
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLatest advances in in situ and operando X-ray-based techniques for the characterisation of photoelectrocatalytic systems
Mariam
Barawi
 a,
Camilo A.
Mesa
b,
Laura
Collado
a,
Ignacio J.
Villar-García
c,
Freddy
Oropeza
a,
Víctor A.
de la Peña O'Shea
*a and
Miguel
García-Tecedor
*a
a,
Camilo A.
Mesa
b,
Laura
Collado
a,
Ignacio J.
Villar-García
c,
Freddy
Oropeza
a,
Víctor A.
de la Peña O'Shea
*a and
Miguel
García-Tecedor
*a
aPhotoactivated Processes Unit, IMDEA Energy, Avda. Ramón de la Sagra, 3, Móstoles, 28935, Spain. E-mail: miguel.tecedor@imdea.org; victor.delapenya@imdea.org
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC, Barcelona Institute of Science and Technology, UAB Campus, 08193 Bellaterra, Barcelona, Spain
cDepartment of Chemistry and Biochemistry, Facultad de Farmacia, Universidad San Pablo-CEU, CEU Universities, Boadilla del Monte, 28668 Madrid, Spain
First published on 25th July 2024
Abstract
In situ and operando X-ray techniques have emerged as powerful tools for unravelling the complex mechanisms underlying photoelectrochemical transformations. These techniques offer real-time insights into the dynamic processes occurring at the electrode–electrolyte interface during solar-driven water splitting and other PEC reactions. The present work aims to summarise the latest advances in in situ and operando X-ray absorption spectroscopy (XAS), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) for the characterisation of photoelectrocatalytic systems and materials for the generation of solar fuels and value-added chemicals. This review highlights the recent advancements achieved using these techniques for elucidating the structural, chemical, and electronic properties of photoelectrocatalytic materials and interfaces during operation. Besides, this review provides technical guidance for performing these measurements, considering the experimental requirements for each of these spectroscopies. Further, we provide an overview of different state-of-the-art synchrotron-based techniques employed for the characterisation of photoelectrocatalytic materials, focusing on the possibilities of the studied techniques, cell designs and more relevant results.
 Miguel García-Tecedor | Miguel García Tecedor (PhD in Physics 2017, Universidad Complutense de Madrid) is a Senior Assistant Researcher at IMDEA Energy. During his PhD, he was focused on the growth and characterisation of semiconducting nanostructures and their applications in optoelectronics and energy. As part of his international PhD, in 2015, he joined the Institute for Energy Technology, located in Kjeller, Norway, to work on the synthesis and characterisation of organic–inorganic composites for silicon solar cells' passivation. In July 2017, he took a position as Research Scientist at the Institute of Advanced Materials at Universitat Jaume I, to develop novel strategies for (photo)electrocatalytic water splitting and CO2 reduction. Lately, in March 2021, Miguel joined the Photoactivated Processes Unit at IMDEA Energy to work on photo(electro)catalytic approaches towards wastewater oxidation, CO2 reduction and N2 fixation. Miguel is currently co-author of 45 scientific publications and he has participated in 14 research projects, being principal investigator in three. |
Introduction
Artificial photosynthesis technologies have been attracting increasing attention, from fundamental understanding to, more recently, the industry, as an alternative source of fuels and value-added chemicals using ubiquitous molecules, such as CO2 and H2O, and sunlight.1–3 These technologies have achieved higher technology readiness levels (TRLs) of development in recent years, and are becoming a real alternative for the generation of solar fuels and chemicals at an industrial level. In the case of photoelectrochemical (PEC) routes, different architectures could be considered: (i) full photoelectrochemical systems, where both anode and cathode contain photoabsorber materials, (ii) electrocatalytic systems coupled to photovoltaic cells (PV-EC) and (iii) photoelectrochemical systems coupled to photovoltaic cells to reduce the overpotential for the target chemical reaction.4 In recent years, all these technologies have increased their maturity towards industrialisation. As explained in the recent work by G. Segev et al.,5 lab-scale photoelectrochemical devices exhibit efficiencies over 10% currently, which has been traditionally considered as the minimum threshold for any industrial application. However, in order to establish the photoelectrocatalytic approach at a relevant industrial level, from both the scientific and economic points of view, there are still many technological questions to be answered and physicochemical mechanisms to be revealed. To overcome this problem, the scientific community is currently highly interested in operando and in situ methods, which allow for the characterisation of the photoelectrocatalytic materials under relevant working conditions.6,7In particular, “in situ” characterization refers to the study of materials, usually catalysts, and reactions in their natural working environment without disrupting the experimental conditions. It is performed within the reactor or reaction cell during the reaction but not necessarily under real operating conditions. However, “operando” characterization refers to the study of materials and catalytic reactions under real operating conditions. Both approaches are crucial for understanding and improving the catalytic systems; however, they are applied in slightly different contexts depending on whether a study is needed under controlled conditions (in situ) or realistic catalytic conditions (operando).8
Spectroscopy-based techniques are powerful tools to investigate the photoelectrode–electrolyte interface where information on the electronic and structural nature of the active sites can be obtained upon careful selection of excitation and probe sources.9 As such, there is a growing tendency in the field to perform an in situ and operando analysis using various techniques such as Raman spectroscopy,10,11 UV-vis spectroscopy,12 Fourier-transform infrared (FTIR) spectroscopy,13 electrochemical impedance spectroscopy (EIS),14 and incident modulated photocurrent spectroscopy (IMPS),15 due to the key importance of understanding the materials under real working conditions. Hence, the combination of in situ and operando synchrotron techniques with photoelectrocatalytic experiments is pivotal to understand the performance of these materials, in terms of stability, efficiency, and selectivity, under working conditions and thus, establishing this technology as an important element in the green energy transition.
In terms of X-ray-based spectroscopy techniques such as XPS, XAS and XRD, the majority of in situ and operando reported PEC studies have employed synchrotron radiation. This is due to the advantages of synchrotron radiation compared with other lab-scale X-ray sources: (i) high brightness and energy resolution, which enables high-speed data acquisition and provides more accurate data, (ii) high spatial resolution, which allows for precise material mapping, (iii) wide energy range, enabling variable energy X-ray techniques (XAS) and conducting experiments at a range of selected optimum wavelengths (XPS and XRD), (iv) polarized nature, allowing specific experiments to probe the magnetic and electronic properties of materials and (v) precise flashing time structure at a very high frequency, which can be used to study physical and chemical processes taking place at very short time scales.16
In situ and operando synchrotron techniques are more mature in the electrocatalytic (EC) field, which can be explained not only by the maturity of the field but also due to several inherent difficulties in order to study photoelectrocatalytic reactions in situ: (i) although many novel oxide and chalcogenide electrocatalysts undergo bulk changes that lead to the actual catalytic species,17–20 the bulk of photoelectrodes are usually stable and most relevant changes are limited to their surfaces involving usually a few tens of nanometres,21,22 which demands surface-sensitive techniques or special analysis modes to make commonly bulk techniques surface sensitive, and both (ii) the need to illuminate the sample during operation,23 and (iii) the usual employment of thick transparent conductive substrates (FTO or ITO, most frequently), instead of thin current collectors, add an extra level of complexity to the experimental set-up. Due to these challenges, only a few works have reported successful in situ or operando synchrotron-based spectroscopies in photoelectrochemical systems.
This review aims to give a general vision of the current state of the art of in situ and operando X-ray-based studies, focusing mainly on X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and X-ray diffraction spectroscopy (XRD), in PEC systems for solar fuel generation (Scheme 1). Besides, this work provides technical guidance for performing these experiments at synchrotron facilities, with the hope of increasing the activity in this field, which has the potential to answer some of the main questions to take the comprehension of PEC systems to the next level.
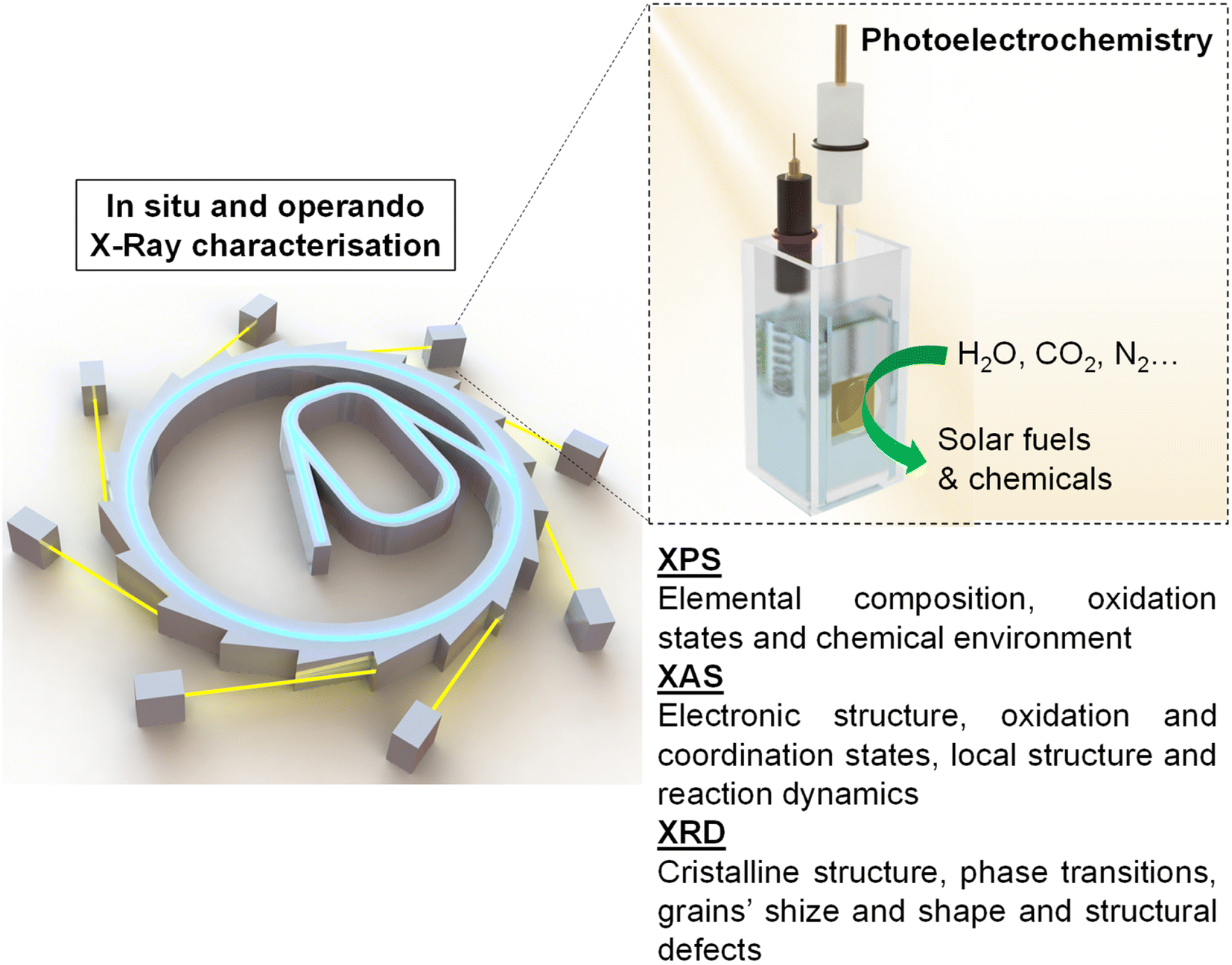 | ||
| Scheme 1 Overview of the addressed topics in the present review. | ||
By synthesizing and critically evaluating these analytical possibilities, this work aims to serve as a pivotal resource for guiding future research efforts and informing the design of more efficient and sustainable photoelectrocatalytic technologies. We believe that the comprehensive nature of this revision, its clear organization of existing knowledge, and its potential to advance the field make it well-suited for energy researchers interested in initiating or advancing their work with operando and in situ experiments in PEC systems.
In situ and operando X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy (XPS) is a commonly used characterisation technique for the analysis of the surface of materials. This technique, also called ESCA (Electron Spectroscopy for Chemical Analysis), provides qualitative and quantitative elemental analysis, except for hydrogen and helium, of the top 1–20 nm (depending on the sample and instrumental conditions) of a surface.24 In XPS, the sample is irradiated with soft X-rays (energies lower than ∼6 keV) and the kinetic energy of the emitted electrons is analysed. When an X-ray of known energy (hν) interacts with an atom, a photoelectron can be emitted via the photoelectric effect. The emitted photoelectron is the result of complete transfer of the X-ray energy to a core level electron. It simply states that the energy of the X-ray (hv) is equal to the binding energy (Eb) of the electron (how tightly it is bound to the atom/orbital to which it is attached) plus the kinetic energy (Ek) of the electron that is emitted and the spectrometer work function (Φsp), a constant value, and hence, Eb can be calculated using the following equation:| Eb = hν − Ek − Φsp | (1) |
Thus, valuable chemical information about the sample can be extracted because binding energies are sensitive to the chemical environment of the atom.25 Hence, XPS provides unique information on the chemical composition and electronic structure of materials, so that it has been traditionally used for the fundamental characterisation of semiconductors with application in the field of photoelectrochemistry. The development of near-ambient pressure (NAP) XPS has extended the applications of XPS to the performance of detailed studies of the chemical state of photoelectrodes and the reaction intermediates under relevant or even operando conditions, which facilitate the understanding of reaction mechanisms. As a result, NAP XPS is nowadays commonly used for the in situ characterisation of catalytic, photocatalytic and (photo)electrocatalytic reactions. The field has evolved exponentially since the development of the new generation of NAP XPS analysers at the beginning of the century.26 The last comprehensive review in 2020 estimates that there are more than 70 NAP XPS systems around the world, including laboratory and synchrotron-based systems.27 Most NAP XPS systems allow the dosing of gases and gas mixtures and heating in order to perform in situ characterisation of heterogeneous catalytic reactions in different gas atmospheres. These systems can be easily adapted for electrochemical systems based on solid electrolytes.28 However, the measurement of electrocatalytic systems based on liquid electrolytes requires the use of especially designed electrochemical cells.29 There are two main types of approaches in the literature for the operando analysis of electrochemical reactions using liquid electrolytes by XPS. One requires the use of graphene windows that separate the liquid and UHV environments, and through which, X-rays and electrons can pass through, allowing XPS measurements. Despite being first established in 2011,30 these cells have been rarely studied, mainly due to the fragility of these 2D windows under operation in UHV chambers.31,32 The other approaches are windowless and require the delicate fine control of the continuous evaporation of the electrolyte in a UHV chamber.33 which requires a fine balance of the operating conditions but has given rise to a more significant amount of contributions in both photoelectrochemical and electrocatalytic studies.28
The performance of in situ measurements of photoelectrocatalytic systems adds an additional complication to the photoelectrochemical approach, as the sample must be properly illuminated. For the illumination, there are two possibilities, direct illumination by the set-up of an UV light within the UHV chamber using electrical feedthroughs or illumination through a window outside the XPS instrument. This port can be one facing directly at the sample in the UHV chamber or the port at the photoelectron analyser that allows directing light down the axis of the spectrometer. Common UHV windows made of borosilicate allow the transmittance of most ultraviolet light down to 300/350 nm wavelengths. If the experiments require the use of more energetic UV radiation, UV windows made of sapphire, fused silica or even MgF2 could be used with absorbance cut-offs that go down to 200 nm wavelengths.34 Specifically, there are different available commercial windows for performing these experiments: (i) OROFLOAT borosilicate windows (Edmund Optics) have a transmission of more than 90% between 350 and 2500 nm; (ii) TECHSPEC sapphire windows (Edmund Optics) are useful in the transmission range of 330–5500 nm; (iii) fused quartz windows (Knight Optical) are a more suitable material choice for UV light; and (iv) any other window materials of the correct size, which satisfies the needs of the users' experiment. User-supplied light sources can also be attached to the main chamber with minimal effort. Moreover, illumination from outside the UHV chamber requires a lens focusing system at the sample distance or a large collimated source. This latter set up adds the advantage of illuminating the sample only in the area of investigation. One also has to consider that depending on the energy range and intensity, illumination can lead to sample heating. Therefore, a way to measure the temperature of the sample or a previous calibration is advisable, in order to discount the temperature effect on the studied reaction.
