 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A perspective on the future of quantum chemical software: the example of the ORCA program package
Frank
Neese
Department of Molecular Theory and Spectroscopy, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: neese@kofo.mpg.de
First published on 4th April 2024
Abstract
The field of computational chemistry has made an impressive impact on contemporary chemical research. In order to carry out computational studies on actual systems, sophisticated software is required in form of large-scale quantum chemical program packages. Given the enormous diversity and complexity of the methods that need to be implementation in such packages, it is evident that these software pieces are very large (millions of code lines) and extremely complex. Most of the packages in widespread use by the computational chemistry community have had a development history of decades. Given the rapid progress in the hardware and a lack of resources (time, workforce, money), it is not possible to keep redesigning these program packages from scratch in order to keep up with the ever more quickly shifting hardware landscape. In this perspective, some aspects of the multitude of challenges that the developer community faces are discussed. While the task at hand – to ensure that quantum chemical program packages can keep evolving and make best use of the available hardware – is daunting, there are also new evolving opportunities. The problems and potential cures are discussed with the example of the ORCA package that has been developed in our research group.
1 Introduction
There can be little doubt that over the past several decades quantum chemistry has enjoyed tremendous success and should now be considered an integral and indispensable component of chemical research. In fact, few papers are being published these days without including the results of quantum chemical calculations. A big part of this success, and the accompanying popularity of quantum chemistry, is the fact that program packages have evolved to the point of being highly efficient and also highly user-friendly. This allows experimental chemists to make use of quantum chemistry without having to be experts in the underlying theory or experts in the advanced use or programming of computers. Calculations can usually be carried out without access to large-scale computing facilities, but rather can be performed on modest computer clusters or even, to some extent, on personal computers or laptops.Concomitant with this undeniable success, there arise many questions that surround the creation, maintenance and also the proper use of these highly complex software packages. In this commentary, I wish to discuss some of these challenges. It is important to point out that the comments made in the manuscript merely reflect the opinions of the author and are not intended in any shape or form to represent undisputable facts. The goal is rather to stir a debate that our community needs to have rather sooner than later.
This article touches on many aspects that were also discussed in a recent perspective by Di Felice et al.,1 as well as the perspective of Lehtola and co-workers on open-source software.2,3 The discussion here should be considered as complementary.
2 The evolution of quantum chemical program packages
It is probably fair to state that none of the quantum chemistry packages in large-scale use were started with a long-term master plan by a large group of individuals. There have been attempts in this direction, but they apparently met with limited success. Rather, it appears that all packages have been initiated by one person, or a very small group of individuals (often close friends). The driving force appeared to be largely scientific curiosity, or, perhaps, the intention to solve a chemical problem for which there was no other solution available.There may be a few noticeable exceptions, for example when a quantum chemical software project was started after a dispute between the developers of another, older package that led a group of people to start all over again and make use of the knowledge that had been gained in the earlier project. Since the author has no inside knowledge about specific cases in which the design was initiated in this way, further comment is deferred.
Hence, most packages that were started in this way have evolved for several (presently up to five) decades. Along the way, typically dozens if not hundreds of programmers have contributed to the project and these individuals, more likely than not, were disjointed in space and time. The code base may have grown to consist of millions of lines of code and may be spread over tens of thousands of source files. At this point no single individual can probably rightfully claim to know and understand every single detail of such a code.
It is an enormously challenging task for the team-leaders of the project to ensure that the code remains streamlined without major duplication of functionality, dead-ends or insurmountable design obstacles along the way. It is questionable whether any project can be legitimately considered as fully successful in this respect.
Importantly, when a large-scale software project is started without a detailed master plan, it is certainly subject to a vulnerable evolution. Every shortcut and every design flaw incorporated into the initial design will tend to amplify and compromise the integrity of code down the line. This is a necessary consequence of the code writing being carried out, at least up to the present day, by humans. While science, in general, intendeds to take the actual observer out of the picture to create what could be considered as “objective facts”, this is impossible or at least highly unrealistic when it comes to software design: software is written by humans and individual skills, taste, discipline and team-spirit varies by many orders of magnitude over a group of a few hundred programmers.
At this point, a few archetypes of individuals may be broadly characterized without the idea that any specific individual purely belongs to any of these categories.
The ideal developer may be characterized as a person that carefully studies the literature and underlying theory of the methods they are going to implement before starting to code. The initial implementation plan would then be discussed with the development team. The code would be written in a clean manner and kept as simple as possible, while being as complex as necessary. The code would be well-tested and well-commented in the source (in English as the smallest common denominator). There would be a comprehensive set of test-jobs created and their results would be archived such that code integrity could always be controlled. Finally, these ideal developers also write a user-friendly manual and see their project to completion, even after the initial excitement may have faded. These idealized developers also always discuss before changing, rewriting or massively extending code of other developers and stay clear of “short-term hacks”.
Unfortunately, not all developers of quantum chemical program packages, the author included, belong to this class of idealized programmers. There are several types of behaviors that render the development and maintenance of large-scale software projects challenging. Among those, some frequently encountered are:
(1) Always taking the shortest shortcut to meet one’s individual goals. This may be characterized as a “no matter how” or “crash and burn” policy.
(2) Rewriting or changing other people’s code without prior notice or consent.
(3) Never documenting their code or leaving a comment as to why something was done a certain way.
(4) Using in-depth knowledge of programming languages to “show-off” a high-level of sophistication. This is frequently combined with an unwillingness to comment the code. Frequently, this results in very difficult, if not plainly unreadable, code that tends to be not understood even by its own creator only a few months later.
(5) Being afraid to break something may lead to uncontrolled “copy-paste” approaches to development. With the best of intentions sometimes thousands of lines of code are copied and pasted only to make a few minor changes at the very end or change a few prefactors or signs.
(6) Implementing a useful improvement only in one part of the code but lacking the commitment to make the feature consistent throughout.
(7) Leaving code parts that are known to be incorrect in the code without warning or comments. This is often motivated by a lack of trust in one’s own abilities or a lack of time or commitment.
(8) Trying to be funny during development. A funny name of a routine that makes reference to, say, a certain movie character or celebrity may be considered to be funny by a given developer at a given moment in time. However, this wears off very quickly and what remains is cryptic function name that confuses later generations of developers.
The result of years of continued accumulation of these problems will be a code base that is deeply incoherent, nearly impossible to decipher and very difficult to maintain. Modern generations of students may even (and rightfully so) refuse to work with such a code. Adapting such a code to the quickly shifting hardware landscape is a daunting prospect and probably not possible with realistic effort.
In recent years artificial intelligence (AI) systems have made impressive progress and have shown some promise in the production of high-level code. Whether such AI tools, such as ChatGPT, will be able to replace humans as developers of quantum chemical software remains to be seen. It seems certain, however, that AI will at least complement human efforts in program development. A thorough discussion of the multitude of legal and ethical questions that arise from these developments is outside the scope of this article.
3 Desirable features of large-scale quantum chemistry packages
A modern quantum chemistry package typically incorporates the collective wisdom of 80+ years of research in the field as well as in the neighboring disciplines of physics, numerical mathematics and computer science. The code base may consist of several million lines of source code and may be spread over thousands of source files. In order to remain manageable, a number of requirements should be met.At this point it is important to point out that the requirements for a large, general-purpose quantum chemistry package are very different than for a purpose- and project-specific piece of software. If an individual needs to solve a specific problem in order to complete a project, then whatever is necessary to reach the finish line is adequate. This can involve mixed language programming, exotic and platform-dependent libraries, no documentation or comments in a language different from English. After the project is finished, the code is likely to disappear or is of no use anymore. It only matters to the individual who created it. On the other hand, a big package that is supposed to live for decades and that is distributed to tens of thousands of computational chemists world-wide must meet far more stringent requirements. There certainly is no consensus in the community as to what these requirements precisely are. Therefore, further comment will be deferred to the section describing our own efforts.
Perhaps the most important requirement for a big program is modularity. It is important to break down the complex workflow that characterizes modern algorithms into digestible pieces. These pieces should exist as independently of each other as possible. This means that global data should be avoided to the largest extent. Cross dependencies should be avoided, code duplication for related tasks should be avoided, object-oriented programming should be used in order to unify data with productive tasks. It will usually be necessary to break down a big package into separate executables with well-defined tasks. The more independent these “task handlers” are of each other, the more readily the whole package will be maintainable.
Modularity also comes in the form of clarity. “Clarity” in this context refers to the transparency with which a given workflow is implemented such that it can be read and understood by future generations of developers. A single 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-line source file containing one monstrous function that accepts 38 arguments is likely very hard to follow and understand. Hence, modularizing such software pieces is important.
000-line source file containing one monstrous function that accepts 38 arguments is likely very hard to follow and understand. Hence, modularizing such software pieces is important.
An equally important subject is documentation. Source code must be extensively documented and commented on in order to stay maintainable. Hence, programmers are encouraged to envision a future developer reading their code and having to add or modify something. It should be possible for this person to understand what the code is doing and how it is doing it in order to make additions and changes with confidence.
Among the many other requirements that could be mentioned, code integrity is another highly important subject. Large programs contain many input options. Not all of these options will lead to valid workflows with correct results. Since programs tends to evolve while features are being added or improved, it is important to maintain a library of reference results for implemented and supported tasks. It should regularly (and preferably automatically) be checked that the current state of the program is still able to regenerate the reference results. Without such a check, it is almost inevitable that certain functionalities in a given program will be lost over time.
In order for a large program project to remain maintainable, it should not incorporate dependencies on specific operating system constructs (e.g. reliance on shell scripts) and the dependence on external pieces of software or libraries that are outside the control of the development team should be minimized. In some cases, for example in the case of basic linear algebra (BLAS), message passing interface (MPI) or the Linear Algebra package (LAPACK) such dependencies are hardly avoidable. However, these are extremely well-established libraries that can be relied on long term. Any reliance on external pieces of software or libraries of uncertain origin or uncertain future, are a potential breaking point for the entire software project. Whether the development team wants to accept the associated risks is an important design decision that should not be made lightly.
4 Challenges associated with maintaining large quantum chemistry packages
4.1 Academic software development
Among the many challenges associated with quantum chemical program development, the circumstances under which this development takes place is probably the biggest. The overwhelming majority of this development takes place in an academic environment. In these environments there are very few permanent positions for software engineers, which means that the majority of the development is carried out by PhD students and postdoctoral fellows that are on termed contracts. Thus, these individuals are under high-pressure to produce publishable results in a relatively short amount of time.These requirements are clearly strongly conflicting with the requirements for sustainable, high-quality quantum chemical software. Such software, ideally, is well conceptualized and largely feature complete. A necessary consequence is that a fair amount of time in quantum chemical software development is spent “re-inventing the wheel”, e.g. implementing methods and features that were previously already available in other program packages. Such a re-invention may sometimes be involved with additional innovation in which case the results are publishable. A simple re-implementation of a given method, even if it takes a substantial amount of time and effort, is only marginally justifiable as an addition to the scientific literature.
To create software that is feature complete, well-tested, highly optimized and well-documented is very difficult, if not impossible under such circumstances. What the community would require is a far larger number of software-oriented quantum chemists on permanent contracts that ensure the continuity and code quality of large-scale program packages.
4.2 The legacy curse
Quantum chemistry is a fast-progressing field. While a few decades ago, the state of the art consisted basically of a closed-shell Hartree–Fock calculation and perhaps a geometry optimization, the array of available methods and properties together with sophisticated analysis tools that can be approached with quantum chemistry now is bewildering. Thus, it is very difficult to see how a single young researcher or a small team could possibly start all over again from scratch to generate the next generation quantum chemistry software. It would probably take at least one decade of highly concentrated work to arrive at a package that would be state-of-the-art today.Finding financial support for such an endeavor, that initially creates nearly no new research, seems to be challenging if not impossible. Since the cycles of performance evaluation by funding agencies or universities tend to get shorter and shorter and future funding depends on being successful in the previous funding period, it seems unlikely that another major quantum chemistry package could emerge from an empty piece of paper anytime soon – if ever again.
What does that mean for future developers? A lot of future development will probably focus on specific tasks. For example, writing a program that only does approximate full-CI calculations, say, together with relativistic corrections to calculate some properties can probably be approached from scratch. This is something that can, will and should be done. However, those researchers who are interested in creating a full featured package for general purpose use by a large audience will probably remain tied to one of the existing major packages. This begs the question of how well-suited these existing packages are to adapt themselves to the quickly evolving hardware landscape? The answer is probably: in general, not very well.
The history of quantum chemistry speaks a clear language: there have been many ingenious methods that were implemented on a specific hardware, say the CRAY or the CYBER 205. After these hardware platforms became hopelessly outdated, these programs disappeared and the hard- and dedicated work that led to the programs in question was lost to the community. Given the difficulty to secure funding, expertise (and also enthusiasm) to re-create existing quantum chemical methods, the community simply cannot afford let history repeat itself and accept the losses that arise from our programs not keeping up with the hardware anymore. Thus bringing the established quantum chemical machinery onto the computers of the future in a way that is efficient and clean, is certainly one of the major challenges that our field faces.
4.3 Leadership
It is obviously very important how project leadership is implemented. Leadership could rest on the shoulders of a single individual or, more commonly, on a small leadership team. It is important that leadership clearly communicates development guidelines and expectations on quality standards as well as procedures to the development team. Regular team-meetings are required to inform developers about emerging new infra-structure or changing policies. Shared “delocalized” responsibility that appeals to the discipline of individual developers is usually not a successful model since it tends to trigger no- or arbitrary actions. Whether open-source projects provide a successful avenue towards reliable, efficient large-scale quantum chemistry program packages (as opposed to open-source libraries discussed below) remains to be seen.Quantum chemical program packages are frequently very closely associated with the names and personalities of the original inventors. This is a necessary consequence of the way that these packages evolve. The question is what happens when these inventors eventually leave the scientific arena? It clearly is a very tall order for a young and upcoming researcher to make the life’s work of another person the central projects of their own careers. While the willingness to “carry on the torch” is commendable, it may be difficult for young researchers to convince the community of their own and independent scientific standing if they “follow the line of succession”. The result is that a number of significant, influential and highly successful program packages have already disappeared and more are likely to disappear in the foreseeable future. Thus, finding a solution to the problem of continued career-spanning program development is an important topic for the future of the quantum chemistry community.
4.4 Community and inter-operability
No matter how large or dedicated a research group is, it will not be possible for a single quantum chemical package to combine all the know-how and achievements of the community in one place. Thus, it is necessary and also desirable that there remains a multitude of software solutions for computational chemists to choose from. It is also not necessary for the progress of the field that every package is as feature-complete as possible. Rather, having specialized programs for specialized tasks appears to be perfectly acceptable.However, in order to guarantee efficient workflows it would be highly desirable for programs to be more interoperable than they currently are. Unfortunately, this would seem to imply that the developer community agrees on certain data formats that are used to exchange data or other information. To the best of this author’s knowledge, all attempts in that direction have failed so far. It would still be desirable for the community to make a joint-effort towards more readily exchangeable data.
Of course, as long as inter-operability means exchanging coordinates and orbitals and similar quantities that are not time-critical, there is no problem as long as a well-defined, stable and well-documented interface exists. Unfortunately, not even this criterion appears to be met by most existing packages. The need for a general, well-designed, well-documented and well-maintained interface can hardly be overemphasized. Our own experience is that custom-made interfaces to other programs are always vulnerable, short-lived and unreliable. Hence, the efforts should not be directed towards “patching something together” that just works for a short moment in time, but to create a more general interface solution. Our own efforts within the ORCA project are discussed below.
The next level of inter-operability would result from using common libraries. If these libraries are well-designed, they have an easy-to-use programming interface and make no demands on specific operating features or create new dependencies on other external libraries. Such libraries could be created in a highly task specific way, for example, the calculation of integrals, the evaluation of density functionals and their functional derivatives, the numerical integration in density functional theory, the solution of large-scale linear equations or eigen-equations, the optimization of molecular structures etc. Very successful examples of this kind are the libint4 and libcint5 integral libraries, the LibXC6 and XCFun7 DFT libraries, the IntegratorXX8 numerical integration library, among many others. Such efforts should definitely be applauded as they represent an invaluable asset to the community and save humungous amounts of development effort. In order for such libraries to be successful, it is important that they do not depend on complicated data structures that require the host program to deeply incorporate these data structures, or even other external libraries (such as, e.g., matrix/vector/tensor libraries) in its own design. This requirement certainly limits the number and nature of tasks that can be “library-field”. Also, the language in which the libraries are written is an important aspect as mixed language programs tend to be more vulnerable than programs that are written in established, stable and truly platform independent languages such as C/C++ or Fortran.
Given that open-source software faces certain challenges (briefly discussed below) that may prevent some developers to making their libraries open-source, there might be a medium ground in which libraries are provided in binary form with a documented Application Program Interface (API). Such libraries can be linked to other existing programs in binary form. Of course, this requires these libraries to be available on a multitude of platforms and operating systems. This avenue seems to remain largely unexplored to date.
4.5 Community and funding aspects
One aspect that can certainly not be ignored in the context of a community effort is money – in the form of research funding, as well as in terms of commercialization. Quantum chemistry went commercial probably around the 1980s. The pros and cons of commercial versus free versus open-source software is a complex subject in itself that is outside the scope of this article. It is self-evident, however, that commercialization does not help the case for open-science as software companies, understandably, want to protect their intellectual property. However, as far as publicly funded, basic research is concerned, the algorithms developed must be available in full in the open scientific literature. This is a non-negotiable requirement for science that should not be violated. Whether this requires the actual source code to be available or not is a complex question with far-reaching legal and ethical ramifications that are outside the scope of the present discussion.The other aspect that limits a more widespread exchange within the scientific community is the competition for research funding and the way that the merit generation system in science works. Both are closely related to citation statistics and, at least subconsciously, also to publication impact factors. Thus, a person that spends years designing, debugging, optimizing and documenting a fantastic library may generate a single paper in a low- to medium impact factor journal. A person taking that library, plugging it into their workflow and running some calculations on “hot en vogue” systems may be able, as a matter of only a few weeks, to generate far more (on paper) impact that then a method developer could in a lifetime. It is then customary to cite the inventor of a given functional and the creator of a given basis set but to not even provide any reference to the people who’s algorithmic development efforts have enabled their study in the first place. It is clear that this system of scientific merit generation is strongly disfavoring long, tedious and complex development tasks. If, as a community, we want to maintain development at a high-level, it will be necessary to create some “safe space” where developers are “protected” from unfair treatment by the scientific merit generation machinery.
5 The case of the ORCA package
In order to make the foregoing discussion more concrete, I will now use the ORCA package as an example and will provide some hints towards the answers that the ORCA team has been pursuing in response to the challenges discussed above. The ORCA program package is free-of-charge for academic researchers (nearly 70000 registered users world-wide turn ORCA into the second-most used quantum chemistry program) and is commercially distributed by the company FAccTs to industrial users. The evolution of the ORCA package is probably a rather typical example of a development that was never intended to turn into a large-scale, general-purpose toolbox. Rather the cradle version of ORCA resulted as a byproduct of my PhD thesis in order to solve a very specific problem in metalloprotein biochemistry.9,10 Space does not permit a description of the program’s evolution. The only thing that should be mentioned is that ORCA underwent a complete rewrite in the years following the release of ORCA 5 (ref. 11) for all the reasons stated in Section 2. That this was possible is a luxury of secure positions and generous base level funding and is therefore not a generalizable strategy.
5.1 Performance progress: driven by software or by hardware?
During rewriting the ORCA code, we stumbled upon an old calculation on one the guinea pigs that was consistently used as a test case: the molecule Vancomycin with 176 atoms. Back in 2003, I had presented a calculation in a meeting of the group of the late Walter Thiel, where I was proud to show that the calculation could finish within 1 hour of elapsed time. Today, the same calculation takes about 1 minute on a contemporary laptop computer. The question arose as to how much of this improvement is due to improved hardware and how much is due to the algorithmic progress made over the course of two decades. In order to address this question, ORCA versions between 2005 and 2023 were taken from the source code repository (Fig. 1). It was pleasing to see that the old code could be compiled, linked with modern libraries and executed without any problems on present day computers – something that would not possible with many other codes or less stable languages (such as Python). | ||
| Fig. 1 Algorithmic speedup over time relative to the early ORCA versions of 2005. The respective versions were all compiled with the same compiler, linked against the same libraries and run on the same computer. The test molecule was Vancomyin with 176 atoms. Left: def2-SVP basis set, right: def2-TZVP basis set. | ||
Since the increase in clock speed of a single processor is well known, we could estimate the algorithmic progress by comparing the timings between the old code and the present code by compiling and running them on the same modern computer under identical conditions. The results very clearly demonstrated that the algorithmic progress led to a speedup approaching 20 for small split-valence basis sets (def2-SVP) while a speedup approaching a factor of 200 was achieved for larger basis sets (def2-TZVP). The hardware related speedup is estimated to be only a factor of seven. While this comparison is not without flaws, it still clearly demonstrates that the algorithmic progress largely outweighs the performance increase due to the hardware. It is not claimed that this is a general and generalizable conclusion. Rather, the result serves to show that investment of time in effort to create better algorithms is well invested and, in many cases, will render the use of super-computing facilities unnecessary.
5.2 Organization of low-level tasks: the case of SHARK
The SHARK integral algorithm is based on the simple observation that the McMurchie–Davidson integral algorithm can be written in terms of matrix multiplications.| (μν|κτ) = (EbraRbra,ketEket)μν,κτ | (1) |
The E-coefficients transform between the spherical harmonics Gaussian basis and the Hermite Gaussian basis and the R-integrals represent the repulsion integrals over pairs of Hermite Gaussian functions. The algorithm is readily extended to different integral kernels such as spin–orbit integrals, spin–spin integrals, range separation or F12-integrals among many others.12 The SHARK algorithm is not only very simple but also highly performant since it relies on the most efficient operations that a modern computer can carry out: matrix multiplications. Given that every present and future computer will be delivered together with a highly optimized BLAS library, the performance of this integral package appears to be guaranteed long-term.
However, a perhaps even more important aspect of the development has been the use of virtual functions for the digestion of the integrals. Many quantum chemical codes rely on loops over shell-pairs, triples or quadruples of the kind shown in Scheme 1.
 | ||
| Scheme 1 Pseudocode showing a typical loop structure to generate batches of two-electron integrals. | ||
Loops of this kind are then repeated over and over again throughout the program for all kinds of tasks such as calculation of Fock matrices, response matrices, direct CI residuals, gradients, Hessians, integral transformations or alternative integral kernels, to name only a few. If an innovation appears that requires changes, for example, more modern pre-screening algorithms,13–17 the changes would need to be implemented in all places that contain such integral loops. Quite typically, the team may have enough resources to implement the innovation in a few places but will leave less frequented parts of the code untouched. This eventually leads to a large functional incoherence of the code that will contribute to its decline.
In order to avoid this trap, the revamped ORCA code only has a single place where integrals are actually generated, e.g. a function called IntegralLoop. This function receives as an argument an object called IntegralConsumer whose task it is to take a batch of integrals and perform some action on it as defined by a virtual function DigestIntegrals. Thus all that is required to trigger the sophisticated integral machinery is to write one small consumer function consisting of a few lines of code and then call the IntegralLoop function that provides a highly optimized way to generate all kinds of integrals over different kernels. We refer to this as the “Loop-Kernel-Consumer” (LKC) concept.
This architecture was, for example, crucial in bringing point group symmetry into ORCA. Traditionally, it was a program made for calculations on biological molecules where there is no symmetry. With the demands on a general-purpose program, it became evident that the proper handling of symmetry via a “petite list” approach is desirable. Using the LKC concept, this the implementation of symmetry was only necessary in a single place in the code in order to be available everywhere in the code. This is a good example of how unnecessary and counter-productive code duplication can be avoided.
5.3 Organization of mid-level tasks: the case of molecular properties
Over time, many different electronic structure methods were implemented in ORCA. Starting from the Hartree–Fock and DFT modules, methods such as MP2, coupled cluster theory with singles, doubles and perturbative triples (CCSD(T)), their domain-based local pair-natural orbital (DLPNO) counterparts, complete active space self-consistent field (CASSCF), multi-reference configuration interaction (MR-CI), N-electron valence perturbation theory (NEVPT2) among many others were added. Each of these methods is typically implemented in its own module that is independent of the other modules and only shares a “Tools” library that is global to ORCA.When it comes to the calculation of molecular properties, it is possible to express a first-order response property in terms of energy derivatives (response to a perturbation λ):18
 | (2) |
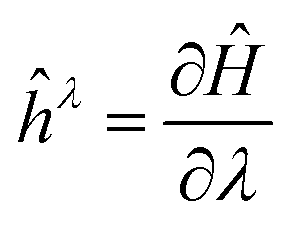 is the first-derivative of the Hamiltonian operator with respect to the perturbation. Likewise, a second order property can be written as:18
is the first-derivative of the Hamiltonian operator with respect to the perturbation. Likewise, a second order property can be written as:18 | (3) |
Traditionally, each module dealt with these properties separately. This led to a large amount of incoherence in the code where the feature set available for different electronic structure methods varied significantly. In addition, a large amount of overhead and redundancy was created by each module calculating and re-calculating the property integrals. In addition, relativistic corrections, such as picture change effects or finite nuclei treatments were handled incoherently by the legacy code.
In the workflow of the re-written code, the commonalities between the property treatments are fully explored. To this end, there is common property integral “container” that collects the integrals required for all property calculations downstream. If there are relativistic picture change effects to be taken into account, they can be incorporated at this point. The electronic structure methods are next triggered in order to create the density and response density electron and spin densities and store them in a “density container”. All that is required now is that a single, common property program (called orca_prop) picks up the property integrals and (response) densities and evaluates the first- and second-order properties. This happens automatically for all “eligible” densities. Thus, if a single run of the program produced Hartree–Fock, MP2 and CCSD(T) densities, then the property program will evaluate and print, say, the requested NMR shielding tensors for all of these methods without any creation of overhead.
SCF response properties are of particular importance since they are the by far most requested in actual application studies. In order to streamline the generation of SCF response densities, a separate module was created called orca_scfresp. This module categorizes all requested response properties into the categories “real”, “imaginary” and “triplet” perturbations. For each category all right hand sides for the coupled-perturbed SCF equations are collected together and treated simultaneously. Thus any overhead relating to the repeated solution of response equations is completely avoided and all response densities become available simultaneously. For example, the nuclear and electric field perturbations both represent “real” perturbations and are treated simultaneously. This allows for the nuclear Hessians, polarizabilities and Raman intensities to be handled simultaneously and without overhead. Likewise, nuclear magnetic perturbations are pooled and treated simultaneously for NMR spin–spin or EPR second-order hyperfine calculations with very large computational benefits over solving for each nuclear perturbation separately.
The generated properties are all stored in the “property” file, which is a fixed-format human as well as machine readable file that is a result of an ORCA run. This file can be used by post-processing tools such as, say, spectra simulation programs in order to generate further analysis.
Excited state properties and properties that do not result from a response treatment (for example properties resulting from quasi-degenerate perturbation theory treatments) are a bit more complicated to handle in a streamlined fashion. However, this has also been accomplished as part of the code redesign.
5.4 Information flow and shell structure design
Taken together, the streamlining of the integral and property codes led to a very large compaction of the of the code base and the near-complete removal of any redundant code. At the same time, the infra-structure became very robust and highly reliable since multiple tasks all run through the same, highly-optimized and well-tested code. The final layer of organization now concerns the separation of organizational and computational tasks. To this end, the revamped code uses a “shell” structure (Fig. 2).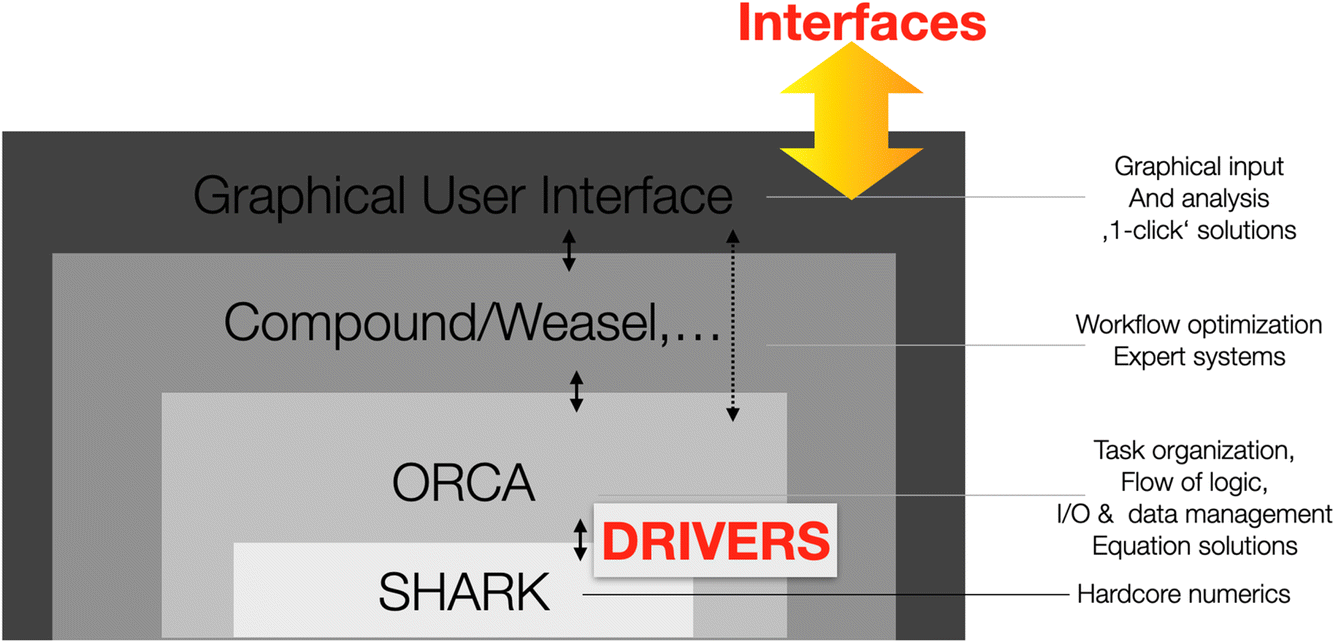 | ||
| Fig. 2 Illustration of the shell structure of the redesigned ORCA program package. | ||
The outer shell consists of the core of the original and now streamlined ORCA code. Its main task is to interact with the user and to organize the overall flow of the computation. Nearly all hard-core computational tasks are handled in the innermost shell by the SHARK interface which is largely independent of the remaining ORCA code base. The missing link between the two shells is provided by so-called “DRIVERS”. These drivers are called from within ORCA, pass on the necessary information to SHARK and calls the appropriate SHARK functions. Importantly, this is the place where all the various integral approximations that can be chosen by the user are differentiated. Thus thousands and thousands of lines of legacy code that simply dealt with figuring out whether, say, the chain-of-spheres (COSX) and Split-RI-J approximations are to be used or not and if yes, how they are going to be used, are eliminated. What remains is a very straightforward call from within ORCA to a given task driver which will then ensure that the necessary tasks are properly executed. For example, the DRIVER for the construction of a Fock matrix will differentiate between Hartree–Fock and DFT and then between RHF, UHF, ROHF and CASSCF, between exact four-index or approximated integrals, within approximated integrals between the RIJCOSX, RI-JK (both Coulomb and exchange handled by the RI approximation) and RI-DX (Coulomb handled by RI, exchange by exact integrals). It will know when to add solvation terms and within solvation differentiate between CPCM, SMD or RISM. The driver also understands when to add external point charge fields or add relativistic corrections.
By structuring the code in this way, the main ORCA code can fully concentrate on setting up and organizing the various computational tasks at hand, the drivers take care of the nitty-gritty logic required to orchestrate the calculations and the SHARK infrastructure executes the hard-core numerical tasks. We believe that this infrastructure is highly streamlined, transparent and reliable and is a solid basis for future development.
5.5 Dealing with complex methods: automatic code generation
The intellectual complexity of quantum chemical methods has been steadily increasing. In particular, in the field of wavefunction theory, one eventually hits a wall of complexity that is beyond the capacity of the human brain. Implementing these high-level ab initio electronic structure theories by hand is a tedious, lengthy and error prone task. At the same time, there is very little merit in yet another implementation of the CCSD(T) method since it has been done so many times. Thus, despite the fact that the effort for the implementation is very high and may take years to accomplish, there is little scientific merit to be gained from these efforts.To ensure that high-level ab initio methods become available in a given package together with their gradients and response properties and lead to code that is maintainable and could be extended to future hardware platforms without years of re-writing effort, we look to the automatic generation of code. This is a complex subject with a long history and hence, only a few aspects will be briefly touched upon that are most relevant in the context of sustainable code development.
Many code generators have been developed over the years, each one with a specific focus and each with a specific set of strengths and weaknesses. Code that has been generated with these code generators may live inside a given host program. However, these code generators tend to be separate from their host programs and once the developer of the generator moves on to a different career, these code generators tend to get lost. This means that if the need arises to change, adapt or extend the generated code, one finds oneself in the position of being unable to do so which eventually means that the generated code and the efforts that went into it, are lost.
In our efforts in the field of automatic code generation, we have insisted on a concept of “deep integration”.24,25 The underlying vision is the following: we envision that in the future, developers only check in the wavefunction Ansatz into the code repository. This Ansatz is then automatically translated into equations, the equations are automatically factorized and the factorized equations are automatically translated into high-level (or even machine level) code. This code is then automatically placed in the appropriate place within a given host program where it is triggered by a, presumably hand-written, wrapper that drives the desired computational task (for example, the generation of a residual vector of some kind). The code generation chain (equation generator, factorizer and code generator) are themselves part of the source repository. Organizing the sequence of actions in the indicated way, ensures that whenever an improvement is made to the generation chain, it will be immediately applied to all generated code. This ensures a perfect coherence of all generated code. Since new hardware only requires an extension of the last step of the chain – the code generator – the concept allows developers to react very quickly and efficiently to future hardware developments.
In our opinion, a development like this is not only desirable but also necessary in order to ensure that high-level wavefunction theories of incredible complexity can not only be implemented in an efficient and reliable manner, but also remain maintainable and adaptable to the quickly shifting hardware landscape. For example, using this concept, we have been able to generate the hierarchy of closed- and open-shell coupled cluster methods together with nuclear gradients as well as their densities and response densities as well as methods like internally contracted multi-reference coupled cluster theory (FIC-MRCC), the implementation of which would be impossible without the use of automatic code generation. Importantly, the code generation chain has been optimized to the point that the generated code, where comparison is possible, performs very similarly to the best hand-written codes that perform the same task. The development of an optimized, coherent and general code generation tool required many years of effort and was only possible because of the existence of generous base level funding in a way that is scarcely available in our community.
5.6 Organization of high-level tasks: compound scripting language
A typical computational chemistry study does not consist of a single or a few calculations, but often of the series of calculations. In addition, computational chemists may use a combination of methods in order to obtain the best possible results in the shortest amount of time. For example, one may want to generate a geometry optimization and frequencies together with thermochemistry by an efficient DFT method, then compute an accurate single point energy using a DLPNO-CCSD(T) calculation and estimate core-correlation effects with DLPNO-MP2, then extrapolate all of this to the PNO and basis sets limits and finally add a correction for solvent effects. This is clearly a multi-step procedure that requires a significant number of individual steps that need to be repeated for each species one might be interested in over the course of the study. Implementing such a workflow is tedious and error prone. Of course, one could resort to shell-scripts to execute such a workflow. However, this then involves some parsing of the output files and collection of information that might not be straightforward to obtain from the electronic structure host program.Of course, the host program could implement complex workflows, such as the indicated one, by hard-cording it into the host programs feature set. However, this would severely limit the creativity of the computational chemist who wants to invent their own workflows and treats them according to their needs and preferences.
In order to organize such high-level computational tasks in an efficient and user-friendly manner, we have created the “compound” functionality. In a nutshell, the compound functionality can be thought of as a shell scripting language with deep integration of ORCA functions and ORCA data structures. Using the compound functionality one can drive jobs like:
(1) Multi-step protocols such as the one indicated above.
(2) Sequences of identical calculations on a series of molecules.
(3) Sequences of calculations stepping through a range of input options or parameters.
(4) Read and process any property created by ORCA from the property file.
(5) Read, write and manipulate molecular structures.
(6) Execute system commands.
(7) Do custom analysis and create customized printouts.
(8) Use programming language constructs such as “for”, “while”, “if” … “else”, “goto”.
(9) …
The compound functionality can be written directly into an ORCA input file alongside other ORCA commands or “compound scripts” can be saved into a common directory and triggered from an ORCA input file. In the latter case, it is possible to assign some variable inside the compound script from the ORCA input file.
For example, a compound script that explores a range of approximate electronic structure methods could look like this (Scheme 2).
 | ||
| Scheme 2 An example for an ORCA compound script that implements the comparison of several quantum chemical approximations. | ||
When it is called from an ORCA input, one could do the following (Scheme 3).
 | ||
| Scheme 3 An example how to trigger an ORCA compound script from the ORCA input. | ||
Clearly, the applications that can be realized with a powerful scripting language are near endless and they can lead to highly streamlined workflows. For example, an entire research group could put down their preferred workflows for different tasks into a series compound script that is then triggered by every researcher individually in their specific applications.
5.7 Inter-operability: property file, JSON interface, plugins
No single program package can ever hope to incorporate all features and methods that a user may need or desires. Hence, it is important that there is a certain amount of inter-operability between different programs. The challenges associated with a lack of standardized data formats were discussed above. In addition, at least for the foreseeable future, there will be no universally accepted and implemented open-source policy for quantum chemical programs. Even if there was one, programs are not automatically inter-operable. Rather complex operations and changes to the respective codes would be necessary that the majority of users that desire inter-operability of different codes neither could nor would want to make themselves.The first layer of interfacing ORCA to outside programs is provided by the property file alluded to in Section 5.3. The property file is a concise summary of the calculation results during a given ORCA run. The file is written in a fixed ASCII format and is designed to be human as well as machine readable. This file is provided as a longer-term stable interface that allows access to calculation results without parsing the output file, the format of which may not only be long but is also subject to change without prior notice. At this point in time over 200 different properties can be accessed including coordinates, various energies, population analysis results, and excitation energies among many other properties.
In addition, the upcoming ORCA version will feature a somewhat detailed interface that uses the Java Script Object Notation (JSON)26 data format. We have chosen the JSON – not for its simplicity or beauty – but for the fact that it is widely used and accepted by a generation of younger researchers and computer scientists. Using JSON, one also obtains files that are human as well as machine readable.
The interface is capable of doing the following:
(1) Provide the entire contents of the property file in JSON format.
(2) Output coordinates, basis sets.
(3) Output molecular orbital coefficients.
(4) Output CIS/RPA/TD-DFT amplitudes.
(5) Output various one- and two-electron integrals.
(6) Run in reverse and let the user create GBW files from JSON files.
In order to exchange integrals and MO coefficients a detailed understanding of ORCA orders and normalized basis functions is required. This subject is explained in detail in the accompanying manual.
Clearly, an interface like this is somewhat limited to types of data sets that do not get overwhelmingly large. For example, while the interface is able to provide two-electron integrals, the size of the files that holds these integrals quickly becomes too large to allow driving calculations on large molecules. For the same reason, coupled cluster amplitudes are not offered. Nevertheless, the ability to use ORCA as an integral generator may prove to be very useful for other developers that want to test their own codes.
In addition to this data exchange, we have also experimented with the concept of a “plugin”. A plugin would be an external, user-provided program that can be called at a strategic place from within ORCA. For example, to make a new Fock matrix using a method that ORCA does not have or provide a new set of orbitals using the solver that ORCA does not have. Time will tell, whether this concept is successful and will be embraced by the community.
6 Discussion
In this article, some of the challenges associated with the development of large-scale, general-purpose quantum chemical program packages were discussed. Possible solutions or partial solutions to some of the challenges were discussed in the context of the ORCA program package that underwent a major re-design and re-write 25 years into its existence.It appears to be clear that it will be a major challenge for our community to ensure that the software packages that drive the success of computational chemistry remain strong, healthy and well-adapted to the rapidly shifting hardware landscape. To keep such large projects, that necessarily involve millions of lines of source-code, up to date and streamlined, while largely having to rely on academic developers is clearly a very tall order, especially while facing the realities of competitive research funding. Whether the now quickly evolving AI tools and/or an open-source policies alone are sufficient to reach this ambitious goal, remains to be seen. After all, to keep a large project coherent over extended periods of time, is not a self-organizing process but requires some form of leadership and agreed-upon policies. The track record of the quantum chemistry community in agreeing on standards or data formats is not overly impressive. Rather localized, individual solutions exist for these problems with no consensus presently in sight. What appears to be more realistic is that the most repetitive of tasks are being implemented in generally available, possibly open-source, libraries that greatly benefit the development of future quantum chemistry packages.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
In this article, I have spoken openly about how I perceive the challenges associated with quantum chemical software development. It has been written with the greatest amount of affection for our discipline. I am deeply indebted to my students, collaborators and long-term co-workers that have shared my passion for and my vision of quantum chemical software. I am also deeply indebted to the Max Planck Society and the German Science Foundation for their continued support of basic science with no strings attached. The author grateful acknowledges financial support of the Max Planck Society as well as the German Science Foundation in the framework of the special research unit 1639 (“Numeriqs”) that is installed at the University of Bonn, Germany.References
- R. Di Felice, M. L. Mayes, R. M. Richard, D. B. Williams-Young, G. K. L. Chan, W. A. de Jong, N. Govind, M. Head-Gordon, M. R. Hermes, K. Kowalski, X. S. Li, H. Lischka, K. T. Mueller, E. Mutlu, A. M. N. Niklasson, M. R. Pederson, B. Peng, R. Shepard, E. F. Valeev, M. van Schilfgaarde, B. Vlaisavljevich, T. L. Windus, S. S. Xantheas, X. Zhang and P. M. Zimmerman, J. Chem. Theory Comput., 2023, 19, 7056–7076 CrossRef CAS PubMed.
- S. Lehtola, J. Chem. Phys., 2023, 159, 180901 CrossRef CAS PubMed.
- S. Lehtola and A. J. Karttunen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1610 CAS.
- E. F. Valeev, 2024, http://libint.valeyev.net/.
- Q. M. Sun, J. Comput. Chem., 2015, 36, 1664–1671 CrossRef CAS PubMed.
- S. Lehtola, C. Steigemann, M. J. T. Oliveira and M. A. L. Marques, Softwarex, 2018, 7, 1–5 CrossRef.
- U. Ekström, XCFun: A Library of Exchange-Correlation Functionals with Arbitrary-Order Derivatives Search PubMed.
- S. Lehtola and M. A. L. Marques, J. Chem. Phys., 2022, 157, 174114 CrossRef CAS PubMed.
- J. A. Farrar, F. Neese, P. Lappalainen, P. M. H. Kroneck, M. Saraste, W. G. Zumft and A. J. Thomson, J. Am. Chem. Soc., 1996, 118, 11501–11514 CrossRef CAS.
- F. Neese, W. G. Zumft, W. A. Antholine and P. M. H. Kroneck, J. Am. Chem. Soc., 1996, 118, 8692–8699 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Neese, J. Comput. Chem., 2023, 44(3), 381–396 CrossRef CAS PubMed.
- D. S. Lambrecht, B. Doser and C. Ochsenfeld, J. Chem. Phys., 2005, 123, 184101 CrossRef PubMed.
- S. A. Maurer, D. S. Lambrecht, J. Kussmann and C. Ochsenfeld, J. Chem. Phys., 2013, 138, 014101 CrossRef PubMed.
- T. H. Thompson and C. Ochsenfeld, J. Chem. Phys., 2019, 150, 044101 CrossRef PubMed.
- D. S. Hollman, H. F. Schaefer and E. F. Valeev, J. Chem. Phys., 2015, 142, 154106 CrossRef PubMed.
- E. F. Valeev and T. Shiozaki, J. Chem. Phys., 2020, 153, 097101 CrossRef CAS PubMed.
- J. Gauss, in Modern Methods and Algorithms in Quantum Chemistry, ed. J. Grotendorst, John von Neumann Institute for Computing, Jülich, 2000, vol. 3, pp. 1–52, NIC Series Search PubMed.
- F. Neese, Chem. Phys. Lett., 2000, 325, 93–98 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2003, 118, 3939 CrossRef CAS.
- F. Neese, J. Am. Chem. Soc., 2006, 128, 10213–10222 CrossRef CAS PubMed.
- F. Neese, Biol. Mag. Res., ed. G. Hanson, 2009, vol. 28, pp. 175–232 Search PubMed.
- F. Neese, in Multifrequency Electron Paramagnetic Resonance. Theory and Applications, ed. S. Misra, Wiley VCH, Weinheim, 2011, pp. 297–326 Search PubMed.
- M. Krupicka, K. Sivalingam, L. Huntington, A. A. Auer and F. Neese, J. Comput. Chem., 2017, 38, 1853–1868 CrossRef CAS PubMed.
- M. H. Lechner, A. Papadopoulos, K. Sivalingam, A. A. Auer, A. Koslowski, U. Becker, F. Wennmohs and F. Neese, Phys. Chem. Chem. Phys., 2024, 26, 15205–15220 RSC.
- F. Pezoa, J. L. Reutter, F. Suarez, M. Ugarte and D. Vrgoc, Foundations of JSON Schema, WWW '16: Proceedings of the 25th International Conference on World Wide Web, ACM, Montreal, CANADA, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |