
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic ruthenium–zinc complexes with bulky N-heterocyclic carbenes: syntheses, structures and reactivity†
Maialen
Espinal-Viguri
,
Victor
Varela-Izquierdo
,
Fedor M.
Miloserdov
,
Ian M.
Riddlestone
,
Mary F.
Mahon
* and
Michael K.
Whittlesey
 *
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: m.k.whittlesey@bath.ac.uk
First published on 19th February 2019
Abstract
The ruthenium–zinc heterobimetallic complexes, [Ru(IPr)2(CO)ZnMe][BArF4] (7), [Ru(IBiox6)2(CO)(THF)ZnMe][BArF4] (12) and [Ru(IMes)′(PPh3)(CO)ZnMe] (15), have been prepared by reaction of ZnMe2 with the ruthenium N-heterocyclic carbene complexes [Ru(IPr)2(CO)H][BArF4] (1), [Ru(IBiox6)2(CO)(THF)H][BArF4] (11) and [Ru(IMes)(PPh3)(CO)HCl] respectively. 7 shows clean reactivity towards H2, yielding [Ru(IPr)2(CO)(η2-H2)(H)2ZnMe][BArF4] (8), which undergoes loss of the coordinated dihydrogen ligand upon application of vacuum to form [Ru(IPr)2(CO)(H)2ZnMe][BArF4] (9). In contrast, addition of H2 to 12 gave only a mixture of products. The tetramethyl IBiox complex [Ru(IBioxMe4)2(CO)(THF)H][BArF4] (14) failed to give any isolable Ru–Zn containing species upon reaction with ZnMe2. The cyclometallated NHC complex [Ru(IMes)′(PPh3)(CO)ZnMe] (15) added H2 across the Ru–Zn bond both in solution and in the solid-state to afford [Ru(IMes)′(PPh3)(CO)(H)2ZnMe] (17), with retention of the cyclometallation.
Introduction
Heterobimetallic complexes featuring a transition metal (TM) in partnership with a Lewis acidic (LA), typically main group element, have been the focus of considerable interest1 because of their potential to bring about the cooperative activation of E–H (E = H, N, Si etc.) bonds.2 The most commonly found heterobimetallic complexes feature a late transition metal (groups 8–10) and an element from group 13 (particularly B and Al) and, in many cases, are readily prepared by salt elimination reaction of a TM anion with a halide of the LA.3 While this approach is very flexible in that there are many possible TM and LA fragments that can be combined in this way, one (if not both) of the partners is typically left coordinatively saturated, reducing the subsequent reactivity for bond activation processes. An alternative approach which has been employed, although less frequently, is an alkane elimination pathway via the reaction of a TM hydride precursor with a LA hydrocarbyl reagent.4 This synthetic approach does come with potential issues (e.g. the use of highly pyrophoric group 13 trialkyls, cost of Ga/InMe3etc.), but does allow access to heterobimetallic complexes with unsaturation at both centres, thereby opening up an opportunity to probe true TM-LA cooperativity.Very recently, we reported that addition of GaMe3, InMe3 and ZnEt2 to the bulky N-heterocyclic carbene (NHC) stabilised cationic ruthenium hydride complex [Ru(IPr)2(CO)H][BArF4] (1; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; BArF4 = [B{C6H3(3,5-CF3)2}4]−)5 resulted in alkane elimination and formation of the Ru–Ga, Ru–In and Ru–Zn complexes 2–4 shown in Scheme 1.6 Of most relevance to this current paper was the ruthenium–zinc complex [Ru(IPr)2(CO)ZnEt][BArF4] (4) which, upon treatment with H2, both coordinated dihydrogen at Ru and added H2 across the Ru–Zn bond to give [Ru(IPr)2(CO)(η2-H2)(H)2ZnEt][BArF4] (5). Dissociation of the dihydrogen ligand from this highly fluxional species took place upon heating under vacuum to give the agostically stabilised dihydride complex, [Ru(IPr)2(CO)(H)2ZnEt][BArF4] (6, Scheme 1).7,8
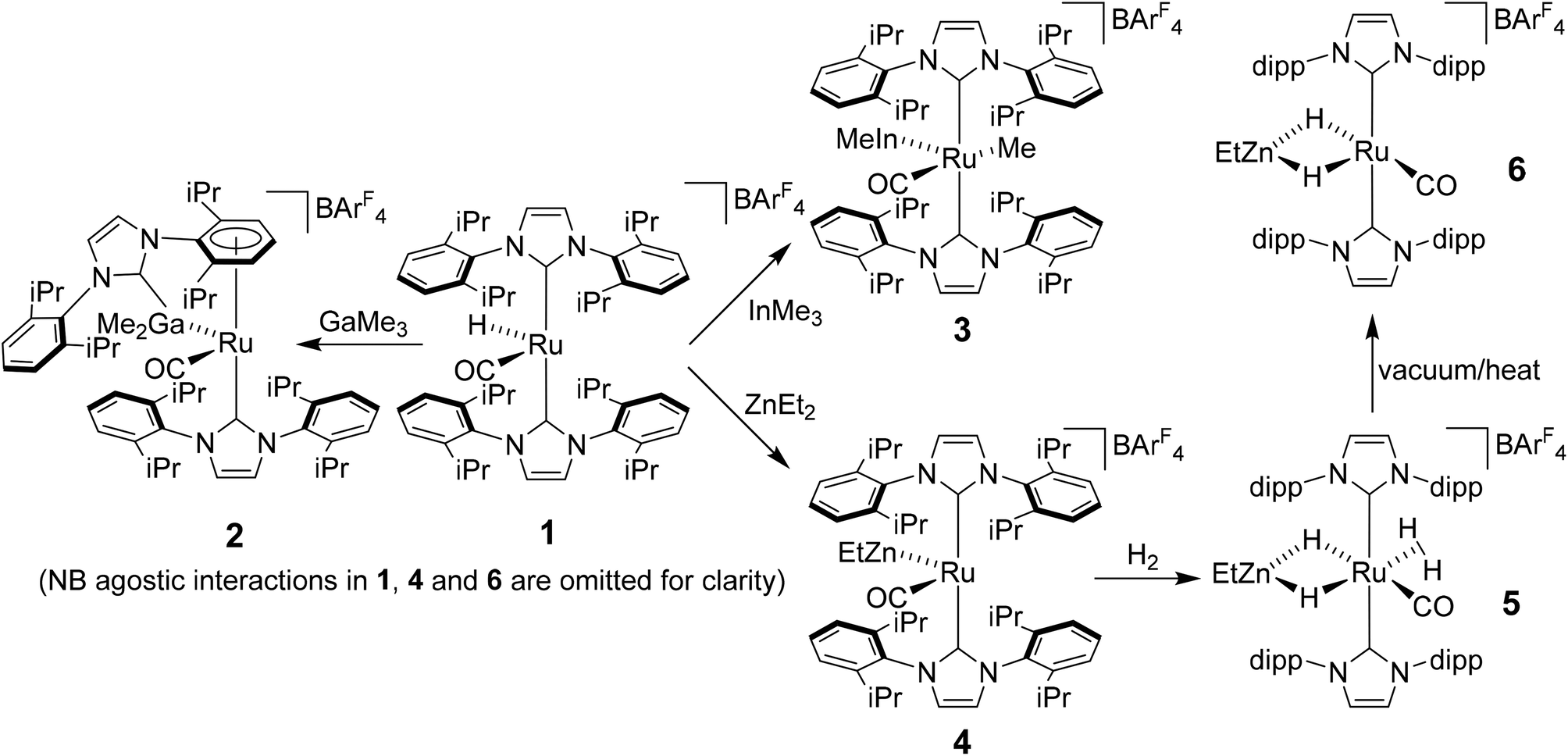 | ||
| Scheme 1 Summary of the reactivity of [Ru(IPr)2(CO)H][BArF4] (1) with ZnEt2 and MMe3 (M = Ga, In). | ||
Structural analysis showed that 4 (as well as 1) was also agostically stabilised, in this case through a bifurcated η3-H2C ξ-agostic interaction involving an iPr substituent of the IPr ligand. Thus, while 1 and 4 appear at first sight to be rare examples of isolable, four-coordinate Ru(II) complexes, the bifurcated agostic interactions impart formally 18-electron configurations. The participation of the bulky IPr ligand in forming agostic interactions seems to play a role in allowing 1, 4 and 6 to be isolated and structurally characterised, given that the less sterically crowded analogue [Ru(IMes)2(CO)H]+ (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) is found only as an oil.9
Herein, we describe efforts to elaborate on the chemistry of 1 and 4–6 through studies in which (i) the reactivity of 1 towards other ZnR2 reagents (R = Me, Ph) is probed and (ii) analogues containing the bulky oxazoline-derived IBiox class of NHC ligands are investigated. We also show that the formation of coordinatively unsaturated and reactive (NHC)Ru–Zn complexes is not limited to just cationic Ru–H precursors.10
Results and discussion
Reactivity of 1 towards ZnR2 reagents
The methyl zinc analogue of 4, [Ru(IPr)2(CO)ZnMe][BArF4] (7), was prepared by subjecting a C6H5F solution of 1 to a slight excess of a toluene solution of ZnMe2. 7 was isolated as a dark red solid in good yield (73%) and exhibited diagnostic low frequency 1H (δ – 0.86) and 13C (δ – 0.7) NMR resonances for the Zn–Me group, along with high frequency 13C signals (δ 200.6 and 188.0) arising from the presence of the carbenic and carbonyl carbons respectively. The structure of 7 was confirmed by X-ray crystallography (Fig. 1), which revealed a Ru–Zn distance of 2.3997(8) Å, comparable to that in 4 (2.4069(7) Å). There was no reaction between 1 and ZnPh2 (even upon heating to 70 °C) presumably due to the unfavourable combination of bulky substituents on the NHC and Zn.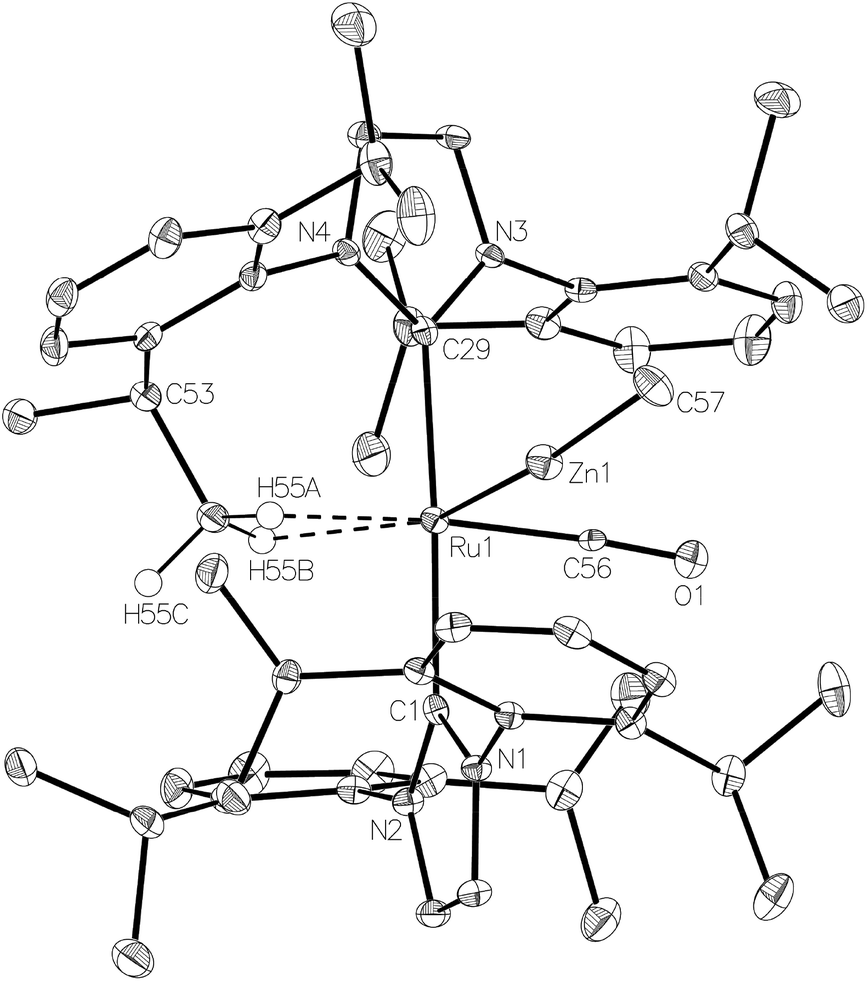 | ||
| Fig. 1 Molecular structure of the cation in [Ru(IPr)2(CO)ZnMe][BArF4] (7). Ellipsoids are shown at 30% probability. All hydrogen atoms (with the exception of those attached to C55) removed for clarity. Selected bond lengths (Å) and angles (°): Ru(1)–Zn(1) 2.3997(8), Ru(1)–C(1) 2.123(5), Ru(1)–C(29) 2.108(5), Ru(1)⋯C(55) 2.462(6), Ru(1)–C(56) 1.837(6), C(1)–Ru(1)–C(29) 175.4(2), C(56)–Ru(1)–Zn(1) 77.1(2). | ||
Upon exposure of a fluorobenzene solution of 7 to 1 atm H2, an instantaneous change in colour from red-orange to colourless was observed, resulting from the formation of the dihydrogen dihydride complex, [Ru(IPr)2(CO)(η2-H2)(H)2ZnMe][BArF4] (8, Scheme 2).11 This showed less fluxional behavior than the ZnEt analogue 5, exhibiting three low frequency hydride signals (δ −5.15, −7.83 and −12.16 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio) at room temperature compared to just two resonances for 5 (δ −5.33 and −12.13 in a 3:1 ratio). Cooling a THF solution of 8 to 238 K led to sharpening of the two lower frequency resonances, whereas that at ca. −5 ppm remained broader than the others even down to 218 K. Based upon the comparable chemical shifts and assignments in 5, the three signals were assigned to Ru(η2-H2), Ru–H–Zn trans to CO and Ru–H–Zn trans to η2-H2 in order of decreasing frequency.
:1:1 ratio) at room temperature compared to just two resonances for 5 (δ −5.33 and −12.13 in a 3:1 ratio). Cooling a THF solution of 8 to 238 K led to sharpening of the two lower frequency resonances, whereas that at ca. −5 ppm remained broader than the others even down to 218 K. Based upon the comparable chemical shifts and assignments in 5, the three signals were assigned to Ru(η2-H2), Ru–H–Zn trans to CO and Ru–H–Zn trans to η2-H2 in order of decreasing frequency.
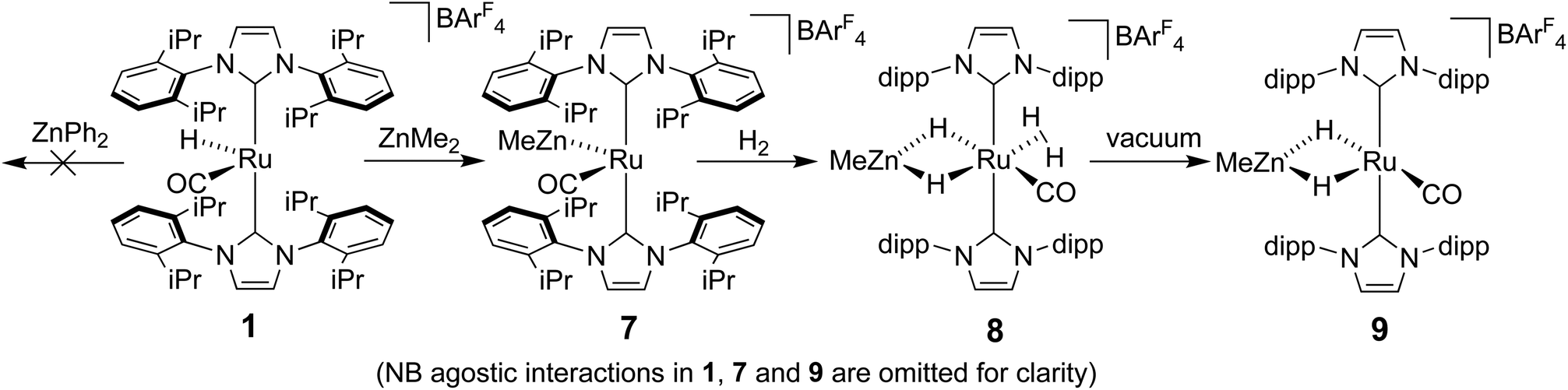 | ||
| Scheme 2 Synthesis and reactivity of [Ru(IPr)2(CO)ZnMe][BArF4] (7). | ||
The η2-H2 ligand in 8 could be removed simply by the application of vacuum to a solid sample of the compound (cf. vacuum and heat for 5, Scheme 1). The resulting product, [Ru(IPr)2(CO)(H)2ZnMe][BArF4] (9), was identified by the appearance of a low frequency (δ −25.77) doublet (2JHH = 7.7 Hz) resonance for Ru–H–Zn trans to the agostic Ru⋯H2C the IPr ligand (vide infra), together with a higher frequency doublet (δ −4.19, 2JHH = 7.7 Hz) arising from the Ru–H–Zn hydride trans to CO.
The Ru–Zn complexes 8 and 9 were characterised crystallographically (Fig. 2). As anticipated, a comparison of these two complexes to their ZnEt analogues 5 and 6 (Scheme 3) shows the same patterns i.e. elongation of the Ru⋯Zn distance relative to 4 and 7, less asymmetry of the Ru–H–Zn distances for H trans to CO and greater association with Ru for H trans to either an agostic interaction (6 and 9) or a dihydrogen ligand (5 and 8).
 | ||
| Fig. 2 Molecular structures of the cations in (left) [Ru(IPr)2(CO)(η2-H2)(H)2ZnMe][BArF4] (8) and [Ru(IPr)2(CO)(H)2ZnMe][BArF4] (right) (9). Ellipsoids are shown at 30% probability with all hydrogen atoms (except hydrides, those agostically bonded and the tentatively assigned dihydrogen ligand) removed for clarity. | ||
 | ||
| Scheme 3 Summary of key distances in [Ru(IPr)2(CO)(H)2ZnR][BArF4] and [Ru(IPr)2(CO)(η2-H2)(H)2ZnR][BArF4] (R = Et, Me). | ||
Synthesis and reactivity of Ru(IBiox) complexes
Given the success of IPr in allowing access to isolable [Ru(NHC)2(CO)H]+ and [Ru(NHC)2(CO)ZnR]+ species, we turned our attention to the IBiox class of NHCs introduced by Glorius,12 on the basis that they are also known to be sterically demanding and flexibly restricted. Moreover, in spite of their use for the preparation of low-coordinate Rh and Ir complexes,13 we were aware of just a single example of a Ru-IBiox complex at the outset of our work.14Employing previous methodology,15 the bis-carbene complexes [Ru(IBiox6)2(CO)HCl] (10; for structure of IBiox6, see Scheme 4) and [Ru(IBioxMe4)2(CO)HCl] (13; for structure of IBioxMe4, see Scheme 5) were isolated in ca. 50–70% yield after heating [Ru(AsPh3)3(CO)H2] with 2.5 equivalents of the free carbenes, followed by addition of dichloromethane. The 1H NMR spectra of these 16-electron species displayed a low frequency hydride resonance (10: δ −24.75; 13: δ −25.14) characteristic of [Ru(NHC)2(CO)HCl] complexes.15,16 Addition of NaBArF4 led to abstraction of the chloride ligand to give [Ru(IBiox6)2(CO)(THF)H][BArF4] (11, Scheme 4) and [Ru(IBioxMe4)2(CO)(THF)H][BArF4] (14, Scheme 5) respectively. The X-ray structures of neither 11 nor 14 (Fig. 3) showed any agostic interactions to the substituents on the IBiox ligands (e.g. shortest C–H⋯Ru in 11 is 3.182 Å).13a,c In order to relieve the electron-deficiency of the Ru(II) centres, a THF ligand resides in the metal coordination sphere of each complex trans to CO.17
 | ||
| Scheme 4 Synthesis and reactivity of Ru(IBiox6) complexes. | ||
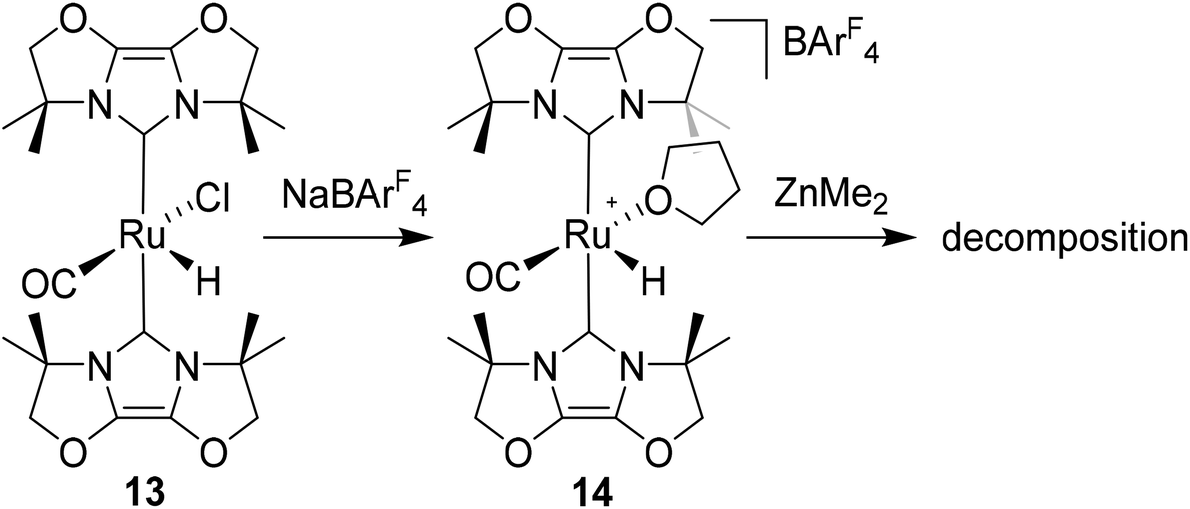 | ||
| Scheme 5 Synthesis and reactivity of Ru(IBioxMe4) complexes. | ||
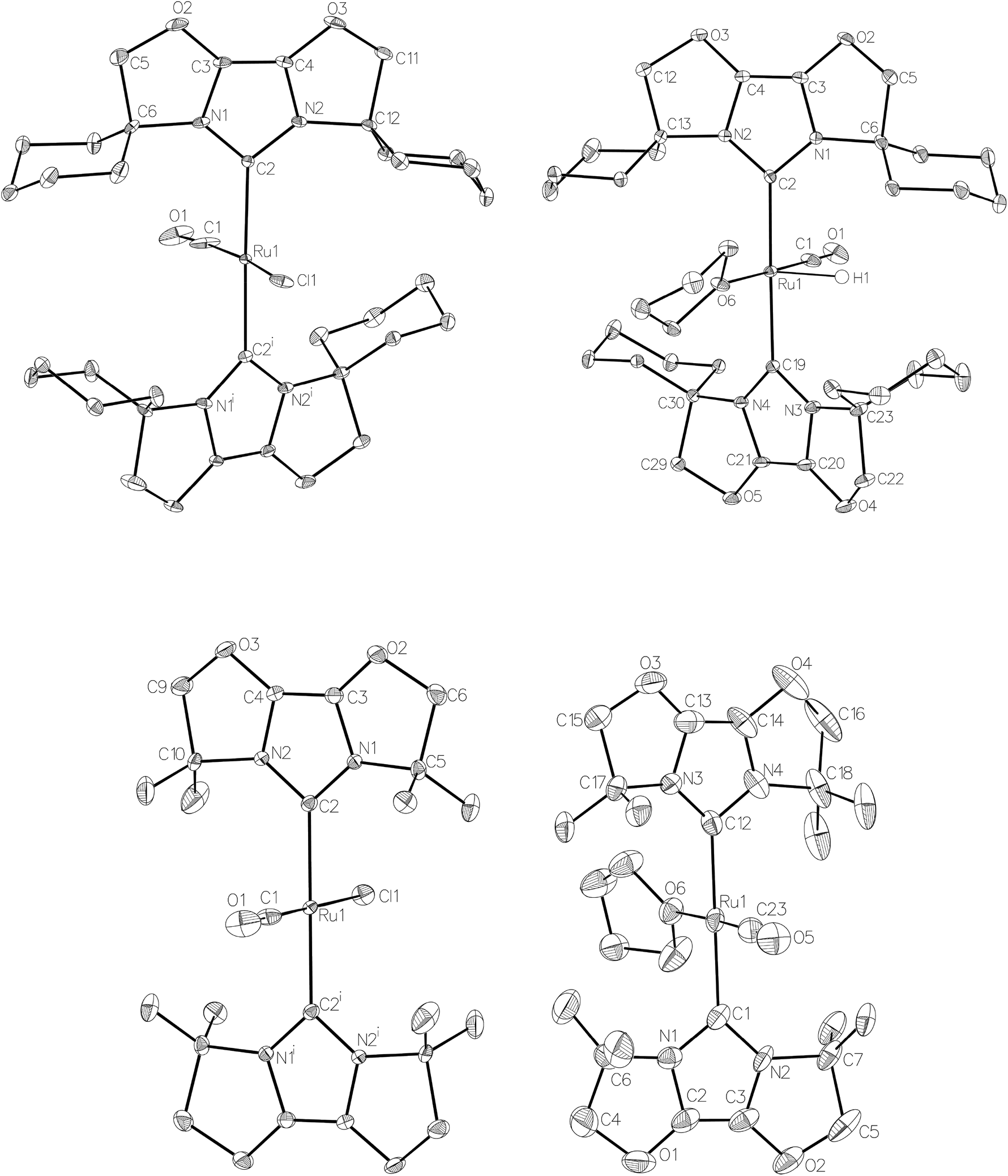 | ||
| Fig. 3 Molecular structures of [Ru(IBiox6)2(CO)HCl] (10, top left), [Ru(IBioxMe4)2(CO)HCl] (13, bottom left) and the cations in [Ru(IBiox6)2(CO)(THF)H][BArF4] (11, top right) and [Ru(IBioxMe4)2(CO)(THF)H][BArF4] (14, bottom right). In all cases, ellipsoids are represented at 30% probability. In 10, solvent hydrogen atoms and disorder have been omitted for clarity. Symmetry operation: i1 − x, y, ½ − z. In 11, hydrogen atoms, with the exception of the hydride ligand, are omitted for clarity. Only one disordered component of the hydride ligand and of C22 is illustrated. In 13, disorder has been omitted for clarity. Symmetry operation: i1 − x, 1 − y, 1 − z. In 14, disorder was rampant and only one component is illustrated, for clarity. | ||
The X-ray crystal structure of 10 and 13, along with those of the cations in 11 and 14, are shown in Fig. 3. A listing of metrical data for the compounds is given in Table 1. As expected, all four compounds exhibited square pyramidal geometries with the hydride ligand in an apical position. Analysis of the NHC tilting angle ΘNHC (Ru–CNHC–centroidNHC)18 revealed angles of >170° in all cases, showing that, despite the coordinative unsaturation at ruthenium, the IBiox ligands remain free of any structural distortions akin to those seen in some [M(IBiox)3]+ (M = Rh, Ir) species.13c
|
|
|
||||
|---|---|---|---|---|---|
| 10 | 13 | 11 (X = H) | 14 (X = H) | 12 (X = ZnMe) | |
| Ru–CIBiox | 2.122(4) | 2.086(3) | 2.112(2), 2.116(3) | 2.093(3), 2.098(3) | 2.120(3), 2.127(3) |
| Ru–CO | 1.782(19) | 1.825(11) | 1.797(4) | 1.795(4) | 1.818(3) |
| Ru–Cl | 2.375(4) | 2.405(3) | — | — | — |
| Ru–O | — | — | 2.168(2) | 2.197(2) | — |
| Ru–Zn | — | — | — | — | 2.3819(4) |
| Ru–CIBiox–IBiox centroid18 | 175.87 | 176.96 | 174.07, 174.01 | N/A due to disorder | 170.84, 171.13 |
| Cl–Ru–CO | 170.0(8) | 179.2(5) | — | — | — |
| O–Ru–CO | — | — | 178.89(17) | 177.99(13) | 154.59(10) |
| Zn–Ru–CO | — | — | — | — | 134.11(6) |

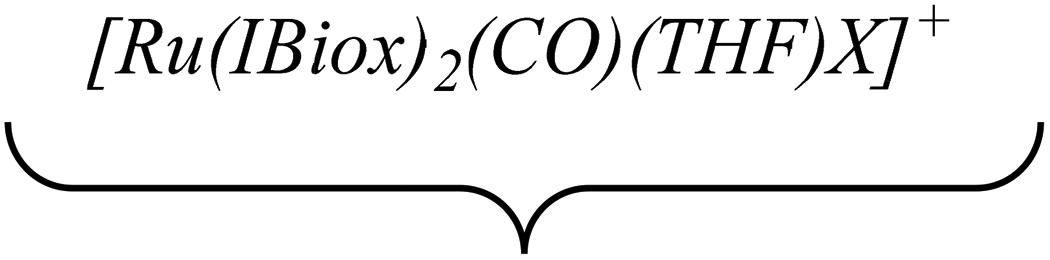
Efforts to generate new Ru–Zn containing complexes through reaction of 11 and 14 with ZnMe2 was successful only in the case of the former,19 which generated [Ru(IBiox6)2(CO)(THF)ZnMe][BArF4] (12, Fig. 4 and Scheme 4). A comparison between the structures of 4 and 12 yield some superficial similarities and some interesting differences. Both structures contain two trans NHC ligands and coordination bonds in the equatorial plane of which two are common, namely, one to a zinc centre and one to a CO ligand. In 4, the remaining site is occupied by a bifurcated agostic interaction, while in 12, there is coordination of a THF molecule. In gross terms, the structures of both cations overlay reasonably well, but the biggest significant difference between them lies in the relative orientations of the NHC ligands. In 4, the angle between the mean planes based on the 5-membered NHC rings is relatively staggered at 102°, while the comparable angle in 12 (32°) reflects a more eclipsed carbene conformation. The arising steric ramifications are that the CNHC–Ru–CNHC angle of 177.32(19) Å in 4 is noticeably more linear than the 170.96(10)° angle observed in 12. It is possible that the significantly shorter Ru–Zn distance of 2.3819(4) Å in 12 (cf. 2.4069(7) Å in 4) may reflect the less encumbered access of the zinc ligand, towards the ruthenium centre, via the opposite face of the cation to the NHC ligand fold.
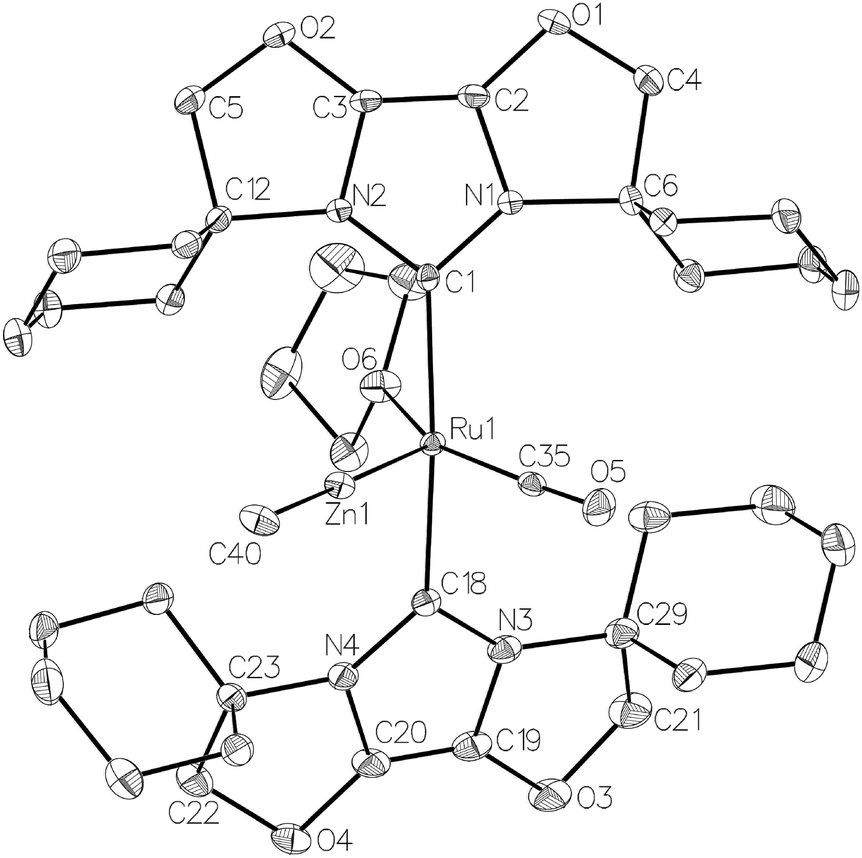 | ||
| Fig. 4 Molecular structure of the cation in [Ru(IBiox6)2(CO)(THF)ZnMe][BArF4] (12). Ellipsoids are shown at 30% probability. Only one component arising from the disordered carbons (C37 and C38) in the THF ligand is illustrated. | ||
Upon exposure to either 1 or 5 atm H2, NMR spectra of fluorobenzene solutions of 12 exhibited signals for free IBiox6 as well as the salt [IBiox6·H][BArF4]. Any products of initial reaction with H2 therefore appear to be of only limited stability.
Ru–Zn bond formation from a neutral Ru–H precursor
The premise behind the initial synthesis of 1 was that addition of ZnR2 to an electrophilic ruthenium hydride complex would result in the facile elimination of an alkane and formation of a new Ru–Zn containing species. In an effort to test whether a cationic (NHC)Ru hydride precursor was necessary, we examined the reaction of the neutral precursor [Ru(IMes)(PPh3)(CO)HCl] with ZnMe2. In the presence of 5 equiv. ZnMe2, a rapid reaction ensued to form the cyclometallated complex [Ru(IMes)′(PPh3)(CO)ZnMe] (15, Scheme 6) as determined by 1H and 31P{1H} NMR spectroscopy. The formation of 15 was accompanied by small amounts of a second product (assigned tentatively as the non-metallated ruthenium chloride complex, [Ru(IMes)(PPh3)(CO)(ZnMe)Cl] (16; see ESI†)), the concentration of which correlated with the rate of addition of ZnMe2. 15 was itself present as major (15a) and minor (15b) forms in solution. As a result, the Ru–CH2 group arising from C–H activation of the IMes ligand gave rise to two sets of diastereotopic signals in the proton NMR spectrum; in THF-d8, 15a showed a broad triplet (2JHH = 3JHP = 5.6 Hz) at δ 2.61 and a doublet of doublets (3JHP = 11.6 Hz, 2JHH = 6.7 Hz) at δ 0.98 (each of integral 1, which both correlated (1H–13C HSQC) to a methylene carbon resonance at δ 31), while 15b displayed a multiplet at δ 1.52 and a doublet of doublets (3JHP = 14.4 Hz, 2JHH = 8.6 Hz) at δ 1.40; both resonances correlated to a 13C NMR signal at δ 32.20 | ||
| Scheme 6 Formation of [Ru(IMes)′(PPh3)(CO)ZnMe] (15) and [Ru(IMes)′(PPh3)(CO)(H)2ZnMe] (17). | ||
The similarity of chemical shifts and J values for both species (e.g. each exhibited a high frequency resonance for the carbenic carbon with a 2JCP value of >80 Hz, indicative of a trans IMes-Ru-PPh3 geometry) suggested that they were most likely conformers. There was a slight solvent dependence on the solution ratio of 15a:15b (88:12 and 82:18 in C6D6 and THF-d8 respectively). The two species were shown to be in exchange in THF-d8 by EXSY, although NOESY measurements failed to divulge any information as to the spatial difference between 15a and 15b. We were unable to establish any difference crystallographically as measurements of a number of different single crystals only ever afforded the same structure as shown in Fig. 5.
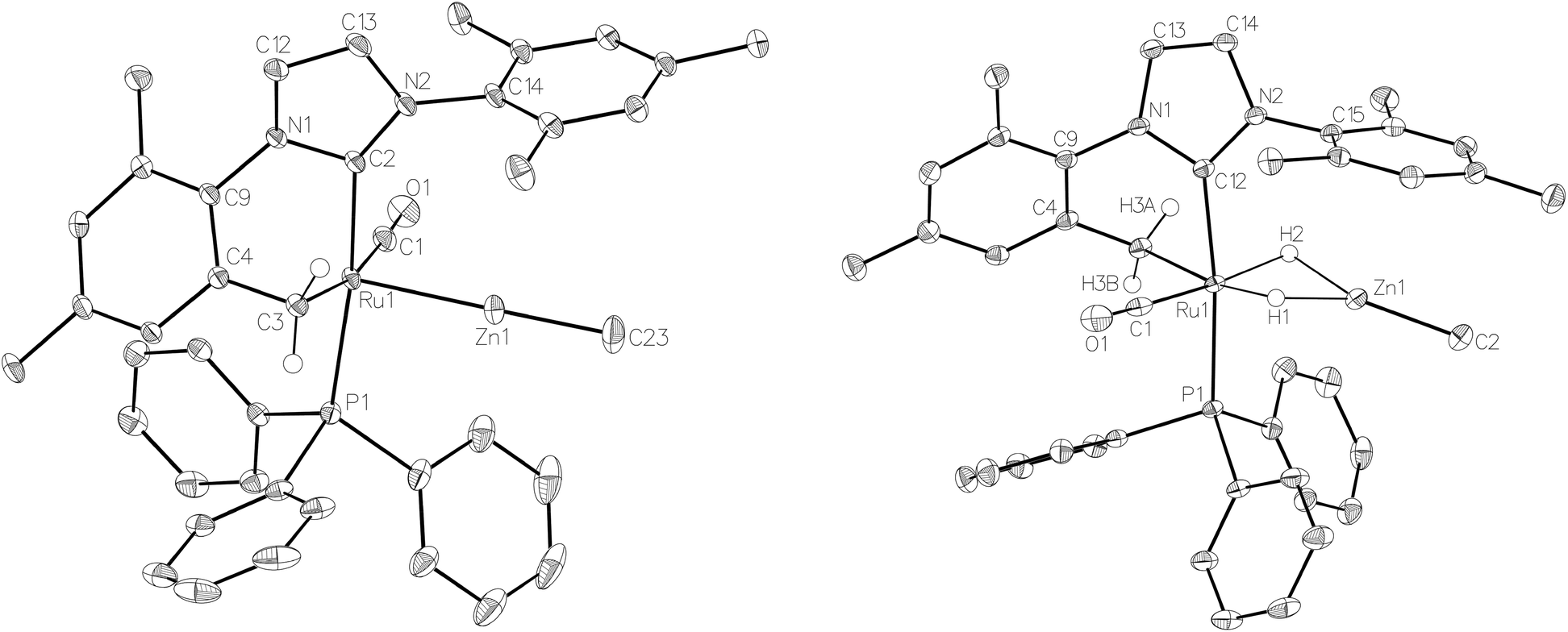 | ||
| Fig. 5 Molecular structures of (left) [Ru(IMes)′(PPh3)(CO)ZnMe] (15) and (right) [Ru(IMes)′(PPh3)(CO)(H)2ZnMe] (17). Ellipsoids are shown at 30% probability with all hydrogen atoms (except Ru–CH2 and Ru–H) removed for clarity. Selected bond lengths (Å) and angles (°) in 15: Ru(1)–C(1) 1.857(2), Ru(1)–C(2) 2.071(2), Ru(1)–C(3) 2.224(2), Ru(1)–P(1) 2.3360(5), Ru(1)–Zn(1) 2.3677(3), C(2)–Ru(1)–P(1) 172.32(6), C(1)–Ru(1)–C(3) 169.63(9), Ru(1)–C(3)–C(4) 83.39(12). 17: Ru(1)–C(1) 1.874(2), Ru(1)–C(12) 1.946(2), Ru(1)–C(3) 2.1971(19), Ru(1)–P(1) 2.3342(5), Ru(1)–Zn(1) 2.4828(3), C(12)–Ru(1)–P(1) 169.10(6), C(1)–Ru(1)–C(3) 98.77(8), Ru(1)–C(3)–C(4) 108.65(13). | ||
Upon addition of 1 atm H2 to a deep red-orange C6D6 solution of 15, an instant colour change to very pale ensued from formation of [Ru(IMes)′(PPh3)(CO)(H)2ZnMe] (17). The 1H NMR spectrum showed the presence of two doublet hydride resonances at δ −6.77 (2JHP = 14.9 Hz) and δ −9.19 (2JHP = 5.0 Hz), alongside a higher frequency doublet at δ 3.22 and doublet of doublets at δ 1.83, consistent with addition of H2 across the Ru–Zn bond rather than reversal of the IMes cyclometallation. This irreversibility contrasts with what we observed previously in the case of the related cyclometallated hydride derivative [Ru(IMes)′(PPh3)2(CO)H], which reacted with H2 to form [Ru(IMes)(PPh3)2(CO)H2].20a Equally surprisingly, monitoring of the reaction with H2 in the solid-state by IR spectroscopy showed complete depletion of νCO for 15 at 1860 cm−1 and appearance of a new carbonyl absorption band at 1941 cm−1 for 17 upon stirring a ground up microcrystalline sample of the former under 1 atm H2 for 2 days at room temperature.
The X-ray crystal structures of 15 and 17 (Fig. 5) show clearly the transformation of 5-coordinate 15 to six-coordinate 17 upon reaction with H2. As anticipated (vide supra), elongation of the Ru–Zn distance from 2.3677(3) Å to 2.4828(3) Å takes place upon H2 addition. Both bridging hydrogens were located and refined without restraints. As in 6 and 9, the hydride trans to CO was more evenly shared between Ru and Zn than that, which in the case of 17, lies trans to the methylene group of the activated IMes ligand. As a result of cyclometallation, neither 15 nor 17 showed a strictly linear CIMes′–Ru–P geometry (172.32(6) and 169.10(6)° respectively). 15 exhibited a particularly noticeable distortion of the angle at the cyclometallated methylene carbon (Ru(1)–C(3)–C(4) = 83.39(12)°).21
Preliminary studies to investigate the mechanism of formation of 17 revealed that exposure of 17 to D2 (1 atm) resulted in slow (1 day, room temperature) deuterium incorporation into both Ru–H–Zn positions, but no H/D exchange at RuCH2. This excludes exchange taking place via a reversible reductive elimination pathway involving both RuH and RuCH2.22 The viability of an alternative pathway through phosphine dissociation was probed by reaction of 17 with 5 equiv. P(p-tolyl)3. Slow PPh3/P(p-tolyl)3 was indeed observed, but the relevance of this to the H/D exchange was complicated by the appearance of other low frequency proton signals arising from the decomposition of 17 that can be seen in solution over 1–2 days.
Conclusions
We have reported that ZnMe2 reacts with both cationic and neutral ruthenium hydride precursors containing bulky N-heterocyclic carbene ligands to afford new heterobimetallic complexes containing Ru–Zn bonds. The IPr complex [Ru(IPr)2(CO)ZnMe][BArF4] (7) proved to be similar in terms of both structure and reactivity towards H2 to the previously reported ZnEt derivative 4. Use of the bulky IBiox carbene ligands met with varying levels of success; the cyclohexyl substituted derivative IBiox6 gave [Ru(IBiox6)2(CO)(THF)ZnMe][BArF4] (12), whereas the analogous tetramethyl IBioxMe4 derivative could not be isolated. Of particular interest was the formation of the neutral complex [Ru(IMes)′(PPh3)(CO)ZnMe] (15), which added H2 across the Ru–Zn bond whilst retaining the cyclometallated NHC ligand. As noted above, this behaviour contrasts with the reversal of cyclometallation that is brought about upon exposure of [Ru(IMes)′(PPh3)2(CO)H] to H2.20a This, together with the fact that 4 reacts with HBcat to bring about dehydrocoupling (and generation of 5)6b in contrast to 1 which reacts with HBcat with loss of H2 and formation of the stable boryl complex [Ru(IPr)2(CO)Bcat][BArF4],5 provides evidence for very different reactivity between Ru–Zn and Ru–H containing species. Indeed, efforts to probe the reactivity of 15 towards a broader range of E–H bonds, as well as prepare derivatives of the complex containing other metallated ligands, are in progress.Experimental
All manipulations were carried out using standard Schlenk, high vacuum and glovebox techniques using dried and degassed solvents. NMR spectra were recorded on Bruker Avance 400 and 500 MHz NMR spectrometers and run either locked in CD2Cl2 (referenced to δ 5.32 (1H); 54.0 (13C)), CDCl3 (referenced to δ 7.26 (1H); 54.0 (13C)), THF-d8 (referenced to δ 3.58 (1H); 67.2 (13C)) or C6D6 (referenced to δ 7.15 (1H); 128.0 (13C)), or unlocked in C6H5F (1H NMR spectra referenced to the centre of the downfield multiplet at δ 7.11). IR spectra were recorded in solution (CH2Cl2, CD2Cl2, CDCl3 or THF) or in KBr discs on a Nicolet Nexus spectrometer. Elemental analyses were performed by Elemental Microanalysis Ltd, Okehampton, Devon, UK. [Ru(IPr)2(CO)H][BArF4] (1),5 IBiox6,12b IBioxMe412b and [Ru(IMes)(PPh3)(CO)HCl]23 were prepared according to the literature.[Ru(IPr)2(CO)ZnMe][BArF4] (7)
Addition of ZnMe2 (25 μL of 1.0 M in toluene, 0.025 mmol) to a solution of 1 (40 mg, 0.023 mmol) in C6H5F (0.6 mL) resulted in an instantaneous change in colour from yellow to deep red. The reaction mixture was layered with hexane, which gave dark red crystals of 7. Yield 30 mg (73%). 1H NMR: δH (500 MHz, CD2Cl2, 298 K) 7.73 (s, 8H, [BArF4]−), 7.56 (s, 4H, [BArF4]−), 7.51 (t, J = 7.8 Hz, 4H, Ar), 7.32 (dd, J = 7.8, 1.4 Hz, 4H, Ar), 7.27 (dd, J = 7.8 1.4 Hz, 4H, Ar), 7.03 (s, 4H, NCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NCH), 2.42 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 2.32 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.10 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.07 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.00 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 0.65 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), −0.86 (s, 3H, ZnCH3). 13C{1H} NMR: δC (101 MHz, CD2Cl2, 298 K, [BArF4]− signals are omitted) 200.6 (s, RuCO), 188.0 (s, RuCNHC), 146.5 (s), 146.3 (s), 135.7 (s), 131.7 (s), 126.3 (s), 126.1 (s), 124.1 (s), 29.8 (s), 29.3 (s, CH(CH3)2), 25.9 (s), 24.7 (s), 24.2 (s), 23.7 (s, CH(CH3)2), −0.71 (s, ZnCH3). IR (CH2Cl2, cm−1): 1919 (νCO). Anal. calcd for C88H87BN4OF24ZnRu: C, 57.14, H, 4.74, N, 3.03. Found: C, 56.79, H, 4.70, N, 2.78.
NCH), 2.42 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 2.32 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.10 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.07 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.00 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 0.65 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), −0.86 (s, 3H, ZnCH3). 13C{1H} NMR: δC (101 MHz, CD2Cl2, 298 K, [BArF4]− signals are omitted) 200.6 (s, RuCO), 188.0 (s, RuCNHC), 146.5 (s), 146.3 (s), 135.7 (s), 131.7 (s), 126.3 (s), 126.1 (s), 124.1 (s), 29.8 (s), 29.3 (s, CH(CH3)2), 25.9 (s), 24.7 (s), 24.2 (s), 23.7 (s, CH(CH3)2), −0.71 (s, ZnCH3). IR (CH2Cl2, cm−1): 1919 (νCO). Anal. calcd for C88H87BN4OF24ZnRu: C, 57.14, H, 4.74, N, 3.03. Found: C, 56.79, H, 4.70, N, 2.78.
[Ru(IPr)2(CO)(η2-H2)(H)2ZnMe][BArF4] (8)
A J. Young's resealable NMR tube was charged with a C6H5F (0.6 mL) solution of 7 (35 mg, 0.020 mmol) and ZnMe2 (22 μL, 1.0 M in toluene, 0.022 mmol) added. The resulting red solution was evaporated to dryness, redissolved in C6H5F (0.3 mL), degassed (freeze–pump–thaw × 3) and placed under 1 atm H2. The solution was layered with H2-purged hexane to afford pale-colourless crystals of 8. Yield 25 mg (69%). Material for elemental analysis was prepared by slow evaporation of a sample of 8 prepared via exposure of a CH2Cl2 solution of 7 to H2. 1H NMR: δH (500 MHz, CD2Cl2, 298 K) 7.73 (s, 8H, [BArF4]−), 7.56 (s, 4H, [BArF4]−), 7.52 (t, J = 8.0 Hz, 4H, Ar), 7.28 (d, J = 8.0 Hz, 8H, Ar), 7.09 (s, 4H, NCHNCH), 2.19 (sept, 3JHH = 6.8 Hz, 8H, CH(CH3)2), 1.10 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.01–0.98 (m, 36H, CH(CH3)2), −0.66 (s, 3H, ZnCH3), −5.15 (br s, 2H, Ru(η2-H2)), −7.83 (br s, 1H, RuHZn), −12.16 (s, 1H, RuHZn). 13C{1H} NMR: δC (126 MHz, CD2Cl2, 298 K, [BArF4]− signals are omitted) 196.8 (s, RuCO), 179.6 (s, RuCNHC), 146.3 (s), 145.0 (s), 137.2 (s), 131.7 (s), 126.4 (s), 126.0 (s), 125.9 (s), 29.5 (s), 29.2 (s, CH(CH3)2), 26.4 (s), 26.2 (s), 22.9 (s), 22.6 (s, CH(CH3)2), 1.38 (s, ZnCH3). IR (CD2Cl2, cm−1): 2005 (νCO). Anal. calcd for C88H91BN4OF24ZnRu·CH2Cl2: C, 55.13, H, 4.83, N, 2.89. Found: C, 55.6, H, 4.64, N, 2.48.
[Ru(IPr)2(CO)(H)2ZnMe][BArF4] (9)
A J. Young's resealable NMR tube was charged with a solution of 7 (25 mg, 0.014 mmol) in C6H5F (0.6 mL), degassed (freeze–pump–thaw × 3) and exposed to 1 atm H2. After 30 min, the solvent was removed in vacuo and the resulting pale orange residue left under vacuum for 3 h. This was then dissolved in C6H5F (0.3 mL) and layered with hexane to yield orange crystals of 9. Yield 13 mg (54%). 1H NMR: δH (400 MHz, CD2Cl2, 298 K) 7.72 (s, 8H, [BArF4]−), 7.56 (s, 4H, [BArF4]−), 7.49 (t, 3 J = 7.8 Hz, 4H, Ar), 7.30 (d, J = 7.8 Hz, 4H, Ar), 7.23 (d, J = 7.8 Hz, 4H, Ar), 7.06 (s, 4H, NCHNCH), 2.35 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 2.19 (sept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.04–0.99 (m, 36H, CH(CH3)2), 0.44 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), −0.72 (s, 3H, ZnCH3), −4.19 (d, 2J = 7.7 Hz, 1H, RuHZn), −25.77 (d, 2JHH = 7.7 Hz, 1H, RuHZn). 13C{1H} NMR: δC (101 MHz, CD2Cl2, 298 K, [BArF4]− signals are omitted) 198.3 (s, RuCO), 182.2 (s, RuCNHC), 146.5 (s), 144.9 (s), 136.4 (s), 131.4 (s), 126.7 (s), 125.8 (s), 125.5 (s), 29.3 (s), 29.3 (s, CH(CH3)2), 26.1 (s), 24.9 (s), 23.1 (s), 22.2 (s, CH(CH3)2), 15.7 (s, ZnCH3). IR (CD2Cl2, cm−1): 2005 (νCO). Anal. calcd for C88H89BN4OF24ZnRu: C, 57.07, H, 4.84, N, 3.03. Found: C, 57.44, H, 4.62, N, 2.90.
[Ru(IBiox6)2(CO)HCl] (10)
[Ru(AsPh3)3(CO)H2] (728 mg, 0.69 mmol) and IBiox6 (500 mg, 1.73 mmol) were dissolved in toluene (10 mL) and stirred in a J. Young's resealable ampoule overnight at 363 K. After addition of 53 μL CH2Cl2, stirring was continued at 383 K for a further 12 h. The solvent was removed under vacuum and the product extracted into toluene (30 mL), reduced to dryness and washed with hexane (7 mL) and EtOH (2 × 10 mL) to give 10 as a yellow powder. Yield 280 mg (54%). 1H NMR: δH (500 MHz, CD2Cl2, 223 K) 4.74 (d, J = 8.5 Hz, 2H, CH2O), 4.59 (q, J = 8.4 Hz, 4H, CH2O), 4.51 (d, J = 8.5 Hz, 2H, CH2O), 3.20–3.14 (m, 2H, Cy), 2.67–2.61 (m, 2H, Cy), 2.50–2.37 (m, 4H, Cy), 2.06–2.03 (m, 2H, Cy), 1.91–1.77 (m, 14H, Cy), 1.67–1.64 (m, 4H, Cy), 1.33–1.10 (m, 12H, Cy), −24.75 (s, 1H, RuH). 13C{1H} NMR: δC (126 MHz, CD2Cl2, 223 K) 204.1 (s, RuCO), 164.9 (s, RuCNHC), 125.3 (s), 123.8 (s, NCO), 83.9 (s), 83.2 (s, OCH2), 65.4 (s), 64.8 (s, CCy), 35.8 (s), 35.3 (s), 34.5 (s), 33.4 (s), 24.5 (s), 24.3 (s), 24.0 (s), 24.0 (s), 23.9 (s), 23.8 (s, CH2Cy). IR (THF, cm−1): 1890 (νCO). Anal. calcd for C35H49N4O5ClRu·CH2Cl2: C, 52.27; H, 6.21; N, 6.77. Found: C, 52.45; H, 6.30; N, 6.62.[Ru(IBiox6)2(THF)(CO)H][BArF4] (11)
Na[BArF4] (426 mg, 0.48 mmol) was added to a C6H5F solution (15 mL) of 10 (324 g, 0.44 mmol) and the suspension stirred for 12 h. After filtration, the solution was evaporated and the oily residue dissolved in THF (4 mL). Addition of hexane and vigorous stirring for 15 min afforded 11 as a yellow solid. Yield: 615 mg (85%). Crystals suitable for X-ray diffraction were obtained by slow diffusion of hexane into a concentrated C6H5F solution of 11 at room temperature. 1H NMR: δH (500 MHz, THF-d8, 298 K) 7.78 (8H, [BArF4]−), 7.57 (4H, [BArF4]−), 4.91 (d, J = 8.7 Hz, 2H, CH2O), 4.88 (d, J = 8.7 Hz, 2H, CH2O), 4.68 (d, J = 8.7 Hz, 4H, CH2O), 2.89–2.83 (m, 2H, Cy), 2.65–2.59 (m, 2H, Cy), 2.24–2.21 (m, 2H, Cy), 2.17–2.11 (m, 4H, Cy), 2.01–1.99 (m, 2H, Cy), 1.96–1.87 (m, 10H, Cy), 1.82–1.80 (m, 4H, Cy), 1.57–1.22 (m, 14H, Cy), −26.11 (s, 1H, RuH). 13C{1H} NMR: δC (126 MHz, THF-d8, 298 K, [BArF4]− signals omitted) 205.9 (s, RuCO), 161.8 (s, RuCNHC), 127.4 (s), 125.9 (s, NCO), 84.9 (s), 83.9 (s, OCH2), 66.9 (s), 66.1 (s, CCy), 37.5 (s), 36.8 (s), 36.7 (s), 35.4 (s), 26.6 (s), 25.8 (s), 25.6 (s), 24.7 (s), 24.6 (s), 24.4 (s, CH2Cy). IR (THF, cm−1): 1929 (νCO). Anal. calcd for C71H69BN4O6F24Ru·C6H5F: C, 53.2; H, 4.29; N, 3.22. Found: C, 52.82; H, 4.22; N, 2.86.[Ru(IBiox6)2(CO)(THF)ZnMe][BArF4] (12)
Addition of ZnMe2 (67 μL of 1.0 M in toluene, 0.067 mmol) to a J. Young's resealable NMR tube containing a C6H5F solution (0.6 mL) of 11 (100 mg, 0.061 mmol) resulted in an instantaneous colour change from yellow to orange. The reaction mixture was evaporated to dryness, redissolved in C6H5F (0.3 mL) and layered with hexane to yield orange crystals of 12. Yield 69 mg (66%). 1H NMR: δH (500 MHz, THF-d8, 298 K) 7.79 (8H, [BArF4]−), 7.57 (4H, [BArF4]−), 4.78 (br s, 8H, CH2O), 2.89 (br s, 4H, Cy), 2.71 (br s, 4H, Cy), 2.08–2.03 (m, 8H, Cy), 1.99–1.92 (m, 8H, Cy), 1.82–1.76 (m, 8H, Cy), 1.54–1.47 (m, 4H, Cy), −0.01 (s, 1H, ZnCH3). 13C{1H} NMR: δC (126 MHz, THF-d8, 298 K, [BArF4]− signals omitted) 209.5 (s, RuCO), 161.8 (s, RuCNHC), 127.5 (s, NCO), 84.2 (s, OCH2), 68.4 (s, CCy), 36.4 (s), 26.6 (s), 25.4 (s), 24.7 (s), 24.5 (s, CH2Cy), −0.28 (s, ZnCH3). IR (THF, cm−1): 1929 (νCO).[Ru(IBioxMe4)2(CO)HCl] (13)
[Ru(AsPh3)3(CO)H2] (1.11 g, 1.06 mmol) and IBioxMe4 (552 mg, 2.65 mmol) were dissolved in toluene (10 mL) and the solution heated in a J. Young's resealable ampoule overnight at 363 K. After addition of 102 μL (1.59 mmol) of CH2Cl2, heating was continued at 393 K for a further 24 h. After that time a solid had formed, which was isolated by cannula filtration and washed with hexane (2 × 10 mL) and EtOH (10 mL) to give 13 as a yellow powder. Yield 424 mg (69%). Yellow crystals suitable for X-ray diffraction were obtained layering a CH2Cl2 solution of the complex with hexane. 1H NMR: δH (500 MHz, CDCl3, 298 K) 4.48 (s, 8H, CH2O), 1.95–1.65 (s, 24H, CH3), −25.14 (s, 1H, RuH). 13C{1H} NMR: δC (126 MHz, CDCl3, 298 K) 203.2 (s, RuCO), 166.6 (s, RuCNHC), 125.4 (s), 124.5 (s, NCO), 88.0 (s), 87.4 (s, OCH2), 61.2 (s), 60.7 (s, C(CH3)2), 27.6 (s), 27.1 (s), 26.3 (s), 26.1 (s, CH3). IR (CDCl3, cm−1): 1895 (νCO). Anal. calcd for C23H33N4O5ClRu·0.5C6H5F: C, 49.56; H, 5.68; N, 8.89. Found: C, 48.97; H, 5.59; N, 8.51.[Ru(IBioxMe4)2(THF)(CO)H][BArF4] (14)
Na[BArF4] (168 mg, 0.19 mmol) was added to a C6H5F solution (15 mL) of 13 (100 mg, 0.17 mmol) and the suspension stirred overnight at room temperature. After filtration, the filtrate was evaporated to dryness and the residue redissolved in THF (4 mL). Addition of hexane and vigorous stirring for 15 min afforded 14 as a brown solid. Yield 190 mg (79%). Crystals suitable for X-ray diffraction were obtained by slow diffusion of hexane into a concentrated THF solution of 14 at room temperature. 1H NMR: δH (400 MHz, THF-d8, 298 K) 7.80 (s, 8H, [BArF4]−), 7.59 (s, 4H, [BArF4]−), 4.71–4.66 (m, 4H, OCH2), 4.61–4.57 (m, 4H, OCH2), 1.90 (s, 6H, CH3), 1.85 (s, 6H, CH3), 1.74 (s, 6H, CH3), 1.59 (s, 6H, CH3), −26.13 (s, 1H, RuH). 13C{1H} NMR: δC (101 MHz, THF, 298 K, [BArF4]− signals omitted) 203.8 (s, RuCO), 161.1 (s, RuCNHC), 126.2 (s), 125.1 (s, NCO), 87.6 (s), 86.8 (s, OCH2), 61.7 (s), 61.0 (s, C(CH3)2), 26.0 (s), 25.8 (s), 25.3 (s), 25.3 (s, CH3). IR (THF, cm−1): 1933 (νCO). Anal. calcd for C55H45BF24N4O5Ru.C4H8O: C, 47.82; H, 3.6; N, 3.78. Found: C, 47.89; H, 3.55; N, 3.14.[Ru(IMes)′(PPh3)(CO)ZnMe] (15)
Rapid addition of ZnMe2 (1.22 mL of 1.2 M solution in toluene, 1.46 mmol) to a J. Young's resealable ampoule containing a THF solution (10 mL) of [Ru(IMes)(PPh3)(CO)HCl] (214 mg, 0.292 mmol) resulted in an instantaneous colour change from yellow to dark orange. The reaction mixture was stirred for 5 min, concentrated to ca. 1 mL and Et2O (15 mL) added. After filtration through a short pad of Celite®, the filtrate was left to stand at room temperature, whereby an initial batch of orange crystals (94 mg, 42% yield) formed. After separation by filtration, the mother liquor was concentrated (ca. 8 mL), left to stand, and a further 40 mg of crystalline product was formed. This was shown by NMR spectroscopy to comprise of ca. 92% 15 and ca. 8% of a second product, which we assign as [Ru(IMes)(PPh3)(CO)(ZnMe)Cl] (16; ESI†). In solution, 15 was found to exist as a mixture of two forms, believed to be the conformers 15a and 15b. 1H NMR of 15a: δH (400 MHz, C6D6, 298 K) 7.57–7.46 (m, 6H, PPh3), 7.06–6.94 (m, 9H, PPh3), 6.84 (s, 1H, Ar), 6.72 (s, 1H, Ar), 6.64 (s, 1H, Ar), 6.38 (d, 3JHH = 1.7 Hz, 1H, NCHNCH), 6.21 (d, 3JHH = 1.7 Hz, 1H, NCHNCH), 6.00 (s, 1H, Ar), 3.17 (br t, 2JHH = 3JHP = 5.9 Hz, 1H, RuCHH), 2.27 (s, 3H, CH3), 2.23 (s, 3H, CH3), 2.08 (s, 6H, CH3), 1.99 (s, 6H, CH3), 1.59 (dd, 3JHP = 11.6 Hz, 2JHH = 6.5 Hz, 1H, RuCHH), −0.57 (s, 3H, ZnCH3). 31P{1H} NMR: δP (162 MHz, C6D6, 298 K) 52.1 (s). 13C{1H} NMR: δC (101 MHz, C6D6, 298 K) 206.7 (d, 2JCP = 11 Hz, RuCO), 196.2 (d, 2JCP = 83 Hz, RuCNHC), 139.1 (s, Ar), 138.9 (d, JCP = 12 Hz, PPh3), 137.5 (s, Ar), 136.6 (s, Ar), 136.3 (s, Ar), 135.2 (s, Ar), 134.4 (d, JCP = 12.0 Hz, PPh3), 133.1 (s, Ar), 131.6 (s, Ar), 130.5 (s, Ar), 130.2 (s, Ar), 129.1 (d, JCP = 1 Hz, PPh3), 128.9 (s, Ar), 128.1 (s, Ar), 125.7 (s, Ar), 122.3 (d, 4JCP = 3 Hz, NCHCHN), 120.3 (d, 4JCP = 3 Hz, NCHCHN), 31.6 (d, 2JCP = 7 Hz, RuCH2), 21.3 (s, CH3), 21.1 (s, CH3), 19.0 (s, CH3), 18.7 (s, CH3), 18.1 (s, CH3), −1.5 (d, 3JCP = 3 Hz, ZnCH3). 1H NMR: δH (500 MHz, THF-d8, 298 K) 7.47–7.13 (m, 17H, Ar + NCHCHN), 7.00 (s, 1H, Ar) 6.99 (s, 1H, Ar), 6.62 (s, 1H, Ar), 5.55 (s, 1H, Ar), 2.61 (br t, 2JHH = 3JHP = 5.6 Hz, 1H, RuCHH), 2.34 (s, 3H, CH3), 2.30 (s, 3H, CH3), 2.25 (s, 3H, CH3), 2.17 (s, 3H, CH3), 1.87 (s, 3H, CH3), 0.98 (dd, 3JHP = 11.6 Hz, 2JHH = 6.7 Hz, 1H, RuCHH), −1.21 (s, 3H, ZnCH3). 31P{1H} NMR: δP (202 MHz, THF-d8, 298 K) 50.1 (s). Selected 13C{1H} NMR: δC (126 MHz, THF-d8, 298 K) 206.4 (d, 2JCP = 11 Hz, RuCO), 195.9 (d, 2JCP = 83 Hz, RuCNHC), 31.1 (d, 2JCP = 7 Hz, RuCH2), −2.4 (d, 3JCP = 4 Hz, ZnCH3). 15b: 1H NMR: δH (500 MHz, THF-d8, 298 K) 7.73 (s, 1H, NCHNCH), 7.47–7.13 (m, 16 H, PPh3 and NCHNCH), 6.95 (s, 1H, Ar), 6.92 (s, 1H, Ar), 6.46 (s, 1H, Ar), 6.03 (s, 1H, Ar), 2.28 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.16 (s, 3H, CH3), 2.12 (s, 3H, CH3), 1.86 (s, 3H, CH3), 1.52 (m, 1H, RuCHH), 1.40 (dd, 3JHP = 14.4 Hz, 2JHH = 8.6 Hz, 1H, RuCHH), −0.90 (s, 3H, ZnCH3). 31P{1H} NMR: δP (202 MHz, THF-d8, 298 K) 57.1 (s). Selected 13C{1H} NMR: δC (126 MHz, THF-d8, 298 K) 203.3 (d, 2JCP = 8 Hz, RuCO), 200.1 (d, 2JCP = 82 Hz, RuCNHC), 31.7 (d, 2JCP = 9 Hz, RuCH2), −4.0 (s, ZnCH3). IR (KBr, cm−1): 1860 (νCO). Anal. calcd for C41H41N2OPRuZn: C, 63.52, H, 5.33, N, 3.61. Found: C, 63.30, H, 5.30, N, 3.69.
[Ru(IMes)′(PPh3)(CO)(H)2ZnMe] (17)
Addition of H2 (1 atm) to a J. Young's resealable ampoule tube containing a Et2O solution (1 mL) of 15 (34 mg, 0.044 mmol) resulted in an instantaneous colour change from red-orange to colourless/pale yellow. After 30 min, the solvent was removed, the solid washed with Et2O (3 × 0.5 mL) and the colourless solid dried under vacuum. Yield 19 mg (55%). 1H NMR: δH (500 MHz, C6D6, 298 K) 7.71–7.65 (m, 6H, Ar), 7.09–6.96 (m, 9H, Ar), 6.89 (d, 3JHH = 1.3 Hz, 1H, NCHCHN), 6.77 (s, 1H, Ar), 6.74 (s, 1H, Ar), 6.68 (s, 1H, Ar), 6.62 (s, 1H, Ar), 6.17 (d, 3JHH = 1.4 Hz, 1H, NCHCHN), 3.22 (d, 2JHH = 9.2 Hz, 1H, RuCHH), 2.26 (s, 3H, CH3), 2.14 (s, 3H, CH3), 2.13 (s, 3H, CH3), 2.07 (s, 6H, CH3), 1.83 (dd, 3JHP = 12.3 Hz, 2JHH = 9.6 Hz, 1H, RuCHH), −1.29 (s, 3H, ZnCH3), −6.77 (d, 2JHP = 14.9 Hz, 1H, RuHZn), −9.19 (d, 2JHP = 5.0 Hz, RuHZn). 31P{1H} NMR: δP (202 MHz, C6D6, 298 K) 55.4 (s).13C{1H} NMR: δC (126 MHz, C6D6, 298 K) 203.0 (d, 2JCP = 14 Hz, RuCO), 194.6 (d, 2JCP = 83 Hz, RuCNHC), 156.0 (s, Ar), 139.9 (s, Ar), 138.7 (d, JCP = 38 Hz, PPh3), 137.8 (s, Ar), 137.1 (s, Ar), 136.7 (s, Ar), 135.4 (s, Ar), 135.1(s, Ar), 134.4 (d, JCP = 11 Hz, PPh3), 130.8 (s, Ar), 130.5 (s, Ar), 129.4 (s, PPh3), 128.8 (s, Ar), 125.2 (s, Ar), 121.8 (d, 4JCP = 2 Hz, NCHCHN), 119.5 (d, 4JCP = 3 Hz, NCHCHN), 21.3 (s, CH3), 21.1 (s, CH3), 19.8 (s, CH3), 18.7 (s, CH3), 18.4 (s, CH3), 7.7 (d, 2JCP = 7 Hz, RuCH2), −5.4 (s, ZnCH3). IR (KBr, cm−1): 1941 (νCO). Efforts to record elemental analyses repeatedly gave low %C values (e.g. Anal. calcd for C41H43N2OPRuZn: C, 63.36, H, 5.58, N, 3.60. Found: C, 61.23, H, 5.40, N, 3.65) which we attribute to the decomposition of the compound with time.
X-ray crystallography
Data for 7, 9, 11 and 12 were collected using an Agilent Xcalibur diffractometer while those for 8, 10, 13, 14, 15 and 17 were obtained using an Agilent SuperNova instrument (Table 2). All experiments were conducted at 150 K, solved using charge-flipping algorithm implemented in Olex224 and refined using SHELXL.25 In structures where disorder was observed in a [BArF4] anion, C–F, F⋯F, C⋯F and ADP restraints were applied, on merit. Otherwise, refinements were largely straightforward. Hence, only points of merit will be detailed hereafter. The asymmetric unit in 7 comprises one cation and one anion. The hydrogens attached to C55 in the former were located and refined subject to having similar C–H bond distances and to being equidistant from each other. F7, F8 and F9 were each disordered over 2 sites in the anion. H1, H2 and H3 in the cationic portion of compound 8 were readily located and, after some effort, an assignment was also made for H4. The associated Uiso values were refined freely, and that for H4 is somewhat higher than one might expect. However, this may well reflect some movement in the ligated dihydrogen, wherein the constituent atoms were refined subject to being equidistant from Ru1 and at a distance of 0.75 Å from each other (the refined H–H distance, on this basis, is 0.75(1) Å). The bridging hydrides were refined without restraints. Residual electron density maxima in this structure are in the region of the anion CF3 groups, five of which merited disorder modelling. In particular, F7–F12 were each refined over 2 positions in a 50:50 disorder ratio while F1–F3 exhibited 70:30 disorder. Moreover, the entire CF3 moieties based on C71 and C80 were refined to take account of 70:30 and 55:45 disorder levels, respectively.
| Identification code | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| Empirical formula | C88H87BF24N4ORuZn | C88H91BF24N4ORuZn | C89H91BCl2F24N4ORuZn | C41H55ClN4O5Ru | C77H74BF25N4O6Ru |
| Formula weight | 1849.86 | 1853.89 | 1936.80 | 820.41 | 1738.28 |
| Crystal system | Orthorhombic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
C2/c |
P |
| a/Å | 16.4346(4) | 12.7837(2) | 13.1638(4) | 22.2537(14) | 14.9600(4) |
| b/Å | 21.1397(5) | 17.1267(3) | 17.7078(6) | 13.5448(8) | 15.8674(5) |
| c/Å | 24.5462(5) | 20.4172(3) | 19.6253(6) | 12.4203(7) | 16.5741(5) |
| α/° | 90 | 84.063(1) | 95.885(2) | 90 | 87.261(3) |
| β/° | 90 | 88.767(1) | 94.337(2) | 93.810(5) | 80.381(2) |
| γ/° | 90 | 85.402(1) | 98.208(2) | 90 | 75.276(2) |
| U/Å3 | 8527.9(3) | 4431.51(12) | 4485.0(2) | 3735.5(4) | 3751.5(2) |
| Z | 4 | 2 | 2 | 4 | 2 |
| ρ calc/g cm−3 | 1.441 | 1.389 | 1.434 | 1.459 | 1.539 |
| μ/mm−1 | 0.559 | 0.538 | 0.593 | 4.463 | 0.327 |
| F(000) | 3784.0 | 1900.0 | 1980.0 | 1720.0 | 1772.0 |
| Crystal size/mm3 | 0.528 × 0.38 × 0.378 | 0.317 × 0.132 × 0.103 | 0.577 × 0.493 × 0.41 | 0.324 × 0.214 × 0.167 | 0.56 × 0.542 × 0.434 |
| Radiation | MoKα | MoKα | MoKα | CuKα | MoKα |
| 2θ range for data collection/° | 6.668 to 54.968 | 5.164 to 61.016 | 6.794 to 54.968 | 10.28 to 145.568 | 6.87 to 54.968 |
| Index ranges | −20 ≤ h ≤ 18 | −18 ≤ h ≤ 18 | −17 ≤ h ≤ 17 | −26 ≤ h ≤ 27 | −17 ≤ h ≤ 19 |
| −27 ≤ k ≤ 27 | −24 ≤ k ≤ 24 | −22 ≤ k ≤ 19 | −16 ≤ k ≤ 16 | −18 ≤ k ≤ 20 | |
| −31 ≤ l ≤ 31 | −29 ≤ l ≤ 28 | −24 ≤ l ≤ 22 | −15 ≤ l ≤ 9 | −21 ≤ l ≤ 21 | |
| Reflections collected | 77177 |
159961 |
40172 |
16638 |
46082 |
| Independent reflections, Rint | 19171, 0.0429 |
26703, 0.0629 |
19792, 0.0362 |
3649, 0.0935 | 16803, 0.0392 |
| Data/restraints/parameters | 19171/67/1136 |
26703/157/1267 |
19792/240/1246 |
3649/0/249 | 16803/334/1188 |
| Goodness-of-fit on F2 | 1.077 | 1.042 | 1.037 | 1.074 | 1.063 |
| Final R1, wR2, I ≥ 2σ(I) | 0.0511, 0.1112 | 0.0503, 0.1188 | 0.0467, 0.1017 | 0.0512, 0.1314 | 0.0617, 0.1407 |
| Final R1, wR2, all data | 0.0680, 0.1206 | 0.0694, 0.1298 | 0.0675, 0.1136 | 0.0531, 0.1326 | 0.0813, 0.1516 |
| Largest diff. peak/hole/e Å−3 | 0.83/−0.86 | 0.76/−1.18 | 0.88/−0.57 | 0.68/−0.64 | 2.80/−0.68 |
| Flack parameter | 0.017(4) | — | — | — | — |
| Identification code | 12 | 13 | 14 | 15 | 17 |
|---|---|---|---|---|---|
| Empirical formula | C72H71BF24N4O6RuZn | C23H32ClN4O5Ru | C59H52BF24N4O6Ru | C41H41N2OPRuZn | C41H43N2OPRuZn |
| Formula weight | 1721.57 | 581.04 | 1480.92 | 775.17 | 777.18 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group |
P |
P21/n |
P |
P21/c |
P |
| a/Å | 12.9608(4) | 10.2772(2) | 9.9024(1) | 10.5880(1) | 9.8428(2) |
| b/Å | 13.1449(4) | 11.4391(2) | 18.4531(2) | 25.6243(2) | 11.4984(3) |
| c/Å | 21.6148(7) | 10.8292(2) | 19.4882(3) | 13.6538(1) | 17.9210(3) |
| α/° | 94.981(3) | 90 | 112.078(1) | 90 | 87.163(2) |
| β/° | 91.296(3) | 99.331(2) | 103.002(1) | 103.780(1) | 81.171(2) |
| γ/° | 97.163(3) | 90 | 96.389(1) | 90 | 65.917(2) |
| U/Å3 | 3637.8(2) | 1256.26(4) | 3138.80(7) | 3597.79(5) | 1829.55(7) |
| Z | 2 | 2 | 2 | 4 | 2 |
| ρ calc/g cm−3 | 1.572 | 1.536 | 1.567 | 1.431 | 1.411 |
| μ/mm−1 | 0.654 | 6.375 | 3.144 | 4.896 | 4.814 |
| F(000) | 1748.0 | 598.0 | 1494.0 | 1592.0 | 800.0 |
| Crystal size/mm3 | 0.561 × 0.375 × 0.27 | 0.072 × 0.058 × 0.019 | 0.283 × 0.229 × 0.094 | 0.471 × 0.095 × 0.092 | 0.116 × 0.08 × 0.05 |
| Radiation | MoKα | CuKα | CuKα | CuKα | CuKα |
| 2θ range for data collection/° | 6.678 to 54.966 | 11.012 to 146.588 | 5.636 to 147.162 | 6.9 to 146.304 | 4.99 to 146.094 |
| Index ranges | −16 ≤ h ≤ 16 | −12 ≤ h ≤ 12 | −9 ≤ h ≤ 12 | −13 ≤ h ≤ 9 | −12 ≤ h ≤ 12 |
| −17 ≤ k ≤ 16 | −14 ≤ k ≤ 11 | −22 ≤ k ≤ 22 | −31 ≤ k ≤ 31 | −14 ≤ k ≤ 14 | |
| −27 ≤ l ≤ 28 | −11 ≤ l ≤ 13 | −24 ≤ l ≤ 24 | −16 ≤ l ≤ 16 | −20 ≤ l ≤ 22 | |
| Reflections collected | 33375 |
13001 |
62311 |
47648 |
21138 |
| Independent reflections, Rint | 16641, 0.0262 |
2531, 0.0392 | 12599, 0.0418 |
7195, 0.0440 | 7307, 0.0340 |
| Data/restraints/parameters | 16641/264/1097 |
2531/0/173 | 12599/2804/1493 |
7195/2/438 | 7307/2/446 |
| Goodness-of-fit on F2 | 1.027 | 1.194 | 1.039 | 1.056 | 1.020 |
| Final R1, wR2, I ≥ 2σ(I) | 0.0441, 0.1015 | 0.0343, 0.0721 | 0.0530, 0.1471 | 0.0279, 0.0729 | 0.0274, 0.0650 |
| Final R1, wR2, all data | 0.0580, 0.1096 | 0.0378, 0.0736 | 0.0573, 0.1523 | 0.0294, 0.0740 | 0.0311, 0.0672 |
| Largest diff. peak/hole/e Å−3 | 0.98/−0.65 | 0.50/−0.55 | 1.46/−1.04 | 1.02/−0.62 | 0.61/−0.52 |
In 9, the asymmetric unit contains one cation, one anion and one molecule of CH2Cl2. H1 and H2 in the cation were located a refined without restraints. The hydrogens attached to C26 were similarly located and refined subject to being located at a distance of 0.98 Å from C26. Fluoride disorder was modelled for two of the [BArF4] CF3 moieties. In particular, F1–F3 were disordered over two sites in a 75:25 ratio while F13–15 were disordered over three sites in a 50:40:10 ratio. The solvent molecule exhibited 55:45 disorder and C–Cl distances were restrained to being similar in both moieties. ADP restraints were included for fractional occupancy atoms. The asymmetric unit in 10 equates to half of one molecule of the complex, and half of a benzene molecule. The chloride and carbonyl ligands within the metal complex are disordered with each other in a 50:50 ratio. The hydride ligand (which is likely to be disordered over 2 sites) could not be reliably located and, hence, was omitted from the refinement. One cation, one anion and two independent fluorobenzene halves constitute the asymmetric unit in 11. The solvent moieties are proximate to crystallographic inversion centres which serve, in each case, to generate the remaining molecule portions. The halides in these solvent moieties are necessarily disordered and, hence, exhibit half site-occupancies. 75:25 disorder was also modelled for C22 in the cation, with chemically similar distances in each disordered component being restrained to being similar. The hydride was also located and is disordered in a 50:50 ratio. The associated metal-hydride distances were refined subject to a 1.6 Å, Ru–H, distance restraint. Anion disorder was limited to the halides in five of the CF3 functionalities. Specifically, the fluorines attached to C46, C54, C55, C63 and C70 exhibited disorder ratios of 65:35, 55:45, 50:50, 75:25 and 75:25, respectively.
Some disorder modelling was necessary in both that cation and the anion present in the asymmetric unit of compound 12. In the cation, this pertained to 55:45 disorder confined to atoms C37 and C38 in the THF ligand. Chemically equivalent distances involving the partial occupancy atoms were restrained to being similar in the final least-squares and some ADP restraints were also included for same. Four of the CF3 groups in the anion were seen to exhibit disorder. In particular, the fluorine atoms attached to C47, C56, C63 and C72 were each modelled over 2 sites, in ratios of 55:45, 60:40, 55:45 and 75:25, respectively. The asymmetric unit in 13 comprises half of a molecule, with the central ruthenium located at a crystallographic inversion centre. This necessarily means that the chloride and carbonyl ligands are disordered in a 50:50 ratio. An exemplary diffraction pattern was observed for crystal of compound 14 where the asymmetric unit was seen to contain one cation and one anion. There was no evident twinning but, yet, the structural motif itself is riddled with disorder. While this was successfully modelled, it has inevitably resulted in the addition of a large number of restraints to the model, as both carbene ligands in the cation were seen to be disordered in a 50:50 ratio. The carbene carbons are, in each ligand, common to both components. In addition, C26 in the THF ligand was also seen to exhibit disorder, which optimally refined to a 60:40 ratio. Distance similarity restraints and ADP restraints were added to the model for the cation, on merit, in the final refinement cycles. In the BArF4 anion, three of the rings were seen to be disordered in an 80:20 ratio. The moiety based on C36 did not exhibit disorder to a level that could be credibly modelled, although the CF3 group based on C42 was treated for 70:30 disorder.
In 15, both H3a and H3b were located and subsequently refined subject to each being a distance of 0.98 Å from C3. In a similar vein, the hydrogens attached to C3 were also readily located in 17, and each refined subject to being situated at distance of 0.95 Å from the parent atom. Finally, the bridging hydride ligands were also located in this compound and refined without restraints.
Crystallographic data for all compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 1882150 (compound 7), 1882152 (8), 1882151 (9), 1882153 (10), 1882154 (11), 1882155 (12), 1882156 (13), 1882157 (14), 1882158 (15) and 1882159 (17).†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EU (Marie Curie Individual Fellowships to ME-V (700189 H2020-MSCA-IF 2015) and FMM (792674 H2020-MSCA-IF 2017)) and EPSRC (grant EP/J009962/1; IMR) for financial support. VV-I thanks MINECO/FEDER (CTQ2017-83421-P) and Gobierno de Aragón (GA/FEDER, Inorganic Molecular Architecture Group E08_17R) for financial support and MINECO/FEDER for a FPI fellowship. We thank Dr Adrian Chaplin for an initial gift of IBiox6 and for subsequent assistance in the syntheses of the IBiox ligands. We are delighted to dedicate this paper to Professor Geoff Cloke FRS on the occasion of his 65th birthday.References
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- (a) W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed; (b) W. H. Harman, T.-P. Lin and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 1081–1086 CrossRef CAS PubMed; (c) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef CAS PubMed; (d) M. A. Nesbit, D. L. M. Suess and J. C. Peters, Organometallics, 2015, 34, 4741–4752 CrossRef CAS; (e) S. N. MacMillan, W. H. Harman and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC; (f) M. Devillard, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 730–732 CrossRef CAS PubMed; (g) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC; (h) K. N. T. Tseng, J. W. Kampf and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 10378–10381 CrossRef CAS PubMed; (i) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed; (j) G. R. Owen, Chem. Commun., 2016, 52, 10712–10726 RSC; (k) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- J. A. B. Abdalla and S. Aldridge, in Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties, ed. S. T. Liddle, Wiley-VCH, Weinheim, 2015, pp. 455–484 Search PubMed.
- (a) F. N. Tebbe, J. Am. Chem. Soc., 1973, 95, 5412–5414 CrossRef CAS; (b) J. N. St. Denis, W. Butler, M. D. Glick and J. P. Oliver, J. Organomet. Chem., 1977, 129, 1–16 CrossRef; (c) P. H. M. Budzelaar, K. H. Denhaan, J. Boersma, G. J. M. Vanderkerk and A. L. Spek, Organometallics, 1984, 3, 156–159 CrossRef CAS; (d) P. H. M. Budzelaar, A. A. H. Vanderzeijden, J. Boersma, G. J. M. Vanderkerk, A. L. Spek and A. J. M. Duisenberg, Organometallics, 1984, 3, 159–163 CrossRef CAS; (e) W. A. Skupiński, J. C. Huffman, J. W. Bruno and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 8128–8136 CrossRef; (f) D. L. Thorn and R. L. Harlow, J. Am. Chem. Soc., 1989, 111, 2575–2580 CrossRef CAS; (g) M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1990, 9, 1359–1360 CrossRef CAS; (h) M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1993, 12, 2152–2161 CrossRef CAS; (i) J. T. Golden, T. H. Peterson, P. L. Holland, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1998, 120, 223–224 CrossRef CAS; (j) C. J. Durango-García, J. O. C. Jiménez-Halla, M. López-Cardoso, V. Montiel-Palma, M. A. Muñoz-Hernández and G. Merino, Dalton Trans., 2010, 39, 10588–10589 RSC.
- I. M. Riddlestone, D. McKay, M. J. Gutmann, S. A. Macgregor, M. F. Mahon, H. A. Sparkes and M. K. Whittlesey, Organometallics, 2016, 35, 1301–1312 CrossRef CAS.
- (a) I. M. Riddlestone, N. A. Rajabi, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Chem. – Eur. J., 2018, 24, 1732–1738 CrossRef CAS PubMed; (b) I. M. Riddlestone, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2016, 138, 11081–11084 CrossRef CAS PubMed.
- For a review that includes discussion of transition metal-H–Zn species, see: M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- For other examples of Ru–H–Zn complexes, see: (a) M. Molon, C. Gemel and R. A. Fischer, Eur. J. Inorg. Chem., 2013, 3616–3622 CrossRef CAS; (b) M. Plois, W. Hujo, S. Grimme, C. Schwickert, E. Bill, B. de Bruin, R. Pöttgen and R. Wolf, Angew. Chem., Int. Ed., 2013, 52, 1314–1318 CrossRef CAS PubMed; (c) M. Plois, R. Wolf, W. Hujo and S. Grimme, Eur. J. Inorg. Chem., 2013, 3039–3048 CrossRef CAS; (d) S. Lau, A. J. P. White, I. J. Casely and M. R. Crimmin, Organometallics, 2018, 37, 4521–4526 CrossRef CAS.
- J. P. Lee, Z. L. Ke, M. A. Ramírez, T. B. Gunnoe, T. R. Cundari, P. D. Boyle and J. L. Petersen, Organometallics, 2009, 28, 1758–1775 CrossRef CAS.
- For a review of Ru–Zn containing complexes, see: T. Bollermann, C. Gemel and R. A. Fischer, Coord. Chem. Rev., 2012, 256, 537–555 CrossRef CAS.
- As the nature of the hydrides in the {Ru(H)2Zn} moiety is ambiguous, we did not adopt the half-arrow formalism of Parkin. J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11841–11503 RSC.
- (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS PubMed; (b) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed.
- (a) A. B. Chaplin, Organometallics, 2014, 33, 3069–3077 CrossRef CAS; (b) A. B. Chaplin, Organometallics, 2014, 33, 624–626 CrossRef CAS; (c) J. N. Luy, S. A. Hauser, A. B. Chaplin and R. Tonner, Organometallics, 2015, 34, 5099–5112 CrossRef CAS; (d) S. A. Hauser, R. Tonner and A. B. Chaplin, Organometallics, 2015, 34, 4419–4427 CrossRef CAS; (e) J.-N. Luy, S. A. Hauser, A. B. Chaplin and R. Tonner, Organometallics, 2016, 34, 5099–5112 CrossRef.
- K. Vehlow, M. Porta and S. Blechert, ChemCatChem, 2010, 2, 803–806 CrossRef CAS.
- V. L. Chantler, S. L. Chatwin, R. F. R. Jazzar, M. F. Mahon, O. Saker and M. K. Whittlesey, Dalton Trans., 2008, 2603–2614 RSC.
- (a) S. L. Chatwin, M. G. Davidson, C. Doherty, S. M. Donald, R. F. R. Jazzar, S. A. Macgregor, G. J. McIntyre, M. F. Mahon and M. K. Whittlesey, Organometallics, 2006, 25, 99–110 CrossRef CAS; (b) T. E. Wang, C. Pranckevicius, C. L. Lund, M. J. Sgro and D. W. Stephan, Organometallics, 2013, 32, 2168–2177 CrossRef CAS; (c) C. Pranckevicius, L. Fan and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 5582–5589 CrossRef CAS PubMed.
- D. Huang, J. C. Bollinger, W. E. Streib, K. Folting, J. V. Young, O. Eisenstein and K. G. Caulton, Organometallics, 2000, 19, 2281–2290 CrossRef CAS.
- NHC centroids were calculated from the central five-membered IBiox ring containing the coordinated carbenic carbon, the two nitrogen atoms and the two backbone carbons.
- Addition of excess ZnMe2 (or ZnEt2) to 14 in C6H5F resulted in complete loss of the hydride resonance of the starting material and a change in the colour of the solution from orange to purple. However, within a few minutes, the solution turned black. Nothing conclusive could be extracted from analysis of the 1H NMR spectrum and efforts to identify products by crystallisation proved unsuccessful.
- (a) R. F. R. Jazzar, S. A. Macgregor, M. F. Mahon, S. P. Richards and M. K. Whittlesey, J. Am. Chem. Soc., 2002, 124, 4944–4945 CrossRef CAS PubMed; (b) T. M. Trnka, J. P. Morgan, M. S. Sanford, T. E. Wilhelm, M. Scholl, T. L. Choi, S. Ding, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 2546–2558 CrossRef CAS PubMed; (c) K. Abdur-Rashid, T. Fedorkiw, A. J. Lough and R. H. Morris, Organometallics, 2004, 23, 86–94 CrossRef CAS; (d) S. P. Reade, A. L. Acton, M. F. Mahon, T. A. Martin and M. K. Whittlesey, Eur. J. Inorg. Chem., 2009, 1774–1785 CrossRef CAS; (e) H. J. Liu, M. S. Ziegler and T. D. Tilley, Polyhedron, 2014, 84, 203–208 CrossRef CAS; (f) H. J. Liu, M. S. Ziegler and T. D. Tilley, Dalton Trans., 2018, 47, 12138–12146 RSC.
- (a) S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961–7968 CrossRef CAS PubMed; (b) G. A. Bailey, J. A. M. Lummiss, M. Foscato, G. Occhipinti, R. McDonald, V. R. Jensen and D. E. Fogg, J. Am. Chem. Soc., 2017, 139, 16446–16449 CrossRef CAS PubMed.
- Unsurprisingly, the reverse reaction (17-d2 + H2) gave 17.
- U. L. Dharmasena, H. M. Foucault, E. N. dos Santos, D. E. Fogg and S. P. Nolan, Organometallics, 2005, 24, 1056–1058 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 467–473 CrossRef CAS; G. M. Sheldrick, SHELXL-97, a computer program for crystal structure refinement, University of Göttingen, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1882150–1882159. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt05023f |
| This journal is © The Royal Society of Chemistry 2019 |