The dip and pull method, shown in Fig. 1, is probably the easier type of electrochemical cell to adapt to photoelectrocatalytic measurements and the method of choice in most photoelectrochemical studies reported in the literature. In particular, the SpAnTeX endstation at the BElChem-PGM endstation in BESSY II offers a dip and pull module specially designed for photoelectrocatalytic measurements.35 Other end stations that offer the dip and pull method such as HIPPIE in Max IV, PHOENIX I at Swiss Light Source (SLS) or BL 9.3.1 at Advanced Light Source (ALS) could be easily adapted to photoelectrochemical experiments building up on their experience on photocatalytic reaction monitoring. Most of the dip and pull measurements are done in Synchrotron stations using tender X-rays (2–7 keV) as photoelectrons of high energy have more probabilities of going through the thin electrolyte meniscus that is created in order to allow electrons coming from the “buried” electrode to reach the analyser. As the probabilities of emitting photoelectrons of most probed orbitals go down significantly with excitation energy, 3 keV seems to be the sweet spot for maximising the signal intensity of the buried electrodes.33 Nevertheless, experiments can be done at lower excitation energies, e.g. the HIPPIE beamline works at a maximum excitation energy of 2.2 keV and laboratory systems have also been used successfully to probe buried liquid interfaces in battery systems, for example.36 Moreover, the use of silver (Ag Kα, 2.9 keV) or chromium (Cr Kα, 5.4 keV) sources can increase the probing depth of laboratory systems, complementing the experiments done with the typical Al Kα sources (1.5 keV). The added advantage of using synchrotron systems is that the variable energy can also be used to differentiate between surface and subsurface signals from the electrode, as surface signals will be more enhanced at lower excitation energies. Moreover, synchrotron light allows performing Auger yield NEXAFS, which provides complementary surface-sensitive XAS information.
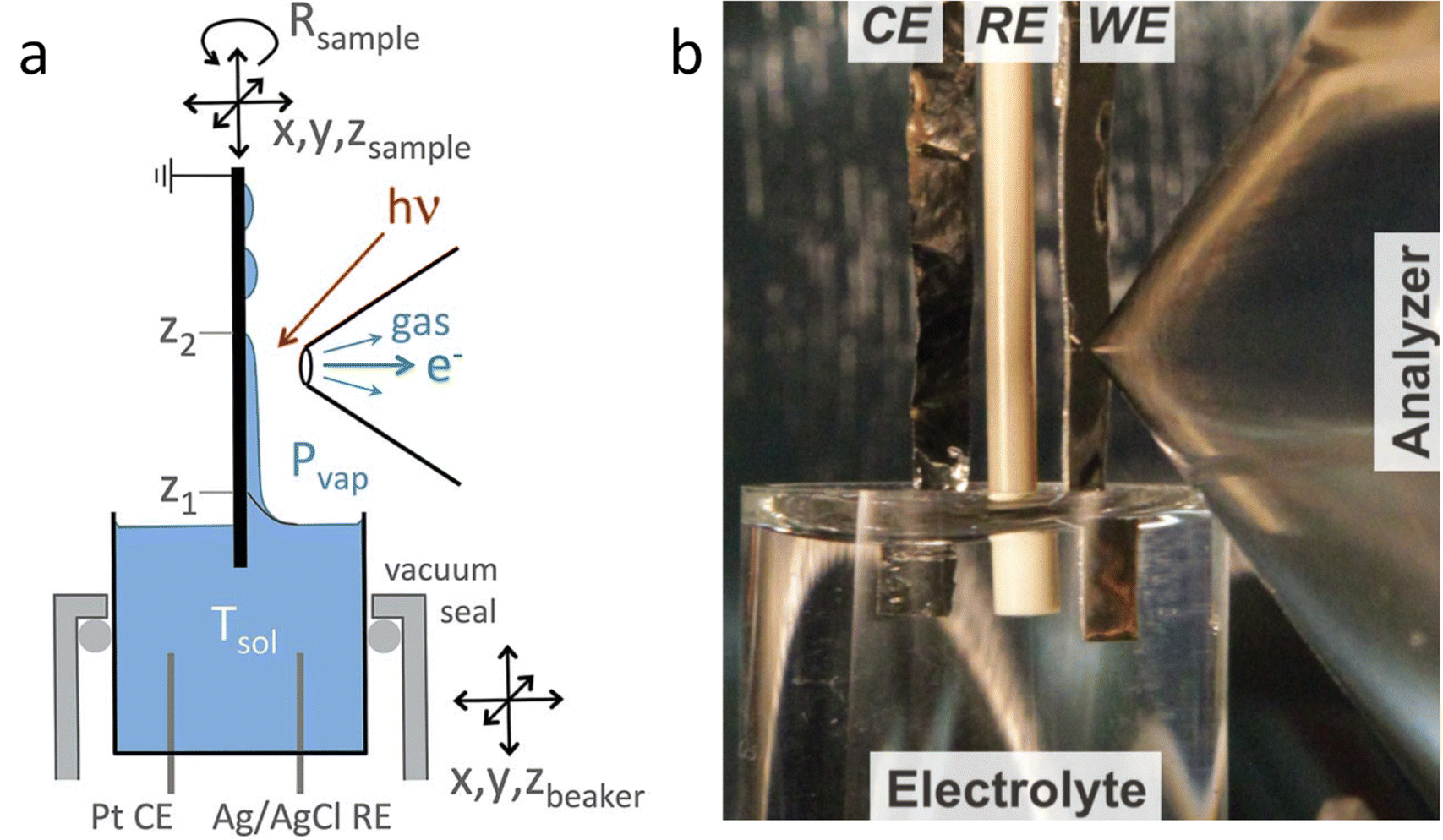 | ||
| Fig. 1 (a) Scheme of the dip and pull method for the preparation of thin liquid films on electrodes. (b) Picture of the dip and pull set up at ALS. Copyright 2015. Reproduced with permission from Springer Nature. Adapted from ref. 33. | ||
The possibility of performing in situ and operando XPS experiments using these setups has advanced the knowledge of the surface chemistry and electronic properties of photoabsorber materials that are used as photoelectrodes under real conditions of illumination and chemical environment. These studies can be divided into three categories based on how close to the actual operando conditions they are performed: (1) XPS under UV-visible illumination in UHV, (2) XPS under UV-visible illumination in NAP conditions, and (3) operando XPS. In the following subsections, we discuss these contributions in terms of the information obtained and the required facilities.
XPS-based SPV analysis offers an advantage over traditional Kelvin probe techniques by delivering extra chemical insights, which make it a valuable tool for semiconductor research.44,45 A study by Oropeza et al.44 examines the effect of surface photoreduction on the PEC properties of CuBi2O4, using XPS to study the surface reduction and its effect on the surface energetics and SPV. They report that irradiation of CuBi2O4 with photons (hν ≥ 2.7 eV) in an inert atmosphere leads to the formation of reduced Cu on the surface. This reduction induces a downward band bending of 0.35 eV, enhancing the charge carrier transport properties, as evidenced by an increase in the characteristic SPV from 0.07 to 0.27 V. This remarkable finding constitutes the first experimental evidence of the beneficial effects of surface photoreduction on the PEC properties of CuBi2O4. Similarly, in their investigation of the InP interphase energetic optimization, Gao et al.45 employed XPS in core and valence band regions to monitor the modulation of the surface energetics caused by the introduction of a buried p–n+ junction, which favourably shifts the valence band edge to promote minority charge carrier's diffusion. They confirmed the enhancement of the charge carrier's diffusion length by XPS-based SPV measurements, using the binding energy shift of the P 2p and In 3d peaks upon in situ illumination, see Fig. 2a. They observed an increase in the SPV from 0.1 V to 0.35 V, and confirmed that the SPV of the n+/p-InP depends on the light intensity with the relationship:
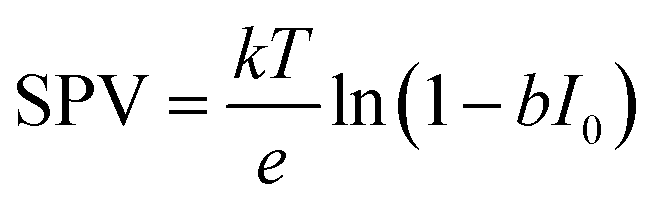 | (2) |
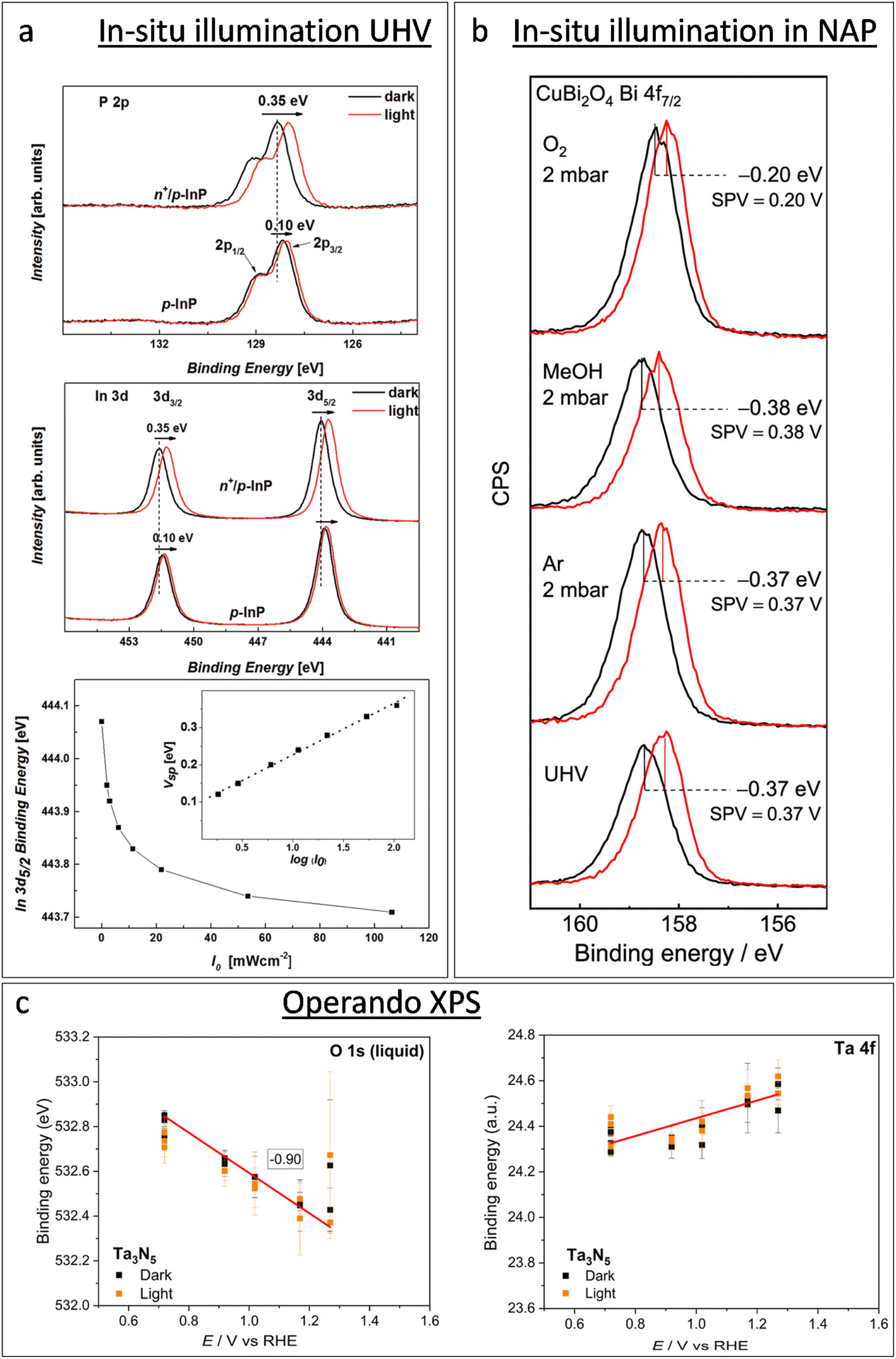 | ||
| Fig. 2 Progress of in situ XPS studies from illumination in UHV operando XPS. (a) From top to bottom: XPS in the P 2p and In 3d regions of p-InP and n+/p-InP in the darkness (black solid lines) and under illumination with an LED hv2 = 1.98 eV (red solid lines), and In 3d5/2 binding energy shift for n+/p-InP as a function of the incident light flux (I0). The inset shows the surface photovoltage (SPV) as a function of log(I0). Copyright 2016. Reproduced with permission from Wiley. Adapted from ref. 45. (b) XPS of a CuBi2O4 photoelectrode in the Bi 4f7/2 region, showing the XPS-based SPV measurement (spectra in black lines were recorded in the darkness, and spectra in red lines were recorded under light with a 405 nm laser) in UHV and in the presence of Ar, methanol vapour, and oxygen. Copyright 2019. Reproduced with permission from the American Chemical Society. Adapted from ref. 44. (c) Binding energy of the Ta 4f7/2 component and liquid contribution in the O 1s spectra during operando AP-XPS measurements for the Ta3N5 photoelectrode. Copyright 2023. Reproduced with permission from the American Chemical Society. Adapted from ref. 46. | ||
Zhang and Ptasinska conducted a series of studies of the water dissociation on model Ga-based photoelectrodes exposing GaP(111) and GaN(0001) to water vapour up to 5 mbar and temperatures from 298 to 773 K.47–49 They found enhanced surface hydroxylation and oxidation of gallium on GaP48 as the water pressure increased, with significant changes at temperatures above 673 K, leading to the formation of non-stoichiometric gallium hydroxide and oxidation of phosphorus atoms. For GaN(0001), water dissociation at room temperature led to the formation of hydroxyl groups and a decrease in intrinsic upward band bending, enhancing its photocathodic properties. However, increased temperatures strengthened the chemical interaction, forming more oxides and hydroxides, which increased upward band bending and negatively affected GaN's photocathodic properties.49 Additionally, their studies on Pt/GaN(0001) revealed that water dissociates readily at the Pt/GaN interface, unlike on pure Pt surfaces.47 This interface showed significant upward band bending, promoting the accumulation of photogenerated holes and enhancing oxidative dissociative adsorption of water, leading to the formation of hydroxyl groups and surface oxides bound to Pt, which are active catalytic sites for the hydrogen evolution reaction (HER).50,51 Jackman et al. studied water adsorption on anatase TiO2(101) using NAP-XPS,52 finding that water adsorbs in a mixed molecular and dissociated state on stoichiometric surfaces. Sputtered surfaces exhibited similar characteristics, indicating that subsurface vacancies do not alter water interaction. The significant amount of adsorbed molecular water on TiO2 suggests limited chemical interaction compared to more reactive surfaces such as GaP, InP, and GaN, where a more dissociative process occurs.47–49
The chemical and electronic interactions between photoelectrodes and adsorbates have also been an important topic of research using NAP-XPS. An early study by Salmeron et al. showed that the adsorption of O2 (with a O2 pressure of 1 torr) on the TiO2 surface leads to a shift in the Ti 2p and O 1s core level binding energies towards a lower binding energy (by 0.4 eV), suggesting that the adsorption process involves the transfer of electrons from TiO2 to O2, resulting in such an upward band bending.53 Mansfeldova et al. studied more specifically the effect of the environment on the surface work function (WF) of the TiO2 photoelectrode.54 Their NAP-XPS data showed a significant decrease in the surface WF of TiO2 (by about 1 eV) upon water vapour exposure at 0.5 mbar, indicating a strong upward shift of the conduction band, which correlates with its photoanode behaviour in liquid electrolytes. The effect of the environment on the SPV of photoelectrodes is also accessible by NAP-XPS. Using in situ illumination under NAP-XPS conditions, Oropeza et al.44 studied the effect of key electron donors and acceptors in the SPV of CuBi2O4 photocathodes, finding that while electron acceptors such as O2 cause a reduction in the SPV, electron donors such as methanol cause an increase in the SPV (see Fig. 2b). Since XPS can simultaneously monitor the surface chemistry of the sample, the authors could correlate the diminution of the SPV with the re-oxidation of reduced Cu surface states, which led to the conclusion that those reduced Cu surface states are key achieving good photoelectrochemical properties of CuBi2O4.
