
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multinuclear solid-state NMR investigation of structurally diverse low-dimensional hybrid metal halide perovskites†
Thomas J. N. Hooper *a,
Benny Febriansyahbc,
Thirumal Krishnamoorthyd,
Walter P. D. Wongd,
Kai Xuee,
Joel W. Agerbf and
Nripan Mathewscd
*a,
Benny Febriansyahbc,
Thirumal Krishnamoorthyd,
Walter P. D. Wongd,
Kai Xuee,
Joel W. Agerbf and
Nripan Mathewscd
aDepartment of Microbial and Molecular Systems (M2S), Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001, Belgium. E-mail: thomas.hooper@kuleuven.be
bBerkeley Educational Alliance for Research in Singapore (BEARS), 1 CREATE Way, Singapore 138602, Singapore
cEnergy Research Institute at NTU (ERI@N), Research Techno Plaza, X-Frontier Block Level 5, 50 Nanyang Drive, Singapore 637553, Singapore
dSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore
eCentre of High Field Nuclear Magnetic Resonance (NMR) Spectroscopy and Imaging, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore
fDepartment of Materials Science and Engineering, University of California at Berkeley, Berkeley, California 94720, USA
First published on 26th July 2024
Abstract
Owing to their synthetic versatility and optoelectronic tunability, low-dimensional hybrid metal halide perovskites (MHPs) provide a key avenue for the design of future optoelectronic materials. Nuclear magnetic resonance (NMR) spectroscopy has emerged as a powerful tool for structural characterisation and molecular dynamics elucidation in MHPs, which are known to control the materials' optoelectronic properties. In this work, we utilise solid state NMR to study structurally diverse hybrid MHPs containing 2D, 1D and 0D inorganic motifs that are templated by a series of xylylenediammonium cations and compare their characteristics with those of archetype 3D perovskites. The highly resolved scalar coupling pattern (J1(207Pb–79/81Br) = 1.98 kHz) in the 207Pb NMR spectrum of 0D meta-xylylenediammonium lead bromide ((mXDA)2PbBr6), reveals that 207Pb NMR of methylammonium lead bromide (MAPbBr3) and formadinium lead bromide (FAPbBr3) is sensitive to local Br positional disorder, associated with the fast reorientation of the MA/FA cations. Variable temperature 1H spin–lattice relaxation quantifies the correlation time of the reorientation of the MA/FA cations at picosecond timescales, in contrast to the slower motion of the bulky cations in the low-dimensional perovskites. Additionally, the study of meta-xylylenediammonium tin halides ((mXDA)2SnX6) provides the first direct detection of tin-halide scalar coupling patterns (J1(119Sn–79/81Br) = 1.51 kHz; J1(119Sn–35/37Cl) = 260 Hz).
Introduction
Metal halide perovskites (MHPs) have been the subject of fervent research over the last ten years due to their exceptional optoelectronic properties and flexible structural characteristics.1,2 The typical three-dimensional (3D) crystal structure of MHPs is given by the chemical formula ABX3, where the metal centre B2+ is surrounded by six halide X− anions (chlorine, bromine or iodine) forming the octahedral repeat unit. Lead halide perovskites have, in particular, garnered the most interest due to their capability to yield high efficiency optoelectronic devices, however less-toxic alternatives have also been actively researched with tin emerging as the most promising non-lead metal centre.3,4 The A+ cations are typically small organic molecules, such as methylammonium (MA) and formadinium (FA), in organic–inorganic “hybrid” perovskites or small inorganic ions (Cs+) in all-inorganic perovskites, all of which act as structural templating and charge balancing agents.In the typical ABX3 perovskite structures, the halometallate octahedral units are connected through corner sharing in three directions, creating 3D inorganic lattices. However, the versatile structural flexibility of the metal halide perovskite system allows different dimensional frameworks to be realised through the use of suitable larger organic cations. In contrast to morphologically controlled species (e.g. 2D nanoplates and 0D quantum dots), the corresponding hybrid materials feature inorganic frameworks that are of reduced dimensionality (i.e. limited connectivity) at the molecular level. 2D perovskites, such as butylammonium lead bromide ((BA)2PbBr4),5 contain sheets, or layers, of typically corner sharing [BX6]4− octahedra, while 1D and 0D congeners contain chains of [BX6]4− octahedra and isolated octahedral [BX6]4− units, respectively. The latter generally follows the chemical formula A4BX6, which is identical to the 0D caesium lead bromide phase Cs4PbBr6.6 Physically, the reduced dimensionality at molecular level leads to various quantum-confinement induced effects, such as tunable band gaps and improved photoluminescence, while also resulting in higher stability than their nanosized analogues. In this article, the use of 0D, 1D, and 2D terminologies shall refer to such low-dimensional hybrid perovskites.
Solid state nuclear magnetic resonance (NMR) spectroscopy has been shown to be a powerful, non-destructive technique in the structural characterisation of perovskites.7–9 It can analyse both amorphous (surface or nanocrystal MHP) and crystalline (bulk MHP) materials, unlike traditional diffraction techniques. In addition to structural information, NMR can probe molecular dynamics and chemical bonding via relaxometry and correlation NMR, respectively. Furthermore, its element-specific nature allows focused investigation on the desired component (metal, ion, ligand, or dopant). The majority of NMR perovskite reports have focused on the study of cation phases, dynamics and molecular rotations in hybrid perovskites using 1H, 2H, 13C, 14N and 133Cs NMR experiments.7,10–44 In addition, 207Pb NMR has been proven to be useful in determining the structure and phase composition of lead halide perovskites with new or mixed cations/anions;31,32,36,44–68 high resolution 207Pb NMR spectra of some perovskites have also revealed scalar coupling patterns.24,31,46,47,69
Scalar coupling (also called J-coupling) refers to the perturbation of the observed nuclei's energy levels due to the polarisation of the covalent bond electron pairs by a secondary nuclei. This creates a splitting in the NMR resonance, with a frequency difference equal to the scalar coupling parameter J1. The energy level is raised or lowered depending on the orientation of the second nucleus's spin sub-level (m) with the external magnetic field. Hence, a bond to a spin S which has 2S + 1 possible spin sub-levels, results in 2S + 1 degenerate energy levels. The splitting of the energy levels is compounded when the observed nucleus is bonded to multiple magnetically equivalent nuclei. The final number of degenerate energy levels is given by 2nS + 1, where n is the number of such bonds. In the NMR spectrum this manifests as a splitting of the resonance into a multiplet of 2nS + 1 resonances, whose intensities follow a binomial distribution. The strength of the scalar coupling, quantified by J1, is dependent on multiple factors including the degree of covalence of the bonds and the degree of orbital overlap. Hence, 207Pb scalar coupling can provide information on the haloplumbate bond lengths, angles and rigidity in perovskite materials.
Recently, Aebli et al. measured the 207Pb NMR of MAPbX3, FAPbX3, CsPbX3 and Cs4PbBr6 (X = Br and Cl) and confirmed an absence of resolved scalar coupling for MAPbBr3 and FAPbBr3 at room temperature. This absence is intriguing as the predicted perfect cubic symmetry of the PbBr6 octahedra in these perovskites rules out merging of the scalar coupling pattern due to structural asymmetry. Conversely, greater structural symmetry explains the more resolved scalar coupling pattern in Cs4PbBr6 than CsPbBr3, which has been observed in several reports.46,47,69 Aebli et al.69 postulated that the lack of scalar coupling resolution at room temperature in MAPbBr3 and FAPbBr3 was result of the fast cation dynamics in both systems. Hence, 207Pb NMR could prove to also be a probe of the fast organic cation dynamics in hybrid perovskites, which, outside of NMR techniques, has relied upon neutron scattering or molecular dynamic simulations for study.18,70,71 For their hypothesis to be proven, Aebli et al. called for comparison with 0D hybrid perovskites. Such materials could provide a symmetric PbX6 octahedral configuration, without the rapid cation dynamics environment, due to the significant steric effect provided by the relatively bulkier organic molecules required to template a 0D perovskite structure.
Therefore, the lead–bromide hybrid perovskite based on the meta-xylylenediammonium cation ((mXDA)4PbBr6) was chosen for this work because it has been shown to feature relatively symmetrical [PbBr6]4− octahedra in an isolated 0D structure.72–74 We further found that by simply changing the position of the methylammonium chain, the resulting inorganic frameworks within the materials crystal lattice can be modulated. In particular, para-xylylenediammonium induces the formation of monolayered 2D lead bromide structure in (pXDA)PbBr4, while ortho-xylylenediammonium templates 1D bromoplumbate chain motifs in (oXDA)2Pb2Br8. As such, the XDA cation series acts as a perfect platform to study not only the scalar coupling properties and how it is related to the cation dynamics, but also to gain insight over how such properties would vary as a function of inorganic structural dimensionality.
This work presents the first 207Pb MAS NMR of the xylylenediammonium lead bromides which, alongside structural data via single crystal X-ray diffractometry (SCXRD), is compared to well-known lead halide perovskites: MAPbBr3, FAPbBr3, CsPbBr3, Cs4PbBr6 and (BA)2PbBr4. The results are corroborated with cation dynamics via 1H/133Cs spin–lattice nuclear relaxation measurements. In addition, we provide the first synthesis and structural characterisations of: ortho-xylylenediammonium lead bromide, (oXDA)2Pb2Br8; meta-xylylenediammonium lead chloride, (mXDA)2PbCl6; meta-xylylenediammonium tin bromide, (mXDA)2SnBr6; and meta-xylylenediammonium tin chloride, (mXDA)2SnCl6. The 0D tin perovskites are also examined via 119Sn MAS NMR experiments.
Methodology
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio total 1 mL) to create 1 M perovskite solutions. The solutions were then slowly heated to 100 °C. The resulting single crystals grown thereof were then harvested, washed and dried under reduced pressure accordingly. Meanwhile, CsPbBr3, Cs4PbBr6 and (BA)2PbBr4 were synthesized through the normal temperature crystallization method with concentrated HBr (48%; 1 mL)) being used as the solvent. Therein, a stochiometric amount of PbO (0.223 g) was dissolved in HBr before CsBr (0.213 g or 0.852 g)) or butylamine (0.200 mL) were added to create 1 M perovskite solutions. The solution was then heated gently to 100 °C with vigorous stirring (50 °C for Cs4PbBr6 with 12 hours of stirring). The solutions were then slowly cooled to room temperature to obtain crystals. The crystals were then washed and dried under reduced pressure, before being ground to fine powder for solid state NMR measurement.
:1 volume ratio total 1 mL) to create 1 M perovskite solutions. The solutions were then slowly heated to 100 °C. The resulting single crystals grown thereof were then harvested, washed and dried under reduced pressure accordingly. Meanwhile, CsPbBr3, Cs4PbBr6 and (BA)2PbBr4 were synthesized through the normal temperature crystallization method with concentrated HBr (48%; 1 mL)) being used as the solvent. Therein, a stochiometric amount of PbO (0.223 g) was dissolved in HBr before CsBr (0.213 g or 0.852 g)) or butylamine (0.200 mL) were added to create 1 M perovskite solutions. The solution was then heated gently to 100 °C with vigorous stirring (50 °C for Cs4PbBr6 with 12 hours of stirring). The solutions were then slowly cooled to room temperature to obtain crystals. The crystals were then washed and dried under reduced pressure, before being ground to fine powder for solid state NMR measurement.Analytical techniques
For single crystal X-ray measurement and study, a Bruker X8 CCD area detector diffractometer was used and the data was collected using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at particular temperatures. Data reduction and absorption corrections were performed using the SAINT and SADABS software packages, respectively.76 All structures were solved by direct methods and refined by full-matrix least squares procedures on F2, using the Bruker SHELXTL-2014 software package.77,78 Non-hydrogen atoms were anisotropically refined before hydrogen atoms were introduced at calculated positions for further refinement of data. The graphical illustrations of crystal structures used throughout were mainly created using the program VESTA.79 Full crystallographic and refinement data can be found in the ESI Tables S1 and S2.†The photoluminescence (PL) spectra were acquired using a WITec alpha 300RAS confocal Raman microscope. The UV line of a linearly polarized CW solid laser (He–Cd, 325 nm) was chosen and the excitation power was kept below 10 μW on sample to avoid photo degradation and saturation of the detector.
Solid-state NMR experiments were completed on a 14.1 T Bruker Advance III HD 600 MHz spectrometer using a 1.9 mm Bruker HXY probe. Single crystal samples were manually ground before packing. The 207Pb NMR (ν0(207Pb) = 125.60 MHz) Hahn-echo experiments utilised an MAS frequency of 12 kHz, a π/2 pulse of 5 μs (determined on Pb(NO3)2(s)), a recycle delay of 5 s, and a rotor synchronised echo delay. The 1H NMR saturation recovery experiments utilised MAS frequencies of 12–40 kHz, a 1H π/2 pulse length of 2.5 μs, and a 200 pulse saturation block. The 119Sn[1H] and 13C[1H] NMR CP experiments utilised an MAS frequency of 12 kHz, a 5000 μs contact pulse length, a 1H π/2 pulse length of 2.5 μs, high power proton decoupling and recycle delays dependent on the 1H saturation recovery results. All spectra were processed using the Topspin software package and referenced to the unified scale using IUPAC recommended frequency ratios relative to the 13C adamantane(s) methylene resonance (δ = 37.77 ppm).80,81 Spectral deconvolution was performed with dmfit.82
Results and discussion
Each of the XDA-based materials crystallize in monoclinic space group P21/c, as determined via SCXRD, and the refined structures are shown in Fig. 1. Fig. 1(a–c) demonstrates how varying the position of the methylammonium functional groups on the phenyl ring of the cations results in lead bromide perovskites of different dimensionalities. In particular, mXDA leads to (mXDA)2PbBr6, a 0D perovskite with isolated octahedral units (Fig. 1(a)), while the isomeric pXDA templates the formation of (pXDA)PbBr4, a 2D perovskite with isolated monolayers of corner-sharing octahedral units expanding in the b–c plane (Fig. 1(c)). The oXDA cation, on the other hand, produces (oXDA)2Pb2Br8, which has a more complex structure, with 1-D chains of edge-sharing octahedral units along the c-axis with additional branches of singular corner-sharing units (Fig. 1(b)). Isovalent substitution of the metal halide components does not typically alter the resulting inorganic framework configuration. This is illustrated by examples shown in Fig. 1(d–f) where hybrid materials (mXDA)2PbCl6, (mXDA)2SnBr6, and (mXDA)2SnCl6 were found to exhibit the same isolated 0D structure as (mXDA)2PbBr6. To confirm the organic cation integrity in the series of low-dimensional hybrid perovskites investigated in this work, 13C CPMAS NMR experiments were conducted and the corresponding spectra can be seen in ESI Fig. S1.†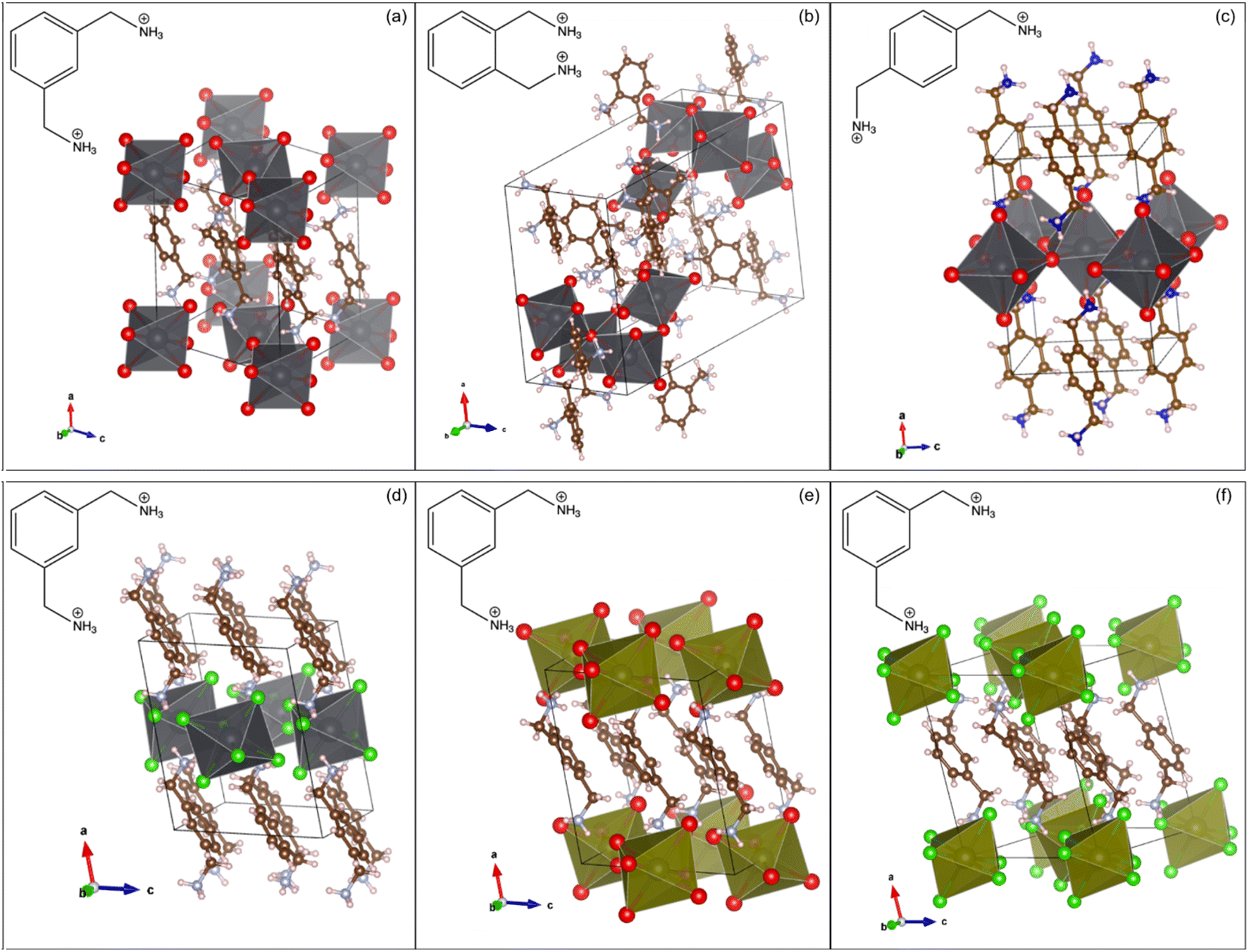 | ||
| Fig. 1 The SCXRD determined structures of (a) (mXDA)2PbBr6, (b) (oXDA)2Pb2Br8, (c) (pXDA)PbBr4, (d) (mXDA)2PbCl6, (e) (mXDA)2SnBr6, and (f) (mXDA)2SnCl6 with molecular drawings of the respective organic cations inset. | ||
Hybrid materials with isolated [PbBr6]4− ions, such as 0D (mXDA)2PbBr6, are relatively rare in occurrence with less than ten compounds being reported so far in the literature.83–89 The organic species responsible for directing such molecular lead-bromide octahedra are diverse in structure, but some similarities can still be drawn. This includes the presence of both dicationic ammonium groups and a bulky core. The former is needed to not only satisfy the charge balancing requirements to the [PbBr6]4− units, but also to drive the lattice formation through H bonding with Br− ions. The relative position of the ammonium groups in the organic molecule dictates the formation of isolated [PbBr6]4− octahedra. For example, a 1D chain was obtained in the case of (oXDA)2Pb2Br8 because the proximity of the methylammonium functionality in oXDA prevents the formation of separate octahedra. On the other hand, the ammonium groups in pXDA are far enough apart to allow one bromoplumbate octahedron to corner-share with four octahedral neighbors along an equatorial plane. Propagation in two directions eventually leads to layered [PbBr4]2− architectures in (pXDA)PbBr4. The bulky core of the organic cation enforces the physical separation between each bromoplumbate octahedron as a result of steric hindering and in the case of XDA, this role is fulfilled by the phenyl ring. In all the investigated mXDA templated perovskites, the phenyl rings are arranged in parallel displaced stacks with centroid–centroid distances of 4.2–4.3 Å and centroid normal-centroid vector angles of 33–38°. The phenyl rings in (pXDA)PbBr4 and (oXDA)2Pb2Br8 are T-stacked and parallel displaced respectively (centroid–centroid distance of 5.8 and 5.4 Å). A more detailed discussion on the specificity of isomeric XDA cations in templating low-dimensional perovskite with different inorganic architectures, in comparison to other reported 0D templating cations, can be found in the ESI.†
Low-dimensional lead halide perovskites tend to demonstrate broadband photoluminescence. Such behaviour has been documented by several studies and the origin is ascribed to the self-trapped excitons formed in the disordered crystal lattice.90–92 Structurally, the broadband emission has also been correlated with different parameters of inorganic lattice distortion within the materials, such as inter-octahedral tilting and intra-octahedral (coordination) deformation.93–95 As expected from the octahedral distortion of the XDA lead bromide perovskites, they produce broadband emission across the visible light region (Fig. S5†).
The 207Pb solid state NMR spectra of each lead bromide perovskite are presented in Fig. 2. Scalar coupling patterns have been resolved in the spectra of (mXDA)2PbBr6, Cs4PbBr6, and CsPbBr3, where coupling of 207Pb nuclei (spin ½) with six 79/81Br nuclei (spin ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) ) results in 19 degenerate resonances with a binomial intensity distribution. Such phenomena have previously been observed for Cs4PbBr6, CsPbBr3, CsPbCl3, MAPbBr3, and MAPbCl3. Our results did not resolve the pattern for the MA perovskites, however previous reports of scalar coupling in the 207Pb spectra of the MA-based perovskites were taken at much lower temperatures and/or lower magnetic fields, which explains the discrepancy.69
) results in 19 degenerate resonances with a binomial intensity distribution. Such phenomena have previously been observed for Cs4PbBr6, CsPbBr3, CsPbCl3, MAPbBr3, and MAPbCl3. Our results did not resolve the pattern for the MA perovskites, however previous reports of scalar coupling in the 207Pb spectra of the MA-based perovskites were taken at much lower temperatures and/or lower magnetic fields, which explains the discrepancy.69
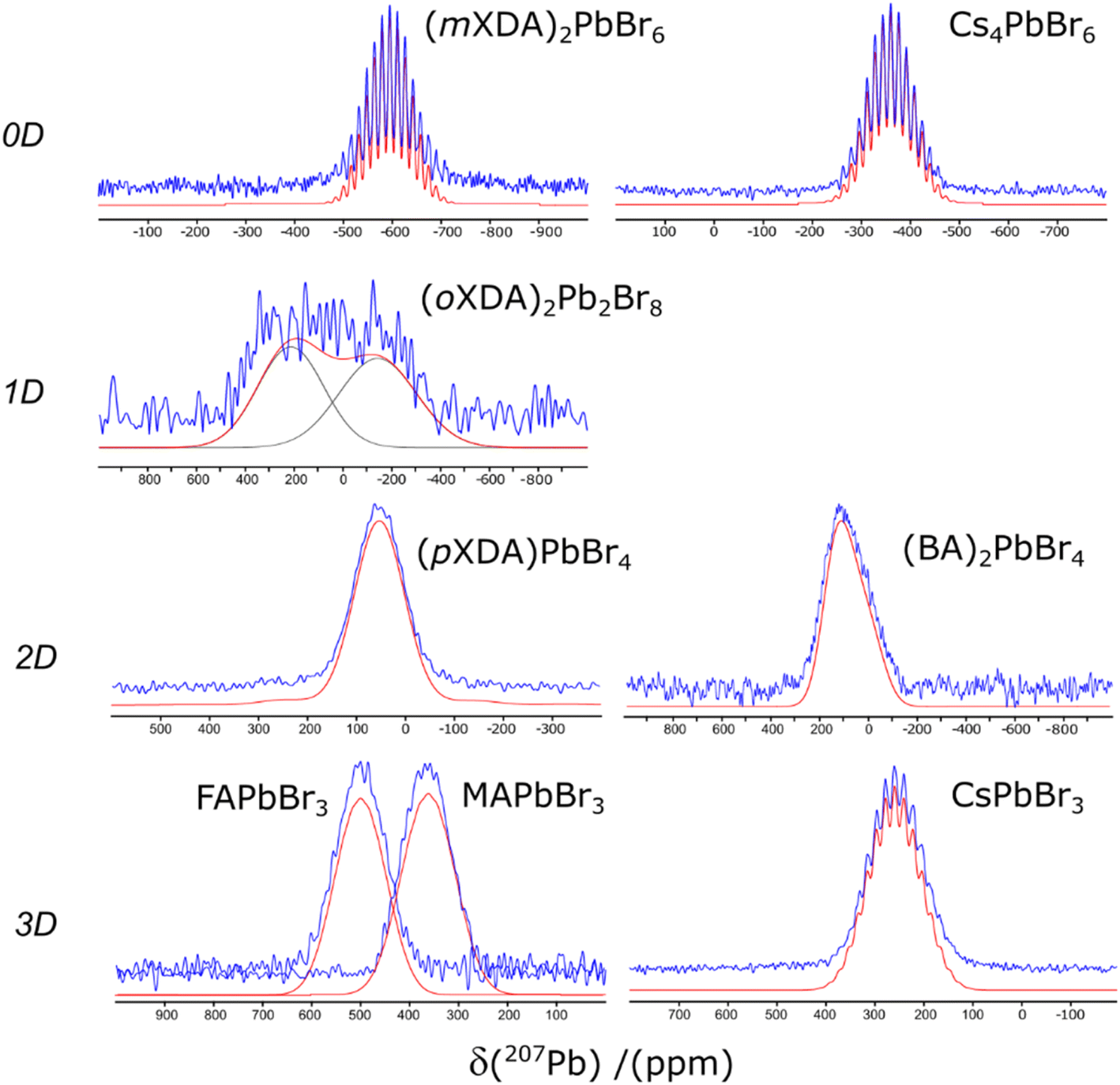 | ||
| Fig. 2 The 207Pb NMR spectra of the lead bromide perovskites. Experimental and simulated spectra are displayed in blue and red respectively. | ||
The 207Pb NMR fitting parameters are detailed in Table 1. The isotropic chemical shift (δiso) varies between 600 and −600 ppm, and has a general dependency on the structure of the PbBr6 octahedra. Fig. 3(a) shows a negative correlation between δiso and the mean Pb–Br bond length, giving a coefficient of determination (R2) of 0.78. As the bond length increases the negative paramagnetic shielding contribution decreases (i.e. the total shielding of the 207Pb nucleus increases), resulting in a decrease in chemical shift frequency. This correlates with the findings of Dimitrenko et al. who utilised density functional theory (DFT) calculations to show that the 207Pb paramagnetic contribution is the dominant variable affecting the 207Pb chemical shielding when varying the electronegativity of the halide bond, achieved by swapping the X halide in Pb(II)X2 compounds.100 In addition, Lee et al. reported a negative correlation between δiso(207Pb) and mean Pb–I bond lengths across a series of 3D/2D lead iodide perovskites.61 Values for the two (oXDA)2Pb2Br8 Pb sites were not included in the linear fit due to the large site disorder. Both sites have effective coordination numbers (ne) below 5.7 (while sites from all other lead bromides have ne > 5.99) which would strongly effect their 207Pb chemical shift. Variations from the predicted trend can be explained by cation differences and distances between octahedral layers. For example, (pXDA)PbBr4 and (BA)2PbBr4 have very similar structures and cations but fall on either side of the trendline. This is due to the greater inter-octahedral Pb–Pb distance in the latter (between both adjacent Pb and Pb in separate layers) resulting in a reduced magnetic shielding and hence a relatively higher chemical shift. Fig. 3(a) demonstrates how 207Pb NMR can differentiate between the structural dimensionalities of the perovskites due to its sensitivity to Pb–Br bond lengths and greater structure.
| Perovskites | Dimensionalities | 207Pb NMR | Cation NMR relaxation | |||
|---|---|---|---|---|---|---|
| δiso (ppm) | Scalar coupling | 1H T1 s | 133Cs T1 s | |||
| J1(207Pb–79/81Br) (kHz) | FWHMa (kHz) | |||||
| a Full-width half maximum (FWHM) of observable scalar coupling resonances. | ||||||
| (mXDA)2PbBr6 | 0D | −595.0(6) | 1.98(7) | 1.17(7) | 1.5 | — |
| (oXDA)2Pb2Br8 | 1D | 215(40), −142(47) | — | — | 0.7 | — |
| (pXDA)PbBr4 | 2D | 53(7) | — | — | 0.7 | — |
| (BA)2PbBr4 | 2D | 83(15) | — | — | 2 | |
| MAPbBr3 | 3D | 358(7) | — | — | 24 | — |
| FAPbBr3 | 3D | 502(7) | — | — | 32 | — |
| Cs4PbBr6 | 0D | −361.1(7) | 2.02(8) | 1.26(8) | — | 159 |
| CsPbBr3 | 3D | 265(1) | 2.38(14) | 2.12(14) | — | 200 |
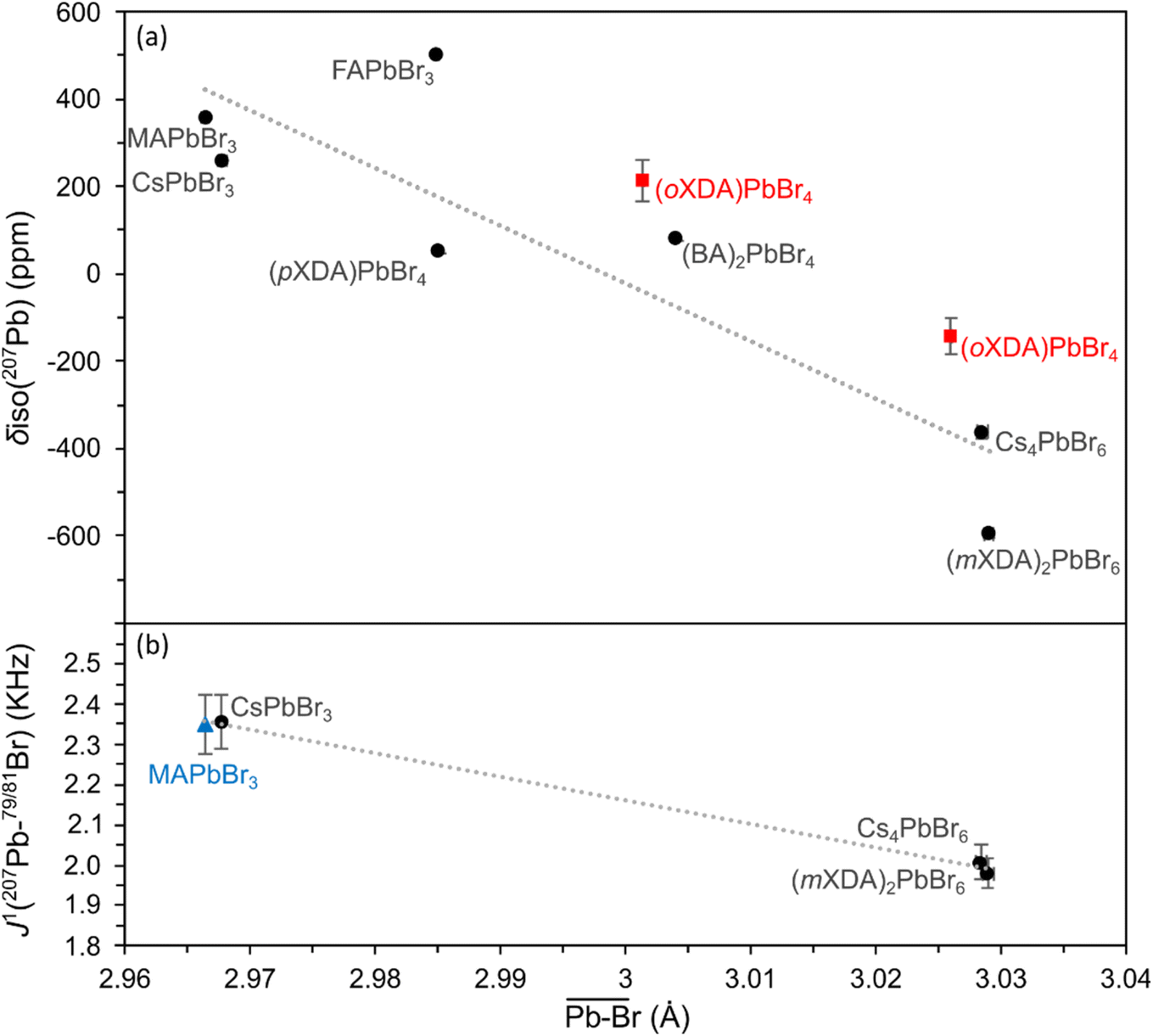 | ||
| Fig. 3 Plots of (a) 207Pb isotropic chemical shift and (b) 207Pb–79/81Br scalar coupling constants vs. octahedral Pb–Br mean atomic distance for the lead bromide perovskite series. Linear fits are presented with R2 values of 0.776 and 0.997 for the isotropic chemical shift and scalar coupling constants respectively. Values taken from the report of Aebli et al.69 are marked with blue triangles. (oXDA)PbBr4 values are considered as anomalous (red squares) and hence are left out of the linear fits. | ||
The scalar coupling parameter J1(207Pb–79/81Br) is the frequency difference between each resonance in the pattern, and is a measure of the intensity of the local magnetic field perturbation created by the shared electrons. It follows that the J1 value for a set pair of nuclei is negatively proportional to the bond length l. Fig. 3(b) confirms the trend for J1(207Pb–79/81Br) in lead bromide perovskites providing the correlation:
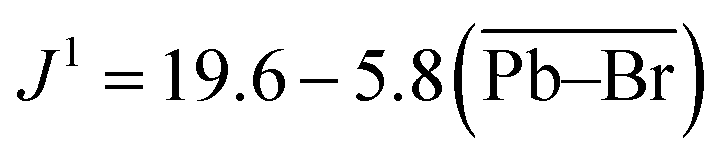 | (1) |
The lead bromide perovskites investigated in this study only provided 3 data points for analysing the scalar coupling, however the J1 value for MAPbBr3 at 100 K by Aebli et al. also fits the trend.69 Furthermore, the same trend can be observed for lead chloride scalar couplings, as seen in ESI Fig. S2.†
For the perovskites whose scalar coupling is not resolved, other interactions must be broadening the individual resonances sufficiently to “smear” the scalar coupling pattern. The most obvious culprit would be distortion in the [PbBr6]4− octahedral units, where asymmetry would result in large 207Pb chemical shift anisotropy broadening. In more symmetrical cases differing bond-lengths and angles could result in mismatched scalar couplings for each bond and smearing of the pattern. Table 2 details the structural parameters of each of the lead bromides investigated here, with several metrics defining distortion of the [PbBr6]4− octahedra.
| Perovskites | Dimensionality | Space group | Pb–Br length | Br–Pb–Br angle variance (°) | [PbBr6]4− nea | Ref. | |
|---|---|---|---|---|---|---|---|
| Mean (Å) | σ (Å) | ||||||
| a Effective coordination number.b Determined in this work. | |||||||
| (mXDA)2PbBr6 | 0D | Pm21/c | 3.0290(4) | 0.0148 | 36.72 | 5.996 | b |
| (oXDA)2Pb2Br8 | 1D | Pm21/c | 3.0259(1), 3.0013(1) | 0.1145, 0.1056 | 99.56, 27.56 | 5.671, 5.688 | b |
| (pXDA)PbBr4 | 2D | Pm21/c | 2.9966(3) | 0.0132 | 6.14 | 5.996 | b |
| (BA)2PbBr4 | 2D | Pbca | 3.0040(3) | 0.0078 | 9.42 | 5.999 | 93 |
| MAPbBr3 | 3D | Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
2.9664(3) | 0.0000 | 0.00 | 6.000 | 96 |
| FAPbBr3 | 3D | Pmm |
2.9849(1) | 0.0000 | 0.00 | 6.000 | 97 |
| Cs4PbBr6 | 0D | Rc |
3.0284(4) | 0.000 | 0.06 | 6.000 | 98 |
| CsPbBr3 | 3D | Pbnm | 2.9677(1) | 0.0120 | 34.84 | 5.997 | 99 |
The 2D and 1D perovskites have 207Pb patterns defined by chemical shift anisotropy (CSA) broadening due to the low symmetry about the [PbBr6]4− octahedral units, which hides any underlying scalar coupling. The large Pb–Br length/angle distortion present in the two distinct lead sites in (oXDA)2Pb2Br8 (see Table 2) results in two very broad overlapping resonances. The 207Pb NMR spectrum is of poor quality as the (oXDA)2Pb2Br8 perovskite was not stable and samples degraded, despite synthesizing under inert conditions and packing quickly. During the long acquisition time necessary for wide-line 207Pb NMR experiments the 207Pb resonance was lost and the sample had changed colour to black. The “step-like” 2D perovskite architecture reported by Hoffman et al., has a similar arrangement of corner sharing [PbX6]4− in shorter chains (2–4 octahedral units) which are linked by edge sharing [PbX6]4− into a larger 2D network.101 The edge sharing octahedral units in these step-like perovskites are similarly distorted as in (oXDA)2Pb2Br8, due to the asymmetrical positioning of neighbouring [PbX6]4− octahedra. They reported no such problems with stability. Instead the organic molecule itself seems to be the source of the instability as the synthesized organic bromide salt degraded when left in open atmosphere. As this study was most interested in comparing the 207Pb NMR of different hybrid perovskite architectures with symmetrical octahedral units, higher quality 207Pb NMR of (oXDA)2Pb2Br8 was not pursued further.
The 2D perovskites (pXDA)PbBr4 and (BA)2PbBr4 have relatively symmetrical [PbBr6]4− units (ne > 5.99) so the source of the asymmetry is less obvious. In contrast, other well-known 2D lead bromide perovskites such as phenylethyl ammonium lead bromide, (PEA)2PbBr4, have more distorted octahedra (ne = 5.76)102 resulting in broad CSA patterns in the 207Pb spectrum (see ESI Fig. S3†), which is why this perovskite was not chosen for further comparison. Instead the 207Pb NMR of (pXDA)PbBr4 and (BA)2PbBr4 is sensitive to the asymmetry beyond their octahedral units, due to their planar arrangement of octahedra. The resulting CSA broadening is evident from the asymmetrical line-shape of the 207Pb resonances, most obvious in the static spectra of (BA)2PbBr4. Their CSA fitting and parameters can be seen in ESI Fig. S3.†
The scalar coupling of CsPbBr3 and Cs4PbBr6 have been reported several times,14,46,69 and the poorer resolution of the pattern in CsPbBr3 is due to its Pb–Br bond length variation, in contrast to the perfect symmetry of the Cs4PbBr6 units. The 0D and 3D hybrid perovskites are more intriguing, as a very well defined scalar coupling pattern is present in the 207Pb NMR spectrum of (mXDA)2PbBr6 but not present in MAPbBr3 or FAPbBr3 (at room temperature). This is despite the perfect symmetry in the [PbBr6]4− units of the cubic 3D perovskites reported by SCXRD. By utilising the linear trend from the resolved scalar coupling patterns we can predict J1 values for the cubic lead bromides as ∼2.3 kHz, which is in agreement with the J1 observed by Aebli et al. for MAPbBr3 at 100 K.69 By simulating the 207Pb NMR line shapes with these J1 values, a minimum scalar coupling resonance broadening required to obscure their patterns is determined as > 3.0 kHz. This broadening is almost triple the width of the (mXDA)2PbBr6 scalar coupling resonances, despite the larger Pb–Br bond length/angle variance (see Table 2) present in the 0D perovskite.
Aebli et al. postulated that the lack of scalar coupling resolution in MAPbBr3 and FAPbBr3 at room temperature was result of the fast cation dynamics in both systems.69 The organic cations in MAPbX3 and FAPbX3 are known to undergo rapid reorientation within the cuboctahedral cage. The MA and FA cation reorientation time have been thoroughly examined in the literature, providing values ranging between 0.2–5 ps for MAPbBr3,16,19,31,70,71,103,104 6 ps for FAPbBr3,70 0.4–14 ps for MAPbI3,16,19,34,103–107 and 2–8 ps for FAPbI3.18,34,108,109
Evidence for Aebli and coworker's hypothesis includes the observation of a scalar coupling pattern in the 207Pb NMR of MAPbBr3 at 100 K, where the fast reorientation of the MA cation is reportedly much reduced.69,70 Here, we utilised variable temperature 1H NMR spin–lattice relaxation measurements to probe the cation dynamics, as reported previously for MA and FA perovskites.16,18,110 Spin–lattice NMR relaxation (measured by the characteristic relaxation time T1) is the return of the nuclear magnetisation to equilibrium parallel with the applied static magnetic field. In solids where the dominant relaxation occurs through the dipole–dipole interaction mediated by molecular motions,111 the relaxation is dependent on the corelation time via eqn (2):
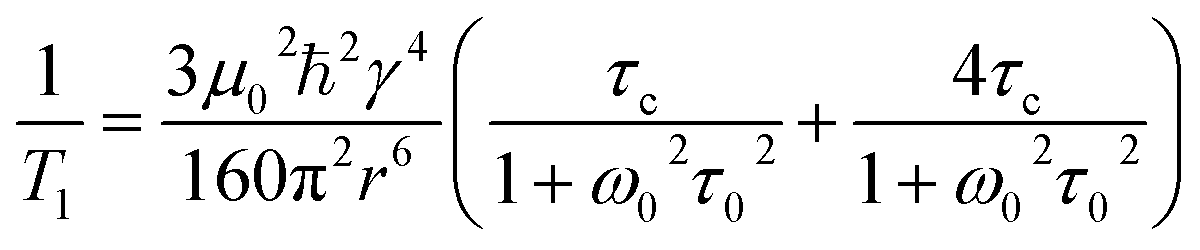 | (2) |
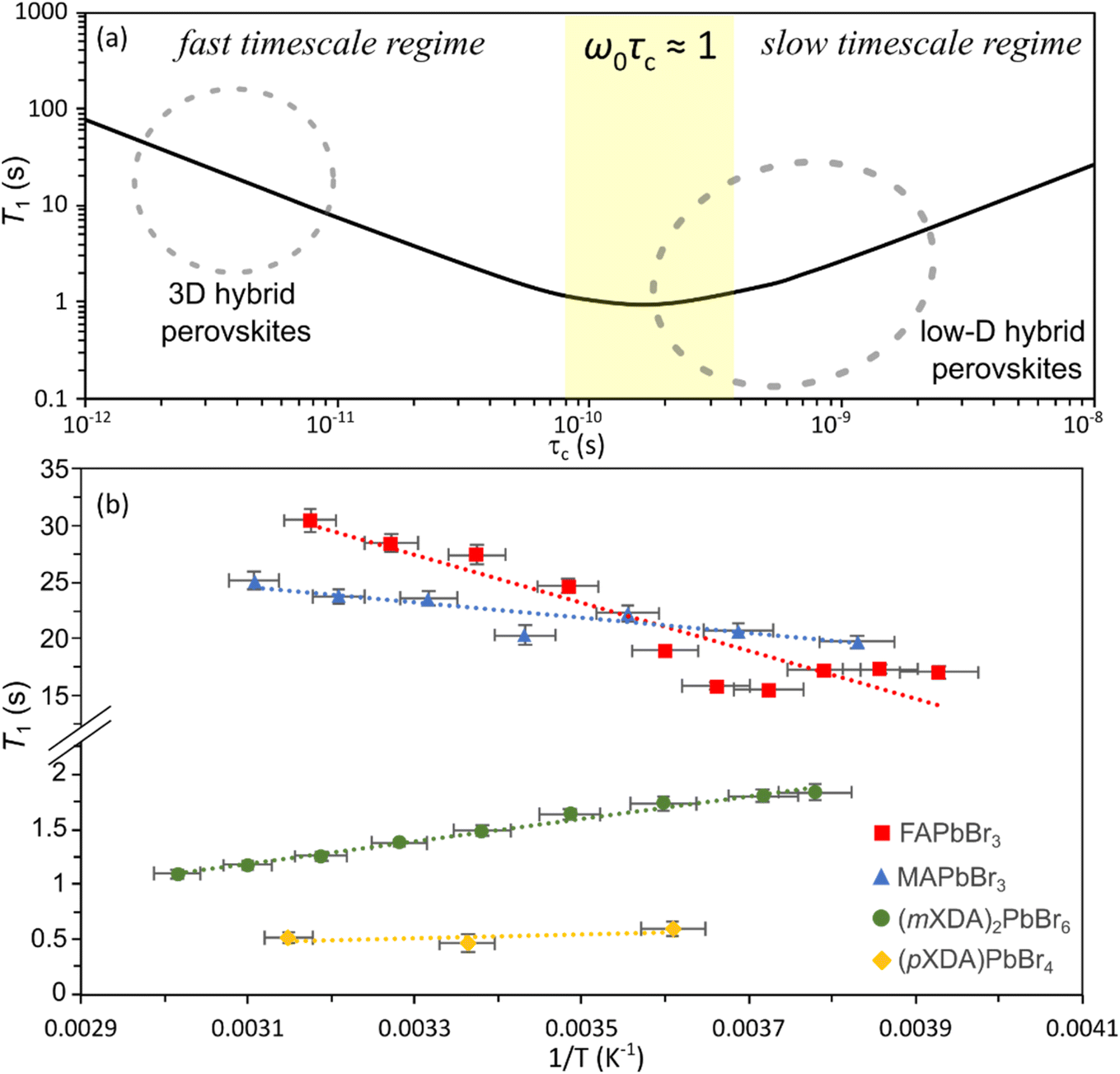 | ||
| Fig. 4 (a) Schematic of the ideal 1H T1-correlation time relationship for dipolar mediated relaxation following eqn (2) at 14.1 T, using a representative inter-moment distance r = 2 Å, to demonstrate the different timescale regimes. (b) Plot of 1H T1 versus inverse temperature for FAPbBr3, MAPbBr3, (mXDA)2PbBr6, and (pXDA)PbBr4. | ||
Fig. 4(b) shows the 1H spin–lattice relaxation times at varying temperatures for the organic cation lead bromides. MAPbBr3 and FAPbBr3 have high 1H T1 relaxation times, which increase with temperature and therefore are in the fast-timescale regime at room temperature, as expected from the reported picosecond reorientation times. In the fast time scale regime (ω0τc ≪ 1) eqn (2) can be simplified to:
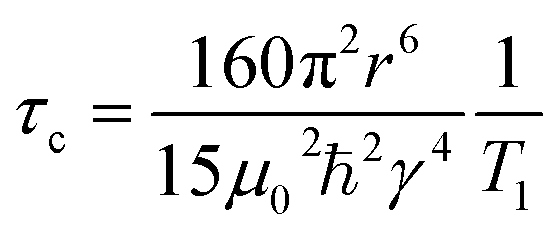 | (3) |
Hence, by utilising the inter-moment distances determined by Fabini et al. for the cations in MAPbI3 and FAPbI3,18 we can determine the MA and FA correlation times as 2 ps and 7 ps respectively in MAPbBr3 and FAPbBr3. These values are based on many assumptions and therefore should be used to judge the scale of the rate of motion, however they are in good agreement with the reported values for MAPbBr3 and FAPbBr3 which utilised a variety of techniques including: 2H and 14N quadrupolar relaxation;19,31 quasi-elastic neutron scattering;70 GHz spectroscopy;103 2D infrared spectroscopy;104 molecular dynamics;71 and 1H nuclear relaxation.106
Conversely, the 0D perovskite (mXDA)2PbBr6 demonstrates 1H T1 relaxation times that become shorter with increasing temperature, putting it within the slow timescale regime (τc ≫ 100 ps). By observing the perovskites 1H relaxation over a wide enough temperature range to cover the entire behaviour shown in Fig. 4(a) and fitting the data with eqn (2), a more exact correlation time could be acquired. Unfortunately, we could not access a large enough temperature range for this study. However, we can confirm the intuitive hypothesis that the bulky cations forming the low dimensional perovskites are much less mobile than the rapidly reorientating cations in the 3D perovskite.
Aebli et al. postulated that the rapid reorientation of the cations, causes a local variation of Pb–Br bond lengths/angles in MAPbBr3 and FAPbBr3, due to the H-bonding between the cation NH3+ groups and the surrounding Br atoms. The resulting distribution of scalar coupling strengths, J, would smear the scalar coupling pattern.69 Our examination of a 0D hybrid lead bromide gives us a lower bound on the necessary Pb–Br distortion, as (mXDA)2PbBr6 presents a well-resolved scalar coupling pattern despite a permanent Pb–Br bond length standard deviation of 0.015 Å. Additionally, the distortion cannot be only present at the picosecond timescales of the cation dynamics, otherwise the 207Pb nucleus would observe an averaged Br position. Two discrete NMR resonances separated by a frequency of Δν will merge when the exchange rate between the two sites becomes proportional to the reciprocal of the frequency difference. Much faster site exchange will result in the detection of a single resonance, as the NMR detection is observing the average of the two sites.112 The necessary broadening to merge the 207Pb scalar coupling patterns is ∼3.0 kHz, hence dynamic disorder would need to occur at slow dynamics (>10−4 s timescales) to cause the required NMR line shape coalescence.
Hence, we believe the 207Pb NMR scalar coupling provides further evidence of permanent short-range disorder in the local structures of MAPbBr3 and FAPbBr3, despite the long-range periodic cubic structure observed by SCXRD. This corroborates previous studies of MAPbBr3 and FAPbBr3 at room temperature showing non-cubic refinements of pair distribution function (PDF) data at <10 Å and large atomic displacement parameters (ADP) for Br perpendicular to the Pb–Br bond.113–118 Worhatch et al. used X-ray PDF refinement to show improved fits in cubic 2 × 2 × 2 supercells of MAPbBr3 and FAPbBr3 with Br atoms displaced transversely to the Pb–Pb line, resulting in a distribution of Br–Pb–Br bond angles below 180°.116 This transverse Br distortion is corroborated by the ab initio molecular dynamics studies of Maity et al. who showed that MA cation dynamics were correlated with Br–Pb–Br scissoring distortions.71 Alternatively, X-ray PDF refinements by Page et al. and extended absorption fine structure (EXAFS) refinements by Nandi et al. both found improved fitting of the local structure of MAPbBr3 via pseudo-cubic orthorhombic structures, with Pb–Br bond length standard deviations of 0.0624 and 0.164 Å respectively.113,115 Clearly the exact local structure of the PbX6 octahedra in hybrid 3D perovskites is still uncertain, but there is agreement on the presence of local disorder in these structures. Hence, our 207Pb NMR comparison with low-dimensional hybrid perovskites confirms the presence of significant local disorder at slow timescales in MAPbBr3 and FAPbBr3, and corroborates the linkage between this disorder and the fast dynamics of their organic cations.
Other possible causes of the 207Pb NMR broadening in MAPbBr3 and FAPbBr3 were investigated. Ionic diffusion in MAPbX3 and FAPbX3 is known to be prevalent and dominated by anion vacancy diffusion with large reported diffusion coefficients.119–121 Despite this, the concentration of halide vacancies and mobile halides, reported to be between 1014–1017 cm−3,121–123 is too low to distort the environment of sufficient Pb nuclei to smear the bulk 207Pb NMR pattern. Additionally, the halide positional exchange in MHPs is predicted to occur at the frequency of the ionic lattice vibrations (ps timescales),120,121,124 which are too fast be the cause of the scalar coupling smearing. Dominant broadening via dipolar or CSA interactions can be ruled out with comparison of 207Pb NMR spectra performed at variable MAS frequencies, which showed no difference in the MAPbBr3 line shape between 24 kHz and static conditions (see ESI Fig. S4†). Previous reports by Rosales et al. and Bernard et al. proposed scalar relaxation due to the fast quadrupolar relaxation of the halides as the source of the 207Pb NMR broadening in the lead bromides.31,125 A spin ½ nuclei I coupled to a fast relaxing quadrupolar nuclei S, will gain a contribution to its relaxation if the relaxation of S is at an equivalent or higher rate than the frequency of the scalar coupling parameter. This relaxation is termed scalar relaxation of the second kind (SC2) by Abragam et al.126 To test this hypothesis, the 207Pb T2 relaxation of MAPbBr3 was measured via CPMG pulse sequence. The T2 of 0.23(3) ms correlates to a transverse relaxation broadening of 1400 Hz, close to the measured FWHM of the scalar coupling resonances in (mXDA)2PbBr6 and Cs4PbBr6. Hence, the SC2 broadening, and other components refocused by a CPMG pulse sequence, cannot be the dominant component of the width of the 207Pb resonances of MAPbBr3.
Fig. 5 compares the 207Pb and 119Sn NMR spectra for the Br and Cl analogues of (mXDA)2PbX6 and (mXDA)2SnX6; the NMR parameters are presented in Table 3. The replacement of Br with Cl in (mXDA)2PbX6 shifts the 207Pb chemical shift by −763 ppm due to a less negative paramagnetic contribution to the chemical shielding and hence a lower chemical shift.100 This mirrors the effect of increasing the Pb–X bond length, which was discussed previously. The same trend in 207Pb δiso when changing from Br to Cl can be seen in previous reports of MAPbX3, FAPbX3, CsPbX3, Cs4PbX6, and (BA)2PbX4.45,56,58,63 Interestingly, the Δδiso is larger in the 3D/2D perovskites (−950 to −1000 ppm), than for the 0D perovskites (mXDA)2PbX6 and Cs4PbX6 (−754 ppm). The difference in the scale of the trend for the 0D perovskites may be due to the longer Pb–X bond lengths, which could dampen the influence of changing the halide on the paramagnetic shielding at the Pb nucleus. This could be explored further via DFT calculations. The 119Sn δiso of (mXDA)2SnCl6 is also shifted negatively from that of (mXDA)2SnBr6 (Fig. 5). Corroborating the 207Pb NMR results, the 119Sn Δδiso for the 0D mXDA perovskites is larger (−228 ppm) than that seen in the literature for 3D MASnX3 perovskites (Br to Cl shift of −82 ppm).127
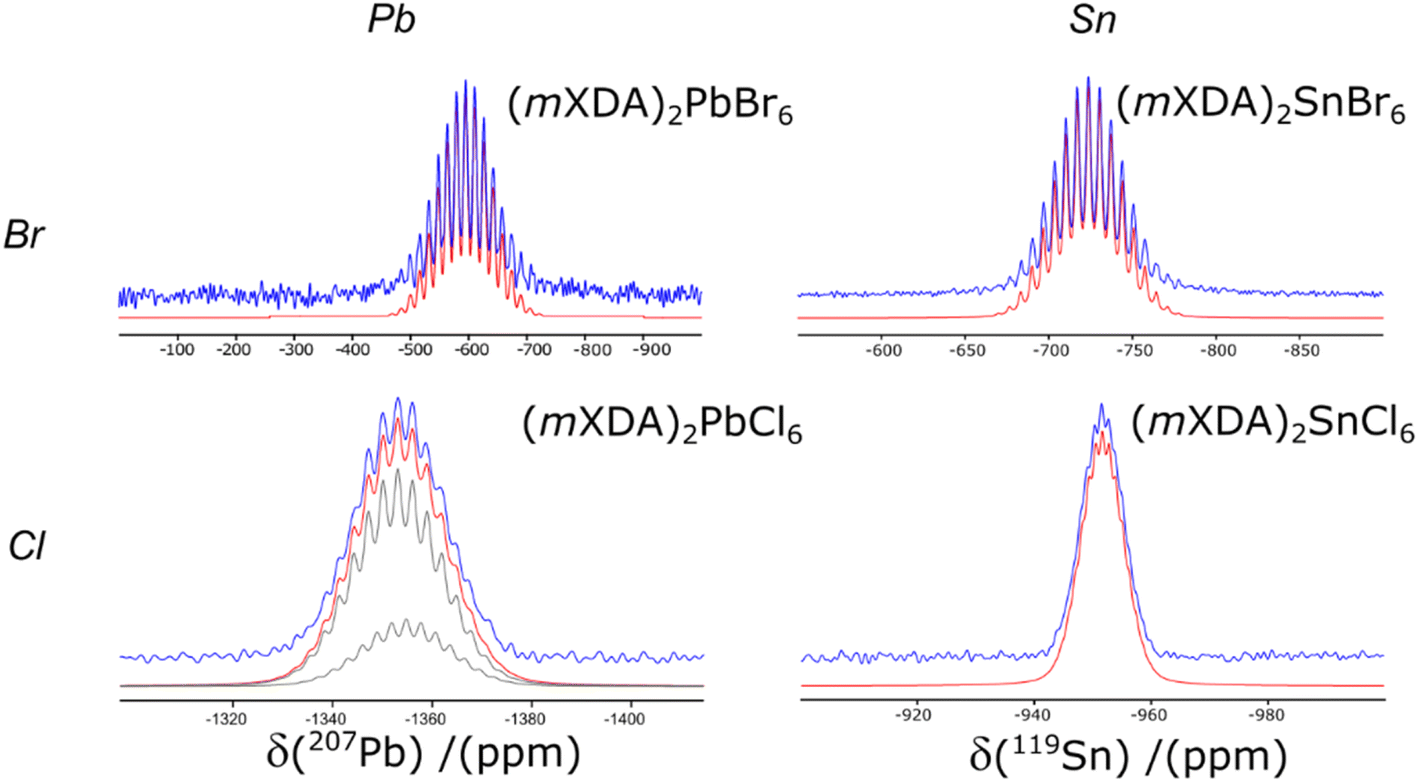 | ||
| Fig. 5 The 207Pb MAS NMR spectra of (mXDA)2PbBr6 and (mXDA)2PbCl6 and the 119Sn CPMAS NMR spectra of (mXDA)2SnBr6 and (mXDA)2SnCl6. Experimental spectra, simulated spectra and deconvoluted resonances are displayed in blue, red and grey respectively. | ||
The 207Pb NMR of (mXDA)2PbCl6 presents with a resolved scalar coupling pattern. The 35/37Cl anions have spin like 79/81Br, resulting in the same pattern as for the lead bromides; however, the scalar coupling constant is smaller at 380 Hz (see Table 3). Furthermore, the 207Pb NMR of (mXDA)2PbCl6 is sufficiently resolved that it may be possible to distinguish slight differences in the resonances of 207Pb coupled to 35Cl and 37Cl isotopes. Fig. 5 shows the two deconvoluted resonances with relative integrals equal to the natural abundance population difference between 35Cl and 37Cl (0.76:0.24); the difference in fitting quality with just one scalar coupling pattern is shown in ESI Fig. S6.† The 119Sn CPMAS NMR of (mXDA)2SnBr6 and (mXDA)2SnCl6 (Fig. 5) demonstrates similar scalar coupling patterns to the lead bromides as 119Sn is also a spin ½ nucleus. The scalar coupling constants are reduced compared to Pb with J1(119Sn–79/81Br) = 1.51 kHz and J1(119Sn–35/37Cl) = 260 Hz. To the authors knowledge this is the first direct experimental observation of J1(119Sn–35/37Cl) and J1(119Sn–79/81Br) coupling, with previous reports providing only indirectly determined or computed values.128,129 The resolution of scalar coupling patterns in all of the 0D perovskites despite B site metal and halide choice, corroborates the relation of the scalar coupling pattern to the rigidity of the perovskite structure. The metal halide octahedra templated by the mXDA cation are all relatively symmetrical with similar metrics of octahedral distortion as (mXDA)2PbBr6 (see ESI Table S3†).
Conclusions
This work presents an SCXRD and solid state NMR structural characterisation of the xylylenediammonium hybrid perovskite series, which form low-dimensional 0D, 1D and 2D perovskites, due to the varying position of the NH3 functional groups about the cations phenyl ring. Improved understanding of these structures is anticipated to assist in the design of future low-dimensional perovskites for optoelectronic perovskite applications. The mXDA cation is shown to form a 0D perovskite structure (mXDA)2BX6 for B = Pb, Sn and X = Br, Cl. Examination of the highly resolved scalar coupling pattern present in the 207Pb NMR of (mXDA)2PbBr6, provides insight into the 207Pb NMR of 3D perovskites MAPbBr3 and FAPbBr3, confirming significant local Br positional disorder coupled to the fast cation reorientation, which is not observed in the long-range averaged model provided by traditional crystallography. The local halide flexibility likely plays a role in the phenomena of halide ion migration and carrier dynamics, which are of importance to the optimal optoelectronic performance of these hybrid materials. Lastly, we further demonstrate the sensitivity of the solid state NMR toolkit to local structure and dynamics in metal halide perovskite material with diverse inorganic architectures and dimensionalities.Data availability
Additional data supporting this article have been included as part of the ESI.† Raw NMR data for this article (Topspin data file format) are available at KU Leuven Research Data Repository at https://doi.org/10.48804/RVHL2D. CIF data for associated crystal structures have been deposited in the Cambridge Crystallographic Data Centre under deposition numbers CCDC 1545198, 2300668–2300671, 2350304.Author contributions
TJNH: conception, drafting, acquisition, analysis, interpretation. BF: conception, revision, acquisition, analysis, interpretation. TK, WPDW, KX: revision, acquisition. JWA: revision. NM: conception, revision.Conflicts of interest
The authors declare the following competing financial interest(s): N. M. is a director of Prominence Photovoltaics Pte Ltd, a perovskite solar cell commercialization company. There are no other conflicts of interest.Acknowledgements
We thank the Singapore Ministry of Education (Academic Research Fund MOE2019-T2-2-032), the Singapore National Research Foundation (Competitive Research Program NRF-CRP14-2014-03; Intra-CREATE Collaborative Grant NRF2018-ITC001-001; Energy Innovation Research Program NRF2015EWT-EIRP003-004; Solar CRP S18-1176-SCRP), and Fonds Wetenschappelijk Onderzoek (Senior Postdoctoral Fellowship 1253824N) for funding this research. We would like to acknowledge the Center of High Field NMR Spectroscopy and Imaging at Nanyang Technological University for the use of their facilities. In addition, TJNH is grateful for valuable discussions about the possible sources of 207Pb NMR broadening with: J. V. Hanna and S. Morris, while at NTU; and D. Sakellariou and R. de Oliveira-Silva, while at KU Leuven.Notes and references
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- R. Chiara, M. Morana and L. Malavasi, ChemPlusChem, 2021, 86, 879–888 CrossRef CAS PubMed.
- E. Aktas, N. Rajamanickam, J. Pascual, S. Hu, M. H. Aldamasy, D. Di Girolamo, W. Li, G. Nasti, E. Martínez-Ferrero, A. Wakamiya, E. Palomares and A. Abate, Commun. Mater., 2022, 3, 1–14 CrossRef.
- D. B. Mitzi, Chem. Mater., 1996, 8, 791–800 CrossRef CAS.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- L. Piveteau, V. Morad and M. V. Kovalenko, J. Am. Chem. Soc., 2020, 142, 19413–19437 CrossRef CAS PubMed.
- D. J. Kubicki, S. D. Stranks, C. P. Grey and L. Emsley, Nat. Rev. Chem, 2021, 5, 624–645 CrossRef CAS PubMed.
- C. J. Dahlman, D. J. Kubicki and G. N. M. Reddy, J. Mater. Chem. A, 2021, 9, 19206–19244 RSC.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS PubMed.
- T. Baikie, N. S. Barrow, Y. Fang, P. J. Keenan, P. R. Slater, R. O. Piltz, M. Gutmann, S. G. Mhaisalkar and T. J. White, J. Mater. Chem. A, 2015, 3, 9298–9307 RSC.
- W. M. J. Franssen and A. P. M. Kentgens, Solid State Nucl. Magn. Reson., 2019, 100, 36–44 CrossRef CAS PubMed.
- A. Kanwat, B. Ghosh, S. E. Ng, P. J. S. Rana, Y. Lekina, T. J. N. Hooper, N. Yantara, M. Kovalev, B. Chaudhary, P. Kajal, B. Febriansyah, Q. Y. Tan, M. Klein, Z. X. Shen, J. W. Ager, S. G. Mhaisalkar and N. Mathews, ACS Nano, 2022, 16, 2942–2952 CrossRef CAS PubMed.
- W. M. J. Franssen, S. G. D. van Es, R. Dervişoğlu, G. A. de Wijs and A. P. M. Kentgens, J. Phys. Chem. Lett., 2017, 8, 61–66 CrossRef CAS PubMed.
- Q. Xu, T. Eguchi, H. Nakayama, N. Nakamura and M. Kishita, Z. Naturforsch. A, 1991, 46, 240–246 CrossRef CAS.
- Q. Xu, T. Eguchi and H. Nakayama, Bull. Chem. Soc. Jpn., 1992, 65, 2264–2266 CrossRef CAS.
- D. H. Fabini, T. A. Siaw, C. C. Stoumpos, G. Laurita, D. Olds, K. Page, J. G. Hu, M. G. Kanatzidis, S. Han and R. Seshadri, J. Am. Chem. Soc., 2017, 139, 16875–16884 CrossRef CAS PubMed.
- R. E. Wasylishen, O. Knop and J. B. Macdonald, Solid State Commun., 1985, 56, 581–582 CrossRef CAS.
- W. T. M. Van Gompel, R. Herckens, G. Reekmans, B. Ruttens, J. D'Haen, P. Adriaensens, L. Lutsen and D. Vanderzande, J. Phys. Chem. C, 2018, 122, 4117–4124 CrossRef CAS.
- W. M. J. Franssen, B. J. Bruijnaers, V. H. L. Portengen and A. P. M. Kentgens, ChemPhysChem, 2018, 19, 3107–3115 CrossRef CAS PubMed.
- C. Anelli, M. R. Chierotti, S. Bordignon, P. Quadrelli, D. Marongiu, G. Bongiovanni and L. Malavasi, Inorg. Chem., 2019, 58, 944–949 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 14173–14180 CrossRef CAS PubMed.
- T. J. N. Hooper, Y. Fang, A. A. M. Brown, S. H. Pu and T. J. White, Nanoscale, 2021, 13, 15770–15780 RSC.
- L. Xie, P. Vashishtha, T. M. Koh, P. C. Harikesh, N. F. Jamaludin, A. Bruno, T. J. N. Hooper, J. Li, Y. F. Ng, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2020, 32, 2003296 CrossRef CAS PubMed.
- D. Prochowicz, P. Yadav, M. Saliba, D. J. Kubicki, M. M. Tavakoli, S. M. Zakeeruddin, J. Lewiński, L. Emsley and M. Grätzel, Nano Energy, 2018, 49, 523–528 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7232–7238 CrossRef CAS PubMed.
- W. Xiang, Z. Wang, D. J. Kubicki, W. Tress, J. Luo, D. Prochowicz, S. Akin, L. Emsley, J. Zhou, G. Dietler, M. Grätzel and A. Hagfeldt, Joule, 2019, 3, 205–214 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Pinon, G. Stevanato, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Mater. Chem. A, 2019, 7, 2326–2333 RSC.
- O. Knop, R. E. Wasylishen, M. A. White, T. S. Cameron and M. J. M. V. Oort, Can. J. Chem., 1990, 68, 412–422 CrossRef CAS.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu, J. M. Buriak and T. Hauger, J. Phys. Chem. A, 2018, 122, 1560–1573 CrossRef CAS PubMed.
- A. Senocrate, I. Moudrakovski and J. Maier, Phys. Chem. Chem. Phys., 2018, 20, 20043–20055 RSC.
- A. Scarperi, N. Landi, A. Gabbani, N. Jarmouni, S. Borsacchi, L. Calucci, A. Pucci, E. Carignani, F. Pineider and M. Geppi, Pure Appl. Chem., 2023, 95, 1031–1042 CrossRef CAS.
- A. Mishra, M. A. Hope, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2023, 145, 978–990 CrossRef CAS PubMed.
- A. Mishra, D. J. Kubicki, A. Boziki, R. D. Chavan, M. Dankl, M. Mladenović, D. Prochowicz, C. P. Grey, U. Rothlisberger and L. Emsley, ACS Energy Lett., 2022, 7, 2745–2752 CrossRef CAS PubMed.
- A. Senocrate, I. Moudrakovski, G. Y. Kim, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2017, 56, 7755–7759 CrossRef CAS PubMed.
- P. Raval, R. M. Kennard, E. S. Vasileiadou, C. J. Dahlman, I. Spanopoulos, M. L. Chabinyc, M. Kanatzidis and G. N. Manjunatha Reddy, ACS Energy Lett., 2022, 7, 1534–1543 CrossRef CAS.
- M. A. A. Kazemi, N. Folastre, P. Raval, M. Sliwa, J. M. V. Nsanzimana, S. Golonu, A. Demortiere, J. Rousset, O. Lafon, L. Delevoye, G. N. M. Reddy and F. Sauvage, Energy Environ. Mater., 2023, 6, e12335 CrossRef CAS.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, E. M. Mozur, E. Morgan, S. Wang, D. Zheng, S. Padgaonkar, G. N. Manjunatha Reddy, E. A. Weiss, M. C. Hersam, R. Seshadri, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 7069–7080 CrossRef CAS PubMed.
- C. J. Dahlman, R. M. Kennard, P. Paluch, N. R. Venkatesan, M. L. Chabinyc and G. N. Manjunatha Reddy, Chem. Mater., 2021, 33, 642–656 CrossRef CAS.
- A. Krishna, M. A. Akhavan Kazemi, M. Sliwa, G. N. M. Reddy, L. Delevoye, O. Lafon, A. Felten, M. T. Do, S. Gottis and F. Sauvage, Adv. Funct. Mater., 2020, 30, 1909737 CrossRef CAS.
- P. Fu, M. A. Quintero, C. Welton, X. Li, B. Cucco, M. C. De Siena, J. Even, G. Volonakis, M. Kepenekian, R. Liu, C. C. Laing, V. Klepov, Y. Liu, V. P. Dravid, G. N. Manjunatha Reddy, C. Li and M. G. Kanatzidis, Chem. Mater., 2022, 34, 9685–9698 CrossRef CAS.
- P. Raval, M. A. Akhavan Kazemi, J. Ruellou, J. Trébosc, O. Lafon, L. Delevoye, F. Sauvage and G. N. Manjunatha Reddy, Chem. Mater., 2023, 35, 2904–2917 CrossRef CAS.
- L. Pan, Z. Liu, C. Welton, V. V. Klepov, J. A. Peters, M. C. De Siena, A. Benadia, I. Pandey, A. Miceli, D. Y. Chung, G. N. M. Reddy, B. W. Wessels and M. G. Kanatzidis, Adv. Mater., 2023, 35, 2211840 CrossRef CAS PubMed.
- A. Karmakar, A. M. Askar, G. M. Bernard, V. V. Terskikh, M. Ha, S. Patel, K. Shankar and V. K. Michaelis, Chem. Mater., 2018, 30, 2309–2321 CrossRef CAS.
- A. Ray, D. Maggioni, D. Baranov, Z. Dang, M. Prato, Q. A. Akkerman, L. Goldoni, E. Caneva, L. Manna and A. L. Abdelhady, Chem. Mater., 2019, 31, 7761–7769 CrossRef CAS PubMed.
- A. Kanwat, N. Yantara, Y. F. Ng, T. J. N. Hooper, P. J. S. Rana, B. Febriansyah, P. C. Harikesh, T. Salim, P. Vashishtha, S. G. Mhaisalkar and N. Mathews, ACS Energy Lett., 2020, 5, 1804–1813 CrossRef CAS.
- M. Jagadeeswararao, P. Vashishtha, T. J. N. Hooper, A. Kanwat, J. W. M. Lim, S. K. Vishwanath, N. Yantara, T. Park, T. C. Sum, D. S. Chung, S. G. Mhaisalkar and N. Mathews, J. Phys. Chem. Lett., 2021, 12, 9569–9578 CrossRef CAS PubMed.
- B. Febriansyah, T. M. Koh, P. J. S. Rana, T. J. N. Hooper, Z. Z. Ang, Y. Li, A. Bruno, M. Grätzel, J. England, S. G. Mhaisalkar and N. Mathews, ACS Energy Lett., 2020, 5, 2305–2312 CrossRef CAS.
- B. Febriansyah, Y. Lekina, J. Kaur, T. J. N. Hooper, P. C. Harikesh, T. Salim, M. H. Lim, T. M. Koh, S. Chakraborty, Z. X. Shen, N. Mathews and J. England, ACS Nano, 2021, 15, 6395–6409 CrossRef CAS PubMed.
- B. Febriansyah, Y. Li, D. Giovanni, T. Salim, T. J. N. Hooper, Y. Sim, D. Ma, S. Laxmi, Y. Lekina, T. M. Koh, Z. X. Shen, S. A. Pullarkat, T. C. Sum, S. G. Mhaisalkar, J. W. Ager and N. Mathews, Mater. Horiz., 2023, 10, 536–546 RSC.
- P. J. S. Rana, B. Febriansyah, T. M. Koh, B. T. Muhammad, T. Salim, T. J. N. Hooper, A. Kanwat, B. Ghosh, P. Kajal, J. H. Lew, Y. C. Aw, N. Yantara, A. Bruno, S. A. Pullarkat, J. W. Ager, W. L. Leong, S. G. Mhaisalkar and N. Mathews, Adv. Funct. Mater., 2022, 2113026 CrossRef CAS.
- K. Fykouras, J. Lahnsteiner, N. Leupold, P. Tinnemans, R. Moos, F. Panzer, G. A. de Wijs, M. Bokdam, H. Grüninger and A. P. M. Kentgens, J. Mater. Chem. A, 2023, 11, 4587–4597 RSC.
- B. A. Rosales, L. Men, S. D. Cady, M. P. Hanrahan, A. J. Rossini and J. Vela, Chem. Mater., 2016, 28, 6848–6859 CrossRef CAS.
- C. Roiland, G. Trippé-Allard, K. Jemli, B. Alonso, J.-C. Ameline, R. Gautier, T. Bataille, L. L. Pollès, E. Deleporte, J. Even and C. Katan, Phys. Chem. Chem. Phys., 2016, 18, 27133–27142 RSC.
- A. Karmakar, M. S. Dodd, X. Zhang, M. S. Oakley, M. Klobukowski and V. K. Michaelis, Chem. Commun., 2019, 55, 5079–5082 RSC.
- A. Karmakar, A. Bhattacharya, G. M. Bernard, A. Mar and V. K. Michaelis, ACS Mater. Lett., 2021, 261–267 CrossRef CAS.
- A. M. Askar, A. Karmakar, G. M. Bernard, M. Ha, V. V. Terskikh, B. D. Wiltshire, S. Patel, J. Fleet, K. Shankar and V. K. Michaelis, J. Phys. Chem. Lett., 2018, 9, 2671–2677 CrossRef CAS PubMed.
- S. Brochard-Garnier, M. Paris, R. Génois, Q. Han, Y. Liu, F. Massuyeau and R. Gautier, Adv. Funct. Mater., 2019, 29, 1806728 CrossRef.
- M. Aebli, B. M. Benin, K. M. McCall, V. Morad, D. Thöny, H. Grützmacher and M. V. Kovalenko, Helv. Chim. Acta, 2020, 103, e2000080 CrossRef CAS.
- J. Lee, W. Lee, K. Kang, T. Lee and S. K. Lee, Chem. Mater., 2021, 33, 370–377 CrossRef CAS.
- N. Landi, E. Maurina, D. Marongiu, A. Simbula, S. Borsacchi, L. Calucci, M. Saba, E. Carignani and M. Geppi, J. Phys. Chem. Lett., 2022, 13, 9517–9525 CrossRef CAS PubMed.
- M. A. Hope, M. Cordova, A. Mishra, U. Gunes, A. Caiazzo, K. Datta, R. A. J. Janssen and L. Emsley, Angew. Chem., 2024, 136, e202314856 CrossRef.
- M.-H. Tremblay, F. Thouin, J. Leisen, J. Bacsa, A. R. Srimath Kandada, J. M. Hoffman, M. G. Kanatzidis, A. D. Mohite, C. Silva, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2019, 141, 4521–4525 CrossRef CAS PubMed.
- O. Nazarenko, M. R. Kotyrba, S. Yakunin, M. Aebli, G. Rainò, B. M. Benin, M. Wörle and M. V. Kovalenko, J. Am. Chem. Soc., 2018, 140, 3850–3853 CrossRef CAS PubMed.
- P. Fu, M. A. Quintero, E. S. Vasileiadou, P. Raval, C. Welton, M. Kepenekian, G. Volonakis, J. Even, Y. Liu, C. Malliakas, Y. Yang, C. Laing, V. P. Dravid, G. N. M. Reddy, C. Li, E. H. Sargent and M. G. Kanatzidis, J. Am. Chem. Soc., 2023, 145, 15997–16014 CrossRef CAS PubMed.
- A. Krishna, V. Škorjanc, M. Dankl, J. Hieulle, H. Phirke, A. Singh, E. A. Alharbi, H. Zhang, F. Eickemeyer, S. M. Zakeeruddin, G. N. M. Reddy, A. Redinger, U. Rothlisberger, M. Grätzel and A. Hagfeldt, ACS Energy Lett., 2023, 8, 3604–3613 CrossRef CAS.
- R. Ji, Z. Zhang, M. Deconinck, Y. J. Hofstetter, J. Shi, F. Paulus, P. Raval, G. N. M. Reddy and Y. Vaynzof, Adv. Energy Mater., 2024, 2304126 CrossRef CAS.
- M. Aebli, L. Piveteau, O. Nazarenko, B. M. Benin, F. Krieg, R. Verel and M. V. Kovalenko, Sci. Rep., 2020, 10, 8229 CrossRef CAS PubMed.
- V. K. Sharma, R. Mukhopadhyay, A. Mohanty, M. Tyagi, J. P. Embs and D. D. Sarma, J. Phys. Chem. Lett., 2020, 11, 9669–9679 CrossRef CAS PubMed.
- S. Maity, S. Verma, L. M. Ramaniah and V. Srinivasan, Chem. Mater., 2022, 34, 10459–10469 CrossRef CAS.
- P. S. Klee, Y. Hirano, D. B. Cordes, A. M. Z. Slawin and J. L. Payne, Cryst. Growth Des., 2022, 22, 3815–3823 CrossRef CAS PubMed.
- T. Krishnamoorthy, Low-dimensional metal halide perovskite phosphors for solid-state lighting, Nanyang Technological University, 2019 Search PubMed.
- R. Chiara, M. Morana, G. Folpini, A. Olivati, B. Albini, P. Galinetto, L. Chelazzi, S. Ciattini, E. Fantechi, S. A. Serapian, A. Petrozza and L. Malavasi, J. Mater. Chem. C, 2022, 10, 12367–12376 RSC.
- M. I. Saidaminov, A. L. Abdelhady, G. Maculan and O. M. Bakr, Chem. Commun., 2015, 51, 17658–17661 RSC.
- SAINT and SADABS, Bruker AXS Inc., 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C, 2015, 71, 3–8 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Concepts Magn. Reson., 2002, 14, 326–346 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- Y. Takeoka, K. Asai, M. Rikukawa and K. Sanui, Chem. Lett., 2005, 34, 602–603 CrossRef CAS.
- Y. Wei, C. Li, Y. Li, Z. Luo, X. Wu, Y. Liu, L. Zhang, X. He, W. Wang and Z. Quan, Angew. Chem., Int. Ed., 2022, 61, e202212685 CrossRef CAS PubMed.
- J.-L. Song, W.-J. Chen, K.-B. Chu and Y.-H. Zhou, Dalton Trans., 2018, 47, 14497–14502 RSC.
- B.-B. Cui, Y. Han, B. Huang, Y. Zhao, X. Wu, L. Liu, G. Cao, Q. Du, N. Liu, W. Zou, M. Sun, L. Wang, X. Liu, J. Wang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 5190 CrossRef PubMed.
- Y. Li, G. Zheng, C. Lin and J. Lin, Solid State Sci., 2007, 9, 855–861 CrossRef CAS.
- Y. Chen, Y.-Y. Zheng, G. Wu, M. Wang, H.-Z. Chen and H. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m417 CrossRef CAS PubMed.
- M.-H. Jung, K. C. Ko and W. R. Lee, Dalton Trans., 2019, 48, 15074–15090 RSC.
- T. Hu, M. D. Smith, E. R. Dohner, M.-J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X.-Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258–2263 CrossRef CAS PubMed.
- A. Yangui, D. Garrot, J. S. Lauret, A. Lusson, G. Bouchez, E. Deleporte, S. Pillet, E. E. Bendeif, M. Castro, S. Triki, Y. Abid and K. Boukheddaden, J. Phys. Chem. C, 2015, 119, 23638–23647 CrossRef CAS.
- X. Wang, W. Meng, W. Liao, J. Wang, R.-G. Xiong and Y. Yan, J. Phys. Chem. Lett., 2019, 10, 501–506 CrossRef CAS PubMed.
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC.
- B. Febriansyah, T. Borzda, D. Cortecchia, S. Neutzner, G. Folpini, T. M. Koh, Y. Li, N. Mathews, A. Petrozza and J. England, Angew. Chem., Int. Ed., 2020, 59, 10791–10796 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
- A. Jaffe, Y. Lin, C. M. Beavers, J. Voss, W. L. Mao and H. I. Karunadasa, ACS Cent. Sci., 2016, 2, 201–209 CrossRef CAS PubMed.
- C. Li, E. J. Juarez-Perez and A. Mayoral, Chem. Commun., 2022, 58, 12164–12167 RSC.
- M. De Bastiani, I. Dursun, Y. Zhang, B. A. Alshankiti, X.-H. Miao, J. Yin, E. Yengel, E. Alarousu, B. Turedi, J. M. Almutlaq, M. I. Saidaminov, S. Mitra, I. Gereige, A. AlSaggaf, Y. Zhu, Y. Han, I. S. Roqan, J.-L. Bredas, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 7108–7113 CrossRef CAS.
- M. Rodová, J. Brožek, K. Knížek and K. Nitsch, J. Therm. Anal. Calorim., 2003, 71, 667–673 CrossRef.
- O. Dmitrenko, S. Bai and C. Dybowski, Solid State Nucl. Magn. Reson., 2008, 34, 186–190 CrossRef CAS PubMed.
- J. M. Hoffman, X. Che, S. Sidhik, X. Li, I. Hadar, J.-C. Blancon, H. Yamaguchi, M. Kepenekian, C. Katan, J. Even, C. C. Stoumpos, A. D. Mohite and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 10661–10676 CrossRef CAS PubMed.
- K. Shibuya, M. Koshimizu, F. Nishikido, H. Saito and S. Kishimoto, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1323–m1324 CrossRef CAS PubMed.
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS.
- O. Selig, A. Sadhanala, C. Müller, R. Lovrincic, Z. Chen, Y. L. A. Rezus, J. M. Frost, T. L. C. Jansen and A. A. Bakulin, J. Am. Chem. Soc., 2017, 139, 4068–4074 CrossRef CAS PubMed.
- A. A. Bakulin, O. Selig, H. J. Bakker, Y. L. A. Rezus, C. Müller, T. Glaser, R. Lovrincic, Z. Sun, Z. Chen, A. Walsh, J. M. Frost and T. L. C. Jansen, J. Phys. Chem. Lett., 2015, 6, 3663–3669 CrossRef CAS PubMed.
- T. Chen, B. J. Foley, B. Ipek, M. Tyagi, J. R. D. Copley, C. M. Brown, J. J. Choi and S.-H. Lee, Phys. Chem. Chem. Phys., 2015, 17, 31278–31286 RSC.
- A. M. A. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kockelmann, C. Law, X. Li, F. Foglia, A. Walsh, B. C. O'Regan, J. Nelson, J. T. Cabral and P. R. F. Barnes, Nat. Commun., 2015, 6, 7124 CrossRef PubMed.
- M. T. Weller, O. J. Weber, J. M. Frost and A. Walsh, J. Phys. Chem. Lett., 2015, 6, 3209–3212 CrossRef CAS.
- M. A. Carignano, Y. Saeed, S. A. Aravindh, I. S. Roqan, J. Even and C. Katan, Phys. Chem. Chem. Phys., 2016, 18, 27109–27118 RSC.
- E. M. Mozur, M. A. Hope, J. C. Trowbridge, D. M. Halat, L. L. Daemen, A. E. Maughan, T. R. Prisk, C. P. Grey and J. R. Neilson, Chem. Mater., 2020, 32, 6266–6277 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- R. G. Bryant, J. Chem. Educ., 1983, 60, 933 CrossRef CAS.
- K. Page, J. E. Siewenie, P. Quadrelli and L. Malavasi, Angew. Chem., 2016, 128, 14532–14536 CrossRef.
- A. Bernasconi and L. Malavasi, ACS Energy Lett., 2017, 2, 863–868 CrossRef CAS.
- P. Nandi, S. Mahana, E. Welter and D. Topwal, J. Phys. Chem. C, 2021, 125, 24655–24662 CrossRef CAS.
- R. J. Worhatch, H. Kim, I. P. Swainson, A. L. Yonkeu and S. J. L. Billinge, Chem. Mater., 2008, 20, 1272–1277 CrossRef CAS.
- G. Reuveni, Y. Diskin-Posner, C. Gehrmann, S. Godse, G. G. Gkikas, I. Buchine, S. Aharon, R. Korobko, C. C. Stoumpos, D. A. Egger and O. Yaffe, J. Phys. Chem. Lett., 2023, 14, 1288–1293 CrossRef CAS PubMed.
- J.-W. Lee, S. Seo, P. Nandi, H. S. Jung, N.-G. Park and H. Shin, iScience, 2021, 24, 101959 CrossRef CAS PubMed.
- A. Osherov, Y. Feldman, I. Kaplan-Ashiri, D. Cahen and G. Hodes, Chem. Mater., 2020, 32, 4223–4231 CrossRef CAS.
- J. M. Frost and A. Walsh, Acc. Chem. Res., 2016, 49, 528–535 CrossRef CAS PubMed.
- L. McGovern, M. H. Futscher, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2020, 11, 7127–7132 CrossRef CAS PubMed.
- L. Bertoluzzi, C. C. Boyd, N. Rolston, J. Xu, R. Prasanna, B. C. O'Regan and M. D. McGehee, Joule, 2020, 4, 109–127 CrossRef CAS.
- H. Xue, G. Brocks and S. Tao, Phys. Rev. Mater., 2022, 6, 055402 CrossRef CAS.
- A. M. A. Leguy, A. R. Goñi, J. M. Frost, J. Skelton, F. Brivio, X. Rodríguez-Martínez, O. J. Weber, A. Pallipurath, M. I. Alonso, M. Campoy-Quiles, M. T. Weller, J. Nelson, A. Walsh and P. R. F. Barnes, Phys. Chem. Chem. Phys., 2016, 18, 27051–27066 RSC.
- B. A. Rosales, M. P. Hanrahan, B. W. Boote, A. J. Rossini, E. A. Smith and J. Vela, ACS Energy Lett., 2017, 2, 906–914 CrossRef CAS.
- A. Abragam and A. Abragam, The Principles of Nuclear Magnetism, Oxford University Press, Oxford, New York, 1983 Search PubMed.
- D. J. Kubicki, D. Prochowicz, E. Salager, A. Rakhmatullin, C. P. Grey, L. Emsley and S. D. Stranks, J. Am. Chem. Soc., 2020, 142, 7813–7826 CrossRef CAS PubMed.
- R. R. Sharp, J. Chem. Phys., 1974, 60, 1149–1157 CrossRef CAS.
- A. Bagno, G. Casella and G. Saielli, J. Chem. Theory Comput., 2006, 2, 37–46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2300670, 2300671, 2300669, 2350304, 2300668 and 1545198. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ta02833c |
| This journal is © The Royal Society of Chemistry 2024 |