unpalatable and bitter taste of the drug.30 Research has revealed that NVP has the propensity to form co-crystals with numerous co-formers.31,32 Five different co-formers (Scheme 2) are selected from the list of compounds which are known to form co-crystals with NVP. Among them, benzoic acid (BA) and salicylic acid (SA) are aromatic acids, glutaric acid (GTA) and maleic acid (MLA) are aliphatic acids, and saccharin (SAC) is widely used as a sweetener. In addition to the variation in chemical structure, the stoichiometric amount between NVP and the co-formers varies: the NVP–BA, NVP–GTA and NVP–MLA co-crystals form in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 while for NVP–SAC and NVP–SA the ratio is 2:1. In this study we aim to systematically investigate the taste masking effect of NVP co-crystals by employing NMR spectroscopy to explore solution aggregation, while assessing the taste through measurements of dissolution rate and conducting an e-tongue test.
:1 while for NVP–SAC and NVP–SA the ratio is 2:1. In this study we aim to systematically investigate the taste masking effect of NVP co-crystals by employing NMR spectroscopy to explore solution aggregation, while assessing the taste through measurements of dissolution rate and conducting an e-tongue test.
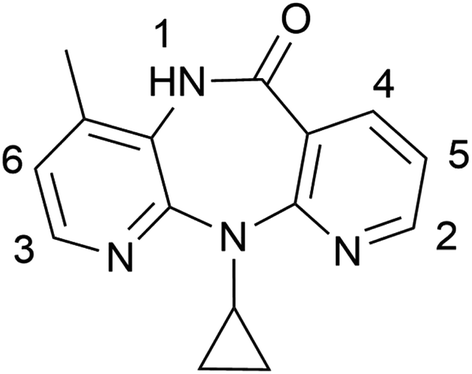 | ||
| Scheme 1 Chemical structure of nevirapine with protons labelled. | ||
 | ||
| Scheme 2 Chemical structures of co-formers (a) benzoic acid, (b) saccharin with protons labelled from upfield to downfield, (c) salicylic acid, (d) glutaric acid and (e) cis maleic acid. | ||
2. Materials and methods
2.1 Materials
NVP and MLA were bought from Fluorochem Ltd (UK), SAC and GTA from Acros Organics (UK), SA from Honeywell Fluka Chemicals (UK), acetone-d6 from Apollo Scientific (UK) and BA, tartaric acid acetone, ethanol, potassium chloride (KCl), and tetramethylsilane (TMS), were obtained from Sigma-Aldrich (UK).2.2 Co-crystallisation
:1 for BA, GTA and MLA and 2:1 for SAC, SA) was added to acetone. The slurry was stirred at room temperature in a sealed vial using a magnetic stirrer bar in the fume hood to equilibrate the system and produce the crystal form through solvent mediated transformation.
:1 for BA, GTA and MLA and 2:1 for SAC and SA) was ground in a MM 400 mixer mill (Retsch, Haan, Germany) in stainless steel grinding jars (V = 25 mL) with one stainless steel ball (2r = 1 cm) and the addition of 50 μL acetone to facilitate co-crystal formation. The grinding experiment was carried out at room temperature at a frequency of 30 Hz for 20 min.
2.3 NMR host–guest titration
1H NMR spectra were recorded on an Avance-III HD 500 spectrometer (Bruker, Karlsruhe, Germany) and calibrated to TMS (0 ppm) as internal standard. The data were analysed using MestReNova (Mnova) software (Mestrelab Research, A Coruña, Spain). A change in chemical shift (Δδ) in all 1H NMR experiments is regarded as significant if the peak shift value is larger than the instrument resolution of 0.002 ppm.For the sample preparation, the host (NVP) stock solution was prepared at a concentration of 0.02 M in acetone-d6. To prepare the guest stock solution, the weighted guest solid (co-former) was added into the host stock solution to ensure the host concentration did not change during titration. The largest guest molar ratio was based on the solubility of the co-former in the deuterated solvent. The molar ratios between 0 and maximum were obtained by mixing different amounts of host and guest stock solution. The apparent binding constant (K) was calculated by BindFit33 and three binding models were employed for fitting, encompassing 1:1, 1:2, and 2:1 host:guest ratios. The best fitting binding model was chosen based on lowest error and chemical logic.18,34,35 The apparent Gibbs free binding energy (ΔGbind) can be calculated by eqn (1):
| ΔG = −RTlnK | (1) |
2.4 NMR dilution
Dilution experiments were conducted on the same NMR spectrometer as described above. For the sample preparation, the concentration of pure NVP stock solution was the solubility limit of the NVP in acetone-d6 with the concentration of 0.024 M. The solution was diluted with acetone-d6 to the point where the proton peaks were no longer detectable in the NMR spectra. The endpoint concentration was 0.001 M. For SAC dilution, the initial stock concentration was 0.1 M in acetone-d6 and the endpoint 0.001 M.2.5 Dissolution rate measurement
In vitro dissolution testing of NVP and the five prepared co-crystals was performed using a Hanson Research SRII 6-flask dissolution tester (California, USA). The USP II paddle set up was used with a rotation speed of 100 rpm at 37.0 ± 0.5 °C.Each dissolution vessel (n = 6) contained 900 mL of phosphate buffer at pH 6.8 prepared according to the British Pharmacopoeia (BP, 2022).36 Powder of the pure drug and co-crystals equivalent to 200 mg NVP was weighed into suitable size test tubes (n = 6) and approximately 400 mg glass beads (r ≥ 100 μm, Merck, Johannesburg, South Africa) were added to each test tube. An aliquot of pre-warmed dissolution medium was added to each test tube, vortexed for 20 seconds and immediately transferred into the corresponding dissolution vessel followed by careful rinsing of the test tube.
The NVP dissolution rate was tested over a period of 60 minutes and sample withdrawals (5 mL) were done at 5, 10, 20, 40 and 60 min. The samples were filtered with 0.22 μm nylon syringe filters into HPLC vials for subsequent HPLC analysis. The dissolution medium was replaced after each withdrawal to maintain sink conditions.
An Azura (Berlin, Germany) HPLC system equipped with a photodiode array detector, column oven and autosampler was used for HPLC analysis. A Phenomenex Kinetex C18 150 mm × 4.6 mm 5 μm column (Torrance, USA) was used as stationary phase. The mobile phase consisted of chromatography grade acetonitrile and ultrapure water in a ratio of 60:40 v/v. The mobile phase flow rate was set as 1.0 mL min−1 at a temperature of 30 °C and a detection wavelength of 282 nm was used. The injection volume was 10 μL for all samples.
2.6 E-tongue taste assessment
The taste of NVP and its five co-crystals was measured using a TS-5000Z (Insent Inc., Atsugi-shi, Japan) equipped with five lipid membrane sensors and two reference electrodes (New Food Innovation Ltd, Loughborough, UK). Positively charged sensors include C00 (acidic bitterness) and AE1 (astringency). Negatively charged sensors include AC0, AN0 and BT0, all responding to basic bitterness at different selectivity and sensitivity levels. A significant limitation of the e-tongue is its restricted ability to detect drugs that cannot be ionized within the measurement medium.37 Since NVP is a weak base,38 a series of NVP solutions with different concentrations were tested with several of the basic and acidic bitterness sensors and the astringency sensor, to assess whether NVP's bitter taste is detectable. If the result is ≥±5 mV, the drug shows the respective taste quality and is considered as detectable for the sensor.39 In our system, NVP shows strong initial taste and aftertaste quality of acidic bitterness with the sensor C00 (Fig. 1) at the concentration from 0.1 mM to 1 mM, while the response of the remaining four sensors is not significant (ESI Fig. S1†). Therefore, C00 sensor with the NVP concentration of 0.1 mM was used as a baseline in the taste assessment of the co-crystals and co-formers. | ||
| Fig. 1 Dose–response curve representing initial taste and aftertaste (CPA) for NVP co as detected by C00 sensor. | ||
A series of NVP solutions with the concentration 0.01, 0.03, 0.06, 0.1, 0.3, 0.6 and 1 mM were prepared in the liquid base of 10 mM KCl in 10% ethanol, which was also used as a blank control. The reference solution (30 mM KCl and 0.3 mM tartaric acid in water) was used in all reference slots. The measurement cycle started with measuring the reference potential (Vr) of the reference solution, followed by the sample potential (Vs) resulting in Vs–Vr being the initial taste. Subsequently, the sensors were washed three times with reference solution and the reference potential (Vr1) was measured again. Vs–Vr1 represented the aftertaste or Change of membrane Potential caused by Adsorption (CPA) of the drug. Between the two sample measurements, the sensors were refreshed in ethanol for 330 s. If the initial taste of pure NVP at a specific concentration showed a larger potential, this concentration was selected for the five co-crystals. Each sample was measured three times and statistical significance was taken at p-value < 0.05. The data was plotted using OriginPro 2019b (OriginLab Corporation, Northampton, MA, USA).
3. Results and discussion
The five NVP co-crystals are easily obtained from both neat grinding (ESI Fig. S2†) and acetone slurry (ESI Fig. S3†), shown in the experimentally obtained PXRD patterns in comparison with those from literature.31,323.1 NMR titration
To investigate the strength of interaction between NVP and co-formers, 1H NMR titration using NVP as host and co-formers as guest were performed in acetone-d6 (Fig. 2 and Table 1). We chose acetone as solvent due to the poor aqueous solubility of NVP and the co-formers BA, SAC and SA. Our discussion focuses on protons 1–6 since the remaining protons in NVP show no significant shift. In addition, NVP protons 3 and 4 in the NVP–SAC titration overlap with SAC protons and are thus omitted. | ||
| Fig. 2 1H NMR proton positions of NVP proton 1 with different co-formers as function of co-former concentration in acetone-d6. | ||
| Co-crystal | Proton 1 | Proton 2 | Proton 3 | Proton 4 | Proton 5 | Proton 6 |
|---|---|---|---|---|---|---|
| NVP–BA | 0.509 | 0.066 | 0.082 | 0.074 | 0.041 | 0.036 |
| NVP–SAC | 0.050 | 0.008 | — | — | 0.006 | 0.007 |
| NVP–SA | 0.459 | 0.12 | 0.161 | 0.134 | 0.087 | 0.093 |
| NVP–GTA | 0.916 | 0.095 | 0.12 | 0.07 | 0.064 | 0.069 |
| NVP–MLA | 0.759 | 0.192 | 0.307 | 0.29 | 0.305 | 0.527 |
| NVP | 0.043 | 0.002 | 0.002 | 0.002 | 0.0004 | −0.0005 |
The shift of proton 1 is much larger than the rest of the protons in all experiments, showing the environment of the NH proton changes most with the addition of any of the five co-formers. When compared to the supramolecular interaction in the solid-state co-crystal structures, this result is not surprising: NVP–GTA and NVP–MLA co-crystals form a hydrogen bond through the cyclic amide (NVP)–carboxylic acid (GTA/MLA) synthon.31 It is thus likely that a similar hydrogen bond interaction with the respective co-formers exists in solution. The large shift of protons 2–6 in NVP–MLA may be the formation of additional interactions on NVP pyridine rings.32 In the co-crystal structure of the aromatic co-formers (BA/SAC/SA) with NVP, NVP forms a homodimer through the amide–amide synthon and the hydrogen bonded dimer tethers the co-former with the hydrogen-bond interaction forming between the NVP nitrogen near proton 3 and the carboxylic hydrogen in BA/SA, or the sulfimide hydrogen in SAC.31 Proton 3 in the titration of NVP–BA and NVP–SA shows the largest shift compared with the rest of the aromatic protons indicating that proton 3 is affected most among all aromatic protons by the co-formers’ addition in solution, which coincides with the interactions in the solid structure. However, proton 1 still shows the largest shift among all protons, indicating the possibility of heterodimer formation between NVP and the three aromatic co-formers. There is another possibility that the NVP homodimer interaction changes, either getting stronger or weaker, with the addition of co-formers. Therefore, the NMR dilution of pure NVP was performed in acetone-d6 to explore the strength of the self-aggregation (Table 1). The largest peak shift is still observed in proton 1, with the shift of 0.043 ppm being much lower than the shift in the titration, suggesting the homomeric interaction is not preferred in solution. It indicates that it is unlikely that the large shift of proton 1 in the titrations is caused by changes in the NVP homodimer interaction. Noticeably, the protons in NVP–SAC titration show the smallest shift compared to the other co-formers, suggesting that the chemical environment of the protons changes little with the addition of SAC and the interaction between the two compounds is small in solution.
To obtain the apparent binding constant of these solution interactions, multiple binding models with different stoichiometries were fitted to the shift of proton 1, and the model with the lowest error and meeting chemical logic was chosen as best fit (Table 2). These fitted parameters represent only apparent binding constants (K) and apparent Gibbs free binding energy (ΔGbind), since it is unlikely that only one specific stoichiometry of complexes exists in solution.34,35 For BA, SA, GTA and MLA, a 2:1 model fits the data best with large K21 and ΔGbind, indicating a strong heteromeric interaction between NVP and co-formers. The result is supported by the 1H NMR dilution experiment of NVP in acetone-d6 (Table 2). The Kdimer and ΔGdimer of the NH proton are 3.41 ± 0.03 M−1 and −2.99 ± 0.02 kJ mol−1, respectively, for the pure compound but the values are smaller than those in the heteromeric binding constants and energy with the four co-formers. The result indicates that NVP is less likely to interact with itself in solution and thus proves that the strong binding interaction through proton 1 is not due to any changes in NVP homodimer but to the heteromeric interaction with the co-formers.
| Co-crystal | [H]:[G] binding model |
K 11 (M−1) | K 21 (M−1) | ΔGbind (kJ mol−1) |
|---|---|---|---|---|
| NVP–BA | 2:1 |
(4.97 ± 0.32) × 10−3 | 1462 ± 26 | −18.1 ± 0.1 |
| NVP–SAC | 1:1 |
0.14 ± 0.002 | — | 4.87 |
| NVP–SA | 2:1 |
(1.43 ± 0.04) × 10−2 | 181.2 ± 2.1 | −12.9 ± 0.1 |
| NVP–GTA | 2:1 |
0.81 ± 0.05 | 7.66 ± 0.89 | −5.05 ± 0.4 |
| NVP–MLA | 2:1 |
2.41 ± 0.14 | 36.96 ± 3.14 | −8.95 ± 0.2 |
| NVP | Dimer aggregation | 3.41 ± 0.03 | — | −2.99 ± 0.02 |
The shift of proton 1 for NVP–SAC is less significant and can only be fitted with a 1:1 model with an extremely small K11 and a positive ΔGbind, showing the formation of NVP–SAC dimers is not favored in the solution phase. It is assumed that there are competing stronger interactions which inhibit direct interaction of NVP and SAC. To gauge this, the dilution experiment of pure SAC has been performed in acetone-d6 (ESI Fig. S4†). Here, proton 9 overlaps with proton 8 so it is not tracked in the experiment (ESI Fig. S5†). The other three SAC protons shift strongly with an apparent dimerisation constant Kdimer = 731 ± 14 M−1 and apparent dimerisation energy ΔGdimer = −16.3 ± 0.1 kJ mol−1. This indicates strong self aggregation of SAC in acetone-d6, hindering aggregation with NVP and causing weak binding parameters in the titration experiment. Since the NVP–SAC co-crystal is easily obtained from solution, it is hypothesised that the SAC dimer must be broken during the co-crystal formation.
According to the result from 1H NMR titration of NVP with different co-formers, BA, SA and MLA could be used as candidate co-formers for taste masking of NVP in the follow-up tests based on the strong heteromeric interaction in solution. For GTA, although the value of ΔGbind in titration is lower than that in NVP dilution, the small difference shows it is possible that part of NVP forms the homodimer rather than interact with GTA, which could influence the effect of taste masking. Therefore, GTA could be regarded as a sub-optimal co-former to improve the bitter taste of NVP. For the NVP–SAC co-crystal, the two compounds are more likely to form homodimers because of the weak interactions between NVP and SAC in solution. Thus, SAC might not be a favorable co-former for taste masking. However, its salts are commonly used sweeteners in daily life40 and in taste masking of ibuprofen syrup and caffeine.41 But since taste masking is more than simply adding sweetness to counteract bitter taste and needs to reduce the interaction between the bitter compound and the taste buds, saccharin as sweetener might not be effective for strongly bitter drugs like NVP.
3.2 Dissolution rate
Since hindering the release of the bitter drug in the oral cavity is useful for taste masking,11 an in vitro dissolution study of NVP and its five co-crystals was conducted at pH 6.8, similar to the pH in saliva (Fig. 3). It is evident that all NVP co-crystals enhance the dissolution rate of pure NVP, and NVP–MLA co-crystal shows the fastest dissolution of all materials. The five co-crystals release between 22–50% of NVP in the first 5 min, which is 2–5 times more than the 10% release of pure NVP. Most of the release rates level off during the remaining 55 minutes. After 60 min, the order of dissolution rate enhancement from high to low is NVP–MLA, NVP–BA, NVP–GTA, NVP–SAC and NVP–SA. Noticeably, NVP–BA dissolves fast within 5 min and exceeds the amount of dissolved NVP in NVP–GTA co-crystal after 20 min without plateauing in 60 min. | ||
| Fig. 3 Dissolution profiles for NVP and its five co-crystals in aqueous solution (pH = 6.8). | ||
The rapid dissolution rate of the co-crystals compared with pure NVP might be due to the presence of highly water-soluble co-formers such as GTA and MLA, which are released from the crystal surface at a faster rate. This process results in greater exposure of NVP molecules to water, increasing their ability to leave the crystal lattice and resulting in a higher apparent solubility of NVP. This has also been described as the “spring and parachute” effect.42 The higher dissolution rate of the co-crystals, and especially the ones with co-formers showing lower water solubility than NVP, may also be caused by the higher co-crystal lattice energy due to less efficient packing compared to the pure NVP.43 From this data it is clear that any taste-masking effect of the co-crystals is not due to the reduction in drug dissolution rate, but it is possible that taste-masking occurs via the supramolecular aggregation of NVP and co-formers in solution.
3.3 E-tongue taste-assessment
To evaluate the taste and acceptability of the co-crystals compared with pure NVP, we assess their taste with the e-tongue. The test of the pure co-formers was performed in addition due to their potential effects on the taste of the co-crystals.The initial taste and aftertaste of the co-crystals and co-formers from the C00 sensor are shown in Fig. 4. Due to the acidic bitterness gauged by the C00 sensor, a bitterer compound gives a more negative response, and conversely a positive response means no bitterness.44 For the initial taste, the pure drug NVP gives the most negative response, indicating the highest bitterness compared with the co-crystals. The result shows the bitter initial taste of NVP can be fully masked with the co-formers BA, GTA and MLA (p < 0.001). Furthermore, the bitter taste can be reduced with the co-former SAC (p = 0.010) but not with SA (p = 0.743). The weaker taste masking exhibited by SAC is likely due to its large negative response and bitterness. Even though SAC is commonly used as a sweetener with sweet taste, it has been found that it shows a bitter, metallic and drying taste when the compound is highly purified.45 The unpleasant taste of SAC limits its use as a sweetener. Even though the NMR titration shows strong interaction between NVP and SA, the bitter taste of SA is dominant in the taste of the co-crystal, which is hence not usable for taste masking. For the co-formers showing less bitterness, such as MLA and BA, the bitterness of NVP could be fully masked. Moreover, the detected response of the co-crystals is not the simple addition of the responses of NVP and the co-former, corroborating the NMR titration result that there is NVP/co-former aggregation in solution contributing to the taste masking. For GTA, although showing only moderate interaction with NVP in solution in the NMR titration experiment, the less bitter taste of the co-former takes over in the co-crystal solution, causing it to be the strongest taste masking among all co-formers.
 | ||
| Fig. 4 Initial taste and aftertaste (CPA) response of NVP, the five co-formers and the five co-crystals detected by the C00 sensor. | ||
The aftertaste of the NVP–SAC and NVP–SA co-crystals is observed slightly stronger than in the pure drug (Fig. 4). It has been reported that SAC has an intrinsic bitter aftertaste;46 therefore, the strong aftertaste in the NVP–SAC is probably due to the co-former. While there is no research on the aftertaste of SA, it is likely that the detected aftertaste is due to the poor solubility of SA in water and its incomplete removal during the washing regime. For the aftertaste of BA, GTA, MLA and their co-crystals, the responses are around −5 mV, which is borderline insignificant but similar to that of the pure drug (p = 0.945, 0.785 and 0.094), showing the bitter aftertaste changes little.
Given that all the co-formers are weak acids, they could protonate NVP in aqueous solution, hence influencing the readings of the e-tongue due to the ionisation state of the drug. The acids rank in terms of pKa in water as follows: saccharin (1.8)47 < maleic acid (1.9)48 < salicylic acid (3)49 < benzoic acid (4.2)50 < glutaric acid (4.3),51 and since the concentration of all co-formers used is identical, their pKa values correspond to the pH and ionisation conditions in the solution.52 If the taste assessment was impacted by the proton transfer, one would expect a decrease in bitterness with increasing pKa of the co-former. However, the initial taste values of the co-formers and co-crystals do not align with the same ranking. Despite maleic acid and saccharin having smaller pKa values than salicylic acid, the latter exhibits the largest initial bitter taste response. Furthermore, none of the NVP readings in the presence of the co-formers is more bitter than the pure drug solution, which would be a further indication that ionisation impacts on the taste assessment. Hence, the results suggest that the e-tongue readings are not dominated by the ionisation of NVP.
Overall, BA and MLA show solution aggregation in NMR and detectable taste-masking results in the e-tongue test and are hence good candidates in the subsequent clinical tests. GTA, as a third alternative, has the best taste-masking effect in the taste test due to its low bitterness, even though the interaction in solution is not strong. Neither SAC nor SA seems to be suitable co-formers for taste masking, as they both show strong bitterness signal, with SAC exhibiting weak interaction with the drug. Even though e-tongue measurements have been widely used as an in vitro method in the assessment of the taste of drugs, it is merely a sensing system containing sensors to mirror the taste buds, unable to precisely replicate the conditions of the oral cavity. Therefore, further in vivo tests are still necessary to check whether the good co-former candidates for NVP work on real taste buds.
From our results, the two aliphatic acids MLA and GTA are effective in taste masking, but the aromatic acids have limited effect on the taste masking result. For the different stoichiometric ratios, the co-crystals with 1:1 ratio (BA/MLA/GTA) seems more effective than the ones in a 2:1 ratio. These are very preliminary results of a very small sample set, so it is important to conduct more experiments using different co-formers and varying stoichiometric ratios to further investigate this effect.
The results indicate the hypothesis that sweeteners as co-formers, such as SAC, are not always good for taste masking and some co-formers without sweetness can help cover the bitter taste. On the contrary, the strong interaction between the drug and co-former in solution is necessary for masking the unpalatable taste, which is one mode of action of how co-crystals can be used in taste-masking. Besides, the taste of co-formers is equally important since the taste-masking effect may be diminished by the bitter taste of a co-former.
4. Conclusion
The co-crystallisation of NVP has shown potential to mask the bitter taste of pure NVP. Interactions in solution were found between NVP and the co-formers BA, SA, MLA and GTA ranging from strong to weak, as demonstrated using 1H NMR spectroscopy, indicating the formation of small clusters. The e-tongue test shows the bitter taste of NVP is fully masked in the presence of the co-formers BA and MLA, coinciding with the result from NMR titration. The NVP–SA co-crystal has the bitterest taste among all five co-crystals, which may be due to the strong bitterness of the co-former. Although the interaction between NVP and the co-former GTA is not strong, this co-crystal shows the best taste-masking effect in the e-tongue test likely due to the least bitterness of GTA. For the co-former SAC, there is no interaction with NVP in solution, and along with the bitterer taste of SAC,45,53 this co-crystal has a weak taste-masking ability based on the e-tongue results. Therefore, BA, MLA and GTA demonstrate promising outcomes and qualify as potential candidates for subsequent clinical trials.The enhanced dissolution rate of all co-crystals compared with pure NVP indicates the taste masking of these co-crystals does not rely on the variation in drug dissolution, while the supramolecular aggregation between NVP and co-former is more important. Since the bitter taste of NVP has been perfectly masked in NVP–BA/MLA/GTA co-crystals, the improved dissolution rate might rather provide better bioavailability and therapeutic effectiveness.
The result indicates that the molecular aggregation of the drug and co-former in solution is a major factor for taste masking by co-crystallisation – the stronger the solution interaction, the better the taste masking effect. Besides, the less bitter taste of co-formers is equally important for a better masking result. From our results, it is not clear what the influence of the structure of co-formers or the stoichiometric ratio of co-crystals on the taste-masking is and further systematic experiments are needed to fully elucidate these factors. In addition, due to the limitation of selectivity and precision in e-tongue sensors, the in vivo testing is still necessary to determine if the promising co-former candidates for NVP are effective in taste masking. Overall, we show that co-crystallisation is a promising technique to cover the bitter taste of drugs with a large variety of co-formers. The investigation of solution aggregation conducted by NMR spectroscopy can be used as a promising technique to screen co-formers for taste-masking before any in vitro or in vivo taste tests in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Royal Society FLAIR Collaboration Grant scheme (GCF/R1/211035). Y. S. thanks the China Scholarship Council (CSC) (202006250018) for funding. We thank Geffery Okello, Sandrine Tanga and Candidah Nephawe for the help in the dissolution rate experiment.References
- J. A. Mennella, A. C. Spector, D. R. Reed and S. E. Coldwell, Clin. Ther., 2013, 35, 1225–1246 CrossRef.
- N. Sofia, K. Elin, L. Franca, L. Birka, C. Joana, N. Begonya, T. Ann Marie and P. Benjamin, Arch. Dis. Child., 2019, 104, 900 CrossRef PubMed.
- S. K. Chay, A. V. Keating, C. James, A. E. Aliev, S. Haider and D. Q. M. Craig, RSC Adv., 2018, 8, 3564–3573 RSC.
- B. Dave, L. Emma, B. Amanda, P. Ann and N. F. Saul, Arch. Dis. Child., 2012, 97, 293 CrossRef.
- M. Behrens and W. Meyerhof, Cell. Mol. Life Sci., 2006, 63, 1501–1509 CrossRef CAS PubMed.
- A. Tripathi, D. Parmar, U. Patel, G. Patel, D. Daslaniya and B. Bhimani, J. Pharm. Sci. Bio-Sci. Res., 2011, 1, 136–142 Search PubMed.
- D. Douroumis, Expert Opin. Drug Delivery, 2007, 4, 417–426 CrossRef CAS.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed.
- C. B. Aakeroy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
- M. F. Arafa, S. A. El-Gizawy, M. A. Osman and G. M. El Maghraby, Drug Dev. Ind. Pharm., 2016, 42, 1225–1233 CrossRef CAS PubMed.
- Y. Maeno, T. Fukami, M. Kawahata, K. Yamaguchi, T. Tagami, T. Ozeki, T. Suzuki and K. Tomono, Int. J. Pharm., 2014, 473, 179–186 CrossRef CAS PubMed.
- S. Aitipamula, A. B. H. Wong and P. Kanaujia, J. Pharm. Sci., 2018, 107, 604–611 CrossRef CAS PubMed.
- T. Ogata, D. Tanaka and T. Ozeki, Drug Dev. Ind. Pharm., 2014, 40, 1084–1091 CrossRef CAS PubMed.
- S. A. Kulkarni, E. S. McGarrity, H. Meekes and J. H. ter Horst, Chem. Commun., 2012, 48, 4983–4985 RSC.
- J. D. Loya, J. C. Qiu, D. K. Unruh, A. F. Cozzolino and K. M. Hutchins, Cryst. Growth Des., 2018, 18, 4838–4843 CrossRef CAS.
- A. Bērziņš, A. Semjonova, A. Actiņš and M. Salvalaglio, Cryst. Growth Des., 2021, 21, 4823–4836 CrossRef.
- R. Bobrovs, L. Drunka, A. A. Auzins, K. Jaudzems and M. Salvalaglio, Cryst. Growth Des., 2021, 21, 436–448 CrossRef CAS.
- Y. Shen, A. J. Cruz-Cabeza, O. Azzouz and K. Edkins, Mol. Pharm., 2023, 20, 1942–1950 CrossRef CAS PubMed.
- J. L. Finney and A. K. Soper, Chem. Soc. Rev., 1994, 23, 1–10 RSC.
- R. C. Burton, E. S. Ferrari, R. J. Davey, J. L. Finney and D. T. Bowron, J. Phys. Chem. B, 2009, 113, 5967–5977 CrossRef CAS PubMed.
- Y. Shen, Y. Xiao, R. M. Edkins, T. G. A. Youngs, T.-L. Hughes, J. Tellam and K. Edkins, Int. J. Pharm., 2023, 647, 123520 CrossRef CAS.
- R. C. Burton, E. S. Ferrari, R. J. Davey, J. Hopwood, M. J. Quayle, J. L. Finney and D. T. Bowron, Cryst. Growth Des., 2008, 8, 1559–1565 CrossRef CAS.
- L. Tavagnacco, U. Schnupf, P. E. Mason, M.-L. Saboungi, A. Cesàro and J. W. Brady, J. Phys. Chem. B, 2011, 115, 10957–10966 CrossRef CAS PubMed.
- C. A. Hunter and M. J. Packer, Chem. – Eur. J., 1999, 5, 1891–1897 CrossRef CAS.
- M. Pein, M. Preis, C. Eckert and F. E. Kiene, Int. J. Pharm., 2014, 465, 239–254 CrossRef CAS PubMed.
- M. Münster, A. H. A. Mohamed-Ahmed, L. I. Immohr, C. Schoch, C. Schmidt, C. Tuleu and J. Breitkreutz, Int. J. Pharm., 2017, 529, 310–318 CrossRef.
- X. Zhao, Q. Li, C. Wang, S. Hu, X. He and C. C. Sun, Int. J. Pharm., 2020, 577, 119089 CrossRef CAS.
- H. A. Hideyuki Akiyama, K. T. Kiyoshi Toko and K. Y. Kaoru Yamafuji, Jpn. J. Appl. Phys., 1996, 35, 5516 CrossRef.
- S. Takagi, K. Toko, K. Wada, H. Yamada and K. Toyoshima, J. Pharm. Sci., 1998, 87, 552–555 CrossRef CAS PubMed.
- R. Lodha and M. Manglani, Indian J. Pediatr., 2012, 79, 1625–1633 CrossRef PubMed.
- M. R. Caira, S. A. Bourne, H. Samsodien, E. Engel, W. Liebenberg, N. Stieger and M. Aucamp, CrystEngComm, 2012, 14, 2541–2551 RSC.
- R. N. Costa, D. Choquesillo-Lazarte, S. L. Cuffini, E. Pidcock and L. Infantes, CrystEngComm, 2020, 22, 7460–7474 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 5922–5923 RSC.
- A. Guerrero-Martínez, G. González-Gaitano, M. H. Viñas and G. Tardajos, J. Phys. Chem. B, 2006, 110, 13819–13828 CrossRef PubMed.
- P. Brocos, N. Díaz-Vergara, X. Banquy, S. Pérez-Casas, M. Costas and Á. Piñeiro, J. Phys. Chem. B, 2010, 114, 12455–12467 CrossRef CAS PubMed.
- British Pharmacopoeia, British Pharmacopoeia Commission Secretariat, London, UK, 2022 Search PubMed.
- Y. Tahara and K. Toko, IEEE Sens. J., 2013, 13, 3001–3011 Search PubMed.
- M. Monschke and K. G. Wagner, Int. J. Pharm., 2019, 564, 162–170 CrossRef CAS PubMed.
- H. E. Abdelhakim, A. Coupe, C. Tuleu, M. Edirisinghe and D. Q. M. Craig, Pharmaceutics, 2021, 13, 1665 CrossRef CAS PubMed.
- R. P. Batzinger, S.-Y. L. Ou and E. Bueding, Science, 1977, 198, 944–946 CrossRef CAS PubMed.
- H. Sohi, Y. Sultana and R. K. Khar, Drug Dev. Ind. Pharm., 2004, 30, 429–448 CrossRef CAS.
- D. D. Bavishi and C. H. Borkhataria, Prog. Cryst. Growth Ch., 2016, 62, 1–8 CrossRef CAS.
- K. Chaturvedi, H. S. Shah, K. Nahar, R. Dave and K. R. Morris, ACS Omega, 2020, 5, 9690–9701 CrossRef CAS PubMed.
- H. E. Abdelhakim, A. Coupe, C. Tuleu, M. Edirisinghe and D. Q. M. Craig, Mol. Pharm., 2019, 16, 2557–2568 CrossRef CAS.
- C. P. Rader, S. G. Tihanyi and F. B. Zienty, J. Food Sci., 1967, 32, 357–360 CrossRef.
- C. Kuhn, B. Bufe, M. Winnig, T. Hofmann, O. Frank, M. Behrens, T. Lewtschenko, J. P. Slack, C. D. Ward and W. Meyerhof, J. Neurosci., 2004, 24, 10260–10265 CrossRef CAS.
- O. H. Azeez, S. Y. Alkass and D. S. Persike, Medicina, 2019, 55(10), 681 CrossRef PubMed.
- S. Mohanty, B. Das and S. Dhara, J. Asian Ceram. Soc., 2013, 1, 184–190 CrossRef.
- E. Robertson, in xPharm: The Comprehensive Pharmacology Reference, ed. S. J. Enna and D. B. Bylund, Elsevier, New York, 2007, pp. 1–7 Search PubMed.
- P. W. G. Smith and A. R. Tatchell, in Aromatic Chemistry, ed. P. W. G. Smith and A. R. Tatchell, Pergamon, 1969, pp. 176–195 Search PubMed.
- N. Pehlivanoǧlu, H. Uslu and Ş. İ. Kırbaşlar, J. Chem. Eng. Data, 2009, 54, 3202–3207 CrossRef.
- N. V. Bhagavan, in Medical Biochemistry, ed. N. V. Bhagavan, Academic Press, San Diego, 4th edn, 2002, pp. 1–16 Search PubMed.
- A. N. Pronin, H. Xu, H. Tang, L. Zhang, Q. Li and X. Li, Curr. Biol., 2007, 17, 1403–1408 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3pm00074e |
| This journal is © The Royal Society of Chemistry 2024 |