For instance, the research conducted by Lichterman et al.55 provides a fundamental perspective on the operational characteristics of semiconductor/liquid junctions, in more realistic conditions than previous gas-phase-based NAP-XPS studies. Using the above-described dip-and-pull technique, they demonstrated how operando ambient-pressure XPS can be used to directly characterize the energetics of semiconductor/liquid junctions, focusing on the dynamics of semiconductor accumulation, depletion, and Fermi level pinning by defect states. In their work, the observed shifts in binding energy with respect to the applied potential directly revealed the ohmic junction nature at the liquid interface on metallized samples, and the rectifying junction behaviour at the liquid interface with semiconductor photoabsorbers. Furthermore, their findings on the behavior of metallized semiconductor/film contacts under varying conditions contribute to clarifying the mechanisms of charge transfers in photoelectrochemical cells. They highlighted that all parameters that can be directly observed by operando XPS are crucial variables for the design and operation of semiconductor–liquid junction devices for the PEC water splitting and CO2 reduction. In a similar dip-and-pull approach, and introducing in situ illumination, Dahl et al.,46 demonstrated that the semiconductor/electrolyte interfacial energetics of a tantalum nitride (Ta3N5) film, with and without a nickel oxide (NiOx) protective layer, was explored under an applied bias under both dark and illumination conditions. Fig. 2c shows the variation in the binding energy of Ta 4f from the sample and O 1s from the electrolyte as a function of the applied potential under dark and illumination conditions. As expected, the O 1s peak position of the electrolyte shifts constantly with the applied potential; however, the Ta 4f peak position is independent of the applied potential under both dark and illumination conditions. The authors interpreted it as a direct evidence of Fermi-level pinning in Ta3N5 at the Ta3N5/electrolyte that effectively prevents band bending toward the Ta3N5/electrolyte. This means that there is no potential drop in the SCR that can promote the separation of photogenerated holes and electrons, which leads to poor PEC performance of bare Ta3N5. However, this work highlighted that surface passivation through the NiOx layer could significantly reduce the recombination of photogenerated carriers, leading to increased surface band bending of Ta3N5, and better PEC performances. They did not find any clear oxidation of the Ta3N5, which correlated with the significantly larger photocurrents and stability of the NiOx-coated materials. These types of operando XPS studies of the interface energetics provide key elements for the development of efficient and durable systems for water photoelectrolysis.
However, Favaro et al.56 focused on bismuth vanadate (BiVO4) electrodes in contact with an aqueous potassium phosphate (KPi) solution at an open circuit potential under both dark and light conditions, observing that visible light illumination triggers the formation of bismuth phosphate on the surface of the BiVO4 photoanode. They suggested that this chemical modification leads to surface passivation that impacts the distribution of ions within the thin electrolyte layer due to the repulsive interaction between the negatively charged surface under illumination and the phosphate ions in the solution, which is observed as an increase in their photoelectron signals. Interestingly, they found that such light-induced changes at the BiVO4/KPi electrolyte interface are fully reversible upon returning to dark conditions. Their findings emphasize the significance of in situ methods to understand the impact of operational conditions on the performance of photoanodes.
These studies collectively illustrate the critical role of operando XPS techniques in advancing the field of photoelectrochemistry. By providing direct insights into the interfacial energetics and chemical states of photoelectrodes under operational conditions, researchers can better understand and mitigate the challenges of photoelectrode degradation and inefficiency. Such advancements not only contribute to the fundamental knowledge of photoelectrochemical mechanisms but also pave the way for the development of more robust and efficient solar fuel production systems. The current availability of several synchrotron stations capable of performing operando photoelectrochemical studies with robust setups is the optimal platform to establish routine analysis of photoelectrochemical systems and quickly advance on these ambitious scientific objectives. The possibility of adapting the new AP XPS instruments such as Polaris in DESY57 and 3Sbar in ALBA (in construction phase), capable of performing XPS analysis at pressures of a few bars using tender X-ray sources, could provide more stable environments to establish routine analysis of photoelectrochemical systems. Moreover, in combination with the recently developed stroboscopic technique in MAX IV,58 it could lead to the performance of time-resolved experiments in the microsecond range and allow the investigation of charge transfer dynamics and reaction mechanics in the microsecond time scale, exploiting this way the maximum potential of operando XPS experiments of photoelectrochemical systems.
In situ and operando X-ray absorption spectroscopy (XAS)
Synchrotron X-ray absorption spectroscopy (XAS) has been proven as a highly effective tool for material characterisation. Specifically, in XAS, the absorption of energy, carried by a photon, promotes an electron from a core-level to an empty orbital, monitoring the unoccupied electronic states, and hence, probing the local chemical environment (4–5 Å), providing information regarding the specific chemical bonds, oxidation states, spin states, site symmetry, and coordination environment.59 In a typical XAS experiment, the monochromatic incident X-ray beam scans the vicinity of the absorption edge of an element, usually requiring a synchrotron X-ray source (with tuneable energy, high monochromatism and high intensity), even though there are currently available lab-scale XAS equipment.60 Specifically, there are two ways of performing XAS experiments: (i) X-ray absorption spectra are recorded by measuring the ratio of the transmitted and incident X-ray beam intensities, known as the transmission mode, or (ii) the yield of secondary particles, fluorescence photons or emitted electrons, as a function of photon energy varying across the absorption edge of an element (total fluorescence yield, TFY, and total electron yield, TEY, respectively).61 In transmission XAS, X-rays undergo attenuation while passing through the matter and intensity drops exponentially with the travelled distance, which is given as follows:| It = I0e−μx | (3) |
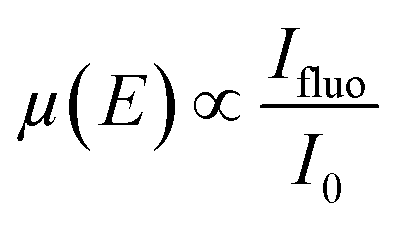 | (4) |
Finally, in analogy to XRF, the TEY measurements are based on the proportionality between the X-ray absorption coefficient and the emitted Auger electrons during the core decay as follows:
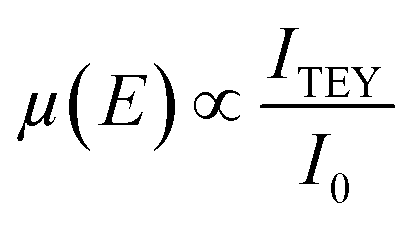 | (5) |
Specifically, this experimental mode consists of measuring the total necessary applied current to compensate the photoinduced drain current in the sample.63 Hence, every XAS spectrum represents the X-ray absorption coefficient, μ(E), of a system as a function of the incident X-ray photon energy. Besides, different regions can be distinguished in a typical XAS spectrum: (i) the pre-edge region, determined by the transition to the lowest unoccupied states, (ii) the X-ray absorption near-edge structure (XANES), also called near-edge X-ray absorption fine structure (NEXAFS), dominated by core transitions to quasi-bound states (multiple scattering resonances) for photoelectrons with kinetic energy in the range from 10 to 150 eV above the chemical potential and (iii) the extended X-ray absorption fine structure (EXAFS) region, associated with a weak scattering cross-section with neighbour atoms.
X-ray absorption spectroscopy (XAS) can be categorized into two measurement methods: hard-XAS, which employs high-energy X-rays to study bulk properties and heavy elements through core-level transitions, and soft-XAS, which utilizes lower-energy X-rays, offering surface sensitivity and probing valence band transitions for studying surface phenomena and organic material. Specifically, hard-XAS experiments are usually conducted in the transmission mode, which provides bulk-sensitive analysis and does not suffer from self-absorption effects.59 However, due to the attenuation length of less than 1 μm, soft-XAS is usually measured through fluorescence and electron yield modes, having different probe depths.64 The TFY technique shows an estimated probing depth of a few hundred nanometres, while TEY measurements are more surface-sensitive, with a probing depth of approximately a few nanometers.65 Additionally, depending on the nature of the sample, different detection modes can work better. For example, sometimes, transmission XAS experiments can suffer from sample damage, caused by inhomogeneous concentrations or densities in the sample. To avoid this problem, transmission experiments require careful sample preparation and relatively concentrated samples, ideally in a light matrix. In contrast, the fluorescence yield mode is more suitable for diluted samples or systems with heavy environments.66
In situ XAS measurements have been demonstrated to be essential for advancing our fundamental understanding of photoelectrocatalysis and accelerating the development of efficient catalytic materials for sustainable energy technologies. This spectroscopy allows monitoring changes in the chemical and electronic states of the catalyst in real time as they undergo photoelectrocatalytic reactions. This real-time monitoring provides information on reaction mechanisms, intermediate species, and reaction kinetics that are difficult to obtain using ex situ techniques. Besides, XANES and EXAFS can provide information about the local coordination environment and electronic structure of catalytic materials. This information helps in identifying the active sites responsible for catalytic activity, facilitating the design of more efficient catalysts.
In general, the experimental requirements for performing in situ and operando (hard)-XAS measurements are easier than those for XPS mainly due to the vacuum requirements, which make the development of in situ electrochemical cells much more complicated. Due to this reason, a considerable higher number of studies have reported in situ and operando (hard)-XAS measurements in electrodes for solar fuel generation compared to in situ and operando XPS.67,68 However, still limited studies have reported in situ XAS on photoelectrodes, as X-ray absorption measurements with photoelectrochemical cells require complex engineering to ensure compatibility and functionality of both systems simultaneously, compared to the in situ study of electrocatalysts.69,70 This involves designing specialized cells that can accommodate both X-ray transparency and photoelectrochemical functionality. Furthermore, most relevant chemical and electronic changes in photoelectrochemistry occur on the surface of the electrode, and not in the bulk, further complicating the collection of reliable data that provide true and reliable information. Thus, due to the bulk nature of the XAS technique compared with the shallow nature of the changes under PEC conditions in the photoelectrodes, there are mainly two ways to perform in situ experiments: (i) to fabricate really thin films of the photoelectrode under study, trying to maximize the surface-to-volume ratio or (ii) to investigate the induced changes in a co-catalyst by the effect of the photogenerated carriers in the absorbing material. Both types of studies are considered in the present review.
Among the characterisations of photoelectrodes for solar fuel generation by in situ and operando XAS, much effort has been devoted to study the oxidation reactions, mainly oxygen evolution reaction (OER), and hence, to characterise n-type semiconductors. However, there have already been some recent works that report in situ XAS in p-type photocathodes for the hydrogen evolution reaction (HER), and the challenging CO2 reduction reaction (CO2RR), as well as N2 fixation (N2RR) to ammonia (NH3).
Regarding photocathode analysis, in 2020 Chen et al.71 investigated bimetallic Ni-based p-Si micropillar photocathodes through in situ XAS for solar-driven HERs. The authors incorporated Ni/Mo, Ni/Co, and Ni/Fe into silicon electrodes and analysed the effect of incorporation of Mo, Co and Fe on the photoelectrochemical properties, finding out that the most active is the Ni/Mo combination (Fig. 3a). To investigate the oxidation state and local atomic environment under photoelectrochemical conditions, they performed in situ XANES measurements in the liquid medium. A custom reaction cell designed for this in situ measurement was used, but the authors do not provide details or schematics about its design. After introducing NiMo/p-Si MPAs in the alkaline electrolyte, no noticeable change was observed in the Ni K-edge spectra (Fig. 3b). However, under cathodic conditions, above 0 V, they found out that Ni XANES edge shifted to a higher energy, indicating an increase in the Ni valence state (Fig. 3c). Interestingly, when the same potential is applied to the NiCo/p-Si and NiFe/p-Si MPAs, negative changes in the absorption energy can be observed, indicating that the reduction of Ni was caused by the cathodic potential during the HER (Fig. 3d and e).
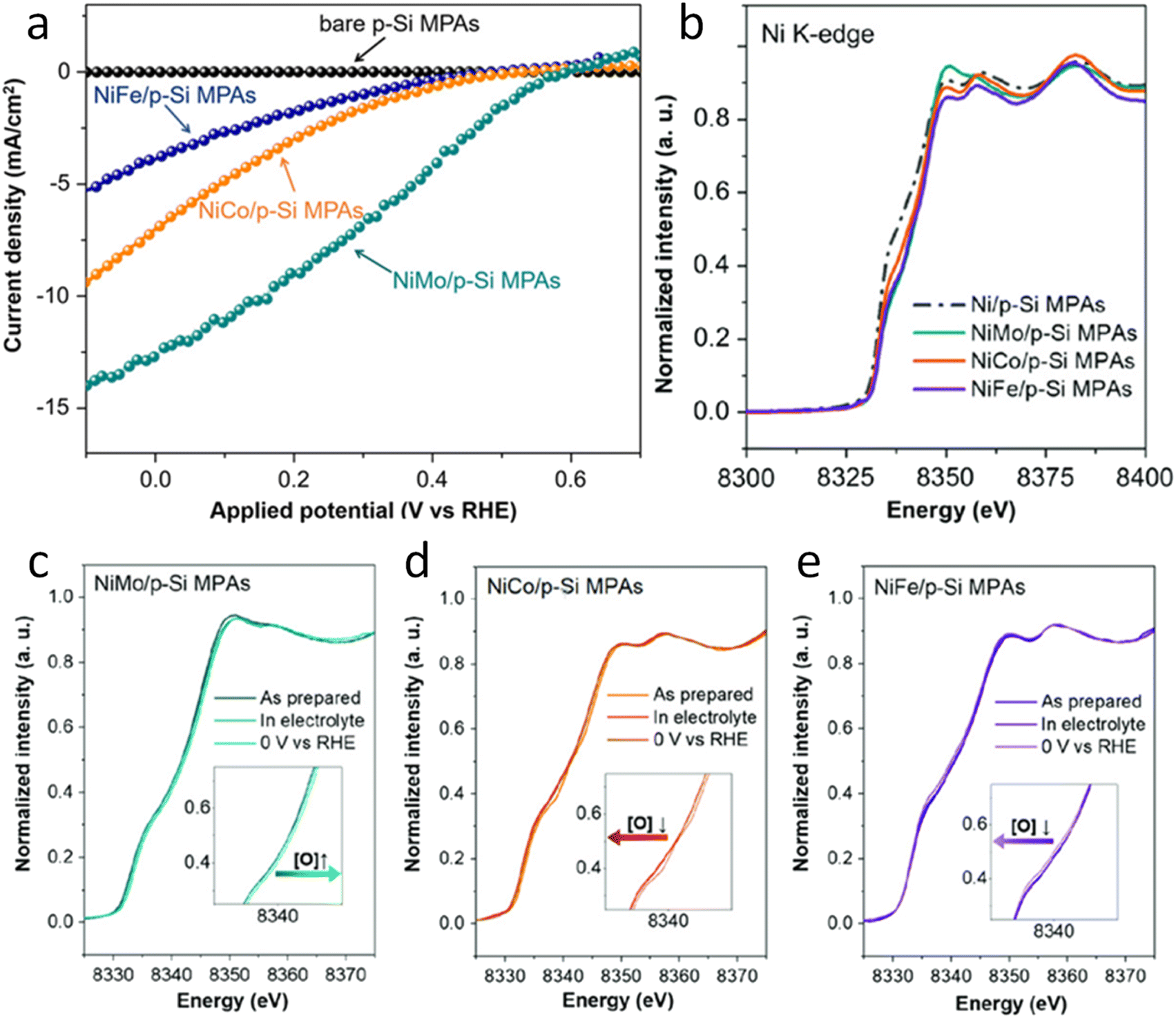 | ||
| Fig. 3 (a) Photoelectrochemical characterisation of the Ni-based alloys/p-Si MPA photocathodes in 1 M KOH under illumination. (b) Normalized Ni K-edge XANES spectra of the p-Si MPAs/alloy photocathodes. (c–e) Ni K-edge of in situ X-ray absorption spectroscopy measurements of the Ni-based alloys/p-Si MPAs during the photoelectrochemical hydrogen production reaction. Copyright 2020. Reproduced with permission from Wiley. Adapted from ref. 71. | ||
Furthermore, by in situ EXAFS, the authors analysed the local atomic environment. Once the HER starts, the peak intensity of the Ni–O scattering path at 1.4 Å in the NiMo/p-Si sample significantly increased. This effect is not observed in the rest of the electrodes. The scattering paths of Ni–Ni were mostly unchanged in all cases. Besides, the intensity of the Ni–O peak in NiCo/p-Si MPAs decreased, indicating that the nickel oxides were changed into the metallic state during the reaction. These in situ-formed Ni-species might be considered as the active intermediates for the high activity of NiMo/p-Si MPAs. Other researchers correlated the HER activity with the NiO introduction on the Ni surface before.72 According to these results, the authors suggest that the growth of the high-valence Ni (hydr)oxides during HERs could favour the photoelectrochemical properties in alloy-based photocathodes.
Furthermore, Wang et al. investigated silicon-based photocathodes for photoelectrochemical (PEC) nitrogen reduction to ammonia, aiming to emulate the natural reaction processes, involving multiple protonation and electron transfer steps. They designed a two-cell electrochemical system to enhance the PEC nitrogen reduction reaction (NRR) efficiency. This system included a stainless-steel mesh cathode modified with encapsulated Fe3C nanoparticles and amorphous carbon. The two cells, physically and electrically separated, allowed the N2 feed gas to pass through the porous electrode of the first cell (C2) before reaching the working photocathode in the second cell (C1). In 2021, they reported significant improvements using this setup on a Au/SiO2/Si photocathode, achieving an NH3 yield rate of 22.0 μg cm−2 h−1 and a faradaic efficiency (FE) of 23.7% at −0.2 V vs. RHE. Without the stainless-steel-based cathode, the yield rate and FE were markedly lower, at 6.6 μg cm−2 h−1 and 7.2%, respectively.73 First, XPS was used to track changes in the oxidation states of the Au/SiO2/Si photocathode before and after illumination. The survey scans showed similar characteristic peaks for Au 4f, Si 2p, and O 1s under both conditions. High-resolution XPS spectra revealed a decrease in the Au1+-to-Au0 ratio under illumination, and a higher Si4+-to-Si0 ratio compared to dark conditions.
Additionally, operando XAS was used to explore the chemical environment under working conditions. The XANES spectra at the Au L3 edge (Fig. 4a) indicated that the oxidation state of Au was related to the intensity of the white line (WL) peak at ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920 eV. The Au L3 edge XANES difference spectra (Fig. 4b) revealed a WL peak at ∼11920 eV, which intensified with the increasing Au oxidation state.74 This peak intensity varied with different photoelectrode potentials and the presence of the modified stainless-steel cathode, correlating with changes in the PEC NRR efficiency. The system with the modified stainless-steel cathode maintained at −0.2 V vs. RHE exhibited the highest oxidation state of Au, suggesting an increase in vacancies at the Au 5d valence level. Besides, Fourier transform k3-weighted EXAFS spectra (Fig. 4c) under reaction conditions showed contributions from Au–Au bonds and a new peak at ∼1.3 Å, attributed to Au–N coordination, which was not present in less efficient PEC NRR reactions. The single-cell system, lacking the modified stainless-steel cathode, showed a lower Au oxidation state and reduced HER performance. The study suggested that the modified stainless-steel cathode first activates N2 molecules at the optimal potential, forming active nitrogen species that adsorb onto the surface of Au NPs, enhancing covalent bonding and improving the PEC NRR performance on the Au/SiO2/Si photocathode.
920 eV. The Au L3 edge XANES difference spectra (Fig. 4b) revealed a WL peak at ∼11920 eV, which intensified with the increasing Au oxidation state.74 This peak intensity varied with different photoelectrode potentials and the presence of the modified stainless-steel cathode, correlating with changes in the PEC NRR efficiency. The system with the modified stainless-steel cathode maintained at −0.2 V vs. RHE exhibited the highest oxidation state of Au, suggesting an increase in vacancies at the Au 5d valence level. Besides, Fourier transform k3-weighted EXAFS spectra (Fig. 4c) under reaction conditions showed contributions from Au–Au bonds and a new peak at ∼1.3 Å, attributed to Au–N coordination, which was not present in less efficient PEC NRR reactions. The single-cell system, lacking the modified stainless-steel cathode, showed a lower Au oxidation state and reduced HER performance. The study suggested that the modified stainless-steel cathode first activates N2 molecules at the optimal potential, forming active nitrogen species that adsorb onto the surface of Au NPs, enhancing covalent bonding and improving the PEC NRR performance on the Au/SiO2/Si photocathode.
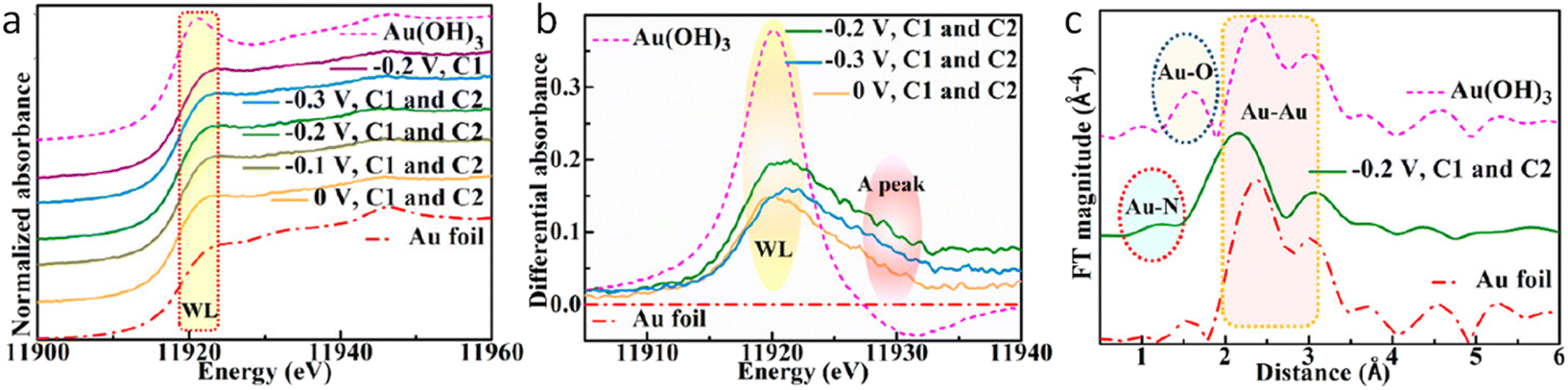 | ||
| Fig. 4 Operando XAS of the Au/SiO2/Si photocathode in the NRR PEC process. (a) Au L3-edge XANES spectra with and without the excitation of a modified stainless-steel cathode at each given photoelectrode potential during the PEC NRR. (b) Comparison between the Au L3-edge XANES difference spectra of the photocathodes with a modified stainless-steel cathode at different photoelectrode potentials. The Au foil as the baseline was chosen to obtain various difference spectra. (c) Fourier transform of the experimental EXAFS spectra of the photocathode with the modified stainless-steel cathode at −0.2 V vs. RHE, Au foil, and Au (OH)3. Copyright 2021. Reproduced with permission from the American Chemical Society. Adapted from ref. 73. | ||
In 2023, the same group investigated the role of the synergy between electron localization and alloying Au, Co and Pd (ACP) in determining the photoelectrochemical conversion of nitrogen to ammonia in the multilayered AuCoPd-CoOx/SiO2/Si photocathodes.75 On the one hand, XPS analysis showed that while Au and Pd maintained their peak positions under illumination, their average oxidation levels decreased. The peak positions associated with Co were unaffected, but the Cox+/Si4+-to-Co0/Si0 ratio increased, differing from Au and Pd trends. These tests were pseudo in situ since no potential was applied. On the other hand, as in their previous work, the XANES spectra of the Au L3 edge indicated that ACP samples had spectra similar to those of the Au foil and different from those of Au(OH)3. The ACP sample performed better than the Au-only sample. At the highest yield potential (−0.3 V vs. RHE), the ACP sample showed the highest Au oxidation state, indicating maximum formation of hybridized chemical bonds during the PEC NRR. EXAFS spectra revealed consistent Au–Au bonds across all bias potentials. However, the intensity of the ∼2.0 Å peak, corresponding to the Au–Pd bonds, increased as the applied potential decreased, peaking at −0.3 V vs. RHE. This increase reflected the enhanced formation of the AuPd alloy due to greater reduction of Ag+/Pd sites under illumination.
In 2021, Sawangphruk et al. analysed a cobalt-based photocathode by in situ XAS.76 Specifically, the authors investigated cobalt hydroxide nanosheets as photoelectrodes and the effect of silver particles on the optical properties and conductivity. This material is usually used as an electrode material for various energy storage and conversion applications,77 and recently, it has been reported as a photoactive material.78,79 Photon energy can cause cobalt hydroxides to generate charge carriers. This process can generate additional photocurrent, leading to higher performance when Co(OH)2 is used as the electrode. However, their intrinsic electronic and electrical conductivities are very poor, and the introduction of AgNPs can be a solution to overcome these problems. In this work, the authors claim that under dark conditions, AgNP-doped α-Co(OH)2 shows a higher capacitance than that of undoped β-Co(OH)2, and the authors attributed this to its higher electrical conductivity and, therefore, faster charge transfer. However, under illumination, while an improvement in the charge storage performance of undoped Co(OH)2 is observed, the opposite phenomenon occurs with the addition of Ag. The excessively narrow band gap and the competitive surface of the resonance process of the plasmon occur in parallel on the AgNP surfaces, leading to less charge capacity performance. In this sense, the change in the oxidation state of Co during the electrochemical process is crucial evidence to investigate the charging mechanism of electrode materials. To gain a deep understanding of the local structural change in Co and its pseudocapacitive behavior when the photoelectrodes are charged and discharged, the authors carried out in situ XAS experiments by analyzing the XANES spectra of the Co K-edge. To do that, the authors used an electrochemical cell developed for a previous work.80 Then, they analysed the Co K-edge XANES spectra of α-Co(OH)2 Ag doped with the AgNP photoactive electrode under light and dark conditions at different potentials. The average oxidation state of Co in Ag–Co(OH)2 is +2.58 at an energy edge of 7719.89 eV. This result refers to mixed valences of Co2+/Co3+. The initial oxidation state of Co in α-Co(OH)2 Ag doped in 1 M NaOH was found to be +2.46 eV. Under −0.4 to 0.1 V versus SCE bias potential, the edge positions change significantly to a higher energy for both conditions, indicating an increasing oxidation state of Co during the charging process. Under dark conditions with the fully charged state of Ag-doped α-Co(OH)2, the oxidation state of Co increases to +3.08, while under light illumination it was +3.14. The higher oxidation number of the Ag–Co(OH)2 electrode under illumination was attributed to the photovoltaic effect, which generates the photoelectrons. However, the oxidation state of Co in Ag–Co(OH)2 under dark conditions is +3.0, while under light irradiation, it decreases to +2.92. The conclusion they reached was that when the Ag-doped α-Co(OH)2 photoelectrode is discharged, the photoelectrons can recombine with the holes generated in the CB, while the photoexcited electrons under dark conditions were transferred to the external system.
In the case of photoanode materials, hematite, Fe2O3, and bismuth vanadate, BiVO4, stand out as the most studied candidates for PEC oxidation reactions and are the most studied semiconductors with in situ and operando synchrotron XAS. Specifically, hematite (α-Fe2O3) photoanodes have attracted increasing attention as photoanode materials for photoelectrochemical applications thanks to their adequate bandgap, abundance and stability.81 Although progress has been made to achieve highly efficient hematite photoelectrodes for the OER,82 understanding the nature of recombination and charge transfer processes using X-ray-based techniques is essential.83 In this sense, XAS measurements have been used to correlate the presence of defects in hematite photoanodes with changes in their photoelectrochemical behaviour. Ex situ FT-EXAFS of the Fe K-edge revealed that the peak intensity corresponds to whether the Fe–O or Fe–Fe bonds can be reduced when oxygen vacancies are present.84 Mesa et al., have recently reported in situ XAS showing that the Fe–Fe bond signal, in the Fe K-edge FT-EXAFS data, reduces when applying a positive potential (1.5 V vs. RHE compared to open circuit potential conditions) in oxygen vacancy-containing hematite photoelectrodes (Fig. 5a).85 The application of an oxidative potential resulted in a slight shift of the absorption edge in the XANES measurement towards positive energies, which was tentatively associated with the Fe2+/Fe3+ oxidation, given that the presence of oxygen vacancies in the hematite structure would generate Fe2+ atoms. Similarly, for M-doped α-Fe2O3 photoanodes (where M = Nb or Ta), an increased X-ray absorption of the Fe L-edge, upon illumination, was observed for the doped hematite photoelectrodes.89 This increased absorption was assigned to enhanced electron transfer in the Fe 3d conduction band of hematite, giving rise to the observed improved photocurrent.
 | ||
| Fig. 5 (a) R-space plot of the Fe K-edge XANES spectra of the hematite (α-Fe2O3) photoanode in the open circuit potential (i.e., no applied potential, blue trace), applying 1.5 V vs. RHE (orange trace). Copyright 2024. Reproduced with permission from Springer Nature. Adapted from ref. 85. (b) O 1s NEXAFS spectra recorded at bias from 100 to 900 mV under dark (left) and light (right) conditions. Copyright 2012. Reproduced with permission from the American Chemical Society. Adapted from ref. 86. (c) X-ray absorption coefficient (μ) at a fixed energy of 8350 eV and photocurrent density at a fixed potential of 1.3 V acquired simultaneously, switching the light on and off. Copyright 2020. Reproduced with permission from the American Chemical Society. Adapted from ref. 87. (d) Valence states of Mn and Fe on α-Fe2O3/Mn3O4+δ photoanodes during the photoelectrochemical OER. Copyright 2018. Reproduced with permission from the American Chemical Society. Adapted from ref. 88. | ||
However, both O 1s NEXAFS and O K-edge measurements have been used to investigate the electronic structure of the photogenerated electrons and holes in hematite photoanodes.90 For example, ex situ time-resolved O K-edge XAS was used to identify the transient (∼0.3 ps) formation of O 2p hole states upon photoexcitation.91 Interestingly, Braun et al. identified two transitions in the pre-edge region of the in situ O 1s NEXAFS as a function of applied potential under light and dark conditions. These two transitions appear to be separated by 1.5 eV and were assigned to photogenerated holes of both Fe 3d and O 2p character (Fig. 5b).86 Although changes in the O 1s NEXAFS due to changes in the filling state of oxygen vacancies have not been ruled out,92 evidence of the electronic and chemical nature of photogenerated holes, in particular those at the surface, is still lacking.
Interfacing photoelectrodes with electrocatalysts to enhance charge transfer to the electrolyte is a highly used engineering strategy in PEC applications. Thus, studying and understanding the interface is crucial for producing highly efficient photoelectrode architectures. For example, Naldoni et al. studied the charge transfer at the interface between hematite and a highly active OER electrocatalyst, NiOx, by means of fixed-energy XAS under both potentiostatic and galvanostatic conditions.87 In this work, the authors probed the Ni K-edge to probe the hole transfer from the hematite photoanode to the NiOx catalyst layer prior to the OER. Interestingly, under photoelectrochemical water oxidation conditions, the differential XANES spectra (light–dark) coincided with the differential XANES spectra of γ-NiOOH–Ni(OH)2 standards. Given that, in NiOx electrocatalysts, the active species towards the OER are reported to be NiOOH. Naldoni et al. assigned their differential XANES spectra to the accumulated OER active species formed after a hole transfer process from hematite. Additionally, the authors performed time-resolved (in the s timescale) X-ray absorption of a fixed energy (8350 eV), thus probing the X-ray absorption coefficient of NiOx during the OER (Fig. 5c). The observed reduction in the X-ray absorption coefficient, measured at 1.3 V, was assigned to the transformation of Ni(OH)2 into NiOOH, which is recovered upon turning off the light.87 A similar system, but with a NiFeOOH electrocatalyst, was studied by Ismail et al., reporting, through a linear combination analysis, that a γ-FeOOH phase is formed at the α-Fe2O3/NiFeOOH interface under illumination at a positive potential (Fig. 6).93
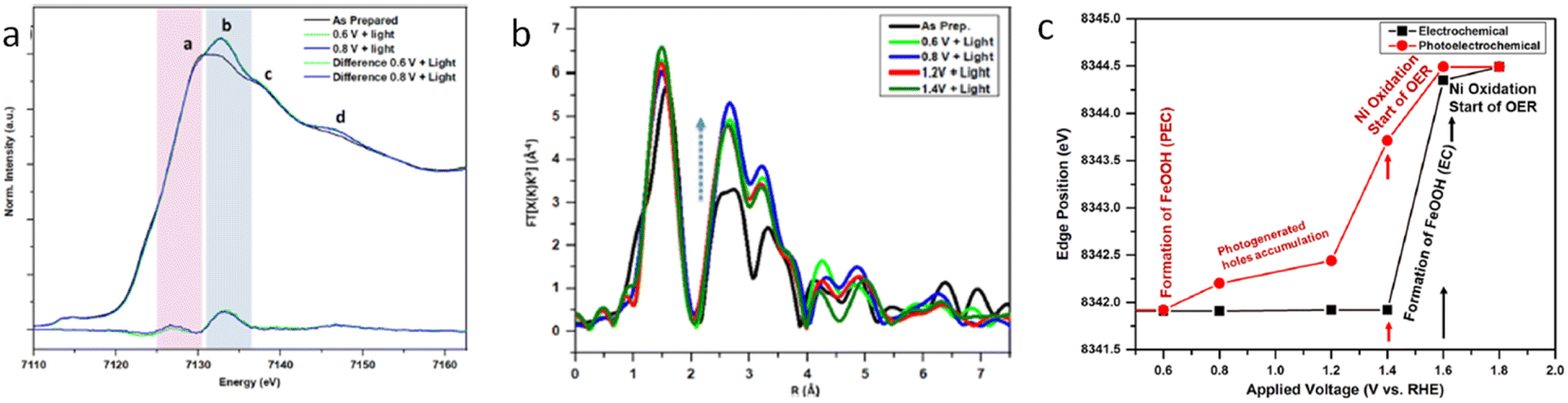 | ||
| Fig. 6 Operando XAS of iron in the α-Fe2O3/Ni0.8Fe0.2OOH photoanode. (a) Iron K-edge XANES at different applied potentials under illumination. (b) FT-EXAFS of Fe at different applied potentials under illumination. (c) Plot of the α-Fe2O3/Ni0.8Fe0.2OOH photoanode at different applied potentials in the darkness (EC) and under illumination (PEC), explaining the OER mechanism. Copyright 2021. Reproduced with permission from the American Chemical Society. Adapted from ref. 93. | ||
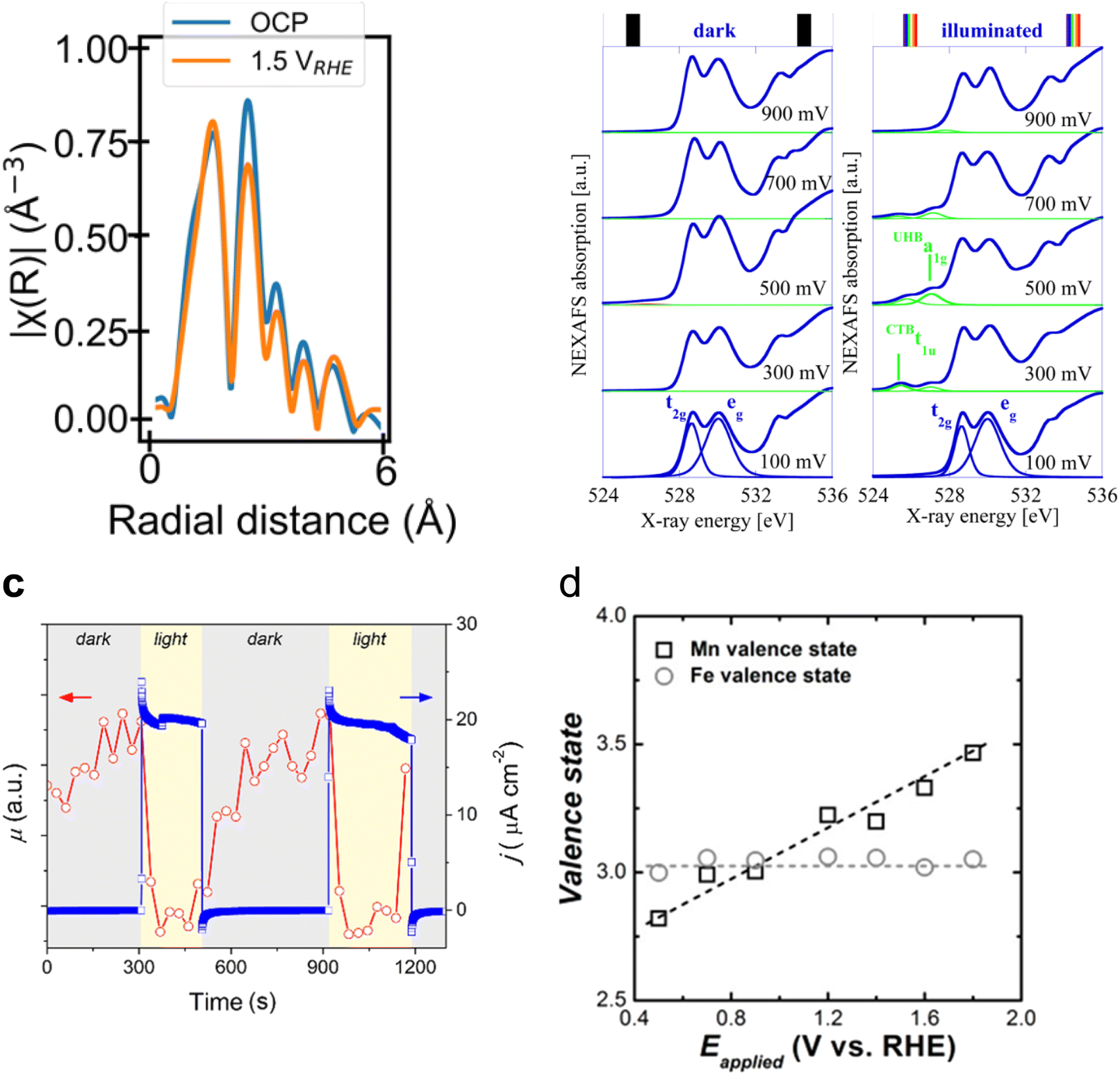
The increase in the PEC water oxidation rate upon the deposition of co-catalysts, based on metals such as Co and Mn, on α-Fe2O3 photoanodes has also been investigated by in situ XAS means. For example, Lange et al. measured the Co K-edge on α-Fe2O3/CoBi (cobalt borate) photoanodes in situ.94 In this work, the authors, through the use of Co standards with precise oxidation states, calculated a slightly higher Co oxidation state when photodeposited on hematite than when grown on FTO. This was assigned to a built-in potential at the α-Fe2O3/CoBi interface. Additionally, in situ measurements revealed that CoBi oxidises by accepting holes from hematite, and the authors suggest that a fraction of them oxidise water. Similarly, Liu et al., reported an increase in the oxidation state of Mn in α-Fe2O3/Mn3O4+δ photoanodes during the photoelectrochemical OER.88 However, in this case, the authors did not observe any change in the oxidation state of Fe and suggested the transfer of holes from hematite to the Mn3O4+δ catalyst (Fig. 5d). It is possible that the authors did not observe any changes in the absorption of the Fe K-edge, given the thickness of the reported photoelectrode.
In general, it is apparent from the reported studies that the oxidation of the metal centers in co-catalyst overlayers can be readily monitored by in situ XAS, most probably due to the low thickness exhibited by such co-catalyst's layers. Once the holes transfer to thin layers of co-catalysts, metal centers oxidise and a change in the X-ray absorption edge is more straightforward to observe and quantify. As such, to further determine the nature of photogenerated holes, in bare hematite photoelectrodes, which are active for the OER, it is key to use thin α-Fe2O3 photoanodes.
Moving to BiVO4, Lifei Xi et al.95 reported the study of a MnPi catalyst layer over a BiVO4 photoanode by in situ soft X-ray absorption spectroscopy (XAS) at the Mn L-edge upon varying the applied potentials and the illumination conditions (Fig. 7). As explained before, in situ NAP-XPS is the most suitable approach for PEC conditions, but it can be challenging to obtain reproducible spectra due to the varying thickness of the electrolyte layer and possible pressure changes inside the experimental chamber.33 Because of that, the authors of this work focused on soft-XAS, because while hard-XAS probes the transition metal K-edge, with soft XAS, the metal L-edge is probed, which is more favourable for deciphering mixtures of oxidation states due to the narrower natural line widths (Mn K-edge, 1.12 eV; Mn L-edge, 0.32 eV).96 To conduct these experiments, the authors developed an in situ and operando cell.97,98 In these works, the authors reported an innovative design for an operando soft X-ray transmission and fluorescence cell that allows rapid membrane replacement and simpler operation compared to earlier designs. Specifically, the cell consists of three chambers (a vacuum chamber and two helium-filled chambers) separated by silicon nitride (Si3N4) membranes. This cell allows for the confinement of a liquid or electrolyte between these membranes and supports the application of both electrochemical techniques and visible light. It includes a gold-coated membrane for conductivity, photodiodes for detecting transmission and fluorescence signals, and LEDs for visible light application. The helium pressure can be regulated to adjust the liquid layer thickness, and the cell can be precisely positioned using a translational and angular stage, facilitating in situ studies of solid/liquid interfaces with X-ray absorption spectroscopy. Specifically, Mn L-edge spectra under different applied potentials and light conditions were studied. The authors found that the photogenerated holes transferred into the MnPi layer and to the electrolyte interface were enhanced by band bending in the semiconductor under illumination, which is related to an increased potential by a built-in photopotential. They also found that the photogenerated holes could oxidize the MnPi co-catalyst layer even under open circuit potential conditions. Then, when increasing the applied bias, the electronic and crystalline structures of the MnOx layer were changing to a birnessite-type layer structure associated with the electron migration from the MnPi film into the BiVO4 photoanode. The formation of birnessite must be accompanied by the incorporation of K+ between the MnO6 layers, leading, for example, to a KMn8O16 phase. These experiments were able to conclude that Mn3+ species, instead of Mn4+, in birnessite were the active sites for the OER.
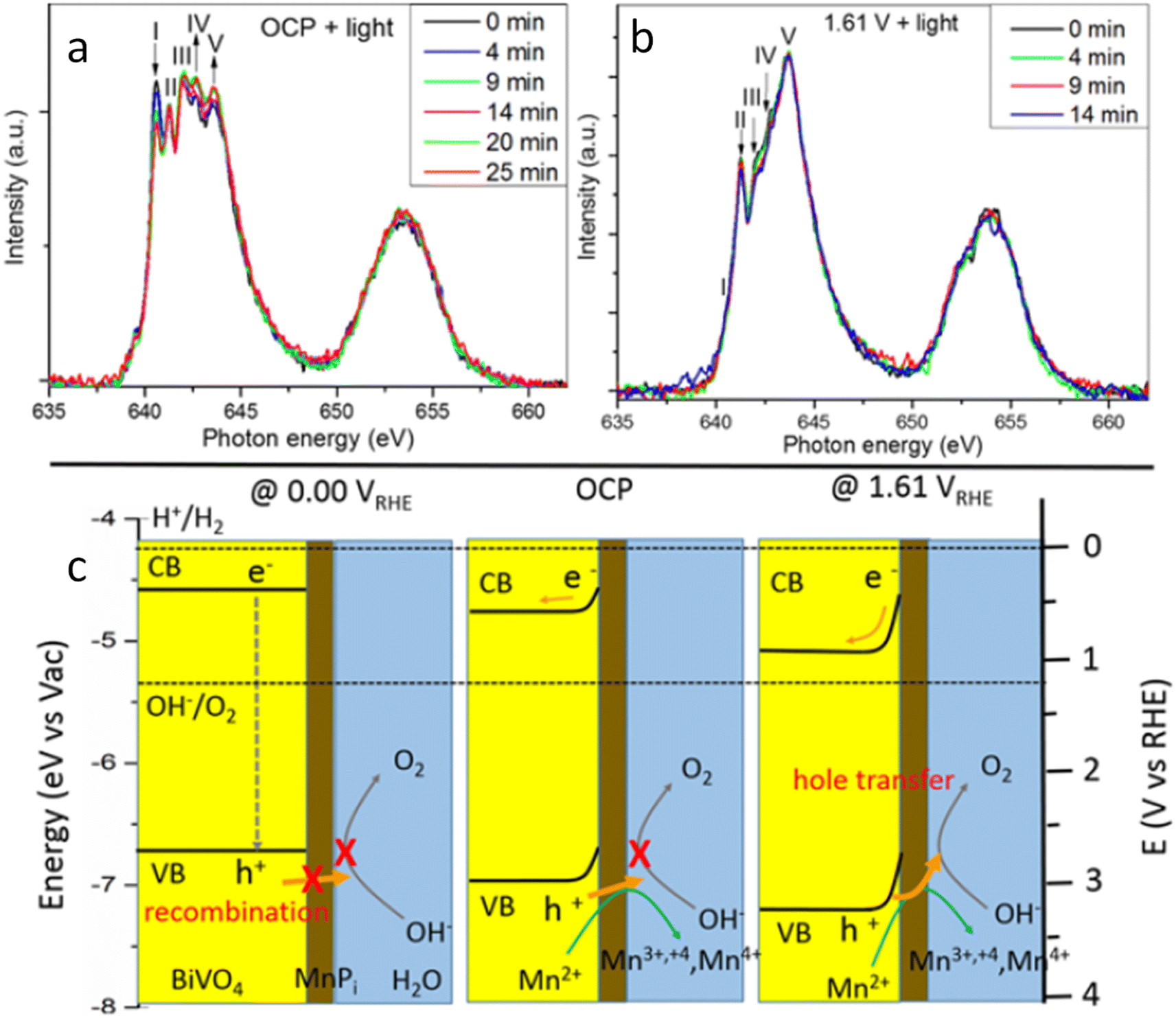 | ||
| Fig. 7 Temporal evolution of XAS spectra of a MnPi-modified BiVO4/Au/Si3N4 photoanode in a 0.1 M KPi buffer solution with KNO3 added at (a) OCP and (b) 1.61 V under light. (c) Schematic illustration of the photogenerated carrier transfer from the BiVO4 photoanode to the MnPi film at 0.00 V, OCP condition, and 1.61 V vs. RHE. Copyright 2017. Reproduced with permission from the American Chemical Society. Adapted from ref. 95. | ||
A couple of years later, some of the team members of a previous work, Lifei Xi and coworkers,99 studied a NiBi co-catalyst over a BiVO4 photoanode by in situ soft and hard XAS. The authors successfully unravel the electronic structure of NiBi-modified BiVO4 photoanodes under working conditions (Fig. 8a) and understand the OER catalytic cycle and the formation of active sites (Fig. 8b). The authors showed that Ni was in +2 state in the electrodeposited amorphous NiBi film and it was readily oxidized up to +4 state under OER conditions. This work reported, for the first time, the direct observation of Ni4+ species during light-driven water oxidation. They also observed that the oxidation of Ni was accompanied by an O K-edge prepeak at 529.17 eV. Specifically, the formation of Ni4+ was associated with the formation of electron-deficient oxygen sites. These sites acted as electrophilic centers leading to the formation of a stable precursor state prior to O–O bond formation. Finally, cyclic voltammetry-XAS results supported that the formation of Ni4+ occurred before the formation of partly electron-deficient oxygen sites.
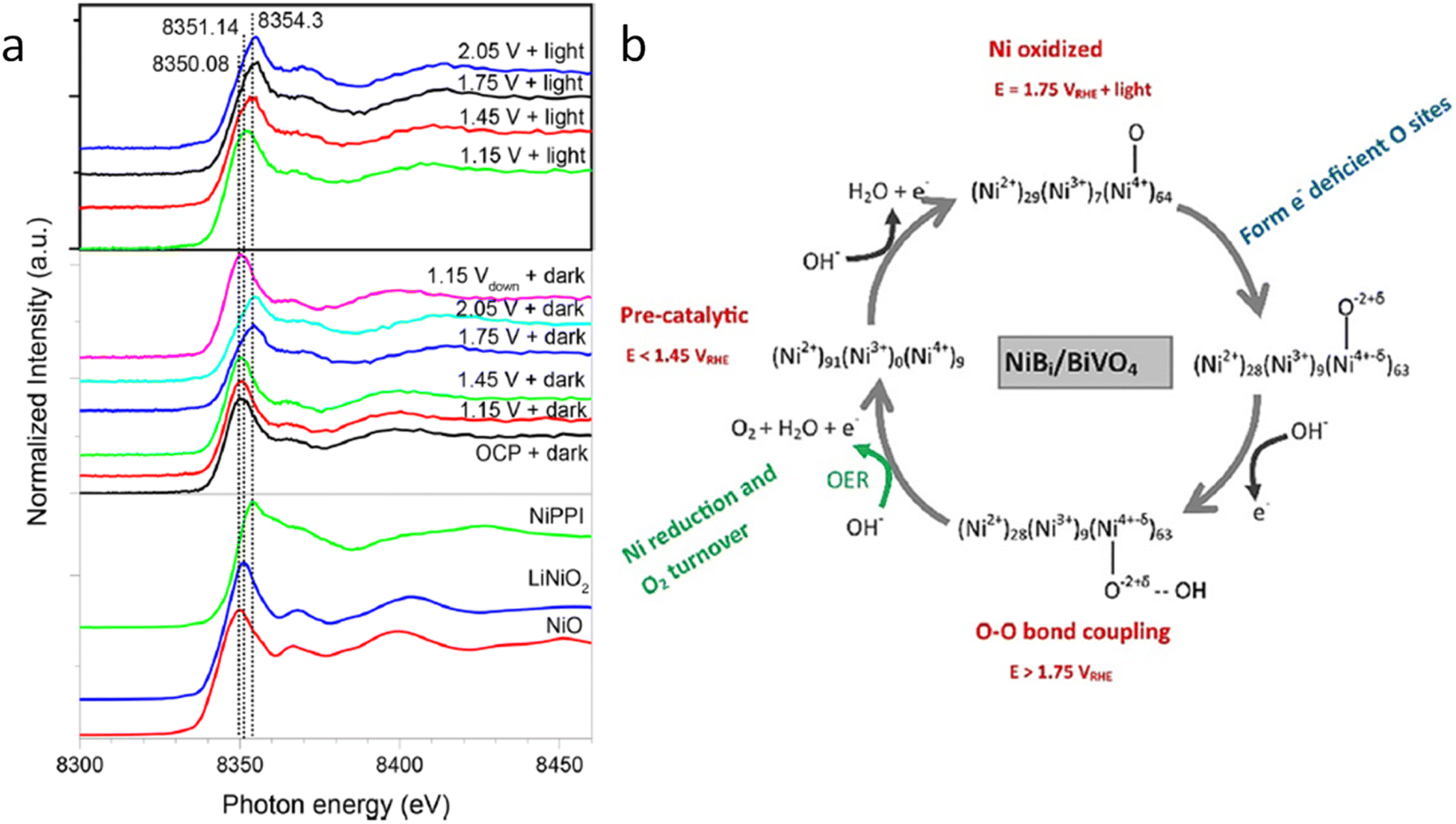 | ||
| Fig. 8 (a) In situ Ni K-edge spectra of the NiBi/BiVO4 photoanode at different potentials under illumination. (b) Schematic showing the NiBi co-catalyst change observed in situ XAS measurements of the NiBi/BiVO4 photoanode. Copyright 2019. Reproduced with permission from the American Chemical Society. Adapted from ref. 99. | ||
All the above-explained works have demonstrated how in situ XAS measurements are a powerful tool to determine the catalytic mechanisms (even catalytic cycles) in photoelectrodes, achieving a true understanding of these materials under relevant working conditions. It is also crucial to develop a correct design for the photoelectrochemical cells, which allows the performance of the in situ and operando experiments with all the above-mentioned requirements. In general, the photoelectrochemical cells found in the reported studies are custom-made, either 3D printed or machined to work in both fluorescence and transmission modes. Fig. 9 displays different PEC cell designs and configurations. First, two examples for in situ XAS measurements in fluorescence mode are shown in Fig. 9a and b. Fig. 9a shows a (photo)electrochemical cell with an X-ray and visible light transparent window, i.e., Mylar, and the sample illumination is from the photoelectrode/electrolyte interface. In this type of photoelectrochemical cell, the thickness of the electrolyte that the X-rays have to travel through is between 0.1 and 1 mm and the sample is illuminated from the same direction of the X-ray irradiation, both to the front of the sample.73,100,101 Similarly, Fig. 9b shows a cell configuration where the photoelectrode is directly prepared on a X-ray transparent material such as Si3N4, thus X-ray irradiation and fluorescence are performed through the back of the photoelectrode. In this case, the electrolyte thickness plays a less relevant role and the illumination is achieved from the front of the photoelectrode.86,102 Alternatively, when the photoelectrode is deposited on a non-X-ray transparent material, such as FTO, the photoelectrode can be placed facing the X-ray transparent window, where Kapton, Si3N4 and Mylar have been used at maximum 1 mm distance. In this configuration, the X-ray irradiation comes from the front (i.e., electrode/electrolyte interface) and the visible excitation is performed through a mica/quartz window at the back of the photoelectrode.87,93 On the other hand, for measurements of soft X-rays, XAS in transmission mode is presented in Fig. 9c and d. Fig. 9c shows the schematic illustration of a tight (photo)electrochemical cell able to support measurements under vacuum by having integrated the illumination system, by LEDs, in the electrochemical chamber.98 Similarly, Fig. 9d presents a cell configuration where the catalyst can be deposited on an X-ray-transparent, Au-coated Si3N4 membrane.
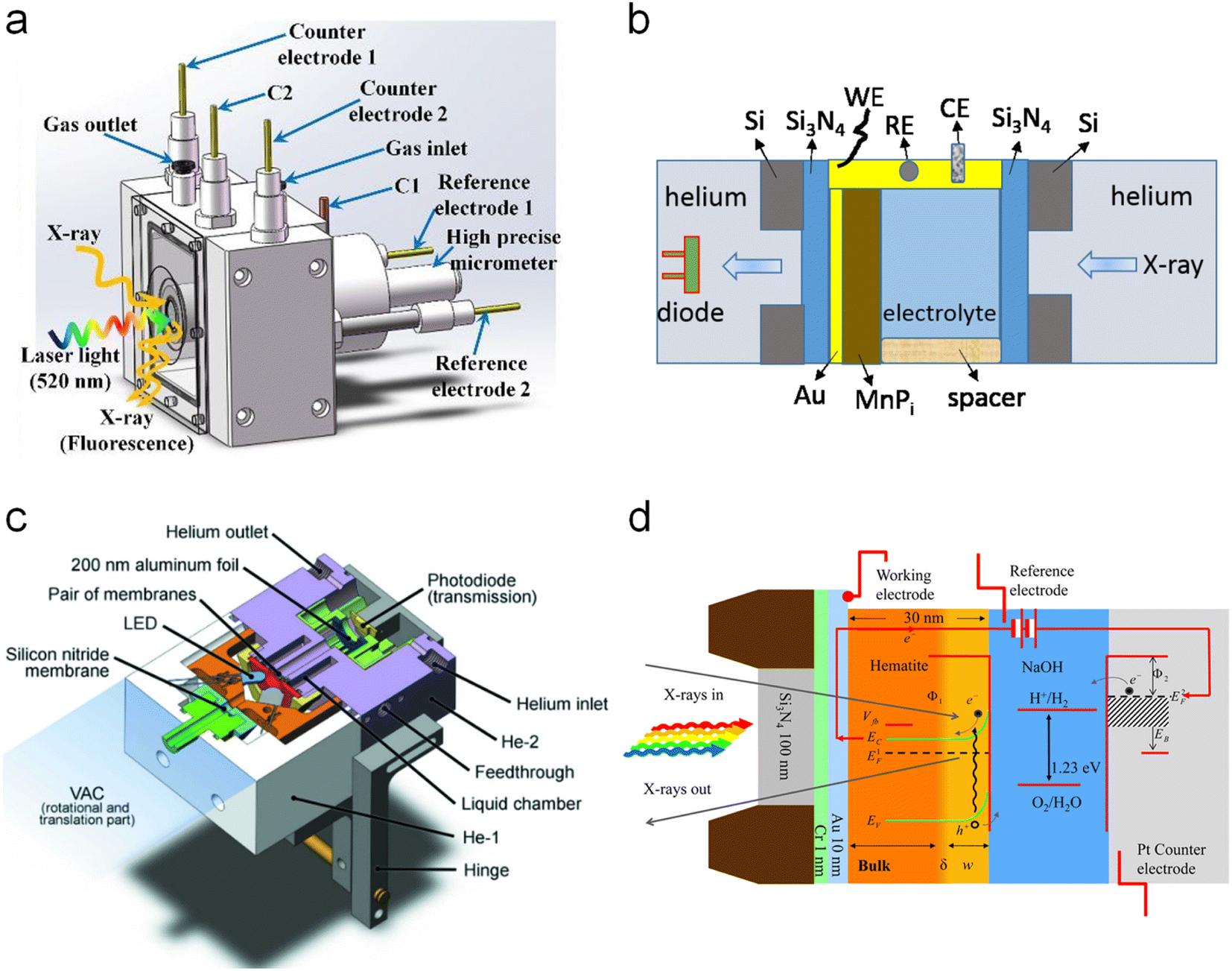 | ||
| Fig. 9 Different cell configurations for in situ/operando XAS experiments. (a) Copyright 2021. Reproduced with permission from the American Chemical Society. Adapted from ref. 73. (b) Copyright 2017. Reproduced with permission from the American Chemical Society. Adapted from ref. 97. (c) Copyright 2016. Reproduced with permission of IUCr. Adapted from ref. 98. (d) Copyright 2012. Reproduced with permission from the American Chemical Society. Adapted from ref. 86. | ||
In situ and operando X-ray diffraction (XRD) and X-ray scattering (XRS)
Diffraction happens with the scattering of incident X-rays by a certain material at different angles. Scattered X-rays are intense in some directions, as a result of constructive interferences, and deficient in other ones, due to destructive interferences. The constructive interference can be explained by Bragg's law, given as follows:| 2dhklsin(θ) = nλ | (6) |
X-ray diffraction (XRD) is a powerful technique to study the atomic crystal structure of bulk materials. This technique is based on the scattering of hard X-rays (photon energies of 5–25 keV) by the crystal lattice of the sample, which, in turn, provides information about the structure of nanoparticles, surfaces, interfaces and defects, and even average structural arrangement within liquids and at solid/liquid interfaces.104In situ XRD has been widely used to correlate X-ray diffraction and electrochemistry. Due to the high energy nature of the employed X-rays for XRD, easily able to pass through the electrolyte, performing successful in situ and operando experiments is easier than XAS and XPS. To be more surface-sensitive, grazing-incidence X-ray diffraction (GIXRD) is employed. In this configuration, the incident X-ray beam is directed onto a sample at a very low angle of incidence (usually < 1°), interacting only with the top few nanometers of the material.105 However, the examples reported so far for PEC applications are very scarce, especially for solar fuel generation. Kang et al.106 used in situ XRD, together with pole figures and HR-TEM, to investigate the formation process and the epitaxial properties of a Bi–WO3 photoelectrode for PEC water oxidation. In situ XRD patterns revealed the process of self-assembly-oriented construction during the temperature-programmed calcination of the film, finding the preferential formation of monoclinic WO3 nanocrystals at high temperatures (T ≥ 470 °C) with dominant orientation along the (002) direction. Very recently, Wang et al.107 have performed quasi in situ GIXRD to study the phase structure and chemical composition of a PdCu/TiO2/Si photocathode surface before and after the Li-mediated PEC nitrogen reduction reaction (NRR). In GIXRD measurements, the incident angle was close to the critical angle for total reflection to increase the X-ray scattering signal.104 By using this technique, the authors showed the appearance of a strong peak associated with Li2N2O3 as the intermediate from the oxidation of Li or Li3N under ambient air. Thus, these results revealed the formation of Li3N from Li and N2 during the PEC NRR, and also showed a higher stability of Li3N and Li2N2O3 in air than Li.107
X-ray scattering (XRS) is an all-atom technique that allows resolving structural parameters across the distance ranging from 10−11 m to 10−6 m,108 whereas XRD describes the scattering of X-rays by a crystal lattice, and other X-ray scattering techniques investigate the atomic-, nano-, and micro-scale structure of less ordered matter, providing structural information of nanoparticles, defects, surface interfaces, and even the average structural arrangement within liquids or at liquid interfaces.104
Small-angle X-ray scattering (SAXS) can be used to study the morphological and structural changes in photoelectrodes and electrolytes, since it allows determining inhomogeneities ranging from the atomic to the nanometer length scales.104,109 Thus, it allows probing local atomic structures and the formation of new phases on the photoelectrode surface.109 More specifically, the grazing-incidence small-angle X-ray scattering (GISAXS) technique is used for studying the nano- and mesoscale evolution of surface structure in the range between 1 and 100 nanometers. This is a powerful technique for time-resolved operando studies during interface reactions, since GISAXS images can be obtained within seconds. Besides, this technique is insensitive to the crystal structure and it can be performed on any sufficiently planar interface, including amorphous and liquid substrates.104
Koziej et al.109 developed an operando grazing-incidence X-ray scattering approach to investigate the photocorrosion mechanisms and the evolution of structural modifications of a CuBi2O4 photoelectrode under illumination and applied bias. A scheme of the employed setup is shown in Fig. 10a. In this study, the total scattering (TS) measurements at the grazing-incidence angle were collected by using a high-energy X-ray probe and a large-area detector. Simultaneously, they also used a second large-area detector to collect the GISAXS signal, which was positioned at a different sample-to-detector distance than TS to acquire both signals at the same time. Specifically, by means of simultaneous and SAXS measurements, they found that CuBi2O4 electrodes quickly underwent a cathodic photocorrosion only few minutes after the beginning of the chronoamperometry, and a decrease of around one order of magnitude in the photocurrents. The authors ascribed these results to the formation of a segregating metallic Bi phase on the surface of the electrode, as shown in Fig. 10b, which significantly reduced the CuBi2O4 surface area in direct contact with the electrolyte. Besides, they observed an additional degradation process at slower timescales, associated with the formation of metallic Cu and subsequent dissolution in the electrolyte as a result of anionic photocorrosion and acidic dissolution. Both Cu2+ and Bi3+ ions released in solution interacted with the phosphate species from the electrolyte forming an insulating BiPO4 layer on the surface of the electrode. This proposed mechanism is schematised in Fig. 10c. These results suggested avoiding the use of a phosphate buffer to prevent the unfavourable formation of BiPO4. They also suggested the employment of a HER co-catalyst to avoid the reduction of CuBi2O4 to metallic Bi and Cu by quickly transferring the photogenerated electrons to the liquid phase.
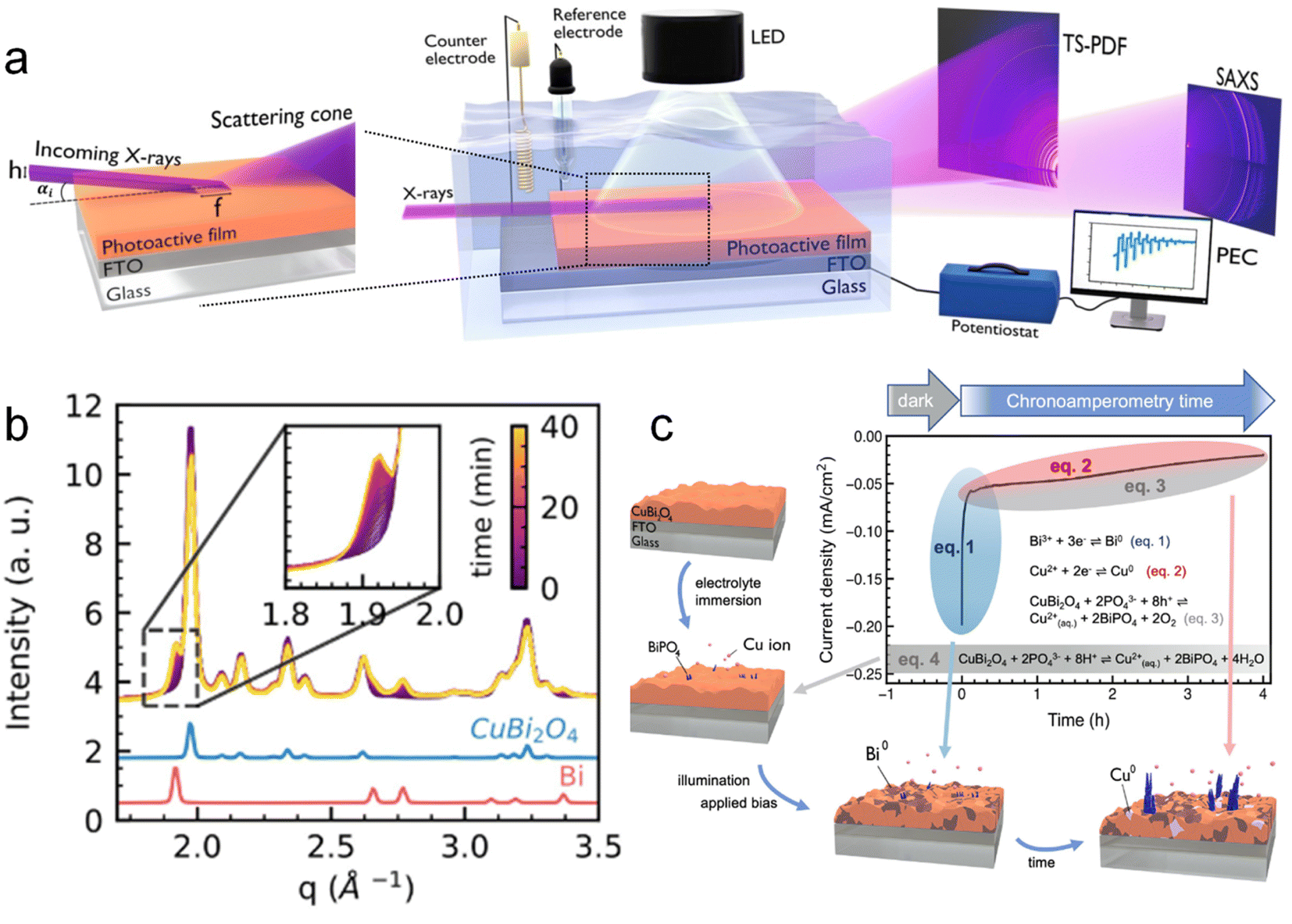 | ||
| Fig. 10 (a) Schematic illustration of the multi-modal experimental set up: PEC properties of a photoactive thin film under illumination and when immersed in an electrolyte solution are probed using a potentiostat in a standard 3-electrode configuration. Two large-area X-ray detectors allow the simultaneous measurement of both TS and SAXS signals from the film surface in a grazing incidence geometry. (b) Time-resolved plot of the operando X-ray scattering signal collected from the surface of a CuBi2O4 electrode under illumination and applied constant bias (0.5 V vs. RHE). In the inset, a zoom into the 1.8–2.0 Å−1 region highlights the formation of metallic Bi with time. (c) Schematic summary of the multiple reaction pathways and relative time scales determining the degradation of CuBi2O4 photocathodes before and during the chronoamperometric test. Copyright 2023. Reproduced with permission from Wiley. Adapted from ref. 109. | ||
Wide-Angle X-ray Scattering (WAXS) can provide signals from the catalyst at high diffraction angles, without any influence from the background or the PEC cell, and with a higher temporal resolution than SAXS.104 Even though we were not able to find any work using in situ or operando WAXS in a PEC system, in 2020, Strasser et al.110 were able to combine electrochemical OER measurements with operando WAXS, XAS data and theoretical calculations (ab initio molecular dynamics simulations and DFT) to track structural transformations during the activation of NiFe and CoFe LDH catalysts. These authors provided the first direct atomic-scale evidence for the oxidation of LDH from their as-prepared precursor state (α-phase) to the catalytically active states (deprotonated γ-phase) at a potential below the onset of the OER. The active γ-phases showed around 8% contractions of the lattice spacing, in both the in-plane lattice constant and the interlayer distance induced by the oxidation of the cations and the dynamics of the intercalated ions. This work demonstrates the high potential of WAXS for the understanding of the structural changes of electrocatalytic and surely photoelectrocatalytic materials under working conditions.
As extensively detailed in this study, (photo)electrochemical cell configurations and the information provided by XPS, XAS and XRD techniques vary significantly. In summary, XPS analyses the surface chemistry by detecting the electrons emitted from a material, revealing the elemental composition and chemical states of the top few nanometres. Furthermore, XAS, including both XANES and EXAFS, examines how a material absorbs X-rays to provide insights into local atomic structures, coordination environments, and oxidation states. Finally, XRD uses diffracted X-rays to determine the crystallographic structure, phase identification, and other structural properties of a material. Then, each technique serves specific purposes, where XPS focuses on surface analysis, XAS on local atomic environments and overall elemental composition and XRD on the crystal structure. Hence, due to these technical differences, the PEC cells for in situ and operando characterisation need to be designed precisely. Table 1 summarises the most relevant technical aspects of the analysed samples in the present study.
| XPS | XAS | XRD | XRS | ||
|---|---|---|---|---|---|
| Soft-XAS | Hard-XAS | ||||
| Principle | Photoemission | Absorption | Diffraction | Scattering | |
| Techniques | UHV-XPS | Total electron yield | Conventional XRD | SAWS | |
| NAP-XPS | Total fluorescence yield | GIXRD | WAXS | ||
| Acquisition depth | 1–10 nm | TEY: 10–20 nm | 1–100 nm | 1–100 nm | |
| TFY: 100 nm–100 μm | |||||
| Detection limit | 0.1–1.0% at. | 10–100 g | 10 μg–100 g | Depends on crystallinity of the sample | Depends on crystallinity and long range order of the sample |
| Advantages | Surface sensitive data acquisition dynamics (seconds) | Easier in situ cell configuration than for XPS | Ease of in situ and operando cell configuration | ||
| Quantitative information | Possibility of obtaining surface sensitive data (GIXRD, GISAWS) | ||||
| Disadvantages | In situ cell complexity (pressure conditions, membranes, etc.) | Complexity of surface sensitive measurements | More qualitative than quantitative | ||
| More qualitative than quantitative | Data acquisition (tens of minutes to hours) | ||||
| Data acquisition (minutes to hours) | |||||
| Cell complexity in comparison to XRD and hard XAS | Difficult to decouple surface and bulk chemical changes | Only for crystalline samples | Complex data analysis (WAXS) SAWS (only for crystalline samples) | ||
Apart from the above-detailed classic X-ray-based techniques (XPS, XAS and XRD), there are other configurations that are currently being developed at synchrotron facilities. Recently, several instruments have allowed performing different techniques using the same camera, known as multi-technique or multi-modal approach, such as NOTOS beamline in ALBA synchrotron, which enables performing XRD and XAS. Moreover, the 3Sbar beamline in ALBA, allows us to perform XPS and SAXS. Furthermore, in Max IV, XPS and IR are possible to be carried out. Besides, NAP XPS in synchrotron allows us to perform Auger yield XAS, which is surface sensitive. The main advantage of this multi-technique approach, summarized in the previous examples, relies on getting information from the exact same point in the sample and under exactly the same conditions. Moreover, there are other techniques such as tomography, for example available at Faxtor beamline in ALBA, that give a 3D image of the sample and could be well adapted to in situ and operando experiments. All these spectroscopies have demonstrated an enormous potential in the characterization of materials in general, and PEC systems in particular, under working conditions.
The future of XPS, XAS, and XRD techniques for in situ and operando characterisation of photoelectrodes is highly promising, where significant advancements are expected. These advances include the development of ambient pressure and time-resolved XPS for realistic and dynamic surface studies, improved resolution and sensitivity in XAS through next-generation synchrotron sources as well as the integration of XAS with neutron scattering for comprehensive structural insights. Additionally, real-time and rapid XRD measurements, along with X-ray tomography, will enhance the understanding of structural changes and phase distribution in photoelectrodes. Additionally, the synergy of multi-modal techniques, as detailed above, and the use of artificial intelligence for data analysis will provide a more comprehensive characterization, driving the discovery and optimization of new materials. Therefore, the enhanced operational conditions during measurements will lead to more precise and relevant studies, ultimately contributing to the development of more efficient and durable photoelectrocatalytic materials.
Conclusions
The present work summarized the most relevant in situ and operando X-ray studies in PEC systems, demonstrating the strength and potential of these spectroscopies for the characterisation of PEC materials under working conditions. Specifically, this work is focused on operando and in situ XPS, XAS and XRD techniques, trying to remark the recent progress using these techniques for elucidating the structural, chemical, compositional and electronic properties of photoelectrocatalytic materials and interfaces. Moreover, this work provides technical guidance for adequately conducting these complex experiments, focusing on the possibilities of the analysed techniques, cell designs and more relevant results.It is apparent that many of the above-discussed X-ray-based techniques have benefited from the developments and upgrades of different synchrotron beamlines. Accordingly, we believe that the success of the in situ and operando characterisation is currently mainly limited by the PEC cell engineering, where film and electrolyte thickness, as well as photoelectrode disposition, play a key role. In this context, making the cell designs publicly available will greatly benefit the field and will promote a deep understanding of PEC processes aiming to design more efficient and stable photoelectrodes for solar-driven fuel production and value-added photoelectrosynthesised compounds.
Data availability
This manuscript is a review article, and as such, it synthesizes and discusses results and findings from previously published studies that have undergone rigorous peer-review and have been published in reputable scientific journals. All data, results, and figures included in this review are sourced from these original publications. We have obtained all necessary permissions for the reproduction of figures and any other content from these studies. Proper citations and acknowledgments are provided to ensure that credit is given to the original authors and sources. No new primary data were generated or analysed in the preparation of this manuscript. Should any additional information be required regarding the sources or permissions for the reproduced content, it is available upon request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/PR23/11980046. Additionally, this work has received financial support through the projects SOLFUTURE (PLEC2021-007906), TED2021-130173B-C41 and TED2021-129999A-C33 funded by MCIN/AEI/10.13039/501100011033 and the European Union NextGenerationEU/PRTR. Also, the authors acknowledge financial support from the EU FETPROACT-EIC-07-2020 programme (HYSOLCHEM 101017928). M. Barawi acknowledges NovaCO2 project (PID2020-118593RB-C22) and the RYC2022-038157-I Grant funded by MCIN/AEI/10.13039/501100011033. L. Collado acknowledges the project ARMONIA (PID2020-119125RJ-I00) funded by MICIU/AEI/10.13039/501100011033. I. J. Villar-García would like to acknowledge the project OSMIBatt (CNS2022-135285) funded by MCIN/AEI/10.13039/501100011033 and European Union NextGenerationEU. F. E. Oropeza thanks to MINECO and European NextGenerationEU/PRTR Fund for the Ramón y Cajal contract (RyC2021-034254-I).References
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- S. D. Tilley, Adv. Energy Mater., 2019, 9, 1802877 CrossRef.
- B. Tumas, J. Dempsey, T. Mollouk, S. Ardo, K. Bren, A. Rappe, W. Shaw, H. Abruna, H. Atwater, K. Ayers, C. Berlinguette, J. Concepcion, V. Cooper, D. Esposito, J. Gregorie, L. Hammarstrom, S. Haussener, F. Houle, S. Linic, H. Shafaat, Y. Shao-Horn, W. Smith, Y. Surendranath, D. Tiede and J. Yang, Report of the Basic Energy Sciences Roundtable on Liquid Solar Fuels, 2019 Search PubMed.
- A. Sharma, T. Longden, K. Catchpole and F. J. Beck, Energy Environ. Sci., 2023, 16, 4486–4501 RSC.
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W.-H. Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K.-S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper, R. Van De Krol and F. Houle, J. Phys. D Appl. Phys., 2022, 55, 323003 CrossRef CAS.
- H. Liang, Z. Yan and G. Zeng, Inorganics, 2022, 11, 16 CrossRef.
- R. Yalavarthi, O. Henrotte, A. Minguzzi, P. Ghigna, D. A. Grave and A. Naldoni, MRS Energy Sustain., 2020, 7, 37 CrossRef.
- A. Gurlo and R. Riedel, Angew. Chem., Int. Ed., 2007, 46, 3826–3848 CrossRef CAS PubMed.
- E. Pastor, Z. Lian, L. Xia, D. Ecija, J. R. Galán-Mascarós, S. Barja, S. Giménez, J. Arbiol, N. López and F. P. García de Arquer, Nat. Rev. Chem, 2024, 8, 159–178 CrossRef PubMed.
- R. R. Rao, S. Corby, A. Bucci, M. García-Tecedor, C. A. Mesa, J. Rossmeisl, S. Giménez, J. Lloret-Fillol, I. E. L. Stephens and J. R. Durrant, J. Am. Chem. Soc., 2022, 144, 7622–7633 CrossRef CAS PubMed.
- Y. AlSalka, S. Schwabe, J. Geweke, G. Ctistis and H. Wackerbarth, Energy Technol., 2022, 11, 2200788 CrossRef.
- R. Arcas, D. Cardenas-Morcoso, M. C. Spadaro, M. García-Tecedor, C. A. Mesa, J. Arbiol, F. Fabregat-Santiago, S. Giménez and E. Mas-Marzá, Sol. RRL, 2022, 6, 2200132 CrossRef CAS.
- A. Venugopal, R. Kas, K. Hau and W. A. Smith, J. Am. Chem. Soc., 2021, 143, 18581–18591 CrossRef CAS PubMed.
- M. García-Tecedor, M. Barawi, A. García-Eguizábal, M. Gomez-Mendoza, F. E. Oropeza, G. Gorni, A. Cibotaru, M. Liras and V. A. de la Peña O'Shea, Sol. RRL, 2024, 8, 2301069 CrossRef.
- A. O. Alvarez, M. García-Tecedor, L. Montañés, E. Mas-Marzá, S. Giménez and F. Fabregat-Santiago, Sol. RRL, 2022, 6, 2200826 CrossRef CAS.
- Advanced Certificate in Powder Diffraction on the Web, Course Material Master Index, School of Crystallography, Birkbeck College, https://pd.chem.ucl.ac.uk/pd/welcome.htm Search PubMed.
- J. Noguera-Gómez, M. García-Tecedor, J. F. Sánchez-Royo, L. M. Valencia Liñán, M. de la Mata, M. Herrera-Collado, S. I. Molina, R. Abargues and S. Giménez, ACS Appl. Energy Mater., 2021, 4, 5255–5264 CrossRef.
- B. Moss, K. L. Svane, D. Nieto-Castro, R. R. Rao, S. B. Scott, C. Tseng, M. Sachs, A. Pennathur, C. Liang, L. I. Oldham, E. Mazzolini, L. Jurado, G. Sankar, S. Parry, V. Celorrio, J. M. Dawlaty, J. Rossmeisl, J. R. Galán-Mascarós, I. E. L. Stephens and J. R. Durrant, J. Am. Chem. Soc., 2024, 146, 8915–8927 CrossRef CAS PubMed.
- X. Ding, D. Liu, P. Zhao, X. Chen, H. Wang, F. E. Oropeza, G. Gorni, M. Barawi, M. García-Tecedor, V. A. de la Peña O’Shea, J. P. Hofmann, J. Li, J. Kim, S. Cho, R. Wu and K. H. L. Zhang, Nat. Commun., 2024, 15, 5336 CrossRef CAS PubMed.
- S. Corby, M.-G. Tecedor, S. Tengeler, C. Steinert, B. Moss, C. A. Mesa, H. F. Heiba, A. A. Wilson, B. Kaiser, W. Jaegermann, L. Francàs, S. Gimenez and J. R. Durrant, Sustainable Energy Fuels, 2020, 4, 5024–5030 RSC.
- Z. Zhang, H. Nagashima and T. Tachikawa, Angew. Chem., Int. Ed., 2020, 59, 9047–9054 CrossRef CAS PubMed.
- H. Wang, Y. Wang, Y. Lin, X. Huang, M. García-Tecedor, V. A. de la Peña O'Shea, C. Murrill, V. K. Lazarov, F. E. Oropeza and K. H. L. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 28739–28746 CrossRef CAS PubMed.
- T. Lopes, P. Dias, L. Andrade and A. Mendes, Sol. Energy Mater. Sol. Cells, 2014, 128, 399–410 CrossRef CAS.
- Surface and Interfacial Aspects of Biomedical Polymer, ed. J. D. Andrade, Plenum Press, New York, 1985 Search PubMed.
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., 2020, 38, 063204 CrossRef CAS.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872–3877 CrossRef CAS.
- J. Schnadt, J. Knudsen and N. Johansson, J. Phys.: Condens.Matter, 2020, 32, 413003 CrossRef CAS PubMed.
- M. Favaro, L. Artiglia and B. S. Mun, J. Phys. D Appl. Phys., 2022, 55, 060201 CrossRef CAS.
- J.-J. Velasco-Vélez, L. J. Falling, D. Bernsmeier, M. J. Sear, P. C. J. Clark, T.-S. Chan, E. Stotz, M. Hävecker, R. Kraehnert, A. Knop-Gericke, C.-H. Chuang, D. E. Starr, M. Favaro and R. V Mom, J. Phys. D Appl. Phys., 2021, 54, 124003 CrossRef.
- A. Kolmakov, D. A. Dikin, L. J. Cote, J. Huang, M. K. Abyaneh, M. Amati, L. Gregoratti, S. Günther and M. Kiskinova, Nat. Nanotechnol., 2011, 6, 651–657 CrossRef CAS PubMed.
- R. S. Weatherup, Top. Catal., 2018, 61, 2085–2102 CrossRef CAS.
- P. Leidinger, J. Kraus, T. Kratky, P. Zeller, T. O. Menteş, F. Genuzio, A. Locatelli and S. Günther, J. Phys. D Appl. Phys., 2021, 54, 234001 CrossRef CAS.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- A. Klyushin, M. Ghosalya, E. Kokkonen, C. Eads, R. Jones, N. Nalajala, C. S. Gopinath and S. Urpelainen, J. Synchrotron Radiat., 2023, 30, 613–619 CrossRef CAS PubMed.
- M. Favaro, P. C. J. Clark, M. J. Sear, M. Johansson, S. Maehl, R. van de Krol and D. E. Starr, Surf. Sci., 2021, 713, 121903 CrossRef CAS.
- A. Benayad, J. E. Morales-Ugarte, C. C. Santini and R. Bouchet, J. Phys. Chem. A, 2021, 125, 1069–1081 CrossRef CAS PubMed.
- H. Sezen and S. Suzer, Thin Solid Films, 2013, 534, 1–11 CrossRef CAS.
- J. E. Demuth, W. J. Thompson, N. J. DiNardo and R. Imbihl, Phys. Rev. Lett., 1986, 56, 1408–1411 CrossRef CAS PubMed.
- S. Teich, S. Grafström and L. M. Eng, Surf. Sci., 2004, 552, 77–84 CrossRef CAS.
- H. Sezen, E. Ozbay and S. Suzer, Appl. Surf. Sci., 2014, 323, 25–30 CrossRef CAS.
- J. E. Demuth, B. N. J. Persson and A. J. Schell-Sorokin, Phys. Rev. Lett., 1983, 51, 2214–2217 CrossRef CAS.
- H. Sezen and S. Suzer, Surf. Sci., 2010, 604, L59–L62 CrossRef CAS.
- O. Ö. Ekiz, K. Mizrak and A. Dâna, ACS Nano, 2010, 4, 1851–1860 CrossRef CAS PubMed.
- F. E. Oropeza, B. T. Feleki, K. H. L. Zhang, E. J. M. Hensen and J. P. Hofmann, ACS Appl. Energy Mater., 2019, 2, 6866–6874 CrossRef CAS.
- L. Gao, Y. Cui, R. H. J. Vervuurt, D. van Dam, R. P. J. van Veldhoven, J. P. Hofmann, A. A. Bol, J. E. M. Haverkort, P. H. L. Notten, E. P. A. M. Bakkers and E. J. M. Hensen, Adv. Funct. Mater., 2016, 26, 679–686 CrossRef CAS.
- Ø. Dahl, M. F. Sunding, V. Killi, I.-H. Svenum, M. Grandcolas, M. Andreassen, O. Nilsen, A. Thøgersen, I. J. T. Jensen and A. Chatzitakis, ACS Catal., 2023, 13, 11762–11770 CrossRef.
- X. Zhang and S. Ptasinska, Appl. Surf. Sci., 2020, 516, 146127 CrossRef CAS.
- X. Zhang and S. Ptasinska, Phys. Chem. Chem. Phys., 2015, 17, 3909–3918 RSC.
- X. Zhang and S. Ptasinska, Sci. Rep., 2016, 6, 24848 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- S. Porsgaard, P. Jiang, F. Borondics, S. Wendt, Z. Liu, H. Bluhm, F. Besenbacher and M. Salmeron, Angew. Chem., Int. Ed., 2011, 50, 2266–2269 CrossRef CAS PubMed.
- V. Mansfeldova, M. Zlamalova, H. Tarabkova, P. Janda, M. Vorokhta, L. Piliai and L. Kavan, J. Phys. Chem. C, 2021, 125, 1902–1912 CrossRef CAS.
- M. F. Lichterman, S. Hu, M. H. Richter, E. J. Crumlin, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain, T. Mayer, B. S. Brunschwig, N. S. Lewis, Z. Liu and H.-J. Lewerenz, Energy Environ. Sci., 2015, 8, 2409–2416 RSC.
- M. Favaro, I. Y. Ahmet, P. C. J. Clark, F. F. Abdi, M. J. Sear, R. van de Krol and D. E. Starr, J. Phys. D Appl. Phys., 2021, 54, 164001 CrossRef CAS.
- P. Amann, D. Degerman, M.-T. Lee, J. D. Alexander, M. Shipilin, H.-Y. Wang, F. Cavalca, M. Weston, J. Gladh, M. Blom, M. Björkhage, P. Löfgren, C. Schlueter, P. Loemker, K. Ederer, W. Drube, H. Noei, J. Zehetner, H. Wentzel, J. Åhlund and A. Nilsson, Rev. Sci. Instrum., 2019, 90, 103102 CrossRef.
- J. Knudsen, T. Gallo, V. Boix, M. D. Strømsheim, G. D'Acunto, C. Goodwin, H. Wallander, S. Zhu, M. Soldemo, P. Lömker, F. Cavalca, M. Scardamaglia, D. Degerman, A. Nilsson, P. Amann, A. Shavorskiy and J. Schnadt, Nat. Commun., 2021, 12, 6117 CrossRef CAS PubMed.
- D. Ketenoglu, X-Ray Spectrom., 2022, 51, 422–443 CrossRef CAS.
- X-Ray Absorption and X-Ray Emission Spectroscopy, ed. J. A. Van Bokhoven and C. Lamberti, Wiley, 2016 Search PubMed.
- N. S. Genz, A. Kallio, F. Meirer, S. Huotari and B. M. Weckhuysen, Chem.: Methods, 2024, 4, e202300027 CAS.
- R. Koningsberger and D. C. Prins, X-Ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, 1987 Search PubMed.
- S. L. M. Schroeder, G. D. Moggridge, T. Rayment and R. M. Lambert, J. Mol. Catal. A: Chem., 1997, 119, 357–365 CrossRef CAS.
- K. Nakanishi and T. Ohta, Surf. Interface Anal., 2012, 44, 784–788 CrossRef CAS.
- W.-S. Yoon, M. Balasubramanian, K. Y. Chung, X.-Q. Yang, J. McBreen, C. P. Grey and D. A. Fischer, J. Am. Chem. Soc., 2005, 127, 17479–17487 CrossRef CAS PubMed.
- P. Zimmermann, S. Peredkov, P. M. Abdala, S. DeBeer, M. Tromp, C. Müller and J. A. van Bokhoven, Coord. Chem. Rev., 2020, 423, 213466 CrossRef CAS.
- J. Deng, Q. Zhang, X. Lv, D. Zhang, H. Xu, D. Ma and J. Zhong, ACS Energy Lett., 2020, 5, 975–993 CrossRef CAS.
- M. A. Soldatov, P. V. Medvedev, V. Roldugin, I. N. Novomlinskiy, I. Pankin, H. Su, Q. Liu and A. V. Soldatov, Nanomaterials, 2022, 12, 839 CrossRef CAS PubMed.
- N. Rossetti, A. Ugolotti, C. Cometto, V. Celorrio, G. Dražić, C. Di Valentin and L. Calvillo, J. Mater. Chem. A, 2024, 12, 6652–6662 RSC.
- P. Ren, T. Zhang, N. Jain, H. Y. V. Ching, A. Jaworski, G. Barcaro, S. Monti, J. Silvestre-Albero, V. Celorrio, L. Chouhan, A. Rokicińska, E. Debroye, P. Kuśtrowski, S. Van Doorslaer, S. Van Aert, S. Bals and S. Das, J. Am. Chem. Soc., 2023, 145, 16584–16596 CrossRef CAS PubMed.
- C.-W. Tung, C.-H. Hou, H.-T. Lin, Y. Zheng, Y.-P. Huang, Y.-F. Liao, J.-J. Shiue and H. M. Chen, Sol. RRL, 2020, 4, 2000028 CrossRef CAS.
- J. Wang, X. Ge, L. Shao, J. Zhang, D. Peng, G. Zou, H. Hou, W. Deng, S. Xu, X. Ji and W. Zhang, Mater. Today Energy, 2020, 17, 100436 CrossRef.
- J. Zheng, Y. Lyu, J.-P. Veder, B. Johannessen, R. Wang, R. De Marco, A. Huang, S. P. Jiang and S. Wang, J. Phys. Chem. C, 2021, 125, 23041–23049 CrossRef CAS.
- Z. H. Lu, T. K. Sham, M. Vos, A. Bzowski, I. V. Mitchell and P. R. Norton, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8811–8814 CrossRef CAS PubMed.
- J. Zheng, Y. Lyu, A. Huang, B. Johannessen, X. Cao, S. P. Jiang and S. Wang, Chin. J. Catal., 2023, 45, 141–151 CrossRef CAS.
- M. Suksomboon, K. Kongsawatvoragul, S. Duangdangchote and M. Sawangphruk, ACS Omega, 2021, 6, 20804–20811 CrossRef CAS PubMed.
- H. Pang, X. Li, Q. Zhao, H. Xue, W.-Y. Lai, Z. Hu and W. Huang, Nano Energy, 2017, 35, 138–145 CrossRef CAS.
- S. Kalasina, P. Pattanasattayavong, M. Suksomboon, N. Phattharasupakun, J. Wutthiprom and M. Sawangphruk, Chem. Commun., 2017, 53, 709–712 RSC.
- J. M. Yu, J. Lee, Y. S. Kim, J. Song, J. Oh, S. M. Lee, M. Jeong, Y. Kim, J. H. Kwak, S. Cho, C. Yang and J.-W. Jang, Nat. Commun., 2020, 11, 5509 CrossRef CAS PubMed.
- P. Iamprasertkun, A. Krittayavathananon, A. Seubsai, N. Chanlek, P. Kidkhunthod, W. Sangthong, S. Maensiri, R. Yimnirun, S. Nilmoung, P. Pannopard, S. Ittisanronnachai, K. Kongpatpanich, J. Limtrakul and M. Sawangphruk, Sci. Rep., 2016, 6, 37560 CrossRef CAS PubMed.
- J. Li, H. Chen, C. A. Triana and G. R. Patzke, Angew. Chem., Int. Ed., 2021, 60, 18380–18396 CrossRef CAS PubMed.
- T. H. Jeon, G. Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- C. A. Mesa, E. Pastor and L. Francàs, Curr. Opin. Electrochem., 2022, 35, 101098 CrossRef CAS.
- M. Li, J. Deng, A. Pu, P. Zhang, H. Zhang, J. Gao, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 6727 RSC.
- C. A. Mesa, M. Sachs, E. Pastor, N. Gauriot, A. Merryweather, M. A. Gomez-Gonzalez, K. Ignatyev, S. Giménez, R. Rao, J. R. Durrant and R. Pandya, Nat. Commun., 2024, 15, 3904 CrossRef PubMed.
- A. Braun, K. Sivula, D. K. Bora, J. Zhu, L. Zhang, M. Grätzel, J. Guo and E. C. Constable, J. Phys. Chem. C, 2012, 116, 16870–16875 CrossRef CAS.
- F. Malara, M. Fracchia, H. Kmentová, R. Psaro, A. Vertova, D. Oliveira de Souza, G. Aquilanti, L. Olivi, P. Ghigna, A. Minguzzi and A. Naldoni, ACS Catal., 2020, 10, 10476–10487 CrossRef CAS.
- Y. Liu, C. Wei, C. K. Ngaw, Y. Zhou, S. Sun, S. Xi, Y. Du, J. S. C. Loo, J. W. Ager and Z. J. Xu, ACS Appl. Energy Mater., 2018, 1, 814–821 CrossRef CAS.
- H.-W. Chang, Y. Fu, W.-Y. Lee, Y.-R. Lu, Y.-C. Huang, J.-L. Chen, C.-L. Chen, W. C. Chou, J.-M. Chen, J.-F. Lee, S. Shen and C.-L. Dong, Nanotechnology, 2018, 29, 064002 CrossRef PubMed.
- Y. R. Lu, Y. F. Wang, H. W. Chang, Y. C. Huang, J. L. Chen, C. L. Chen, Y. C. Lin, Y. G. Lin, W. F. Pong, T. Ohigashi, N. Kosugi, C. H. Kuo, W. C. Chou and C. L. Dong, Sol. Energy Mater. Sol. Cells, 2020, 209, 110469 CrossRef CAS.
- Y. Uemura, A. S. M. Ismail, S. H. Park, S. Kwon, M. Kim, H. Elnaggar, F. Frati, H. Wadati, Y. Hirata, Y. Zhang, K. Yamagami, S. Yamamoto, I. Matsuda, U. Halisdemir, G. Koster, C. Milne, M. Ammann, B. M. Weckhuysen and F. M. F. de Groot, J. Phys. Chem. Lett., 2022, 13, 4207–4214 CrossRef CAS PubMed.
- A. Braun, Y. Hu, F. Boudoire, D. K. Bora, D. D. Sarma, M. Grätzel and C. M. Eggleston, Catal. Today, 2016, 260, 72–81 CrossRef CAS.
- A. S. M. Ismail, I. Garcia-Torregrosa, J. C. Vollenbroek, L. Folkertsma, J. G. Bomer, T. Haarman, M. Ghiasi, M. Schellhorn, M. Nachtegaal, M. Odijk, A. van den Berg, B. M. Weckhuysen and F. M. F. de Groot, ACS Catal., 2021, 11, 12324–12335 CrossRef CAS.
- L. Xi, C. Schwanke, D. Zhou, D. Drevon, R. van de Krol and K. M. Lange, Dalton Trans., 2017, 46, 15719–15726 RSC.
- L. Xi, F. Wang, C. Schwanke, F. F. Abdi, R. Golnak, S. Fiechter, K. Ellmer, R. van de Krol and K. M. Lange, J. Phys. Chem. C, 2017, 121, 19668–19676 CrossRef CAS.
- S. P. Cramer, F. M. F. de Groot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, K. Chan, W. H. Armstrong, E. Libby, G. Christou, J. C. Fuggle, S. Brooker, V. McKee and O. C. Mullins, J. Am. Chem. Soc., 2002, 113, 7937–7940 CrossRef.
- L. Xi, C. Schwanke, J. Xiao, F. F. Abdi, I. Zaharieva and K. M. Lange, J. Phys. Chem. C, 2017, 121, 12003–12009 CrossRef CAS.
- C. Schwanke, L. Xi and K. M. Lange, J. Synchrotron Radiat., 2016, 23, 1390–1394 CrossRef CAS PubMed.
- L. Xi, M. Schellenberger, R. F. Praeg, D. Gao, D. Drevon, P. Plate, P. Bogdanoff, R. van de Krol and K. M. Lange, ACS Appl. Energy Mater., 2019, 2, 4126–4134 CrossRef CAS.
- R. A. Vicente, S. P. Raju, H. V. N. Gomes, I. T. Neckel, H. C. N. Tolentino and P. S. Fernández, Anal. Chem., 2023, 95, 16144–16152 CrossRef CAS PubMed.
- R. A. Vicente, I. T. Neckel, S. K. R. S. Sankaranarayanan, J. Solla-Gullon and P. S. Fernández, ACS Nano, 2021, 15, 6129–6146 CrossRef CAS PubMed.
- P. Jiang, J.-L. Chen, F. Borondics, P.-A. Glans, M. W. West, C.-L. Chang, M. Salmeron and J. Guo, Electrochem. Commun., 2010, 12, 820–822 CrossRef CAS.
- P. Sedigh Rahimabadi, M. Khodaei and K. R. Koswattage, X-Ray Spectrom., 2020, 49, 348–373 CrossRef CAS.
- O. M. Magnussen, J. Drnec, C. Qiu, I. Martens, J. J. Huang, R. Chattot and A. Singer, Chem. Rev., 2024, 124, 629–721 CrossRef CAS PubMed.
- P. Müller-Buschbaum, in Applications of Synchrotron Light to Scattering and Diffraction in Materials and Life Sciences, Springer Berlin Heidelberg, Berlin, 2009, pp. 61–89 Search PubMed.
- J. Y. Zheng, A. U. Pawar, C. W. Kim, Y. J. Kim and Y. S. Kang, Appl. Catal., B, 2018, 233, 88–98 CrossRef CAS.
- X. Zhang, Y. Lyu, H. Zhou, J. Zheng, A. Huang, J. Ding, C. Xie, R. De Marco, N. Tsud, V. Kalinovych, S. P. Jiang, L. Dai and S. Wang, Adv. Mater., 2023, 35, 2211894 CrossRef CAS PubMed.
- K. L. Mulfort, A. Mukherjee, O. Kokhan, P. Du and D. M. Tiede, Chem. Soc. Rev., 2013, 42, 2215–2227 RSC.
- D. Derelli, F. Caddeo, K. Frank, K. Krötzsch, P. Ewerhardt, M. Krüger, S. Medicus, L. Klemeyer, M. Skiba, C. Ruhmlieb, O. Gutowski, A. Dippel, W. J. Parak, B. Nickel and D. Koziej, Angew. Chem., Int. Ed., 2023, 62, e202307948 CrossRef CAS PubMed.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |