
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecision design of covalent organic frameworks for cathode applications
Zhiwei
Zhao
,
Di
Liu
and
Yang
Wang
 *
*
Department of Materials Science, State Key Laboratory of Molecular Engineering of Polymers, Laboratory of Advanced Materials, Fudan University, 2005, Songhu Road, Shanghai 200438, China. E-mail: yangwang@fudan.edu.cn
First published on 25th March 2025
Abstract
The urgent demand for sustainable energy storage solutions has positioned covalent organic frameworks (COFs) as promising alternatives to conventional inorganic cathodes. With their programmable architectures, high theoretical capacities, and elemental sustainability, COFs hold transformative potential for next-generation energy storage devices. Despite their promise, the practical implementation of COFs has been impeded by limitations such as low conductivity and lower-than-anticipated practical capacities. This review explores recent advances in molecular and structural engineering strategies designed to overcome these challenges. The discussion encompasses ion-storage mechanisms, innovative chemical design strategies, and composite material synergies that enhance the performance of COF cathodes (COFCs). Looking to the future, breakthroughs in multi-electron redox chemistry, scalable synthesis, and advances in in situ characterization techniques will be critical to unlocking the full potential of COFCs. This review aims to provide valuable insights and guidance for the design of novel COFC materials, thereby advancing the development of next-generation high-performance secondary batteries.
 Zhiwei Zhao | Zhiwei Zhao graduated from Zhejiang Sci-Tech university in 2021 with a Bachelor of engineering degree. He then joined the Department of Materials Science of Fudan University as a PhD student under the supervision of Prof. Yang Wang. His research interests mainly focus on the design and construction of Covalent Organic Frameworks (COFs) and their applications in electrochemical storage devices. |
 Di Liu | Di Liu is an assistant professor in the Laboratory of Advanced Materials at Fudan University. She earned her PhD from Pusan National University in South Korea in 2020. Following her postdoctoral research under the supervision of Prof. Dongyuan Zhao, she joined his group at Fudan University as a faculty member. Her research primarily focuses on the scale-up synthesis of porous carbon materials, encompassing both fundamental studies and their industrialization for energy applications. |
 Yang Wang | Yang Wang is an associate professor in the Department of Materials Science at Fudan University. He received his PhD degree under the supervision of Prof. T. Michinobu at Tokyo Institute of Technology, Japan, in 2017. After finishing the JSPS postdoctoral research under the guidance of Prof. K. Takimiya (RIKEN, Japan), he joined Prof. Yunqi Liu's group at Fudan University as a faculty member. His main research interests are molecular materials and devices, including conjugated polymers, covalent organic frameworks and multifunctional devices applications. |
1 Introduction
The urgency of mitigating fossil fuel dependence and climate change has intensified the global shift toward renewable energy systems.1 Central to this transition is the development of advanced energy storage technologies, which are critical for managing the intermittent and geographically uneven supply of renewables. While lithium-ion batteries (LIBs) have revolutionized modern industry, they still face escalating demands for improved longevity, safety, and adaptability to dynamic operating environments.2 Conventional cathode materials, such as transition-metal oxides and sulfides, remain constrained by intrinsic limitations. The lattice distortion and irreversible dissolution during cycling, leading to structural collapse and electrochemical degradation.3 Furthermore, parasitic side reactions at electrode–electrolyte interfaces, coupled with sluggish charge-transfer kinetics and inherently low theoretical capacities, collectively impair energy density and cycle stability.4 These challenges underscore the need for innovative cathode materials that circumvent the limitations plaguing traditional inorganic compounds.Organic molecules—composed of abundant and lightweight elements (C, H, O, N, P, S)—offer compelling advantages as cathode materials, including high theoretical capacities, tunable structure, and sustainability. Through molecular engineering, their electrochemical behavior (e.g., redox potential, electron transfer kinetics) can be precisely tailored, directly influencing device metrics such as energy density, voltage, and cycle life. Reported studies classify organic electrodes into small molecules and polymers. Small molecules, while synthetically accessible, suffer from poor conductivity, dissolution in electrolytes, and parasitic side reactions with metal-ions, leading to rapid capacity fade.5 Polymers, by contrast, exhibit enhanced stability in aqueous or organic electrolytes. Extended π-conjugated systems improve electron transport, yet excessive polymerization reduces mass energy density.6 Furthermore, disordered molecular stacking and structural defects in polymers hinder ion diffusion and active-site accessibility. Microstructural optimization offers a pathway to mitigate these limitations. Mesoporous designs—featuring interconnected pore networks and maximized exposed reaction sites—can enhance ion transport while preserving volumetric energy density.7 Such strategies narrow the gap between theoretical and practical performance, underscoring the critical role of porous architecture in advancing organic cathodes.
The critical role of electrode microstructure in ion transport and active-site accessibility has spurred interest in porous materials. Since the 2005 synthesis of the first crystalline covalent organic framework (COF-5), researchers have developed diverse COF derivatives with open pore channels, high surface areas, and robust stability (Fig. 1).8 The strategic design of building blocks and linkers enables precise control over structural parameters, such as pore geometry, topology, and functionalization, thereby enhancing their electrochemical energy storage performance. Redox-active groups grafted in COFs enhance charge storage and ion mobility, yet their ordered structures introduce complexity: multiple active sites within repeating units lead to intricate electron-transfer pathways, particularly in multi-ion systems (Fig. 1). A comprehensive understanding of how functional groups govern ion storage remains elusive, hindering rational design. To bridge this gap, systematic studies elucidating structure–mechanism relationships are essential. Concurrently, hybrid strategies integrating COFs with conductive porous materials (e.g., graphene, MXenes, CNTs, VN, MOFs etc.) can mitigate limitations such as poor conductivity and inaccessible active sites.9 Such composites leverage synergistic effects by combining tailored porosity with excellent conductivity to narrow the disparity between theoretical and practical capacities, advancing cathodes toward scalable energy storage applications.
 | ||
| Fig. 1 Timeline of COF cathodes in batteries. Reprinted with permission from ref. 10, Copyright 2015–2024 Royal Society of Chemistry, Springer Nature, Wiley-VCH, American Chemical Society. | ||
In this review, we systematically summarize the design of COF-based cathodes (COFCs), focusing on both molecular design and composite engineering. We also examine the ion storage mechanisms of COFCs and discuss the advantages and limitations of various energy storage sites, such as imide groups, carbonyl groups, and conjugated systems. Finally, we address the current challenges facing COFCs and explore future directions for the advancement of this field. We hope this review will provide valuable guidance for designing high-performance cathode materials and contribute to the development of next-generation energy storage devices.
2 Ion storage mechanism
The unique structural advantages of COFs have shown strong potential for energy storage, with applications spanning cathodes, electrolytes, separators, anode coating, and more.11 Initially, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in donor–acceptor COFs (D–A COFs) enabled two-electron reversible redox processes for lithiation and de-lithiation. Since then, a wide range of active ligands and linking groups have been shown to exhibit ion-storage properties. A systematic summary of the electron gain/loss, charge state changes, and ion storage mechanisms of recent COF-based materials can help clarify the structure–property relationship. COFCs can be categorized by ion-storage type: cation (Li+, Na+, Zn2+, K+, H+, Mg2+, et al.), anion (OTf−, TFSI−, PF6−, et al.), or bipolar (Fig. 2). The number of electrons involved in reactions must maintain system neutrality. The stability and theoretical capacity of different energy storage sites vary significantly, and strategically Integrating multiple redox-active groups within COF repeating units can enhance theoretical capacity (Ct), calculated as:12
O group in donor–acceptor COFs (D–A COFs) enabled two-electron reversible redox processes for lithiation and de-lithiation. Since then, a wide range of active ligands and linking groups have been shown to exhibit ion-storage properties. A systematic summary of the electron gain/loss, charge state changes, and ion storage mechanisms of recent COF-based materials can help clarify the structure–property relationship. COFCs can be categorized by ion-storage type: cation (Li+, Na+, Zn2+, K+, H+, Mg2+, et al.), anion (OTf−, TFSI−, PF6−, et al.), or bipolar (Fig. 2). The number of electrons involved in reactions must maintain system neutrality. The stability and theoretical capacity of different energy storage sites vary significantly, and strategically Integrating multiple redox-active groups within COF repeating units can enhance theoretical capacity (Ct), calculated as:12where n is the number of electrons transferred, F is Faraday's constant (96

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 486 C mol−1), and M is the molecular weight of the organic molecule. A deviation often exists between the specific capacity (Cm, mA h g−1) and the theoretical value. Cm can be calculated from Galvanostatic charge/discharge (GCD) curves using the following equations:
486 C mol−1), and M is the molecular weight of the organic molecule. A deviation often exists between the specific capacity (Cm, mA h g−1) and the theoretical value. Cm can be calculated from Galvanostatic charge/discharge (GCD) curves using the following equations:where I, Δt, and m represents the current density (A g−1), discharge time (s) and loading mass of cathode materials on the current collector, respectively. Accordingly, the real number of electron transfers (n) involved in the experimental redox reaction can be calculated by the following formula:

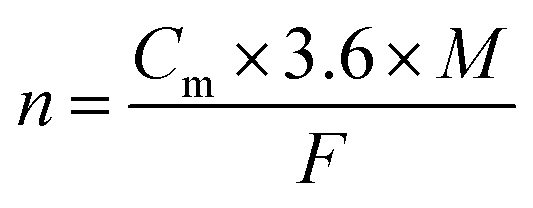
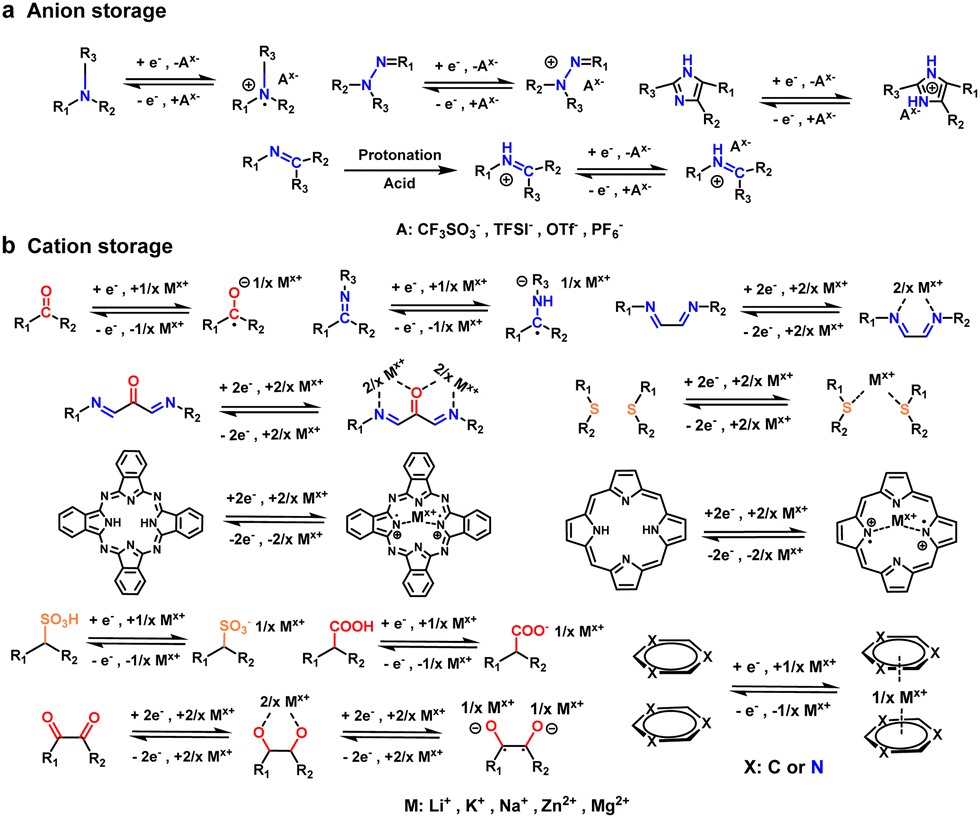 | ||
| Fig. 2 The structure of redox active sites and reversible ion storage mechanism of COFCs. (a) Anion storage; (b) cation storage. | ||
In crystalline frameworks, electrochemical activity is primarily localized at discrete sites within the specific bonds, owing to the non-participation of certain structural motifs in reversible ion storage processes. This inherent limitation necessitates a focus on the relative theoretical capacity of individual repeating units during molecular design. Experimental validation through device assembly, while critical, remains time-intensive and impractical for rapid material screening. To maximize energy density, molecular engineering should prioritize architectures where redox-active components constitute the largest possible proportion of the repeating unit's molecular mass (Table 1). By minimizing inert structural “scaffolding,” researchers can enhance the practical capacity of COF cathodes while preserving structural integrity—a design philosophy critical to advancing high-performance energy storage systems.
| Sample | Active sites | Battery type | Capacity/mA h g−1/(current density) | Ref. |
|---|---|---|---|---|
| TPAD-COF | CO, PhN+H– |
NIBs | 186.4 (0.05 A g−1) | 13 |
| TPDA-DHTA-COF | CO, hydrazone |
LIBs | 308 (0.2 A g−1) | 14 |
| 2DBBL-TP | CO |
ZIBs | 51 (200 A g−1) | 15 |
| BTT-PTO-COF | Thiophene, CO |
LIBs | 275 (0.2 A g−1) | 16 |
| Cu-TH-COF | CO, CN, Cu2+ |
LIBs | 300 (0.05 A g−1) | 17 |
| S@NiS4-TAPT | Ni-bis(dithiolene) | LSBs | 966.3 (1C) | 18 |
| S-DUT-177 | –S–S | LSBs | 720 (0.1 A g−1) | 19 |
| DAQ-HATN | CO, CN |
SAOBs | 80 (5 A g−1) | 20 |
CN and CO groups dominate as ion-storage sites in COF cathodes. CN active centers and their derivatives (such as pyridine, pyrimidine, pyrazine, triazine) typically undergo single-electron reversible reactions.21 During discharge, the imine group is first reduced to –C–N−–, which then combines with the cation to form –C–N−–(Mx+), and is oxidized back to –CN during charging. In addition to imine-based constructs, the –NN– linkage can undergo a two-electron reaction to form a –N–N−– state for energy storage. Carbonyl groups and their derivatives, such as para-quinones, ortho-quinones, and diketones further increase the number of electrons involved in redox processes, with their C–O−–(Mx+) and –O−–(Mx+)–O− states exhibiting higher theoretical capacities. However, carbonyl reduction products are less stable compared than imine groups. In aqueous electrolytes, the monovalent ionic reduced state (–C–O− with Li+, Na+, or K+) is prone to oxidation by dissolved oxygen, leading to significant self-discharge. In contrast, discharge products of multivalent metal-ions coordinating with CO groups show enhanced stability, offering a capacity advantage in aqueous multivalent metal batteries. However, multivalent metal-ion batteries typically face slow kinetics due to more difficult desolvation and stronger binding energies with redox sites. Electrostatic repulsion of multivalent metal-ions in the Helmholtz layer at the solid–liquid interface also increases the ion diffusion energy barrier.22 Recent studies have demonstrated that COFs can co-store H+/Zn2+ in Zn2+ organic cathodes; however, further research is needed to fully understand the competition between these ions and the source of the capacity.
Introducing electron-withdrawing groups (such as sulfonic and carboxylic acids) into the COF backbone can provide additional energy storage sites, though this design is less common due to disruption of conjugation in the main chain, which lowers discharge voltage. The role of sulfur-containing groups and ionic binding within conjugated stacks is still in the early stages of research, requiring further study to quantify the energy storage mechanisms and electron transfer. Additionally, anions in the electrolyte can also participate in energy storage, being oxidized to cations and then combining with the positively charged centers. The p-type functional groups (hydrazone, amino) lose electrons and are oxidized to form positively charged centers, which then combine with the anions from the electrolyte (TFSI−, OTf−, PF6−, CF3SO32−) to maintain the electrical neutrality of the system. Depending on the electrolyte type, anions often exhibit lower-than-theoretical capacities due to steric hindrance and electronegativity, primarily because of slow ionic diffusion and migration kinetics. p-Type sites are typically located at the intersections or linkage of the structures, and their structural fragility during the reversible process results in a low overall cycle lifetime. A common strategy to improve energy storage capacity and cycle stability is to introduce both p and n-type active groups to the system.
Strategic integration of active functional moieties via post-synthetic modification significantly enhances the energy storage capabilities. For example, acid-induced protonation of COFs, can enhance energy storage by introducing anionic binding sites and strengthening π-conjugation in two-dimensional conjugated polymers (2DCPs).23 Introducing metal ions into the structure through coordination expands π-conjugation, which can increase electron density and enhance the conductivity of the material. However, there are few reports on how post-treatment reactions can improve energy storage sites, and this area requires further investigation. Additionally, while molecular layer arrangements in powdered COF structures (e.g., AA-type, AB-type in 2D COFs) are common, the mechanism of ion intercalation energy storage has received limited attention. This represents a gap in understanding the source of energy storage capacity in COF anode materials, necessitating new electrochemical tests and instrumental characterization for further analysis.
Electrolytes play a pivotal role in the reversible transformation of ‘‘rocking chair batteries’’. The mass transfer rate in the liquid phase is the decision rate of the electrode reaction and the key factor affecting the stability.24 Materials with the same channel size cannot show excellent performance in different energy storage devices. There is an essential difference in solvation radius of ions in liquid, which affects the accessibility of desolvated ions to active sites. Electrolyte viscosity is positively correlated with mass transfer impedance. The non-homogeneous mass transfer process in the electrolyte containing complex multiphase additives is worthy of further exploration. Currently, most COF cathodes are used with inorganic electrolyte systems, with few studies exploring organic cathodes in relation to electrolyte systems and additives. This is largely because these groups undergo dynamic cycling processes with varying charged states (positive, negative, intermediate, half-filled, and transition states), resulting in significant differences in solubility and stability. The dissolution-redeposition phenomenon is common in organic battery systems.25 For instance, small-molecule electrodes often show poor cycle stability in LIBs but perform better in aqueous systems. The amorphous-to-crystalline transformation in cathode materials is closely linked to capacity decay. Researchers must integrate molecular engineering of redox-active motifs with electrolyte optimization, prioritizing compatibility between charge states, ion mobility, and interfacial dynamics. Such synergies will bridge the gap between theoretical predictions and practical performance, accelerating the development of robust, high-capacity energy storage systems.
3 Design of chemical structure
The previous paragraphs provided an overview of organic functional groups with energy storage capabilities and their bonding modes. As an emerging energy storage material, COFCs show promising application potential, but several challenges must be addressed before they can transition from the laboratory to industry. (1) Low density: although COFCs offer high capacity, their energy density per unit volume is significantly lower than that of inorganic materials, due to the lightweight elements used. Volume density is a key factor in evaluating device performance, and a smaller value means a higher energy storage capacity. (2) Low electrical conductivity: most COFs are synthesized through solvent-thermal methods, which introduce numerous grain boundaries and defects. This leads to electron trapping during transport, resulting in low conductivity. To compensate, excessive conductive agents are often added during slurry preparation, but this can lead to underestimation of energy density per unit mass. (3) Lack of stability: while the solvent resistance of COFs is a key advantage for wide application, some molecules with covalent-linked energy storage sites suffer from poor reversibility. The ion dissociation/insertion process can break covalent bonds, causing structural collapse and partial dissolution in the electrolyte. A rational structural design is needed to improve the energy storage performance. The following sections will focus on recent progress in the molecular design of COFCs, including chemical and composite structures, and highlight recent work to inspire the development of novel structures.3.1 New active center
In 2015, Jiang et al. reported DTP–ANDI-cathodes by combining COF with CNTs for LIBs, demonstrating their potential for redox reactions.10i Since then, the application of COFs in alkali metal batteries has been extensively explored. Different molecular structures exhibit significant variations in electrochemical properties, such as capacity,10g lifespan,26 and voltage plateau.27 Improving the energy storage capacity of COFCs remains a primary research focus. The energy storage capacity of COFCs arises from their active sites, which mainly originate from linkages and building blocks. However, the design of linkages is constrained by controlled synthesis methods, making it difficult to create multiple active linking sites through improvements in synthesis techniques.For monomer design, the energy storage capacity is measured by the ratio of electrons transferred to the molecular mass of the active group. Active site design has explored single-electron, two-electron, and multi-electron transfer reactions. In small organic molecules, multi-electron transfer groups, such as –NO2, significantly enhance the energy storage capacity of a single active site.28 Notably, multi-electron reactions require special electrolytes. Xu et al. reported a nitroaromatic cathode, 1,5-dinitronaphthalene, where each nitro group undergoes a six-electron reduction, achieving a record energy storage of 1338 mA h g−1, surpassing inorganic electrodes (Fig. 3(a)). This six-electron reduction occurs in an electrolyte containing fluoroethylene carbonate (FEC), which, during discharge, undergoes defluorination to generate HF and vinylene carbonate (VC).29 Protons in HF participate in the reduction, while VC is further reduced to vinyloxy radical anions and CO2. The CO2 reacts with water to form H2CO3, and protons dissociated from H2CO3 also contribute to the reduction of 1,5-DNN (Fig. 3(b)). However, the nitro group requires protons for six-electron reduction, limiting its broader application in COFCs. Further work is needed to integrate such multi-electron active sites into the COF structure to fully harness their theoretical potential.
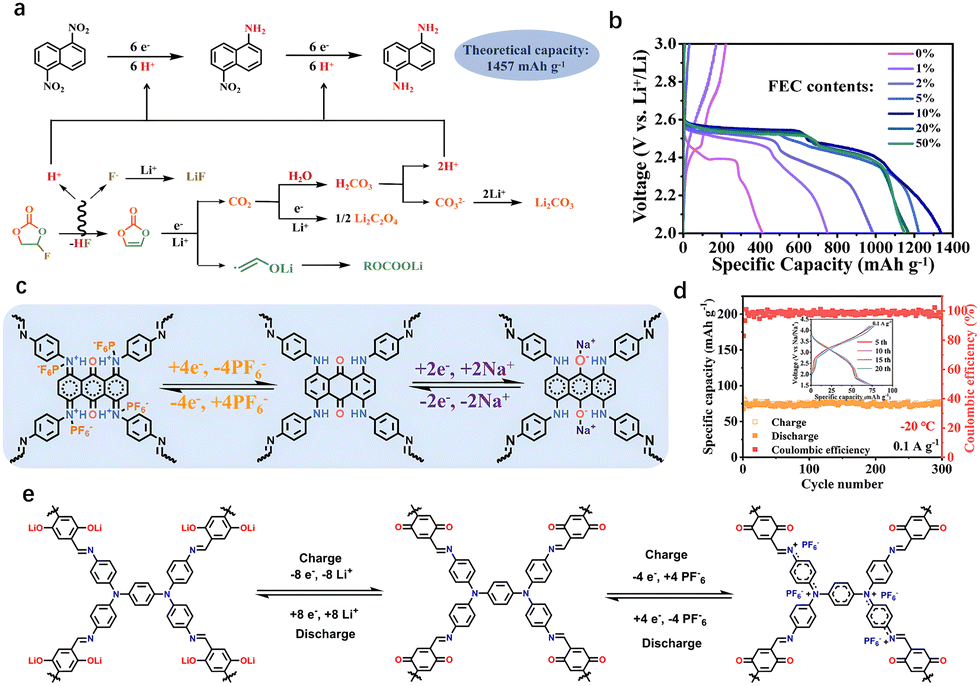 | ||
| Fig. 3 (a) Schematic diagram of six-electron reduction of a single nitro group. (b) Effect of electrolytes with different FEC content on nitro reduction capacity. Reprinted with permission from ref. 28a, Copyright 2022 National Academy of Sciences of the United States of America. (c) Schematic diagram of bipolar redox energy storage of aniline skeleton and quinone groups. (d) The TAB-COF positive electrode is used for the energy storage performance of sodium-ion batteries at low temperatures. Reprinted with permission from ref. 13, Copyright 2025 Wiley-VCH. (e) Schematic diagram of TPDA-DQTA-COF bipolar positive reversible energy storage. Reprinted with permission from ref. 14, Copyright 2024 Chinese Chemical Society. | ||
Most organic monomers used for COF synthesis consist of five-membered rings, six-membered rings, and extended polycyclic molecules. Due to the relatively inactive nature of C–C and CC groups, a common design strategy involves constructing larger rings by replacing some connections with heteroatoms (N, O, S). Recently, Wang et al. attributed the high ion conductivity, abundant redox-active sites, and attractive bipolar properties of TPAD-COF to the unique combination of a conductive aniline skeleton and a quinone redox center (Fig. 3(c)).13 The TPAD-COF cathode for sodium-ion batteries exhibited a higher specific capacity (186.4 mA h g−1 at 0.05 A g−1) and excellent cycle performance (over 2000 cycles at 1.0 A g−1 with a decay rate of 0.015% per cycle). Impressively, the TAG-COF showed a high specific capacity of 101 mA h g−1, even at −20 °C (Fig. 3(d)).
Site-specific atomic substitution along the backbone enables precise regulation of structural conformation and systematic optimization of HOMO, LUMO and electronic band gaps.30 In particular, it can be doped to form electron concentration in the molecular chain, which further enhances the electronic conductivity.31 Functional groups formed by n-type and p-type atomic substitution can be divided into electron substitution-type withdrawal groups (EWGs) and electron donor groups (EDGs). They can affect the valence electricity, change the binding energy with metal ions, and affect the rate of ion diffusion.32 However, partially inactive atomic substitution may add additional molecular mass, thereby reducing theoretical capacity. In the process of molecular design, there is a trade-off between conductivity and capacity. Studies have shown that inactive sites can be transformed into energy storage sites through post-processing reactions based on the established framework.33 The introduction of new active groups into the limited substitution sites on the ring also contributes to increasing the theoretical capacity. Chen et al. synthesized crystalline TPDA-DMTA-COF from 2, 5-dimethoxy p-phenyldiformaldehyde and N,N,N′,N′-4(4-aminophenyl)-1,4-phenylenediamine under suitable solvent conditions. Subsequently, TPDA-DHTA-COF was synthesized by introducing new redox groups into the skeleton. In situ oxidation reaction transformed inactive C–OH into CO site, showing n-type energy storage capacity (Fig. 3(e)). The modified TPDA-DHTA-COF exhibited a reversible capacity of 308 mA h g−1 in LIBs, leveraging dual storage of Li+ cations and PF6− anions to achieve an energy density of 800 W h kg−1. This approach highlights how post-processing can unlock latent functionality in COF frameworks without compromising structural integrity. By coupling targeted atomic substitution with tailored post-synthetic reactions, researchers can optimize both electronic and ionic pathways in COFCs.
Current research on redox-active sites primarily focuses on imine, hydrazone, and carbonyl groups, with new COFCs being designed by altering the position, number, and connectivity of these groups. While sulfur-containing groups have also been studied, their primary application has been in inhibiting polysulfide shuttling in lithium–sulfur batteries (LSBs). When multi-electron groups are introduced into the skeleton of small molecules in positive electrodes, the expected electron gain and loss patterns are often not fully realized. In polymer materials, ion shuttle, diffusion, and migration can be influenced by factors such as binding energy, electrostatic shielding, steric hindrance, pore environment, and structural defects, which reduce the reactivity of ions with the active sites.34 The introduction of new multi-electron active groups could elevate the development of organic positive electrodes. Additionally, the passivated ring skeleton of COFs can also serve as an effective energy storage site. Enhancing the utilization of this part of the structure, while preserving the integrity of the overall framework, could help overcome the limitations of theoretical capacity.
3.2 Conjugate system regulation
The design and synthesis of cathodes with high electronic conductivity remains a key goal in energy storage development. Ion insertion/extraction is driven by an external effective potential, determined by both the potential applied to the surface of a particle and the voltage drop caused by surface resistance. As surface resistance increases, the effective potential decreases, hindering ion insertion and reducing the ability to maintain high capacity under large currents.35 The rate property is heavily influenced by surface conductivity. In addition to recombination with highly conductive substances, for organics, the tunability of their chemical structure offers a distinct advantage in advanced materials science. Charge transfer along the main chain is more efficient than hopping between chains.36 Minimizing the band gap, eliminating electronic defects, and suppressing vibrational modes can promote effective band-like conduction in COFs. Additionally, the extension of π-conjugation along the main chain enhances electron delocalization, which lowering the effective electron mass and amplifying both intramolecular and intermolecular interactions. This cascade of electronic and structural modifications optimizes charge transfer kinetics and ion transport.37The optimization of conjugation system includes π–π conjugation and π–d conjugation.38 By designing large conjugated monomers and modifying the covalent bonding patterns within their molecular structures, it is possible to expand π–π conjugation. Monomers that consist of linearly fused aromatic subunits with enhanced parallel p-orbital interactions exhibit strong π-electron delocalization. These monomers, with their unique molecular shapes, also possess redox-active sites. Two primary types of redox-active sites have been successfully incorporated into COF frameworks: nitrogen-containing groups, such as pyrazine and its derivatives (including phenazine and triazine),39 and conjugated carbonyl groups (like quinone, naphthoquinone and phenanthrenequinone).40 For instance, Feng and colleagues incorporated a perinone-based benzimidazobenzophenanthroline (BBL) into a ladder-type 2D conjugated COF, which has demonstrated excellent proton storage capabilities in both mild aqueous Zn-ion electrolytes and strong acid environments (Fig. 4(a)).15 Furthermore, the ring-fused perinone unit within the BBL-based COF reduces the basicity of the C–O− group when the perinone is in its reduced or discharged state. This leads to a strong C–O− coordination ability for H+, rather than Zn2+ (Fig. 4(b)). The good delocalization ability of pyranone greatly improves the rate performance of the devices. Even at the current density of 200 A g−1, the device still maintains 51 mA hg−1, corresponding to capacity retention of 67% to that at 1 A g−1 (Fig. 4(c)). As a result, the perinone-based 2D BBL-TP exhibits unique proton storage behavior in both weakly acidic (ZnCl2 in water) and strongly acidic (H2SO4) electrolytes.
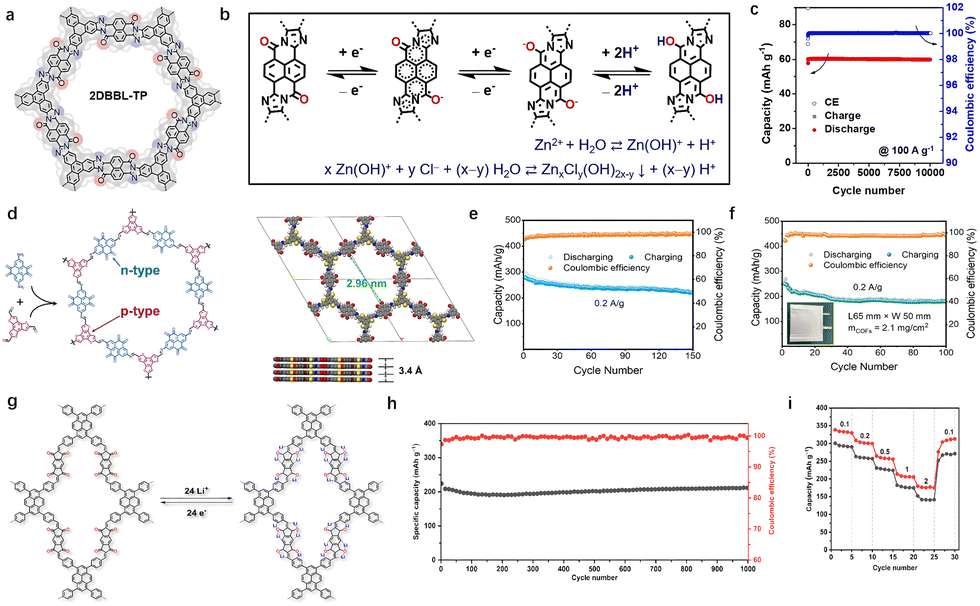 | ||
| Fig. 4 (a) Chemical structure of 2DBBL-TP. (b) The two-electron reaction process of 2DBBL-TP cathode during discharge. (c) The cycling stability of 2DBBL-TP. Reprinted with permission from ref. 15, Copyright 2023 Wiley-VCH. (d) The preparation routine and AA eclipsed model of BTT-PTO-COF. (e) Cyclic stability of BTT-PTO-COF at 10 Ag−1 for 10000 cycles. (f) Cycling performance of the pouch cell at 0.2 Ag−1 for 100 cycles. Reprinted with permission from ref. 16, Copyright 2024 Wiley-VCH. (g) Proposed reversible electrochemical redox mechanism of TFPPy-ICTO-COF during the lithiation/delithiation process. (h) Long cycle stability of full cell. (i) Rate performance of TFPPy-ICTO-COF cathodes at current densities of 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1. Reprinted with permission from ref. 41, Copyright 2023 American Chemical Society. | ||
The donor–acceptor (D–A) structure boasts a narrow band gap and high conductivity, facilitating efficient charge transport and accelerating redox kinetics, thereby contributing to exceptional rate performance. The design of novel covalent frameworks and thiophene derivatives utilizing planar and π-extending monomers, such as porphyrins and pyrene, can effectively enhance π-conjugation and π–π interactions, which in turn promotes charge transfer.42 However, reactive hydrogen derivatives and passivated benzene ring structures can detract from the theoretical capacity. To address this challenge, Yao et al. integrated highly planar, polarized, and electron-rich thiophene units into the backbone of the material. As a result, COFCs generally exhibit improved charge-carrier mobility along the polymer chain.16 The large thiophene/benzene fused π-conjugated BTT structure significantly enhances electron transport (Fig. 4(d)). Furthermore, the star-conjugated plane structure augments the π–π stacking interaction in COF, fostering efficient intermolecular charge transfer through efficient hopping mechanisms. The cathode prepared using benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene (BTT) as the p-type material and pyrene-4,5,9,10-tetraone (PTO) as the n-type material (BTT-PTO-COF) demonstrates impressive performance. It exhibits a high specific capacity of 275 mA h g−1 in a carbonate-based electrolyte at 0.2 A g−1 and an excellent rate capacity of 124 mA h g−1 at 10 A g−1 (Fig. 4(e) and (f)). Additionally, under high pressure conditions, electron depletion occurs in the electron-rich thiophene conjugated structures, promoting anion storage while increasing the discharge platform and specific capacity.
The full conjugation strategy employed in the linkage of COFs plays a crucial role in enhancing their conductivity. Certain linking groups, such as imide, borate, and hydrazone, disrupt the main chain conjugation, resulting in polarization and electron traps that increase the band gap and hinder in-plane charge transport. To address this issue, the conjugated system can be effectively extended by converting unconjugated groups into ethylene, imidazole, or thiazole units. Chen et al. developed a novel Janus-based diketone COF, fully conjugated by olefin unit sp2 carbon linkages, which achieved impressive conductivity and carrier mobility values of 10−3 s cm−1 and 7.8 cm2 V−1 s−1, respectively.41 The ordered structure, excellent porosity, high conductivity, and abundant redox-active carbonyl units of this COF make it a promising candidate for high-performance cathode materials in LIBs (Fig. 4(g)). The TFPPy-ICTO-COF exhibited a remarkable capacity of up to 338 mA h g−1 at a discharge rate of 0.1C, setting a new capacity record for COFCs-based lithium-ion batteries. Moreover, after 1000 cycles, the material retained 100% of its capacity, demonstrating the exceptional stability of these Janus ditone based COFs (Fig. 4(h)–(i)).
In conclusion, prolonging the conjugated system in COFCs can significantly enhance the intermolecular and intramolecular forces, leading to improved structural stability. This stability is crucial for facilitating the transport of electrons and ions, which in turn boosts the actual capacity and rate performance of the device. To further enhance the conjugation effect, several strategies can be employed: 1. Planarity: by carefully designing the covalent bonding mode and monomer structure, the coplanarity of the molecules can be improved. This is because planarity helps to maximize the overlap of π-orbitals, which is essential for effective electron transport. Additionally, covalent interlocking effects between adjacent parts of the framework can further enhance coplanarity, contributing to a more stable and efficient electron transport pathway. 2. Substituents: The incorporation of substituents in the active monomer design can further improve electron delocalization. By carefully selecting and positioning substituents, the conjugation effect can be optimized, leading to improved conductivity and performance of the COF-based device.
3.3 Metal ion coordination
The periodic porous structure and inherent stability of COFs render them an exceptional platform for loading and hosting various species. The abundance of coordination sites within the molecule enables the formation of metal-coordinated organic frameworks (M-COFs), where supramolecular ligands and carriers play a key role. Metal–organic linkers such as porphyrins,43 phthalocyanines,44 2,2′-bipyridine,45 and metallosalen are frequently employed to modulate the environment, type, and quantity of metal coordination.46 Current synthesis methods for M-COFs can be broadly classified into two categories: the first involves the condensation of monomers to form stable crystalline materials, followed by metallization and coordination with metal salts. The second approach is a one-step synthesis in which the monomer and metal salt react simultaneously within the same reaction system to produce M-COFs. Additionally, metal-exchange reactions enable the post-synthesis modification of the metal center, providing extra flexibility in adjusting the properties and functionality of the M-COFs material.The coupling of metals with π-orbitals further extends the π–d conjugation effect. This is particularly evident when transition metal d-orbital electrons interact with π-electrons in the aromatic rings of the monomer units. Such interactions facilitate electron delocalization within the framework, effectively narrowing the band gap and boosting the overall conductivity of the M-COFs. Building on this principle, Wu et al. designed a metallosalen M-COFs with integrated organic–inorganic active sites for LIBs.17 Density functional theory (DFT) analysis revealed that the majority of the density of states near the Fermi level in Cu-TH-COF is primarily contributed by the Cu, N, and O orbitals (Fig. 5(a)). A strong overlap between the d-orbitals of Cu and the p-orbitals of N and O indicates robust π–d conjugation, which facilitates extensive electron delocalization and results in a reduced band gap, thus enhancing electrical conductivity (≈1.0 × 10−3 S m−1).
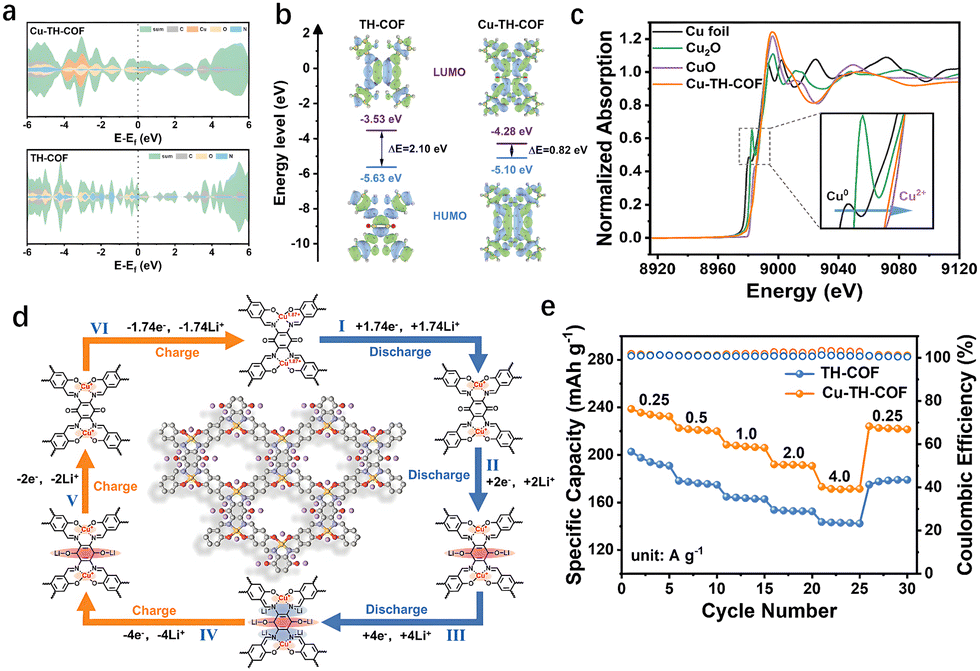 | ||
| Fig. 5 (a) PDOS for Cu-TH-COF and TH-COF. (b) HOMO/LUMO energy levels and energy gaps (ΔE) of Cu-TH-COF and TH-COF. (c) Normalized XANES spectra and spectra at the Cu K-edge of Cu-TH-COF. (d) Schematic diagram of the structural transformation of Cu-TH-COF as a positive electrode during the charge/discharge. (e) Rate performance of Cu-TH-COF cathode. Reprinted with permission from ref. 17, Copyright 2025 Wiley-VCH. | ||
The Cu2+ coordination also lowers the lowest unoccupied molecular orbital (LUMO) level and increases electron affinity, which is beneficial for providing a higher reduction potential (Fig. 5(b) and (c)). As a result, the magnification performance of the Cu-TH-COF cathode is significantly improved, with a retained capacity of 174 mA h g−1 at a high current density of 4 A g−1 (Fig. 5(e)). In situ Fourier transform infrared spectroscopy (in situ FTIR) and ex situ X-ray photoelectron spectroscopy (ex situ XPS) were employed to investigate the lithium storage mechanism and the role of Cu2+ in the electrochemical properties. The high density of Cu2+ functions as an inorganic redox active site for Li+ insertion/extraction, while synergistically interacting with the organic active sites (CO and CN) to facilitate multi-electron transfer redox reactions (Fig. 6(a)). This dual enhancement of the magnification performance and capacity in M-COF cathodes offers a promising strategy for the next generation of LIBs.
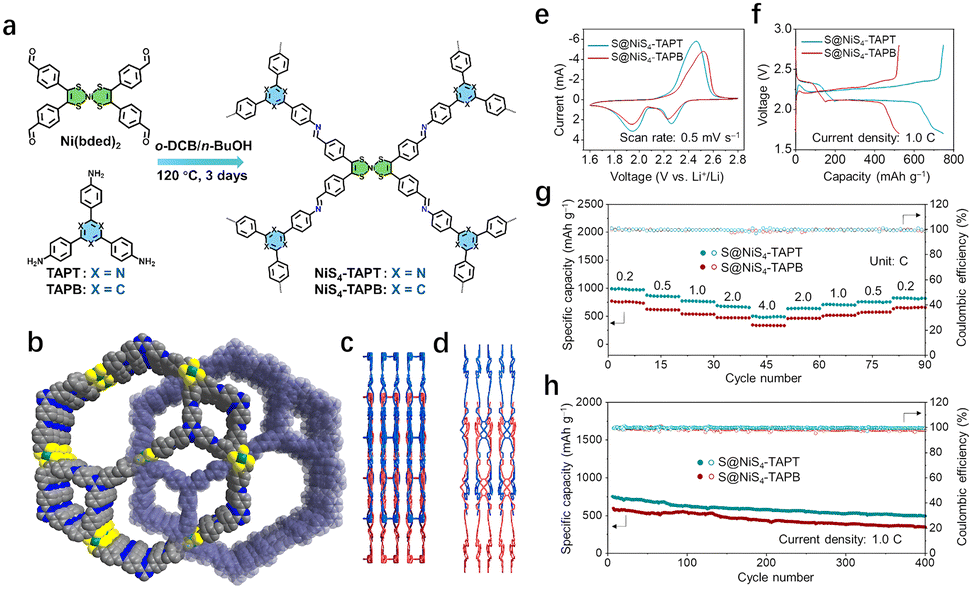 | ||
| Fig. 6 (a) Schematic diagram of the synthesis processes of NiS4-TAPT and NiS4-TAPB. (b) Interpenetrated networks, (c) view along the b-axis, and (d) view along the c-axis of two NiS4-COFs. (e) CV curves of the S@NiS4-TAPT and S@NiS4-TAPB cathodes at a scan rate of 0.5 mV s−1. (f) Galvanostatic discharge/charge profiles of S@NiS4-TAPT and S@NiS4-TAPB cathodes. (g) Rate capabilities of S@NiS4-TAPT and S@NiS4-TAPB cathodes at various current rates from 0.2 to 4.0C. (h) Long-term cycling performances of the S@NiS4-TAPT and S@NiS4-TAPB cathodes at 1.0C for 400 cycles. Reprinted with permission from ref. 18, Copyright 2024 American Chemical Society. | ||
In addition to the enhancement of conjugation effect, the abundant electrocatalytic activity of MCOFs can promote the conversion of compounds. Jin et al. synthesized a three-dimensional (3D) COF, NiS4-TAPT, by incorporating Ni-bis(dithiolene) and nitrogen-rich triazine centers.18 This innovative material has been successfully applied to both the cathode and anode of LSBs. On the anode side, the modified nanochannel walls of NiS4-TAPT promote host–guest interaction, promote Li+ transport, and prevent dendrite formation (Fig. 6(b)–(d)). On the cathode side, the intrinsic pores and abundant N sites in the framework effectively trap polysulfides through physical confinement and adsorption, which migrates shuttle effects and improving coulomb efficiency. Furthermore, the uniform distribution of electrochemical activity Ni within NiS4-TAPT skeleton promotes the transformation of polysulfide during cycling. When assembled into a full battery with NiS4-TAPT serving as the primary material for the sulfur cathode and lithium anode, the device demonstrates a high cathode specific capacity of 966.3 mA h g−1 at a rate of 1.0 C, along with stable cyclability. Notably, the capacity decay rate is low, at just 0.055% per cycle, highlighting the promising potential of NiS4-TAPT for advanced LSBs (Fig. 6(e)–(h)).
Reports on M-COFs are still in its nascent stages, with significant room for advancements in structural design and exploration of their application performance. While structural predictions suggest that the introduction of metal sites into the framework can extend π-conjugation and enhance conductivity, experimental evidence this hypothesis remains scarce. To date, most studies have focused on improving the catalytic performance of M-COFs, with limited in-depth research on their energy storage applications and redox mechanisms. Growing high-quality single crystals of M-COFs presents a formidable challenge, making it difficult to accurately determine their structures. Neither computer simulations nor powder X-ray diffraction (PXRD) tests can fully describe the intricate stacking arrangements of synthesized two-dimensional M-COFs. Consequently, many of the underlying topological networks within M-COFs remain unexplored, and the relationships between these uncharted network structures and their performance are unclear.
3.4 Substituent group
The study of substituents has long been a pivotal area within organic chemistry. By fine-tuning substituents on the side chain, researchers can manipulate molecular packing, charge distribution, and band gap, ultimately influencing the electronic conductivity and reaction kinetics of organic electrodes.47 Substituents play a crucial role throughout the synthesis process and in determining the properties of the resulting materials. Therefore, a systematic investigation of substituents is essential. The electronic effect of substituents and the length of substituent chains both impact the synthesis and reaction kinetics of COFs. For example, incorporating cyano EWGs into amino monomers can weaken their nucleophilic nature.48 This reduction in reactivity can hinder the formation of crystalline structures, ultimately preventing the achievement of a well-defined crystalline lattice. On the other hand, longer chain substituents can enhance the solubility of COF precursors in mixed solvents, thereby facilitating synthesis. These substituents also strengthen intermolecular forces, leading to improved crystallinity of the resulting COFs. Furthermore, the extension of substituent chain length affects the morphology and pore structure of COFs, with specific surface area and pore size decreasing as substituent volume increases.In traditional conjugated polymers, the introduction of solubilizing substituent side chains can convert insoluble polymers into solution-processable materials. The effectiveness of this solubilization is influenced by factors such as the degree of branching, chain length, and polarity of the substituents. Recently, Zhang et al. incorporated methyl polyethylene glycol (mPEG) into the COF structure, leading to the development of a soluble COF known as CityU-29.19 The mPEG side chains were strategically grafted to enhance the solubility of CityU-29 in DMSO, achieving a concentration of up to 7 mg mL−1 (Fig. 7(a)).
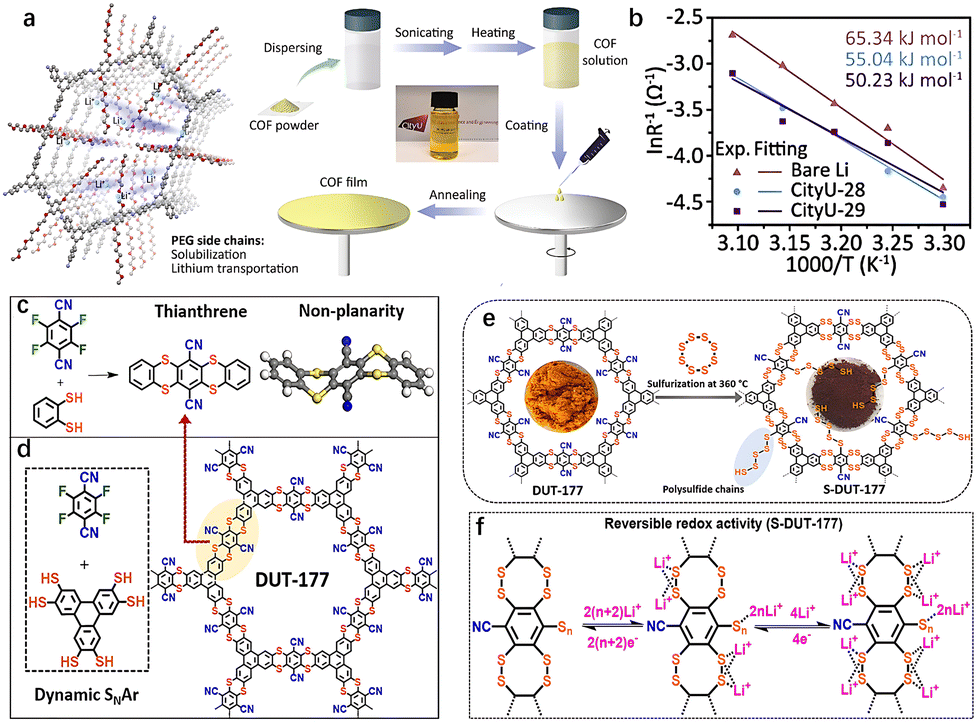 | ||
| Fig. 7 (a) Schematic illustration of PEG enabled solubilization and lithium-ion transportation and Solution preparation process of mPEG-grafted COFs. (b) Arrhenius curves and comparison of activation energies of symmetric cells.19 Copyright 2025 Wiley-VCH. (c) Synthetic route toward the thianthrene model compound via an irreversible SNAr reaction. (d) Synthetic route toward the thianthrene-based COF named DUT-177. (e) Schematic illustration of the sulfurization of DUT-177 resulting in the formation of S-DUT-177 with covalently linked sulfur. (f) Illustration of a plausible reversible interaction mechanism of lithium ions with covalently anchored sulfur in S-DUT-177.49 Copyright 2022 American Chemical Society. | ||
The economic and efficient solution processing method allowed for the production of thin-film materials by spin coating from the COF solution. Theoretical calculations were conducted to investigate the transport pathways and mechanisms of lithium ions (Li+) within the COF. Notably, the oxygen atoms of the mPEG chains and the nitrogen atoms of the imine bonds served as coordination sites for Li+ (Fig. 7(b)). The out-of-plane path near the oxygen atoms exhibited a minimal energy barrier of 0.36 eV during migration, which aligns with the one-dimensional pores. Consequently, Li+ can be transported along these one-dimensional channels within the COF via the mPEG chains. This solubilization strategy not only enhances the processing of COFs but also offers a promising approach for developing solubilized cathode materials in COFCs.
Substituents not only influence the electron density of the aromatic ring but also play a crucial role in regulating the electrochemical reactivity during charge and discharge processes. Kaskel et al. synthesized a dithiine-linked compound, DUT-177, using a dynamic and self-correcting SNAr reaction (Fig. 7(c) and (d)).49 After post-synthetic sulfurization of DUT-177, nitrile substituents were partially replaced by polysulfide chains via the SNAr pathway, resulting in the conversion of the linkage to dithianthrene after sulfurization (Fig. 7(e)). This transformation is similar to a mechanism reported by Thompson and Huestis.50 At high temperatures (360 °C), polysulfides can form strong basic radical dianions (Sn2−·), which may replace some nitrile groups, covalently anchoring polysulfide chains in a similar manner. This process increased the percentage of sulfur covalently bound to the S-DUT-177 skeleton from 38 wt% to 85 wt% (based on elemental analysis), exceeding the theoretical sulfur content in the converted dithianthrene COF. After partial replacement of nitrile groups by covalently linked polysulfides, these groups began to react with Li+ from LiTFSI (lithium bis(trifluoromethanesulfonyl)imide), exhibiting reversible redox peaks within the potential window required for LSBs (1.8 to 2.8 V). By anchoring polysulfides with smaller organic moieties in S-DUT-177, the shuttle effect during cycling was suppressed, resulting in the retention of 76.6% of the initial capacity even after 500 cycles at a current density of 500 mA g−1 (Fig. 7(f)). This sulfurization of thianthrene COF demonstrates the potential for functionalizing inactive substituents in aromatic rings.
Currently, the strategy of modifying COFC performance through the design of substituents remains relatively underexplored. While the integration of active substituents into the framework can significantly enhance capacity, multi-electron transfer functional groups found in small molecule electrodes can also be applied to COFCs. However, the effects of substituents are often multifaceted. For example, the introduction of EWGs lowers the LUMO level of the molecule, potentially reducing electrical conductivity while also increasing the voltage platform. This necessitates a careful balance between enhancing magnification performance and managing the voltage platform.
On the other hand, the incorporation of EDGs such as alkyl, alkoxy, amino, and carboxyl groups increases the nucleophilicity of the active site, strengthens its interaction with cations, and can influence reaction kinetics. Existing reports suggest that optimizing substituents can be a double-edged sword—improvements in one parameter often come at the cost of another.
Given the current lack of extensive experimental data correlating substituent modifications with performance improvements, it is crucial to design experiments that verify the effectiveness of these modifications. Assembling devices to test the electrochemical performance of COFCs with various substituents will provide valuable insights into the rationality of designs. This experimental approach, coupled with the potential for large data sets, is essential for advancing research in this field and accelerating the development of improved materials.
4 Design of composite material
Although molecular structure design can significantly enhance the performance of COFCs, the development of polymerization methods for these new precursors remains energy-intensive. As previously discussed, COFs not only offer promising energy storage capabilities but also serve as ideal carriers and reaction platforms due to their intrinsic structural advantages. Consequently, integrating COFs with other materials—such as carbon-based substances,51 conductive polymers,52 metal–organic frameworks (MOFs),53 MXenes,54 and metal oxides55—holds great potential for improving performance compared to single-material electrodes. Composite COFs often exhibit morphologies similar to their substrates, with fibrous, sheet-like, spherical, mesoporous, and macroporous structures having been reported.56 The morphology of cathode materials influences ion transport pathways and diffusion kinetics. Composite methods can be broadly classified into two categories: physical and chemical methods.57 Physical methods, such as blending (through grinding or ultrasonic oscillation), diffusion, and self-assembly, are relatively simple and efficient. However, they may result in lower composite material stability. Chemical methods involve the chemical bonding of monomers and substrate materials through chemical modification, which can affect the diffusion and adsorption of precursors and thus influence in situ polymerization. While numerous reports have been published on the types, methods, and improvement measures for composite materials, this paper focuses on the latest advancements in this field, particularly in the context of battery cathodes.4.1 Composite with carbon materials
Low electrical conductivity remains a significant challenge for COFs as electrode materials. While certain COFs, such as 2D-COFs, can form electron conduction pathways through in-plane π-conjugations and interlayer π–π stacking, the presence of linkages—such as imines, hydrazones, amides, and borates—can hinder the free movement of electrons.58 These linkages restrict delocalization and long-range electron transport, leaving the framework largely insulative and neutral, without free electrons or holes to serve as charge carriers. Furthermore, the lack of reversibility in dynamic covalent bonds or unfavorable reaction conditions during synthesis can result in structural defects and disordered regions. These defects interrupt electron conduction paths, increasing the overall resistance of the material.Various conductive additives have been used to improve the conductivity of the COFCs. In 2015, Jiang et al. pioneered the in situ growth method to combine COFs with carbon nanotubes (CNTs). This approach constructed a dynamic framework on the surface of CNTs, enhancing the redox active site and demonstrating high coulomb efficiency. More recently, jiang et al. integrated a COFCs with double redox active centers onto CNTs via π–π interactions, which improved both the accessibility of active sites and the charge transfer kinetics (Fig. 8(a)).59 The TAPPPz-COF-40%CNT composite maintained a high capacity of 151 mA h g−1 (88% retention) after 10000 cycles at a high current density of 10 A g−1. In cases where the 2D COF layer is tightly packed, Li+ may struggle to penetrate internal active sites buried deep within the layers (Fig. 8(b) and (c)). Sun et al. achieved controlled growth of 2D COF with fewer layers on CNTs, achieving a reversible capacity of 1536 mA h g−1 and sustaining 500 cycles at 100 mA g−1 (Fig. 8(d)–(f)).60 In another approach, Shen et al. combined COF with graphene, preparing a full COF/graphene battery.20 This aqueous symmetric all-organic battery exhibited excellent cycle life and high rate performance, sustaining over 15000 cycles with a capacity of 80 mA h g−1 at a high current density of 5 A g−1 in punch cells (Fig. 8(g)–(i)).
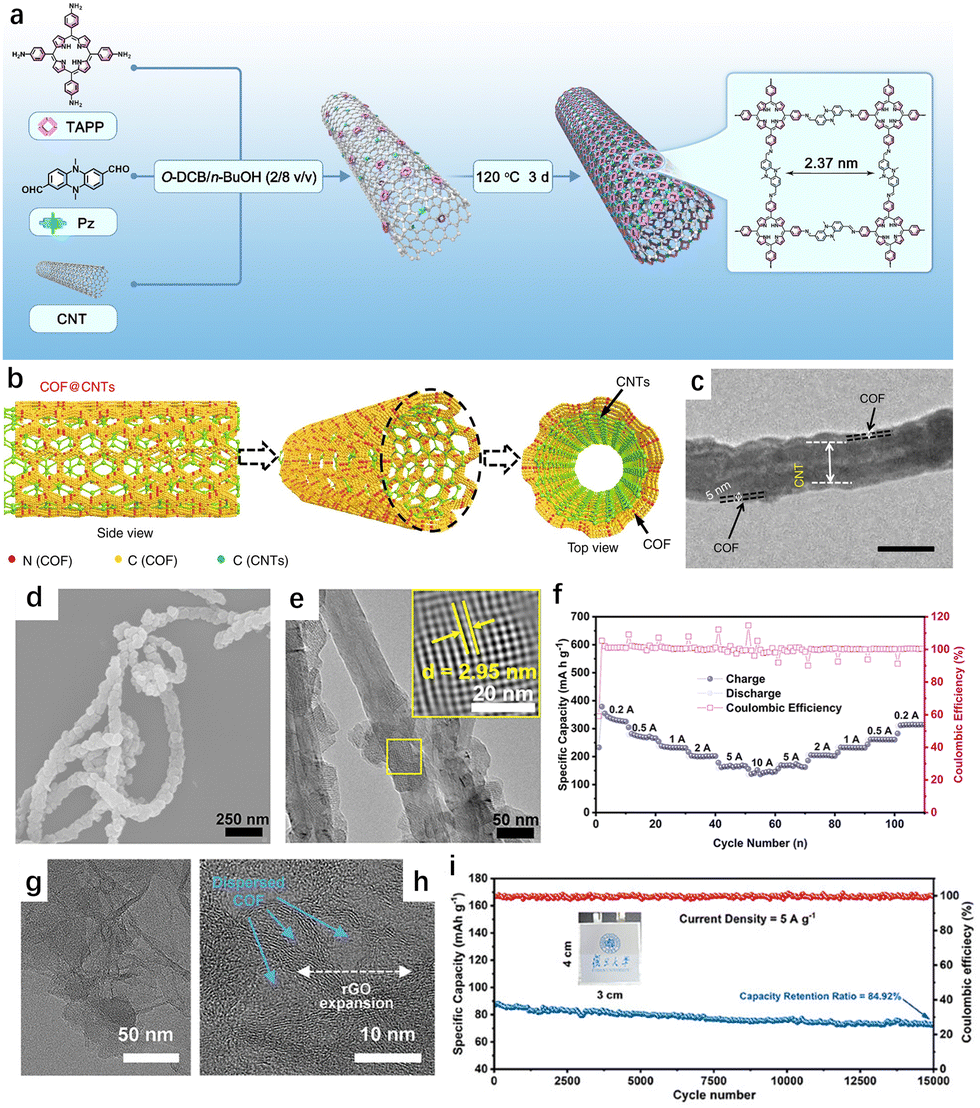 | ||
| Fig. 8 (a) Schematic of the synthesis of TAPP-Pz-COF-40%CNTs.59 Copyright 2024 Royal Society of Chemistry. (b) SEM image and (c) HR-TEM image of TAPP-Pz-COF-40%CNTs.60 Copyright 2018 springer Nature. (d) Rate capability of TAPP-Pz-COF-40%CNTs from 200 to 10000 mA g−1. (e) and (f) TEM images of GDAQ. (g) Cycling stability of the GDAQ//GDAQ-R pouch-type SAOB at a current density of 5 A g−1. (h) Graphical representation of COF@CNTs with few COF layers covered on the exterior surface of CNTs. (i) TEM image of few layers COF@CNTs.20 Copyright 2024 Wiley-VCN. | ||
In general, combining carbon materials with COFs to enhance the electron transport capacity of the positive electrode is a common and effective modification strategy. The in situ polymerization method, based on the solution phase, allows the precursor to polymerize along the carbon material skeleton, with the morphology of hosts further influencing the micro and nano structures. However, this approach has limitations. Excessive incorporation of carbon materials does not contribute significantly to the overall capacity, and the addition of inactive materials can decrease the relative volume capacity of the device.61 Researchers should focus on optimizing this approach, using minimal amounts of carbon additives to achieve high-rate performance, thereby bridging the gap between theoretical designs and practical applications.
4.2 Composite with MOF
Porous materials with well-defined topologies and inherent porosity are highly desirable for electrochemical energy storage applications. The nano-scale porous structure enhances the specific surface area and improves access to reactive sites. Additionally, the unique pore structure creates heterogeneous interfaces that can influence charge separation and carrier migration, making them highly promising for energy storage,62 catalysis,63 and environmental applications. In 2016, Ben et al. demonstrated for the first time the growth of metal–organic frameworks (MOFs) on covalent organic framework (COF) films, resulting in CoF-MOF composite films. These composite films exhibited enhanced selectivity for separating H2/CO2 gas mixtures.64 Following this, researchers further explored integrating MOFs and COFs to combine their unique advantages, aiming to create a composite that not only provides self-complementary properties but also outperforms other well-established MOF or COF-based composites.In addition to the simpler core–shell or coating configurations, researchers have also designed flower-like MOF@COF composites by manipulating the coordination interactions between the two components during the synthesis process (Fig. 9(a)).65 This molecular-level interconnection, which drives the morphological transformation, is attributed to the coordination between the nitrogen atom of the COF imide group and the manganese center in Mn-MOF (Fig. 9(b)–(d)). While M–N coordination bonds (M = Cu, Fe, Ni, Co, etc.) are commonly found in MOFs with nitrogen-based organic ligands, achieving an efficiently interconnected hybrid structure from MOFs and COFs in Fe or Co centers is challenging.66 This difficulty is likely due to their saturated six-coordination and small ionic radii. To address this, MnS composites with either hollow microspheres (MnS@NS-C-g) or core–shell microspheres (MnS@NS-C-l) were synthesized via vapor-phase (using sulfur powder as a source) or liquid-phase vulcanization (using thioacetamide as a source). The COF/Mn-MOF hybrids, with tunable morphology and microstructure, demonstrate impressive lithium storage capacity.
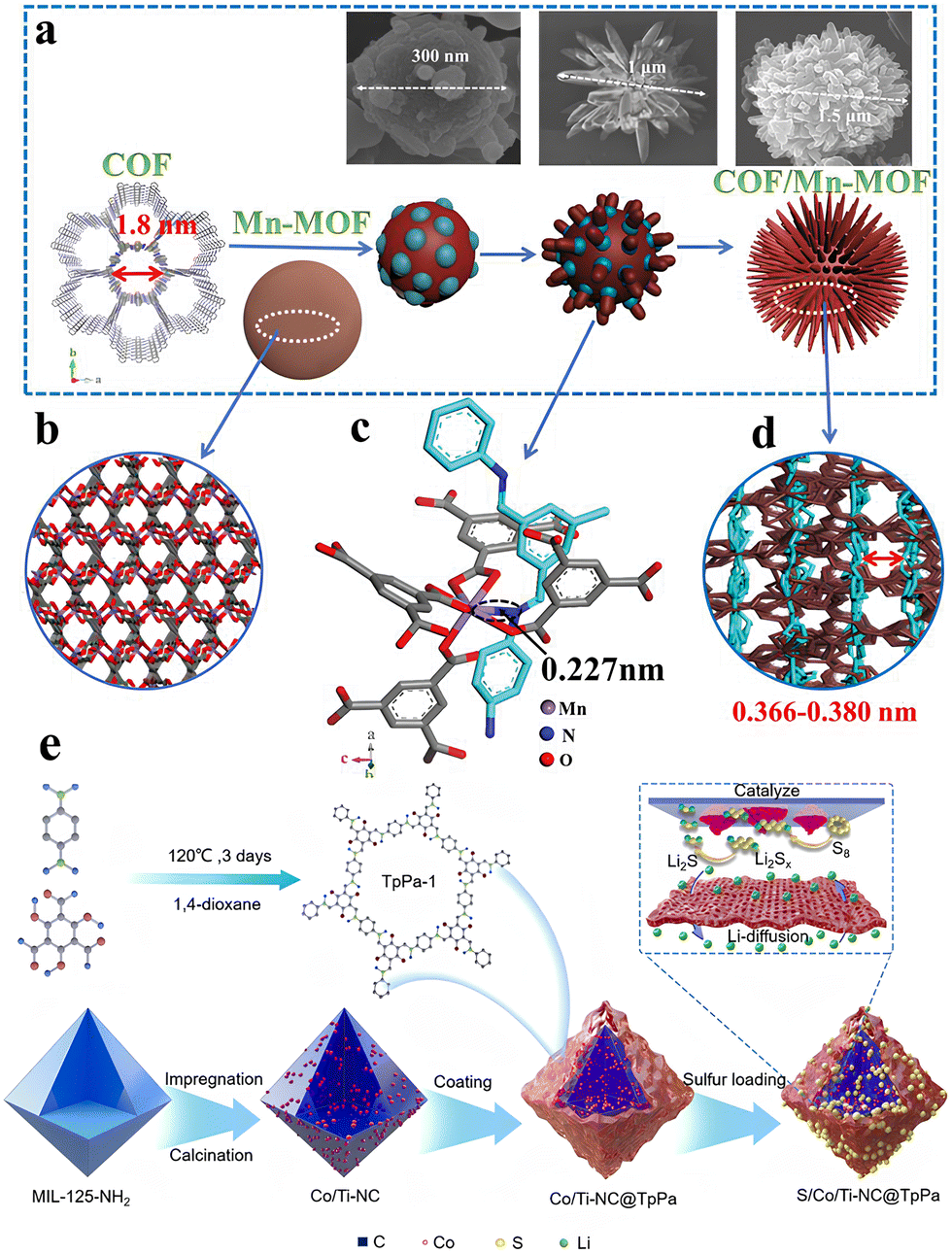 | ||
| Fig. 9 (a) The schematic diagram of the growth process of COF/Mn-MOF hybrid, the inset SEM images indicate the morphology evolution of the COF/Mn-MOF hybrid along with the increased reaction time. (b) The 3D structure of Mn-MOF. (c) The Mn–N interaction along the c-direction with the bonding distance of ≈0.227 nm. (d) Side views of the COF/Mn-MOF hybrid with the calculated interlamellar distance of adjacent COF layers in the range of 0.366–0.380 nm.65 Copyright 2019 Wiley-VCH. (e) Schematic diagram of S/Co/Ti-NC@TpPa composites.67 Copyright 2023, Elsevier. | ||
Metal centers in MOF structures, such as Fe, Co, Ni, Cu, and V, present promising candidates as metal sources for cathode materials. Upon calcination in air, these metals can be converted into high-value metal compounds, including oxides, sulfides, fluorides, and phosphates,68 while retaining a unique porous structure. Yang et al. integrated transition metal-based, porous carbon composites derived from MOFs into COF frameworks, proposing an advanced design strategy for multifunctional sulfur cathodes with core–shell structures.67 In this composite, the internal carbon/metal cores serve dual functions: they capture sulfur and act as catalysts to enhance sulfur reaction kinetics. The COF shell, rich in micropores and polar functional groups, effectively mitigates the lithium polysulfide (LiPS) shuttle via its sieving effect (Fig. 9(e)). The resulting S/Co/Ti-NC@TpPa cathodes demonstrate excellent cycling stability, with an impressive initial specific capacity of 1135 mA h g−1 at 0.2C, and a minimal capacity decay rate of 0.05% per cycle at 1C over 500 cycles.
The development of MOF–COF composites is still in its early stages, with challenges arising in the design and controlled synthesis of these hybrids compared to other composite methods. One of the primary difficulties lies in predicting the bond formation between organic–organic and organic–inorganic components when introducing the second phase.69 This unpredictability is crucial for the subsequent polymerization of the precursor. Additionally, there is a significant gap in understanding the growth mechanism of MOF–COF composites, particularly regarding interface compatibility. Complex factors like nucleation, crystallization processes, lattice mismatch, and lattice strain or bending complicate the controllable synthesis of MOF@COF composites.70
Currently, most MOF@COF composites are constructed using –NH2-derived interactions, which limits the types of MOFs and COFs used in the hybrid materials to those functionalized with –NH2 groups and imine-based COFs.71 There is a growing expectation that new types of composite materials, incorporating different functional groups or linker systems, will be introduced into the combination of MOFs and COFs. This would not only enhance the structural flexibility of the composites but also broaden their application capabilities in various fields.
4.3 Composite with metal oxides
While metal oxides have long been utilized for energy storage due to their cost-effectiveness and high volume capacity, they suffer from issues related to surface parasitic reactions, structural degradation, and collapse, particularly during deep discharge cycles. Especially in the deep discharge state, the anisotropic contraction of the lattice leads to local stress concentration along grain boundaries, which compromises the structural stability and reduces the overall cycle life of the device.72 Although shell cladding strategies have been explored to enhance the robustness of metal oxides, they often result in electrochemical passivation, leading to reduced conductivity and performance over time. By integrating COFs with metal oxides in composite structures, the stability and electrochemical performance of metal oxides can be enhanced without significantly compromising the density or capacity of the device.The rigid framework of COFs offers structural support to metal oxides, mitigating volume changes during cycling. Furthermore, the active functional groups within the COF structure provide ion attachment sites, while the multifunctional coordination environment facilitates ion transport and electron transfer. Saleem et al. functionalized LiNi0.78Mn0.12Co0.1O2 (SCNMC) cathode material with nitrogen-doped 2D COF (N2D-COF) through a simple co-precipitation and scalable solvothermal process (Fig. 10(a) and (b)).73 The inclusion of pyridine-N and pyrrole-N within N2D-COF generates vacancies, improving both conductivity and Li+ diffusion, which enhances the electrochemical performance of SCNMC@COF (Fig. 10(c) and (d)). Additionally, the multilayer porous structure of the composite helps prevent volume expansion and facilitates electrolyte penetration. Therefore, SCNMC@COF retains a specific capacity of 117.1 mA h g−1 after 500 cycles, demonstrating excellent rate performance and cycling stability (Fig. 10(e)).
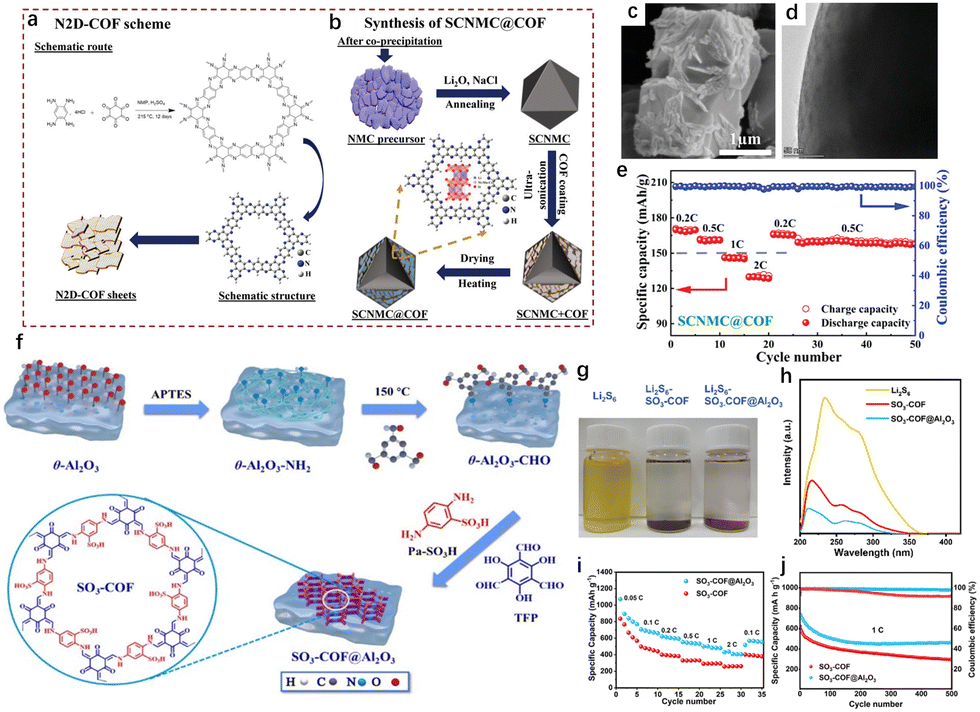 | ||
| Fig. 10 (a) Schematic illustration of the designed route to synthesize N2D-COF and (b) SCNMC@COF. (c) SEM images and (d) cross-sectional TEM image of SCNMC@COF. (e) Rate performances of SCNMC@COF electrodes. (f) Synthesis diagram of SO3-COF@Al2O3.73 Copyright 2022 Wiley-VCH. (g) LiPS (Li2S6) adsorption test using SO3-COF and SO3-COF@Al2O3 as adsorbents. (h) UV-visible absorption spectra and adsorption properties of SO3-COF and SO3-COF@Al2O3 in Li2S6 solution. (i) Rate performances of two cathodes. (j) Long-term cycling performance of the batteries with SO3-COF and SO3-COF@Al2O3 as the cathodes.74 Copyright 2024 Royal Society of Chemistry. | ||
The ordered single-crystal structure of metal oxides typically lacks a large specific surface area, limiting their efficiency when used solely for adsorption and catalysis. By integrating COFs with metal oxides, the hybrid material can leverage the high specific surface area of COFs, enhancing adsorption capabilities. Xu et al. employed a one-step method to in situ load a sulfonic acid-functionalized COF (SO3-COF) onto the surface of Al2O3, resulting in the SO3-COF@Al2O3 composite, which was used as a sulfur carrier for LSBs (Fig. 10(f)).74 The large surface area of SO3-COF enables effective adsorption and encapsulation of sulfur, reducing active substance loss and ensuring high sulfur utilization and stable cycling performance (Fig. 10(g) and (h)). The Al2O3 component adsorbs polylithium sulfides and promotes the conversion of long-chain to short-chain polysulfides through Lewis acid–base interactions, thus mitigating the polysulfide shuttle effect. The SO3-COF@Al2O3 electrode demonstrates a high initial specific capacity of 1141 mA h g−1 at 0.05C and retains a capacity of 466 mA h g−1 after 500 cycles, with a capacity decay rate of just 0.08% per cycle (Fig. 10(i) and (j)). This COF-metal oxide composite design offers a promising strategy for developing high-performance lithium-ion batteries, with significant potential for energy storage applications.
The combination of metal oxides and COFs aims to leverage the physical and chemical strengths of both components, enhancing the structural stability of inorganic materials. However, weak interactions at the organic–inorganic interface often undermine the durability of hybrids, and research on the interface stability is limited. Furthermore, the synergistic mechanisms between the components remain poorly understood. The development of innovative synthesis methods, coupled with advanced theoretical calculations, is essential for creating composite materials with improved stability, recyclability, and robustness.
5. Conclusions and future perspectives
This review provides a comprehensive overview of recent advancements in covalent organic framework cathodes (COFCs). Initially, we analyze the ion storage properties of organic functional groups within COFs, which are fundamental to their theoretical capacity. Subsequently, we discuss cutting-edge developments in molecular structure design, elucidating the structure–property relationship of COFCs through systematic optimization of redox-active centers, conjugation systems, metal ion coordination, and substituent engineering. Finally, we explore strategies for integrating COFs with other materials to address their intrinsic limitations. Leveraging their inherent porosity, chemical stability, and structural tunability, COFCs exhibit exceptional cycle life, rate capability, and high capacity, positioning them as environmentally friendly alternatives to conventional inorganic electrodes.Despite their potential, COFCs currently face significant challenges that limit their widespread adoption compared to inorganic materials. Key limitations include poor electrical conductivity, low capacity density, high production costs, complex synthesis processes, and the absence of a well-established energy storage theory. To address these challenges, we propose the following research directions:
1. Molecular design offers immense opportunities but also presents significant challenges for organic cathode materials. While molecular engineering enables precise control over material performance, discrepancies between theoretical predictions and experimental results persist, largely due to synthesis defects that hinder the full utilization of redox-active sites. Developing scalable, defect-free fabrication methods is critical. Additionally, key scientific questions remain unresolved, including the relationship between electron push–pull effects and voltage profiles, the activation mechanisms of passivated functional groups, and the balance between conjugation extension and capacity enhancement. Addressing these knowledge gaps is essential for unlocking multi-electron redox chemistries.
2. The solvothermal synthesis method commonly used for COFs is characterized by low efficiency and high energy consumption. The production of high-performance organic monomers often involves complex multi-step reactions with low yields, posing significant barriers to large-scale manufacturing. Transitioning from lab-scale coin-cell evaluations to practical pouch-cell prototypes is crucial for validating performance metrics and accelerating commercialization.
3. The electrolyte in COFC systems requires optimization to enhance compatibility with multi-electron transfer reactions in functional groups. Developing non-toxic, thermally stable electrolytes with high ionic conductivity is essential for improving fast-charging capabilities and suppressing dendrite formation.
4. Combining COFs with other materials offers a promising strategy to overcome the limitation of low electron conductivity. By optimizing the type, morphology, and composite structure, electron transfer can be significantly enhanced. However, the use of inactive components in composites must be carefully considered, as the low volumetric density of such approaches may hinder the development of high-performance cathodes. Furthermore, ensuring the stability of the material interface is critical for maintaining structural integrity over long cycling lifetimes. Looking ahead, integrating COFs with other organic molecules could enable the development of binder-free, flexible cathodes, paving the way for applications in wearable electronics.
5. Elucidating the energy storage mechanisms of COFCs requires the development of novel methodologies. The diversity of organic molecules and the unclear movement of ions within or between molecules during cycling pose significant challenges. Additionally, the sensitivity of organic materials to high-energy electron beams limits large-scale characterization. Therefore, advancing in situ characterization techniques that can monitor the electrode–electrolyte interface under operating conditions is essential. Such advancements will provide deeper insights into the energy storage mechanisms of organic electrodes and guide the rational design of future materials.
Overall, the advancements and insights outlined in this review underscore the promising potential of covalent organic framework cathodes as sustainable and high-performance alternatives to conventional inorganic electrodes. Addressing the challenges through molecular design optimization, scalable synthesis methods, advanced characterization techniques, and material integration strategies will be pivotal in unlocking their full potential for next-generation energy storage applications.
Author contributions
Conceptualization, Z. Z. and Y. W.; methodology, Z. Z., D. L. and Y. W. writing – original draft preparation, Z. Z.; writing – review and editing, Z. Z., D. L. and Y. W.; supervision, project administration, and funding acquisition, D. L. and Y. W. All authors have read and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 22375051, and 52203216). D. L. acknowledges the support from the Shanghai Rising-Star Program (24YF2702800).References
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CAS.
- W. Huang, T. Liu, L. Yu, J. Wang, T. Zhou, J. Liu, T. Li, R. Amine, X. Xiao, M. Ge, L. Ma, S. N. Ehrlich, M. V. Holt, J. Wen and K. Amine, Science, 2024, 384, 912–919 CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CAS.
- H. Y. Asl and A. Manthiram, Science, 2020, 369, 140–141 CAS.
- (a) T. Sun, W. Zhang, Z. Zha, M. Cheng, D. Li and Z. Tao, Energy Storage Mater., 2023, 59, 102778 Search PubMed; (b) Z. Li, Q. Jia, Y. Chen, K. Fan, C. Zhang, G. Zhang, M. Xu, M. Mao, J. Ma, W. Hu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207221 CrossRef CAS PubMed.
- (a) Z. Yang, X. Huang, P. Meng, M. Jiang, Y. Wang, Z. Yao, J. Zhang, B. Sun and C. Fu, Angew. Chem., Int. Ed., 2023, 62, e202216797 CrossRef CAS PubMed; (b) S. Wang, Z. Xie, D. Zhu, S. Fu, Y. Wu, H. Yu, C. Lu, P. Zhou, M. Bonn, H. I. Wang, Q. Liao, H. Xu, X. Chen and C. Gu, Nat. Commun., 2023, 14, 6891 CrossRef CAS PubMed.
- X. Yang, L. Gong, X. Liu, P. Zhang, B. Li, D. Qi, K. Wang, F. He and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202207043 CrossRef CAS PubMed.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed; (b) J. Sun, Y. Fei, H. Tang, J. Bao, Q. Zhang and X. Zhou, ACS Appl. Energy Mater., 2024, 7, 7592–7602 CrossRef CAS; (c) Y. Su, B. Li, H. Xu, C. Lu, S. Wang, B. Chen, Z. Wang, W. Wang, K.-i Otake, S. Kitagawa, L. Huang and C. Gu, J. Am. Chem. Soc., 2022, 144, 18218–18222 CrossRef CAS PubMed.
- (a) D. N. Ampong, E. Effah, E. A. Tsiwah, A. Kumar, E. Agyekum, E. N. A. Doku, O. Issaka, F. O. Agyemang, K. Mensah-Darkwa and R. K. Gupta, Coord. Chem. Rev., 2024, 519, 216121 CrossRef CAS; (b) B. Wang, L. Shen, Y. He, C. Chen, Z. Yang, L. Fei, J. Xu, B. Li and H. Lin, Small, 2024, 20, 2310174 CrossRef CAS PubMed; (c) Q. Liu, Q. Li, Y. Li, T. Su, B. Hou, Y. Zhao and Y. Xu, Angew. Chem., Int. Ed., 2025, e202502536 Search PubMed.
- (a) X. Yang, B. Dong, H. Zhang, R. Ge, Y. Gao and H. Zhang, RSC Adv., 2015, 5, 86137–86143 RSC; (b) Q. Zhang, H. Wei, L. Wang, J. Wang, L. Fan, H. Ding, J. Lei, X. Yu and B. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 44352–44359 Search PubMed; (c) R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS PubMed; (d) R. Sun, S. Hou, C. Luo, X. Ji, L. Wang, L. Mai and C. Wang, Nano Lett., 2020, 20, 3880–3888 CrossRef CAS PubMed; (e) H. Lu, F. Ning, R. Jin, C. Teng, Y. Wang, K. Xi, D. Zhou and G. Xue, ChemSusChem, 2020, 13, 3447–3454 CrossRef CAS PubMed; (f) W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed; (g) S. Zhang, Y.-L. Zhu, S. Ren, C. Li, X.-B. Chen, Z. Li, Y. Han, Z. Shi and S. Feng, J. Am. Chem. Soc., 2023, 145, 17309–17320 CrossRef CAS PubMed; (h) Q. Chen, M. Lin, X. Li, Z. Du, Y. Liu, Y. Tang, Y. Yan and K. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202407575 Search PubMed; (i) F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 Search PubMed.
- (a) H. Lu, S. Meng, T. He, C. Zhang and J. Yang, Coord. Chem. Rev., 2024, 514, 215910 CAS; (b) Y. Cao, M. Wang, H. Wang, C. Han, F. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CAS.
- P. Ruan, S. Liang, B. Lu, H. J. Fan and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202200598 CAS.
- L. Cheng, X. Yan, J. Yu, X. Zhang, H.-G. Wang, F. Cui and Y. Wang, Adv. Mater., 2025, 37, 2411625 CAS.
- Z. Xiong, L. Gu, Y. Liu, H. Wang, L. Shi, X. Wu, L. Liu and Z. Chen, CCS Chem., 2024, 6, 2835–2844 CAS.
- M. Wang, G. Wang, C. Naisa, Y. Fu, S. M. Gali, S. Paasch, M. Wang, H. Wittkaemper, C. Papp, E. Brunner, S. Zhou, D. Beljonne, H.-P. Steinrück, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202310937 CAS.
- C. Li, A. Yu, W. Zhao, G. Long, Q. Zhang, S. Mei and C.-J. Yao, Angew. Chem., Int. Ed., 2024, 63, e202409421 Search PubMed.
- H. Zhang, X. Ma, D. Guo, M. Jiao, Y. Liu, Y. Fang, M. Wu and Z. Zhou, Adv. Funct. Mater., 2025, 2419764 Search PubMed.
- S. Lv, X. Ma, S. Ke, Y. Wang, T. Ma, S. Yuan, Z. Jin and J.-L. Zuo, J. Am. Chem. Soc., 2024, 146, 9385–9394 CrossRef CAS PubMed.
- S. Xu, T. Naren, Y. Zhao, Q. Gu, T. Wai Lau, C.-S. Lee, F.-R. Chen, J. Yin, L. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2025, e202422040 Search PubMed.
- P. Yi, Z. Li, L. Ma, B. Feng, Z. Liu, Y. Liu, W. Lu, S. Cao, H. Fang, M. Ye and J. Shen, Adv. Mater., 2024, 36, 2414379 CAS.
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 RSC.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef CAS.
- (a) H. He, R. Shen, Y. Yan, D. Chen, Z. Liu, L. Hao, X. Zhang, P. Zhang and X. Li, Chem. Sci., 2024, 15, 20002–20012 RSC; (b) G. C. Thaggard and N. B. Shustova, ACS Cent. Sci., 2025 DOI:10.1021/acscentsci.5c00357; (c) Y. Su, B. Li, Z. Wang, A. Legrand, T. Aoyama, S. Fu, Y. Wu, K.-I. Otake, M. Bonn, H. I. Wang, Q. Liao, K. Urayama, S. Kitagawa, L. Huang, S. Furukawa and C. Gu, J. Am. Chem. Soc., 2024, 146, 15479–15487 CrossRef CAS PubMed.
- H. Zhang, Y. Geng, J. Huang, Z. Wang, K. Du and H. Li, Energy Environ. Sci., 2023, 16, 889–951 RSC.
- X. Huang, X. Qiu, W. Wang, J. Li, Z. Li, X. Yu, J. Ma and Y. Wang, J. Am. Chem. Soc., 2023, 145, 25604–25613 Search PubMed.
- Y. Lin, H. Cui, C. Liu, R. Li, S. Wang, G. Qu, Z. Wei, Y. Yang, Y. Wang, Z. Tang, H. Li, H. Zhang, C. Zhi and H. Lv, Angew. Chem., Int. Ed., 2023, 62, e202218745 CrossRef CAS PubMed.
- (a) W. Li, Q. Huang, H. Shi, W. Gong, L. Zeng, H. Wang, Y. Kuai, Z. Chen, H. Fu, Y. Dong and C. Zhang, Adv. Funct. Mater., 2024, 34, 2310668 CrossRef CAS; (b) J. Sun, Y. Xu, Y. Lv, Q. Zhang and X. Zhou, CCS Chem., 2023, 5, 1259–1276 CrossRef CAS.
- (a) Z. Chen, H. Su, P. Sun, P. Bai, J. Yang, M. Li, Y. Deng, Y. Liu, Y. Geng and Y. Xu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116775119 CrossRef CAS PubMed; (b) Q. Xue, H. Li, P. Jin, X. Zhou and F. Wang, Angew. Chem., Int. Ed., 2025, e202423368 Search PubMed.
- X. Yi, Y. Guo, S. Chi, S. Pan, C. Geng, M. Li, Z. Li, W. Lv, S. Wu and Q.-H. Yang, Adv. Funct. Mater., 2023, 33, 2303574 CrossRef CAS.
- D. Tomerini, C. Gatti and C. Frayret, Phys. Chem. Chem. Phys., 2016, 18, 2442–2448 RSC.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- (a) X. Tang, Y. Yang, X. Li, X. Wang, D. Guo, S. Zhang, K. Zhang, J. Wu, J. Zheng, S. Zheng, J. Fan, W. Zhang and S. Cai, ACS Appl. Mater. Interfaces, 2023, 15, 24836–24845 CrossRef CAS PubMed; (b) C.-X. Liu, S. Hwang, Y. Lee, Y. H. Ko, S. S. Park and E. Lee, ACS Appl. Mater. Interfaces, 2024, 16, 48203–48210 CrossRef CAS PubMed.
- (a) W. Du, X. Du, M. Ma, S. Huang, X. Sun and L. Xiong, Adv. Funct. Mater., 2022, 32, 2110871 CrossRef CAS; (b) Z. Li, H.-Y. Zhou, F.-L. Zhao, T.-X. Wang, X. Ding, B.-H. Han and W. Feng, Chin. J. Polym. Sci., 2020, 38, 550–557 CrossRef CAS.
- S. Xu, X. Tan, W. Ding, W. Ren, Q. Zhao, W. Huang, J. Liu, R. Qi, Y. Zhang, J. Yang, C. Zuo, H. Ji, H. Ren, B. Cao, H. Xue, Z. Gao, H. Yi, W. Zhao, Y. Xiao, Q. Zhao, M. Zhang and F. Pan, Angew. Chem., Int. Ed., 2023, 62, e202218595 CrossRef CAS PubMed.
- (a) K. Müllen, Nat. Rev. Mater., 2016, 1, 15013 CrossRef; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- S. Ghosh, Y. Tsutsui, T. Kawaguchi, W. Matsuda, S. Nagano, K. Suzuki, H. Kaji and S. Seki, Chem. Mater., 2022, 34, 736–745 CrossRef CAS.
- C. Wang, Energy Environ. Mater., 2020, 3, 441–452 CrossRef CAS.
- (a) Q. Peng, H. Dong, H. Yan, Y. Xiao, Y. Wang, S. Chou and S. Chen, Energy Storage Mater., 2024, 73, 103849 CrossRef; (b) J. Hou, H. Liu, M. Gao, Q. Pan and Y. Zhao, Angew. Chem., Int. Ed., 2025, 64, e202414566 CAS.
- (a) L. Zhong, C. Wang, J. He, Z. lin, X. Yang, R. Li, S. Zhan, L. Zhao, D. Wu, H. Chen, Z. Tang, C. ZHi and H. Lv, Adv. Mater., 2024, 36, 2314050 CAS; (b) J. Liu, Y. Zhou, G. Xing, M. Qi, Z. Tang, O. Terasaki and L. Chen, Adv. Funct. Mater., 2024, 34, 2312636 CrossRef CAS; (c) R. Iqbal, A. Badshah, Y.-J. Ma and L.-J. Zhi, Chin. J. Polym. Sci., 2020, 38, 558–564 CAS.
- X. Xu, S. Zhang, K. Xu, H. Chen, X. Fan and N. Huang, J. Am. Chem. Soc., 2023, 145, 1022–1030 CAS.
- S.-L. Cai, Y.-B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S.-R. Zheng, W.-G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 CAS.
- B. Wen, Y. Huang, Z. Jiang, Y. Wang, W. Hua, S. Indris and F. Li, Adv. Mater., 2024, 36, 2405440 CAS.
- M. Wang, S. Fu, P. Petkov, Y. Fu, Z. Zhang, Y. Liu, J. Ma, G. Chen, S. M. Gali, L. Gao, Y. Lu, S. Paasch, H. Zhong, H.-P. Steinrück, E. Cánovas, E. Brunner, D. Beljonne, M. Bonn, H. I. Wang, R. Dong and X. Feng, Nat. Mater., 2023, 22, 880–887 CAS.
- S. J. Kang, M. Hong, J. H. Won, B. Han, J. K. Kang and H. M. Jeong, Adv. Energy Mater., 2025, 15, 2402651 CAS.
- W. Zhou, W.-Q. Deng and X. Lu, Interdiscip. Mater., 2024, 3, 87–112 CAS.
- (a) L. Wei, S. Wu, C. Li, C. Liu, H. Chen, Y.-B. Zhang, F. Zheng, Y. Ma and Y. Zhao, J. Am. Chem. Soc., 2024, 146, 31384–31390 CAS; (b) Y. Su, Z. Chen, X. Tang, H. Xu, Y. Zhang and C. Gu, Angew. Chem., Int. Ed., 2021, 60, 24424–24429 CrossRef CAS PubMed.
- S. Jiang, H. Niu, Q. Sun, R. Zhao, N. Li and Y. Cai, J. Mater. Chem. A, 2024, 12, 11416–11423 CAS.
- S. Haldar, M. Wang, P. Bhauriyal, A. Hazra, A. H. Khan, V. Bon, M. A. Isaacs, A. De, L. Shupletsov, T. Boenke, J. Grothe, T. Heine, E. Brunner, X. Feng, R. Dong, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2022, 144, 9101–9112 CAS.
- A. D. Thompson and M. P. Huestis, J. Org. Chem., 2013, 78, 762–769 CAS.
- B.-Q. Li, S.-Y. Zhang, B. Wang, Z.-J. Xia, C. Tang and Q. Zhang, Energy Environ. Sci., 2018, 11, 1723–1729 CAS.
- J. Shen, R. Zhang, Y. Su, B. Shi, X. You, W. Guo, Y. Ma, J. Yuan, F. Wang and Z. Jiang, J. Mater. Chem. A, 2019, 7, 18063–18071 CAS.
- (a) L. Garzón-Tovar, J. Pérez-Carvajal, A. Yazdi, J. Hernández-Muñoz, P. Tarazona, I. Imaz, F. Zamora and D. Maspoch, Angew. Chem., Int. Ed., 2019, 58, 9512–9516 Search PubMed; (b) Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 Search PubMed.
- D. Guo, F. Ming, D. B. Shinde, L. Cao, G. Huang, C. Li, Z. Li, Y. Yuan, M. N. Hedhili, H. N. Alshareef and Z. Lai, Adv. Funct. Mater., 2021, 31, 2101194 CAS.
- C.-C. Li, M.-Y. Gao, X.-J. Sun, H.-L. Tang, H. Dong and F.-M. Zhang, Appl. Catal., B, 2020, 266, 118586 CAS.
- (a) N. He, Y. Zou, C. Chen, M. Tan, Y. Zhang, X. Li, Z. Jia, J. Zhang, H. Long, H. Peng, K. Yu, B. Jiang, Z. Han, N. Liu, Y. Li and L. Ma, Nat. Commun., 2024, 15, 3896 CAS; (b) L. Huang, W. Li, F. Wei, S. Ke, H. Chen, C. Jing, J. Cheng and S. Liu, Chem, 2024, 10, 3100–3113 CAS.
- Y. Liu, W. Zhou, W. L. Teo, K. Wang, L. Zhang, Y. Zeng and Y. Zhao, Chem, 2020, 6, 3172–3202 CAS.
- D. Divya, H. Mishra and R. Jangir, Chem. Commun., 2025, 61, 2403–2423 CAS.
- Q. Xu, Z. Liu, Y. Jin, X. Yang, T. Sun, T. Zheng, N. Li, Y. Wang, T. Li, K. Wang and J. Jiang, Energy Environ. Sci., 2024, 17, 5451–5460 CAS.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 Search PubMed.
- B. Sun, Z. Sun, Y. Yang, X. L. Huang, S. C. Jun, C. Zhao, J. Xue, S. Liu, H. K. Liu and S. X. Dou, ACS Nano, 2024, 18, 28–66 CAS.
- Z. Li, Y. Wang, J. Wang, C. Wu, W. Wang, Y. Chen, C. Hu, K. Mo, T. Gao, Y.-S. He, Z. Ren, Y. Zhang, X. Liu, N. Liu, L. Chen, K. Wu, C. Shen, Z.-F. Ma and L. Li, Nat. Commun., 2024, 15, 10216 CAS.
- (a) J. Feng, J. Duan, C.-T. Hung, Z. Zhang, K. Li, Y. Ai, C. Yang, Y. Zhao, Z. Yu, Y. Zhang, L. Wang, D. Zhao and W. Li, Angew. Chem., Int. Ed., 2024, 63, e202405252 CAS; (b) J. Ferrando-Soria and A. Fernandez, Nano-Micro Lett., 2024, 16, 88 CAS.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CAS.
- W. Sun, X. Tang, Q. Yang, Y. Xu, F. Wu, S. Guo, Y. Zhang, M. Wu and Y. Wang, Adv. Mater., 2019, 31, 1903176 Search PubMed.
- (a) Y. Deng, Y. Wang, X. Xiao, B. J. Saucedo, Z. Zhu, M. Xie, X. Xu, K. Yao, Y. Zhai, Z. Zhang and J. Chen, Small, 2022, 18, 2202928 CAS; (b) S. Zhou, Y. Kuang, H. Yang, L. Gan, X. Feng, C. Mao, L. Chen, J. Zheng and G. Ouyang, Angew. Chem., Int. Ed., 2024, 63, e202412279 CrossRef CAS PubMed.
- T. Lin, R. Qiao, Y. Luo, Z. Zhang, Y. Yang and J. Li, J. Alloys Compd., 2023, 936, 168184 CrossRef CAS.
- (a) L. Zhang, S. Ji, L. Yu, X. Xu and J. Liu, RSC Adv., 2017, 7, 24004–24010 RSC; (b) Z. Zhang, Y. An, J. Feng, L. Ci, B. Duan, W. Huang, C. Dong and S. Xiong, Mater. Lett., 2016, 181, 340–344 CrossRef CAS.
- B. Cui and G. Fu, Nanoscale, 2022, 14, 1679–1699 RSC.
- L. Ye, W. Cen, Y. Chu and D. Sun, Nanoscale, 2023, 15, 13187–13201 RSC.
- M.-X. Wu, Y. Wang, G. Zhou and X. Liu, Coord. Chem. Rev., 2021, 430, 213735 CrossRef CAS.
- J. C. Stallard, L. Wheatcroft, S. G. Booth, R. Boston, S. A. Corr, M. F. L. De Volder, B. J. Inkson and N. A. Fleck, Joule, 2022, 6, 984–1007 CrossRef CAS.
- A. Saleem, M. K. Majeed, R. Iqbal, A. Hussain, M. S. Naeem, S. Rauf, Y. Wang, M. S. Javed and J. Shen, Adv. Mater. Interfaces, 2022, 9, 2200800 CrossRef CAS.
- B. Jia, W. Liu, C. Yao, W. Xie and Y. Xu, Chem. Commun., 2024, 60, 6435–6438 RSC.
| This journal is © The Royal Society of Chemistry 2025 |