
Exploring the interaction between a fluorescent Ag(I)-biscarbene complex and non-canonical DNA structures: a multi-technique investigation†
Francesca
Binacchi‡
 *a,
Ester
Giorgi‡
*a,
Giacomo
Salvadori‡
*a,
Damiano
Cirri
a,
Mariassunta
Stifano
a,
Aurora
Donati
a,
Linda
Garzella
a,
Natalia
Busto
b,
Begona
Garcia
c,
Alessandro
Pratesi
a and
Tarita
Biver
a
*a,
Ester
Giorgi‡
*a,
Giacomo
Salvadori‡
*a,
Damiano
Cirri
a,
Mariassunta
Stifano
a,
Aurora
Donati
a,
Linda
Garzella
a,
Natalia
Busto
b,
Begona
Garcia
c,
Alessandro
Pratesi
a and
Tarita
Biver
a
aDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy. E-mail: francesca.binacchi@dcci.unipi.it; ester.giorgi@phd.unipi.it; giacomo.salvadori@phd.unipi.it
bDepartamento de Ciencias de la Salud, Universidad de Burgos, Paseo de los Comendadores s/n, 09001 Burgos, Spain
cDepartamento de Química, Universidad de Burgos, Plaza Misael Bañuelos s/n, 09001 Burgos, Spain
First published on 10th May 2024
Abstract
Silver compounds are mainly studied as antimicrobial agents, but they also have anticancer properties, with the latter, in some cases, being better than their gold counterparts. Herein, we analyse the first example of a new Ag(I)-biscarbene that can bind non-canonical structures of DNA, more precisely G-quadruplexes (G4), with different binding signatures depending on the type of G4. Moreover, we show that this Ag-based carbene binds the i-motif DNA structure. Alternatively, its Au(I) counterpart, which was investigated for comparison, stabilises mitochondrial G4. Theoretical in silico studies elucidated the details of different binding modes depending on the geometry of G4. The two complexes showed increased cytotoxic activity compared to cisplatin, overcoming its resistance in ovarian cancer. The binding of these new drug candidates with other relevant biosubstrates was studied to afford a more complete picture of their possible targets. In particular, the Ag(I) complex preferentially binds DNA structures over RNA structures, with higher binding constants for the non-canonical nucleic acids with respect to natural calf thymus DNA. Regarding possible protein targets, its interaction with the albumin model protein BSA was also tested.
Introduction
N-heterocyclic carbenes (NHCs) are potent tools demonstrating application in both catalysis and coordination chemistry with p-block elements and transition metals.1 The latter has facilitated the use of NHCs in the pharmaceutical environment with antimicrobial and anticancer applications. Among the different metal options, silver(I) and gold(I) NHC complexes have received significant interest. Silver compounds are usually studied as antimicrobial agents,2 but some candidates, such as the monocarbene studied by Citta and co-workers, demonstrate promising properties as anticancer drugs surpassing their gold analogues.3 The study by Streciwilk and co-workers also indicated the efficacy of an Ag(I)–NHC against Gram-negative bacterial strains.4 The interest in NHCs started with the discovery of the antitumor ability of the gold complex auranofin.5 This drug, which was originally used to treat rheumatoid arthritis, completed different clinical trials in the United States as an anticancer agent, although, to date, none of the trials continued after phase II and some were withdrawn.6 The active moiety was found to be the one with a gold metal centre coordinated with a phosphine ligand. This evidence spurred the investigation of several new drugs with modified phosphine ligands.7,8 Interestingly, NHC ligands share similar electron donor properties with phosphine ligands and can strongly stabilise their coordination with transition metal ions.9,10 The review by Aher and colleagues highlighted that NHC metal complexes can play a crucial role in treating all types of cancer.12 All the metal complexes considered in this review showed good biological activity, but Au, Pt and Ag made an important contribution and showed surprisingly better cytotoxicity profiles compared to cisplatin. Silver complexes have demonstrated great versatility in their biological actions ranging from DNA interactions and mitochondrial membrane depolarisation to ROS production, as described in the review by Raju and colleagues.11However, various metal–NHCs may have different mechanisms of action. Often, charged metal–NHCs show selectivity for cancer cells, whereas cisplatin does not show this selectivity.12 Hence, research on new pharmaceuticals needs to extend beyond the assessment of cellular effects, where a thorough analysis of their binding mechanism and structure–activity relationship (SAR) will enable the development of drugs with improved profiles. In this frame, the binding to biosubstrates (proteins and nucleic acids) needs to be carefully studied. Recently, the interest in non-canonical DNA or RNA structures has increased.13,14 Among them, G-quadruplexes (G4s) and i-motifs have emerged as promising targets for anticancer drugs.15,16 The higher presence of these secondary DNA structures in cancerous cells is associated with their involvement in transcription and replication processes, which are known to be overexpressed.17 Thus, targeting these types of structures may lead to new anticancer drugs with better properties. Metal complexes that can bind G4 structures are mainly formed by planar ligands containing a π-delocalized system, which form π stacking interactions with G-quartets.18 In general, to the best of our knowledge, no silver(I) carbenes that can bind non-canonical structures of DNA have been described in the literature to date. Instead, in the case of gold(I) NHCs, only a few examples have been reported. One of the main examples is a caffeine-based gold(I) biscarbene, selective for ovarian cancer cells, which can selectively bind to and stabilise G4s over the DNA double helix due to its planar geometry and similarity of its ligand with the guanine base.15,19 Several molecules have been tested as binders of i-motif structures. However, there was no evidence of their selectivity for i-motif structures over other DNA structures.20,21 Also, metal complexes with terbium or ruthenium were tested and showed low binding affinity and no structure stabilisation.22–24 The only example of i-motif selectivity over duplex and G4 is a derivative of the antibiotic heliomycin.25
Recently, some of these authors published interesting results on the reactivity of some similar Ag(I) and Au(I) anthracenyl carbenes, demonstrating that they could bind nucleic acids but not G4s.26 Herein, anthracenyl complexes with a longer carbon chain on the imidazole were developed, which could both enhance the lipophilicity and act as an arm to grab the G4 structure.
Results and discussion
Here, first the results of the synthesis and characterisation of [Ag(BIA)2]Cl are presented together with the preliminary experiments indicating the interesting properties of this metal complex towards cells and common biosubstrates (albumin and natural DNA). Then, this work was extended to unveil its mechanism of interaction, focusing on more peculiar polymers (RNAs in single, double and triple helix forms) and non-canonical oligos (i-motif and G-quadruplex).Synthesis and characterisation
The [Ag(BIA)2]Cl complex was synthesised following the procedure described in Scheme 1. The synthesis of the BIA ligand (1-(anthracen-9-ylmethyl)-3-butyl-1H-imidazolium chloride, 1) was carried out according to a modified procedure reported in the literature.27 Silver biscarbene (2) was obtained by mixing 1 with 0.75 eq. of Ag2O in a polar solvent such as MeOH for 24 h. We also synthesised gold biscarbene (3), which was obtained by the transmetallation reaction with 1.0 eq. of [AuCl{S(CH3)2}] (Scheme 1). Compounds 2 and 3 were characterised through 1H and 13C NMR spectroscopy (Fig. S1–S7†) and CHN analysis. 1H–1H COSY NMR was carried out for 1 to highlight the coupling between H1 and H2 of anthracene, and consequently correctly assign the signals in its 1H spectrum (Fig. S3†).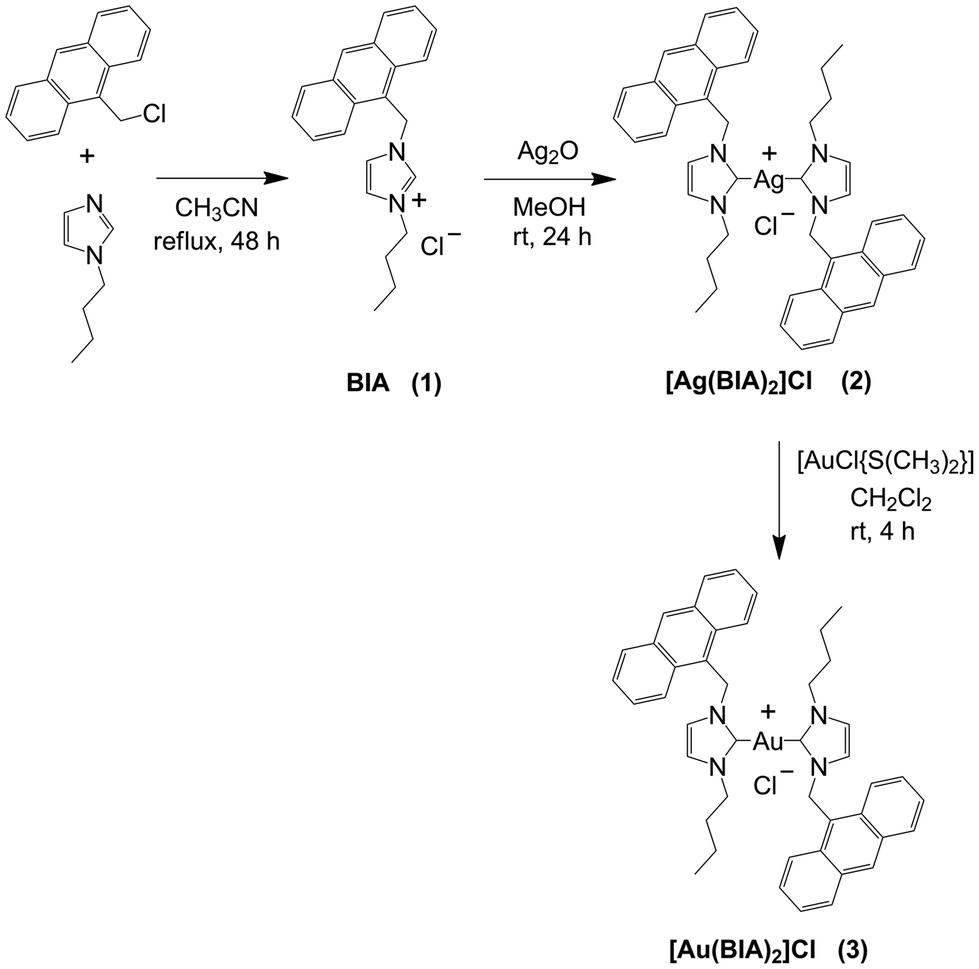 | ||
| Scheme 1 Synthesis of bis(1-(anthracen-9-ylmethyl)-3-butylimidazol-2-ylidene) silver chloride [Ag(BIA)2]Cl (2) and bis(1-(anthracen-9-ylmethyl)-3-butylimidazol-2-ylidene) gold chloride [Au(BIA)2]Cl (3). | ||
The solubility and stability of the complexes in buffer solution (NaCl 0.1 M, NaCac 2.5 mM, pH = 7.0) with time (0–3 h) and as a function of temperature (25–90 °C) were assessed by UV-Vis experiments. The experimental details are presented in the ESI (Fig. S8–S11†). Overall, differently from [Ag(BIA)2]Cl, which was stable over the investigated temperature range and time, [Au(BIA)2]Cl showed non-negligible stability problems depending on the buffer used, namely solubility issues due to aggregation phenomena. Therefore, it was impossible to perform a comprehensive study on the reactivity of [Au(BIA)2]Cl. However, with due care, a few tests were feasible and provided information to compare the features driven by the two different metal centres. In the case of [Ag(BIA)2]+, absorbance and fluorescence experiments were performed to ensure the direct proportionality between their signal and molar concentrations employed to analyse the data (Fig. S12–S14†). The same experiments indicated no auto-aggregation of [Ag(BIA)2]+ when present alone in buffer in the concentration range from 0 to 3.5 × 10−5 M.
Cellular assays
| A549 | SW480 | A2780 | A2780cis | RFa | HEK293 | SIb | |
|---|---|---|---|---|---|---|---|
| a RF (resistance factor) = IC50,A2780cis/IC50,A2780. b SI (selectivity index) = IC50,HEK293/IC50,A2780. | |||||||
| 2 | 10 ± 2 | 10 ± 2 | 6.0 ± 0.4 | 4.0 ± 0.5 | 0.7 ± 0.1 | 15.0 ± 0.2 | 2.5 ± 0.2 |
| 3 | 1.2 ± 0.1 | 3.3 ± 0.4 | 1.0 ± 0.5 | 1.4 ± 0.5 | 2 ± 1 | 2.2 ± 0.9 | 2 ± 1 |
| CDDP | 39 ± 2 | 35 ± 2 | 8 ± 1 | 30 ± 3 | 4 ± 1 | 15 ± 2 | 1.9 ± 0.5 |
To assess if the cell death mechanism is apoptotic or necrotic, a flow cytometry experiment was performed on A549 cells with Annexin V-FITC/PI double staining (Fig. S16†). The sum of cells in early and late apoptosis was higher compared to the number of cells in necrosis for both complexes (Fig. S17†). These values confirm that the cell death mechanism is apoptosis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) Po/w.
The cellular uptake was tested on the A549 cancer cell line and the obtained values (mean of three experiments) are reported as μmol of metal in 106 cells. These data were compared to the lipophilicity of the complexes, which was evaluated using the octanol/water partition coefficient, log
Po/w.
The cellular uptake was tested on the A549 cancer cell line and the obtained values (mean of three experiments) are reported as μmol of metal in 106 cells. These data were compared to the lipophilicity of the complexes, which was evaluated using the octanol/water partition coefficient, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w (Table S1†). Both compounds exhibited increased lipophilic character and cellular uptake compared to CDDP. However, although [Au(BIA)2]Cl showed the same uptake level as [Ag(BIA)2]Cl, it was 10-times more cytotoxic. Overall, lipophilicity alone does not explain the cytotoxicity differences.
Po/w (Table S1†). Both compounds exhibited increased lipophilic character and cellular uptake compared to CDDP. However, although [Au(BIA)2]Cl showed the same uptake level as [Ag(BIA)2]Cl, it was 10-times more cytotoxic. Overall, lipophilicity alone does not explain the cytotoxicity differences.
Proteins: bovine serum albumin (BSA) interaction
:1 metal complex to protein binding model ratio was found to be sufficient to reproduce the fluorometric results. It was found that the KBSA value is of a magnitude order of 106 (2.9 × 106 M−1 at 37.0 °C) and increased with an increase in temperature (entropically-driven exothermic process). Given that the thermodynamic signature indicates the type of interaction that occurs between a small molecule and the protein,28 here the positive ΔH value indicates that for the [Ag(BIA)2]+/BSA system, hydrophobic interactions are dominant, in agreement with logPo/w = 1 and hinting at the predominant contribution of the anthracenyl residue.29 To determine the preferential (if any) binding site in BSA for the metal complex, a standard procedure was followed, which involved performing displacement titrations. Firstly, the protein was saturated with a marker that specifically binds to one of the two hydrophobic BSA binding sites, where phenylbutazone (for site I) and ibuprofen (for site II) were selected.30 Secondly, displacement experiments were performed by adding increasing amounts of the metal complex to marker + BSA mixtures.
The equilibrium constants related to these displacement titrations (HypSpec® software) are presented in Table S3,† where a significant decrease in KBSA occurred in the presence of ibuprofen (from 19.2 × 105 M−1 to 5.8 × 105 M−1), while the variation was significantly limited in the case of phenylbutazone. This suggests that [Ag(BIA)2]+ and ibuprofen compete for the same site, i.e. site II.
938 and 67448 Da, respectively (Fig. S19B†). It should be noted that the protonation state of the protein alone and in the presence of the metal complexes did not change, suggesting that the interaction does not cause denaturation of the protein by altering its folding (Fig. S20†).
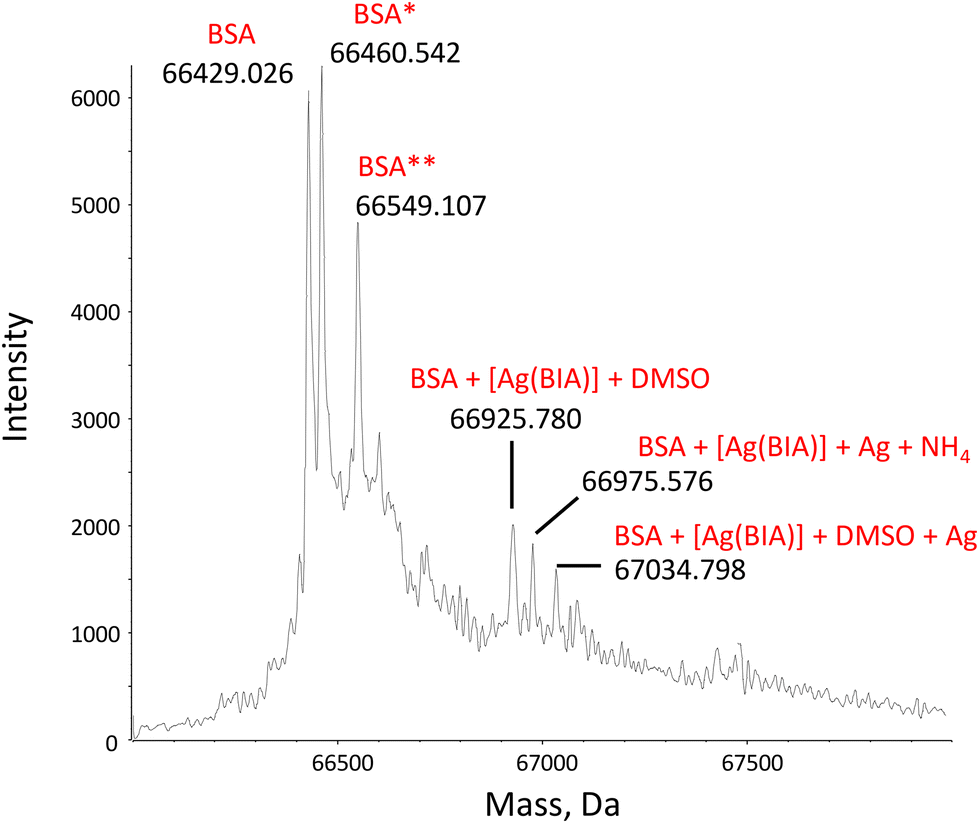 | ||
| Fig. 1 Deconvoluted mass spectrum of BSA, CBSA = 1.00 μM, in the presence of 2.00 μM of [Ag(BIA)2]+ in 20 mM NH4OAc solution, pH = 6.8. 0.1% v/v of formic acid was added just before injection into the mass spectrometer. BSA* = sulfinylation on Cys34; BSA** = cysteinylation on Cys34.35 | ||
Overall, the binding mode seems two-pronged, where at least [Ag(BIA)2]+ is found to both covalently bind Cys34 and reversibly enter the site II pocket.
Nucleic acids: natural DNA interaction
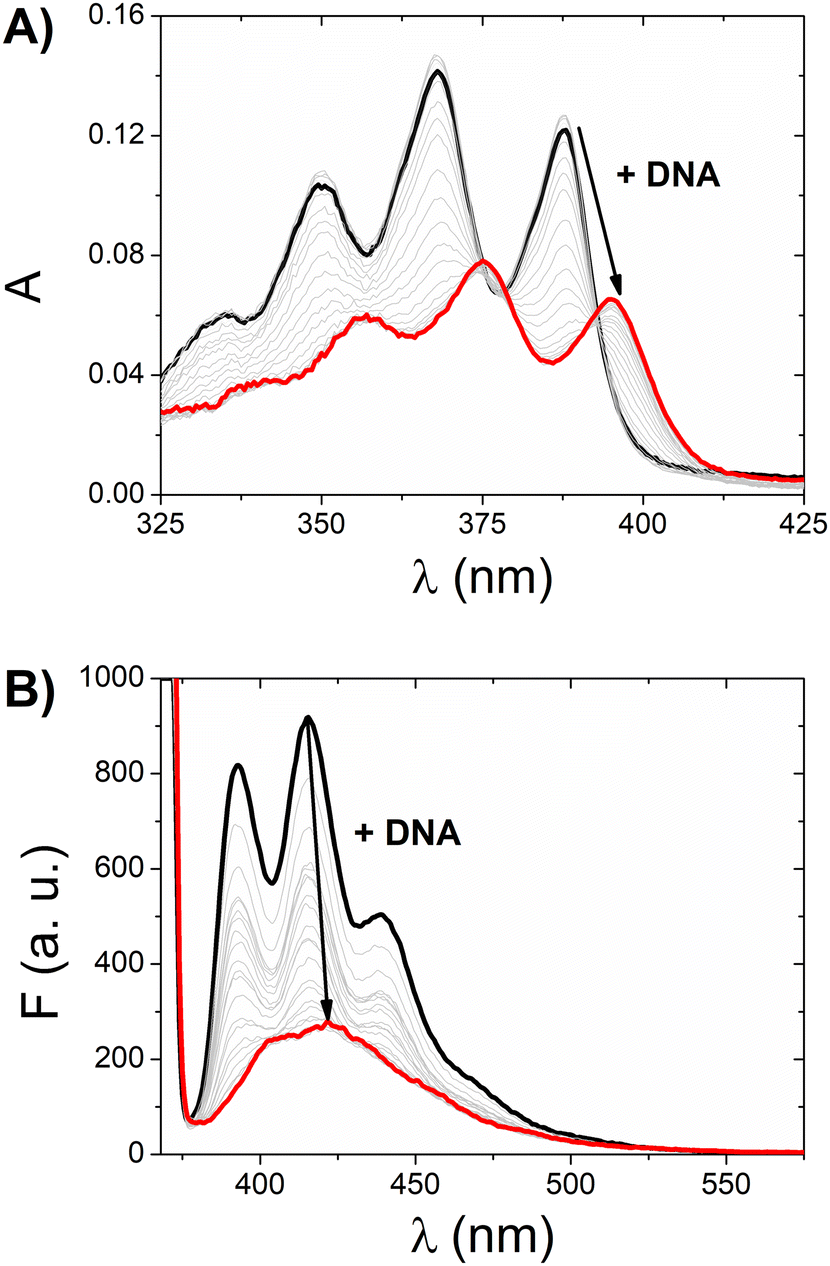 | ||
Fig. 2 (A) Absorbance spectra corrected for the dilution of [Ag(BIA)2]+ (CAg = 10.9 μM) at increasing amounts of CT-DNA (CDNA) from 0 (___) to 227 μM ( ). (B) Emission spectra (λex = 368 nm, slit 5 nm) corrected for the dilution of [Ag(BIA)2]+ (CAg = 0.216 μM) at increasing amount of DNA from 0 (___) to 59.6 μM ( ). (B) Emission spectra (λex = 368 nm, slit 5 nm) corrected for the dilution of [Ag(BIA)2]+ (CAg = 0.216 μM) at increasing amount of DNA from 0 (___) to 59.6 μM ( ). NaCac 2.5 mM, pH = 7.0, T = 25.0 °C; CDNA in base pairs. ). NaCac 2.5 mM, pH = 7.0, T = 25.0 °C; CDNA in base pairs. | ||
Given that both complexes bear an anthracenyl fluorophore, we also evaluated their interaction with DNA via spectrofluorimetric titrations. The high fluorescence of [Ag(BIA)2]+ allowed us to use lower concentrations of this complex (0.216 μM), while in the case of the gold complex, its quantum yield was much lower using the same conditions for the spectrophotometric titrations. The emission of [Ag(BIA)2]+ decreased upon the addition of an increasing amount of DNA (Fig. 2B), in agreement with the strong quenching of the anthracenyl moiety and intercalation between base pairs. Alternatively, in the case of [Au(BIA)2]+, the double behaviour in its emission spectra (Fig. S22B†) was confirmed, with an initial decreasing phase, followed by an increase in emission (with a redshift of 9 nm). Again, aggregation is a possible explanation for these results; overall, the metal centre changes the binding mode, which seems to be an outer-helix nature and of low pharmaceutical significance in the case of the gold compound.26 Regarding [Ag(BIA)2]+/CT-DNA, this system was further investigated by performing absorbance and fluorescence titrations at 25.0 °C at different ionic strengths (concentration of NaCl from 0 to 0.1 M, 2.5 mM NaCac buffer). Here, it was necessary to apply the best data analysis procedure to obtain a reliable evaluation of the binding constant, KDNA, for these complicated polynucleotide interactions. This system and our data were employed to conduct a critical test on how the numerical evaluations depend on the calculation method employed. This severe issue is frequently underestimated, although the blind application of a non-deeply understood equation may lead to incorrect values. The Scatchard plot42 was first used to get information on the site dimensions in the [Ag(BIA)2]+/DNA system, i.e. the number (n) of base pairs involved in the binding of one metal complex (Fig. S23†). It was found that n ≈ 1, and thus one base pair is involved. Once this check was done, it was possible to consider a simple 1:1 metal complex to DNA base pair reaction and use two equations derived from the Hildebrand–Benesi model,43 as follows:
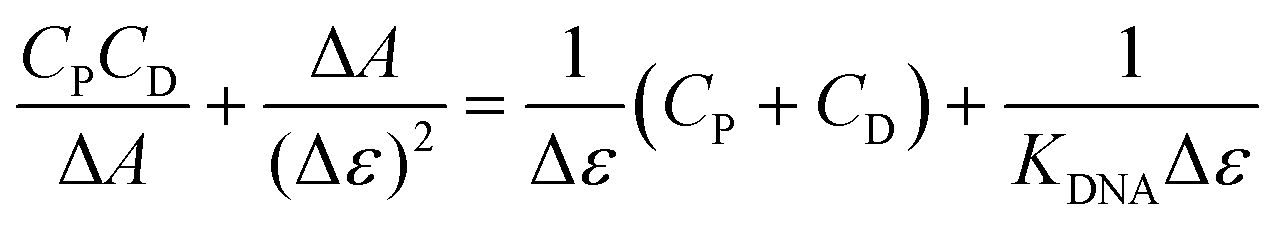 | (1) |
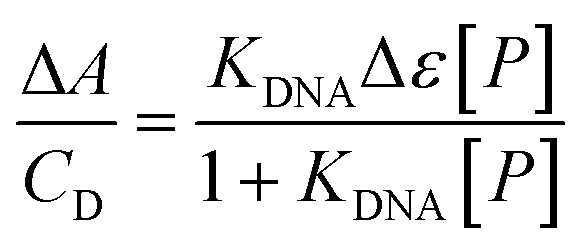 | (2) |
:1 binding but where the DNA concentration is expressed in reaction sites, with CDNA(sites) = CDNA(base pairs)/n. All the obtained values are reported in Table S4† and shown in Fig. 3.
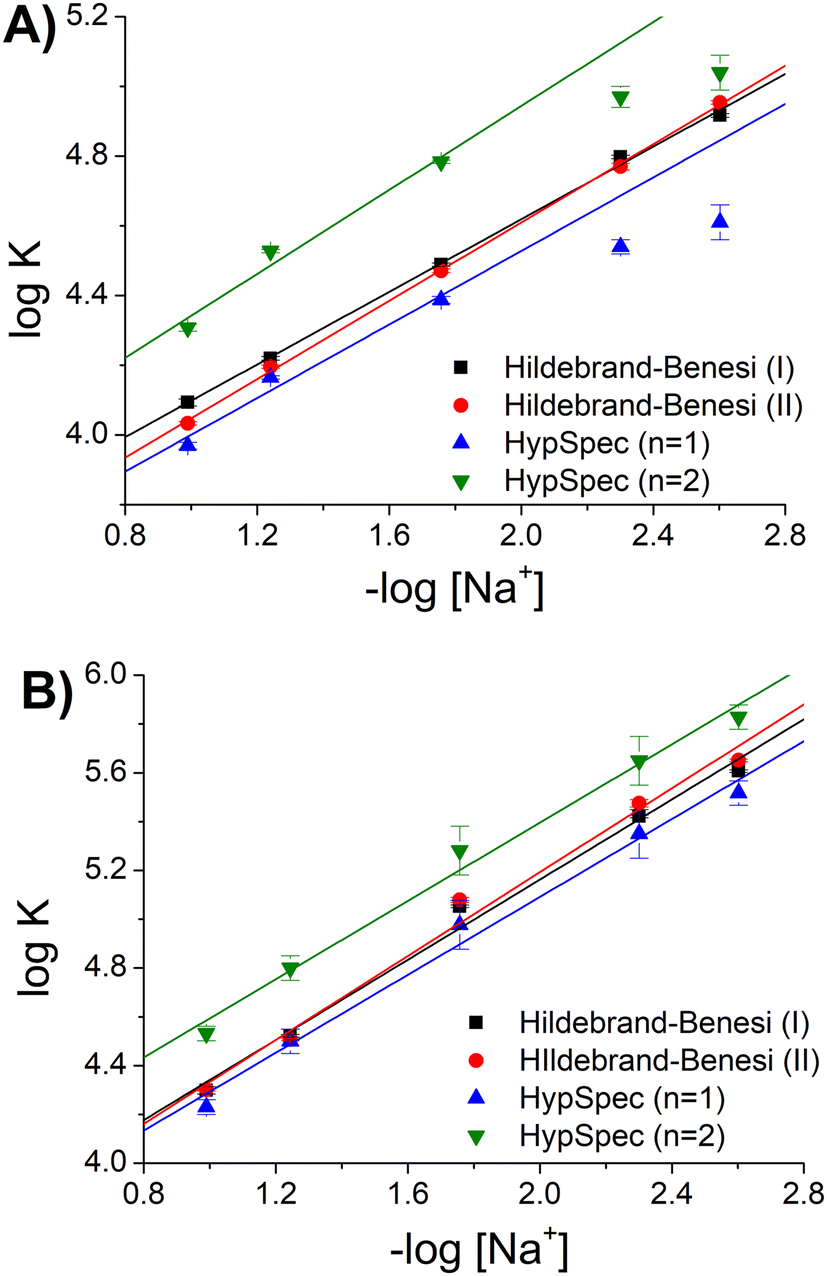 | ||
| Fig. 3 Binding constants (KDNA) for the [Ag(BIA)2]+/DNA system as a function of [Na+], pH = 7.0, T = 25.0 °C: (A) absorption and (B) fluorescence. | ||
Considering the homogeneous set eqn (1)–eqn (2)–HypSpec(n = 1), the agreement was found to be very good, where the dispersion of data (standard deviation) was <1.5% (in log units). The data of −log[Na+] >2 by the HypSpec analysis were neglected, which were found to show some deviation due to DNA-induced aggregation effects. The latter may become more important when using the whole spectral range at low ionic strength and for higher concentrations used in the absorbance mode. It should be noted that interestingly, the absorbance or fluorescence experiments produced completely different datasets. This behaviour is frequently found in the case of DNA when two binding modes are operational. Under these circumstances, KDNA becomes an apparent constant, originating from two contributions that are differently weighted under different experimental conditions. In agreement with the Record theory,45 the slope of the plots shown in Fig. 3 corresponds to m′ψ, where m′ represents the number of phosphodiester residues occupied by a binding molecule and ψ = 0.88 is a shielding factor for DNA. m′ also represents the number of sodium ions displaced by a complex molecule and is correlated with the degree of penetration of a small molecule into the helix.46 The m′ values were found to be reproducible, also considering HypSpec(n = 2): m′ = 0.62 ± 0.04 (ABS), m′ = 0.93 ± 0.03 (FLUO). It can be supposed that in the case of fluorescence, the low concentration conditions suppressed the dye–dye interaction, which stabilised groove binding and intercalation was favoured. This picture was confirmed by the higher penetration degree obtained for fluorescence (m′ FLUO > m′ ABS).
Nucleic acids: RNA interaction
Spectrophotometric titrations and melting tests were repeated with the single-stranded RNA poly(rA), and the double and triple helixes, poly(rA)poly(rU) and poly(rA)2poly(rU). Fig. S27† shows the starting spectrum of [Ag(BIA)2]+ and the final spectra for the [Ag(BIA)2]+/RNA mixtures. In the case of all the polynucleotides, no significant signal shifts as that in Fig. 2A were observed. In general, in the case of RNAs, the interaction of [Ag(BIA)2]+ seems impractical and the result is due to the lack of strong interaction with RNA molecules, which can lead to major spectral changes. The melting experiments (Table S6†) indicate that there is indeed some interaction between the metal complex and double-stranded RNA, but it is weak (ΔTm of a few degrees from +4.4 °C to −3.5 °C with an increase in the ionic strength). The silver complex demonstrated a major affinity for DNA double strands over RNA structures.Non-canonical oligos: i-motif interaction
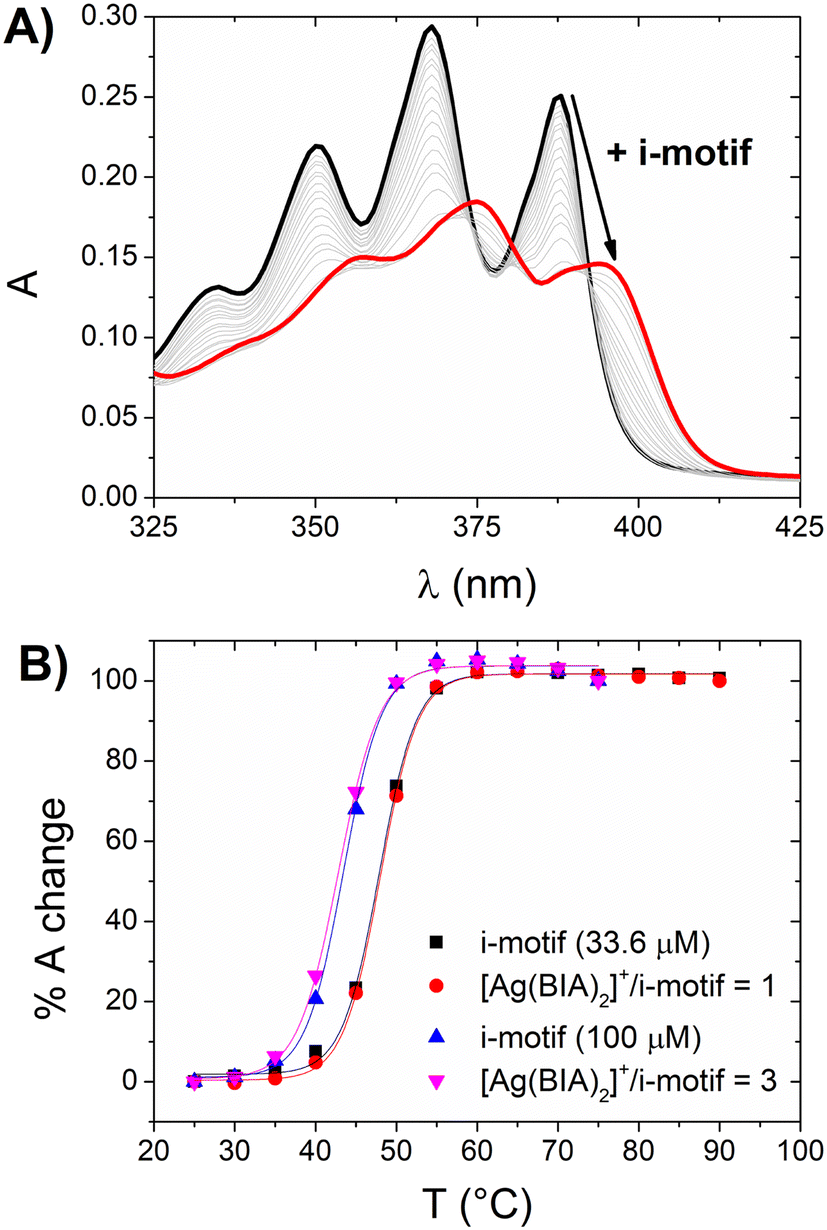 | ||
| Fig. 4 (A) Absorbance spectra of [Ag(BIA)2]+ (corrected by dilution) at increasing amounts of i-motif from 0 to 102 μM (in strands). CAg = 21.8 μM, NaCac = 50 mM, pH = 5.5, and T = 25.0 °C. (B) Melting curves for i-motif alone (black squares 33.6 μM; blue up triangles 100 μM with 7% of DMSO) and the [Ag(BIA)2]+/i-motif mixture at C[Ag(BIA)2]+/Ci-motif = 1 (red circles) or 3 (pink down triangles). | ||
The experimental results demonstrated the hypochromic and bathochromic effects on the spectrum of silver (similar to that observed for CT-DNA), which indicate strong i-motif interactions (Fig. 4A). Alternatively, there was no significant stabilisation of the [Ag(BIA)2]+/i-motif adduct (Fig. 4B). The latter was also observed at higher concentrations of i-motif and silver complex, where even at a concentration in the order of 10−4 M with a metal complex/i-motif ratio = 3, no stabilisation of the DNA structure was noticed. This behaviour suggests a type of interaction involving the groove/external part of the motif. The equilibrium constant was evaluated with the HypSpec® software and a 1:1 binding mode was found to be applicable, showing the value of K = 3.2 ± 0.7 × 104 M−1.
Non-canonical oligos: G-quadruplex interaction
:1 adduct metal complex:G4 is sufficient to describe the data. Table 2 presents the obtained numerical values for the binding constants.
| G4 | Type | K G4 (105 M−1) | T m G4 (°C) | T m [Ag(BIA)2]+/G4 (°C) | ΔTm (°C) |
|---|---|---|---|---|---|
| Tel-23 | Hybrid | 18 ± 5 | 62.8 ± 0.3 | 63.0 ± 0.3 | 0.2 ± 0.6 |
| CTA-22 | Antiparallel | 8.7 ± 0.8 | 62.6 ± 0.2 | 66.2 ± 0.3 | 3.7 ± 0.5 |
| c-myc | Parallel | 7.2 ± 0.8 | 74.0 ± 0.4 | 78.4 ± 0.3 | 4.4 ± 0.7 |
The G4 melting temperature was measured in the absence and presence of the [Ag(BIA)2]+ metal complex (Fig. S29†).
The obtained data are also presented in Table 2. Overall, the data suggest that Tel-23 (hybrid G4) behaves differently from CTA-22 (antiparallel G4) and c-myc (parallel G4), which are similar. Although [Ag(BIA)2]+/Tel-23 is favoured (high KG4, Table 2), the interaction does not produce stabilization of the quartet, which opens as easily as G4 alone. It can be speculated that the binding site is external to the quartet similar to the groove of the G4 structure.51 Depending on the geometry, the metal complex seems to bind to two different sites, where one binding mode affects the quartet more, while the other does not. It was already noticed in the literature that hybrid conformations are less stabilised by the interacting compounds.51 The situation in CTA-22 and c-myc is the opposite, where the interaction is overall slightly less favourable but affects and stabilizes the quartet.
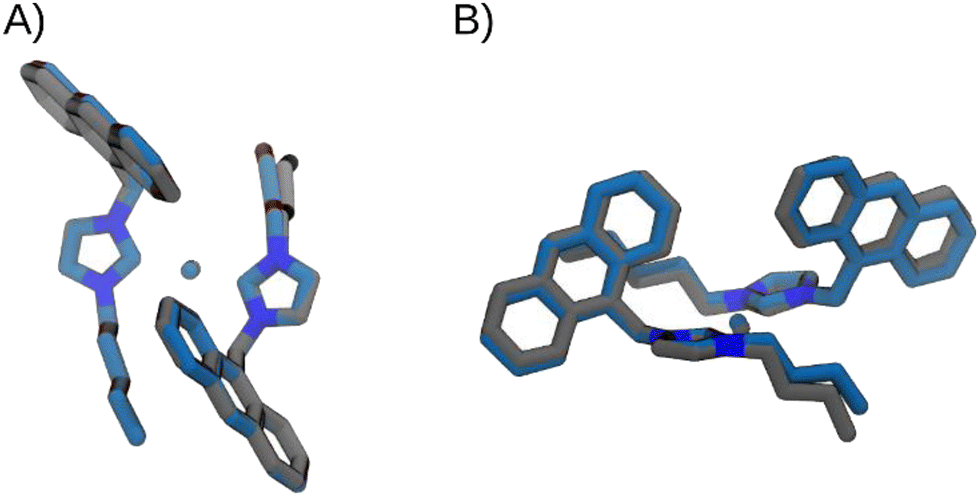 | ||
| Fig. 5 Superimposition of the optimized structures of [Ag(BIA)2]+, in grey, and [Au(BIA)2]+, in blue, in two different views: frontal (A) and lateral (B). | ||
The energetic profile is very similar for both complexes. The main differences can be attributed to the equilibrium value (−9° and −7°, for Ag and Au complexes, respectively), and to the torsional barrier, which is slightly lower in the [Ag(BIA)2]+ complex.
According to the docked models (Fig. 6), a more stable groove binding mode is predicted suggest for the [Au(BIA)2]+ complex in c-myc. In the case of [Ag(BIA)2]+, we identified a stacking interaction with the thymine residue of the loop associated with c-myc and CTA-22. Specifically, the interaction occurs with one thymine of c-myc and one thymine of CTA-22, which are marked in bold in the 5′-TGAGGGTGGGTAGGGTGGGTAA-3′ and 5′-AGGGCTAGGGCTAGGGCTAGGG-3′ sequences, respectively. The same interaction occurs for [Au(BIA)2]+ with CTA-22.
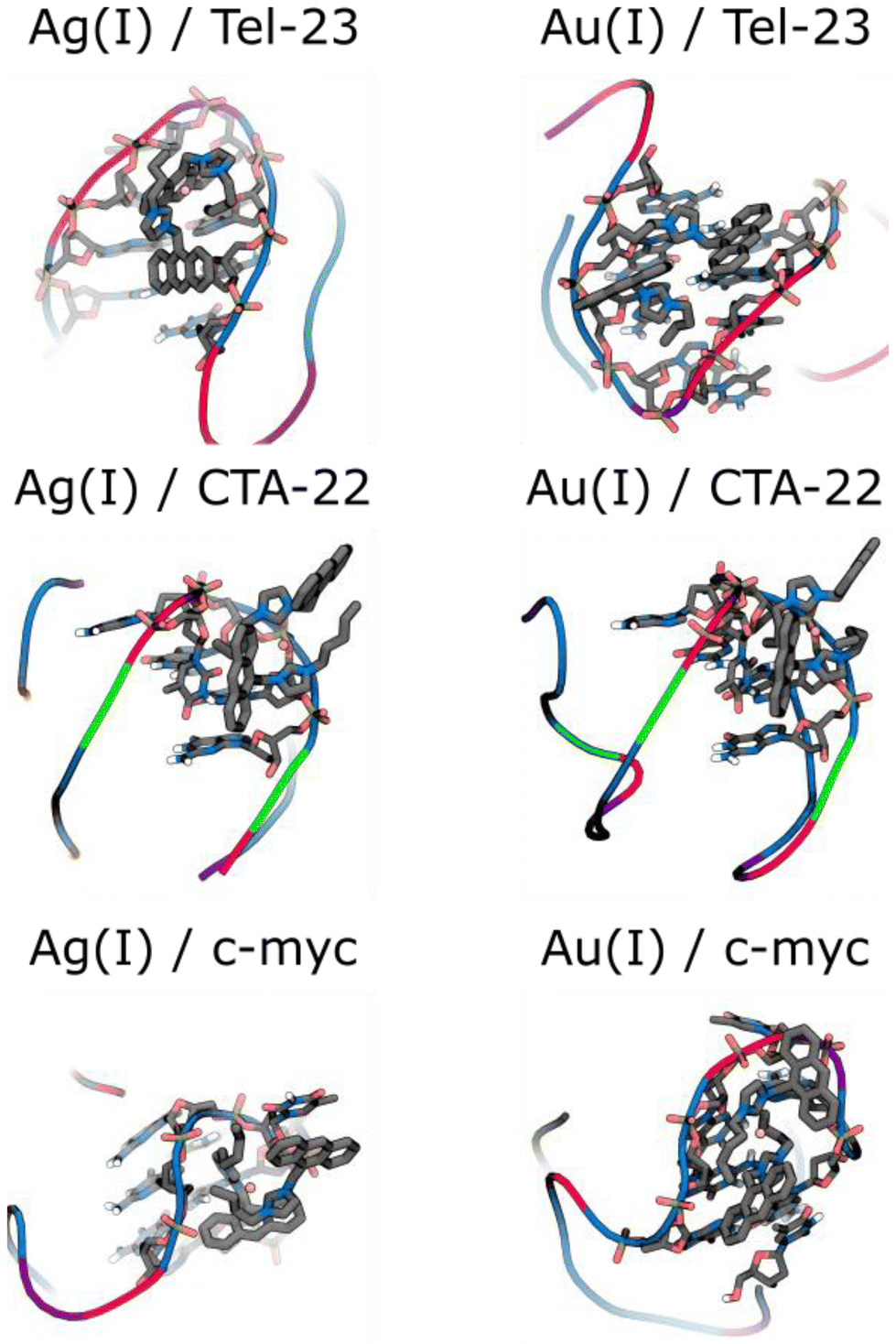 | ||
| Fig. 6 Most stable binding poses for the two ligands in G4s. Adenine, guanine, cytosine, and thymine residues are coloured in violet, blue, green, and red, respectively. Residues within 4 Å from the complex are shown. | ||
Regarding the interaction between c-myc and [Ag(BIA)2]+, an additional intramolecular interaction between the anthracene and the central ring can also be observed, which contributes to the decrease in the scoring value. There are cases with a major contribution of one of the carbene ligands with the other one facing outwards. Even in these cases, the presence of a second ligand is supposed to play a role considering that the overall metal complex: (i) will become positively charged, a feature that can drive the binding mode;55 and (ii) will acquire a given geometrical form, which again is important for pushing more favourable binding matches at the different oligos.
Finally, both complexes show a groove interaction in Tel-23, as the most stable binding modes. In particular, the silver complex interacts with the sugar of the thymine and guanine residues marked bold: 5′-TAGGGTTAGGGTTAGGGTTAGGG-3′.
Experimental
Metal complexes
Stock solutions of the metal complexes were prepared by dissolving a known weight of the compound (≈2 mg) in dimethylsulfoxide (DMSO) to obtain a final solution concentration of 2–20 mM. The stock solution of cisplatin (CDDP) was prepared in water to obtain a final concentration of 3 mM.Biosubstrates
The BSA solution was prepared by weighing freeze-dried BSA purchased from Sigma-Aldrich (MW ≈ 66 kDa) (lyophilized powder, essentially fatty acid-free, ≥98% agarose gel electrophoresis) by dissolving it in 0.1 M NaCl, 2.5 mM NaCac, and pH = 7.0 buffer. The effective concentration was determined by spectrophotometry (λ = 278 nm, ε = 45000 cm−1 M−1),56 obtaining a solution of 80 μM BSA (CBSA). Calf thymus DNA (CT-DNA), natural DNA from calf thymus, lyophilised sodium salt from Sigma-Aldrich (double strand in the B-helix form when in the buffer) was dissolved in ultrapure water and previously sonicated to have about 500 base pairs. Its concentration in the stock was about 2.5 mM expressed in base pairs (CDNA). Before its use, its exact concentration was checked by measuring its absorption spectrum (λ = 260 nm, ε = 13200 cm−1 M−1).57
Cell lines and E. coli DH5α were purchased from ATCC.
Natural DNA, plasmid pUC18 (2686 base pairs in length), was extracted from E. coli DH5α and purified using an HP Plasmid Midi kit (Omega Biotek, VWR). The plasmid was used to perform the agarose gel electrophoresis experiments.
Poly(rA) and poly(rU) were purchased from Sigma-Aldrich as model polynucleotides of RNA structures. Poly(rA) was used as a single filament in 2.5 mM NaCac pH = 7.0 buffer, whose concentration was measured by spectrophotometry (λ = 257 nm, ε = 10110 cm−1 M−1),58 obtaining a concentration expressed as phosphate groups (CA) of 8.4 mM. In the case of the double helix, a poly(rU) solution was prepared in the same buffer whose concentration was measured considering the peak at 260 nm (ε = 8900 cm−1 M−1),58 obtaining a concentration of 14 mM. Subsequently, a solution of poly(rA) with poly(rU) in a 1:1 ratio in buffer was prepared and left to stand overnight in the dark at room temperature to obtain the double helix poly(rA)poly(rU) concentration in base pairs (CAU) of 2.0 mM. In the case of the triple helix, a solution was prepared by combining poly(rA)·poly(rU) with poly(rU) in a 1:1 ratio in buffer and left to stand overnight at room temperature to allow the integration of a third filament in the structure, thus obtaining a solution of poly(rA)2poly(rU) of concentration in the base triplet (CA2U) of 0.4 mM.59
The i-motif used was obtained by dissolving the (CCCTTT)4 telomere in 50 mM NaCac, pH = 5.5 buffer solution at a concentration of 1 mM. The procedure for its annealing is the same as described below for G4.
The DNA G-quadruplexes (G4) used were formed by different telomeres to have different types of conformations, as follows: 5′-TAGGGTTAGGGTTAGGGTTAGGG-3′ (Tel-23 – hybrid); 5′-AGGGCTAGGGCTAGGGCTAGGG-3′ (CTA-22 – antiparallel); and 5′-TGAGGGTGGGTAGGGTGGGTAA-3′ (c-myc – parallel) were dissolved in 0.1 mM KCl, 2.5 mM LiCac, pH = 7.0, obtaining a 1 mM solution of G4 (CG4). The telomere annealing procedure to form the quaternary structure involved placing an Eppendorf tube containing the telomere solution in a thermostatic water bath by slowly heating from a temperature equal to that of the environment up to about 90–95 °C. The maximum temperature was maintained for 10–15 min, and then the heating was turned off and the solution was allowed to cool slowly in the water bath until the next morning.60
Buffers
The used buffer solutions were prepared by dissolving a known amount of the desired salts in MilliQ water, obtained through an “AriumPro SARTORIUS” device, and the pH was possibly corrected by performing micro additions of HCl or concentrated NaOH. The salts used to prepare the buffer solutions were sodium chloride (NaCl), sodium cacodylate (NaCac, caution: cacodylate is toxic if swallowed or inhaled), potassium chloride (KCl), lithium cacodylate (LiCac), and ammonium acetate (NH4OAc). The working solutions were prepared by dilution of the metal complex or biosubstrates in the correct buffer, and thus the DMSO content can be considered negligible (<1%).Synthesis of BIA (1)
453 mg of 9-chloromethylantracene and 248 mg of 1-butylimidazole were dissolved in 40 mL of acetonitrile. The reaction was stirred for 46 h at 80 °C. Subsequently, the solvent was evaporated under reduced pressure and a yellow solid was obtained. The latter was dissolved in a few millilitres of methanol, and then diethyl ether was slowly added to precipitate the final product, which was obtained as a fine yellow powder. (Yield 85%) 1H-NMR (CDCl3, 400 MHz) δ = 11.18 (s, 1H, imidazole H2′), 8.49 (s, 1H, anthracene H10), 8.30 (d, 2H, anthracene H1 and H8, J = 8,8 Hz), 7.99 (d, 2H, anthracene H4 and H5, J = 8,4 Hz), 7.60–7.56 (m, 2H, anthracene H2 and H7), 7.47–7.43 (m, 2H, anthracene H3 and H6), 7.18 (m, 1H, imidazole H4′), 6.64 (m, 1H, imidazole H5′), 6.60 (s, 2H, anthracene-CH2-imidazole), 4.23 (t, 2H, imidazole-CH2CH2CH2CH3, J = 7.6 Hz), 1.86–1.79 (m, 2H, imidazole-CH2CH2CH2CH3), 1.34–1.25 (m, 2H, imidazole-CH2CH2CH2CH3), 0.87 (t, 3H, imidazole-CH2CH2CH2CH3, J = 7.2 Hz) ppm. 13C (CDCl3, 400 MHz) δ = 137.75, 131.33, 130.94, 130.62, 129.59, 128.36, 125.62, 122.85, 122.10, 121.64, 121.07, 50.04, 45.61, 32.09, 19.53, 13.46 ppm.Synthesis of [Ag(BIA)2]Cl (2)
527 mg of ligand BIA (1) was dissolved in 8 mL of methanol in a 50 mL flask with 263 mg (0.750 eq.) silver oxide Ag2O. The mixture was stirred at room temperature for 24 h. Then dichloromethane was added, and the suspension was filtered through Celite to remove the unreacted Ag2O and the eventual unreacted ligand. The solvent was removed under reduced pressure, obtaining a red amorphous solid. Subsequently, the solid was dissolved in 8 mL of methanol and the solution was kept stirring at room temperature for 24 h and protected from light. Subsequently, the suspension was filtered through Celite to remove AgCl. The solvent was evaporated, and crystallisation was carried out using THF/Et2O. The light-orange granular solid was filtered and washed with hexane. (Yield 12%) 1H-NMR (CD3OD, 400 MHz) δ = 8.56 (s, 2H, anthracene H10), 8.14 (d, 4H, anthracene H1 and H8, J = 7.2 Hz), 8.04 (d, 4H, anthracene H4 and H5, J = 6.8 Hz), 7.53–7.44 (m, 8H, anthracene H2, 3, 6, 7), 7.30 (s, 2H, imidazole H4′), 7.19 (s, 2H, imidazole H5′), 5.90 (s, 4H, anthracene-CH2-imidazole), 3.47 (b, 4H, imidazole-CH2CH2CH2CH3), 1.71 (b, 4H imidazole-CH2CH2CH2CH3), 1.20 (b, 4H imidazole-CH2CH2CH2CH3), 0.89 (b, 6H imidazole-CH2CH2CH2CH3) ppm. 13C (CD3OD, 400 MHz) δ = 170.27, 133.02, 132.11, 130.62, 128.45, 127.04, 126.45, 124.60, 123.69, 123.40, 121.98, 53.09, 47.76, 34.41, 20.28, 13.74 ppm. Elemental C, H and N analysis for C44H44N4AgCl: calculated C = 68.44%, H = 5.74%, N = 7.26% and experimental C = 68.24%, H = 5.62%, N = 6.89%.Synthesis of [Au(BIA)2]Cl (3)
To synthesise the [Au(BIA)2]Cl complex, the transmetalation of [Ag(BIA)2]Cl was carried out using [AuCl{S(CH3)2}]. To 38.0 mg of [Ag(BIA)2]Cl, 5 mL of CH2Cl2 and 15.6 mg (1.00 eq.) of [AuCl{S(CH3)2}] were added. The mixture was left stirring at room temperature for 3 h and protected from light. Subsequently, the suspension was filtered over Celite to remove the AgCl formed, and the filtrate was concentrated under vacuum. Precipitation of the metal complex was carried out using hexane. (Yield 87%) 1H-NMR (CD3OD, 400 MHz) δ = 8.57 (s, 2H, anthracene H10), 8.28 (d, 4H, anthracene H1 and H8, J = 7.2 Hz), 8.05 (d, 4H, anthracene H4 and H5, J = 6.8 Hz), 7.56–7.46 (m, 8H, anthracene H2, 3, 6, 7), 7.21 (s, 2H, imidazole H4′), 7.06 (s, 2H, imidazole H5′), 6.07 (s, 4H, anthracene-CH2-imidazole), 3.80 (m, 4H imidazole-CH2CH2CH2CH3), 1.67–1.59 (m, 4H imidazole-CH2CH2CH2CH3), 1.181–1.088 (m, 4H imidazole-CH2CH2CH2CH3), 0.78–0.74 (m, 6H imidazole-CH2CH2CH2CH3) ppm. 13C (CD3OD, 400 MHz) δ = 182.74, 131.49, 130.81, 129.30, 129.09, 126.90, 125.11, 124.93, 123.06, 121.16, 121.09, 50.94, 46.25, 32.87, 19.11, 12.37 ppm. Elemental C, H and N analysis for C44H44N4AuCl: calculated C = 61.22%, H = 5.37%, N = 6.49% and experimental C = 61.33%, H = 5.25%, N = 6.16%.Cell culture
A549 (lung adenocarcinoma) and SW480 (colon adenocarcinoma) cells were cultured in Dulbecco's modified Eagle's medium (DMEM), whereas A2780 (ovarian carcinoma) and A2789cis (ovarian carcinoma cisplatin-resistant) cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 2 mM L-glutamine, and HEK293 (embryonic kidney) cells in Eagle's Minimum Essential Medium (EMEM) supplemented with 1% of non-essential amino acids. All media were supplemented with 10% fetal bovine serum (FBS), which is fundamental for cell growth, and 1% amphotericin–penicillin–streptomycin solution (all from Sigma Aldrich). The cells were incubated in a humid atmosphere at 37 °C under a 5% CO2 atmosphere.MTT assay
Approximately 5 × 103 A549, 1 × 104 SW480, 2 × 104 A2780, A2780cis and HEK293 cells per well were seeded in 200 μL of culture medium in 96-well plates and incubated for 24 h at 37 °C under a 5% CO2 atmosphere. Then, the cells were treated with different concentrations of the complexes under study for 24 h. Cisplatin was used as a positive control and the maximum percentage of DMSO as the vehicle control. Dilution of the stock solutions was done in the corresponding culture medium. A negative control (without any treatment) and blanks were also included. Afterwards, the medium was removed, and the cells were incubated with 100 μL of MTT (3-(4,5-dimethyltiazol-2-yl)-2,5-diphenyltetrazoliumbromide) (Sigma Aldrich) dissolved in culture medium (500 mg mL−1). The reduction of the yellow salt, due to mitochondrial dehydrogenase, forms the blue formazan in living cells. After 3 h of incubation, the formazan crystals were dissolved by overnight incubation with 100 μL of solubilizing solution (10% SDS and 0.01 M HCl). Finally, the absorbance was recorded at 590 nm on a microplate reader (Cytation 5 Cell Imaging Multi-Mode Reader, Biotek Instruments, USA). Four replicates per dose were included and at least three independent experiments were performed for the calculation of the half-maximal inhibitory concentration (IC50) values employing GraphPad Prism Software Inc. (version 6.01) (USA).Apoptosis
Apoptosis was evaluated by flow cytometry using an Annexin V-FITC Assay Kit (BioRad) according to the manufacturer's instructions. 2 × 105 A549 cells were seeded in 1 mL of cell culture medium in 12-well plates. After 24 h of incubation, the cells were treated with the half-maximal inhibitory concentration of each complex for 24 h. Cisplatin was used as the positive apoptotic control, while TRITON 1% was used as the death (necrotic) control and added 10 min before the harvesting of the cells. Afterwards, the cells were washed with cold PBS, harvested, and resuspended in binding buffer. Then, the cells were doubly stained with the Annexin V-FITC conjugate and propidium iodide (PI). Immediately after the addition of PI, the cells were injected in a NovoCyte Flow cytometer (ACEA Biosciences, Inc., USA). 10000 events were counted and analysed by using the NovoExpress 1.4.0 Software. Two independent experiments were performed.
Cellular uptake
1.5 × 105 A549 cells were seeded in 2 mL of cell culture medium in 6-well plates for 24 h. Afterwards, the cells were treated with a concentration of 5 μM for each metal complex for 24 h. CDDP was used as the positive and reference control. Subsequently, the cells were harvested and washed twice with PBS to remove the metal complex that was not internalised to obtain the cellular pellet. The cells were resuspended in 500 μL of PBS and 10 μL was used to count the cells in an automated cell counter (TC-20, BioRad). 2 mL of a solution of HNO3 Suprapure (dilution 1:1 with MilliQ water) for Ag and Au complexes, or 2 mL of 3HCl:HNO3 (aqua regia) Suprapure solution (dilution 1:1 with MilliQ water) for the Pt complex was added to mineralise the samples (90 °C overnight). Dilution with MilliQ water to obtained a final volume of 6 mL per sample was performed, and ICP-OES analysis was performed as already described.61,62 The amount of metal internalised by the is expressed in μmol of metal per 106 cells.
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) Po/w
Po/w
The octanol–water partition coefficients were determined as follows. Water (500 mL, distilled after MilliQ purification) and n-octanol (500 mL) were shaken together for 72 h to allow saturation of both phases (one week for the full separation of the two phases). Approximately 0.5 mg of the complexes was placed in a Falcon tube, and 2 mL of octanol–saturated water and 2 mL of water–saturated octanol were added into the same Falcon of the compound and shaken until the complexes were fully dissolved and divided into the two phases. The biphasic solutions were mixed for 10 min, and then centrifuged for 5 min at 6000 rpm to allow separation. Two aliquots of 500 μL for both phases were placed into metal-free PE tubes and 4 mL of a solution of HNO3 Suprapure (dilution 1:1 with MilliQ water) for Ag and Au complexes or 4 mL of 3HCl:HNO3 (aqua regia) solution Suprapure (dilution 1:1 with MilliQ water) for CDDP was added to the aliquots to obtain 4 samples for each complex (2 for the water phase and 2 for the octanol phase). The solutions were mineralised at 90 °C overnight and 1.5 mL of MilliQ water was added at the end to obtain a final volume of 6 mL for each sample. The metal concentration in both phases was determined by ICP-OES.61,62
Titrations
A Shimadzu UV-2450 or a PerkinElmer Lambda 35 double-beam UV-vis spectrophotometer was used for absorbance experiments. Both instruments were equipped with a tungsten lamp for visible light and a deuterium lamp for the UV range. The fluorescence data were recorded with an LS55 PerkinElmer spectrofluorometer. The excitation light was provided by a pulsed Xenon lamp (50 Hz). In the spectrophotometric titrations, a solution of metal complex (D) of around 20 μM (CD) was placed in a cuvette with a path length of 1 cm. After measuring the spectrum of the metal complex alone, increasing amounts of the biosubstrate (P) were added directly to the cuvette containing the metal complex solution and its spectrum was recorded upon each addition. The titration was followed by plotting the binding isotherm (y-axis: ΔA/CD where ΔA = A − CDεD and εD is the molar absorption coefficient of the metal complex; x-axis: CP). Once the binding isotherm reached the plateau, the titration was concluded. All the spectra shown are corrected for dilution factors. The fluorescent titrations followed the changes in the emission of the fluorescent species upon increasing the addition of the quencher. The procedure is similar to that described for the absorbance experiments, where instead of ΔA, ΔF = F − CDφD, and φD is a proportional constant similar to ε. Finally, the data were treated with the appropriate equations or software to obtain the desired thermodynamic parameters. The HypSpec® software (https://www.hyperquand.co.uk) enabled the simultaneous fit over a range of wavelengths through a least squares procedure and according to different models of multiple equilibria. Three replicates for each titration were done.Mass spectrometry
For the mass spectrometry measurements, all samples were prepared in LC-MS grade solvents or solutions. The high-resolution ESI mass spectra were recorded using an AB SCIEX Triple TOF 5600+ (Sciex, Framingham, MA, USA), equipped with a DuoSpray® interface operating with an ESI probe. All ESI mass spectra were acquired through direct injection at 5 μL min−1 flow rate. The BSA solutions for mass spectrometry experiments were prepared in 20 mM NH4OAc, pH = 7.0, and finally diluted in LC-MS grade H2O containing 0.1% v/v of formic acid (FoA), obtaining a BSA concentration of approximately 0.5 μM. Solutions of BSA containing the studied metal complexes were similarly prepared in various concentration ratios from 1:1 to metal complex excess of 3:1, incubated at 10 μM for 4 h at 37.0 °C. The ESI source parameters were optimized for proteins as follows: positive polarity, ion spray voltage floating of 5500 V, temperature of 25 °C, ion source gas 1 (GS1) of 45 L min−1; ion source gas 2 (GS2) of 0 L min−1; curtain gas (CUR) of 12 L min−1, declustering potential (DP) of 150 V, collision energy (CE) of 10 V; and acquisition range of 1000–2600 m/z. For acquisition, the Analyst TF software 1.7.1 (Sciex) was used and deconvoluted spectra were obtained using the Bio Tool Kit micro-application v.2.2 embedded in the PeakView™ software v.2.2 (Sciex).
Native PAGE gel electrophoresis
Native polyacrylamide gel electrophoresis (PAGE) tests were performed by incubating BSA (3 μM) overnight with different concentrations of metal complexes for [complex]/[protein] concentration ratios of 5, 10 and 20 in 0.1 M NaCl, 2.5 mM NaCac buffer pH = 7.0 and T = 37.0 °C. Vehicle-treated BSA samples were included as a negative control with different percentages of DMSO used. After that, 6 μL of sample buffer 2× (0.01% bromophenol blue and 20% glycerol in 0.5 M Tris HCl buffer, pH = 6.8) was added to 6 μL of the sample solutions (final CBSA = 1.5 μM) and loaded onto 10% polyacrylamide gels. Before loading the samples, a pre-run was done at 200 V for 1 h to remove any possible impurity from the gel. Gels were run in native PAGE buffer of 250 mM Tris base, 1.92 M glycine, pH = 8.3 at 6.6 V cm−1 for 4 h at 4 °C to avoid denaturation of the protein. Finally, the gels were stained with Coomassie Brilliant Blue R-250.Melting
The thermal denaturation process was followed by spectrophotometry using a Shimadzu UV-2450 spectrophotometer and a Peltier thermostat. Melting experiments were performed in triplicate following the absorbance changes (260 nm for CT-DNA and RNA structures, 290 nm for G4 and i-motif) at increasing temperatures ranging from 25 °C to 90 °C (no incubation time). The scan rate was +5 °C min−1 every 7.5 min, and the solution was kept at the current temperature for 5 min before the spectrum was recorded (1.5 min for recording). To reach the following temperature, 1 min was needed. The parameters on the thermostat were as follows: star T = 25 °C; hold time = 360 s; speed T = 5 °C; next T = 37 °C; pre control = 390 s; and repeat time = 14. Cuvettes with a path length of 1.0 cm, 2.0 mm or 1.0 mm were used depending on the experiment. A short path length was needed in particular for the oligos (DNA G4s and i-motif) melting tests. The percentage absorbance change (%A change = 100 × (A(T) − A0)/(A∞ − A0), where A(T) is the absorbance read at each temperature T (°C), A0 the absorbance corresponding to the initial plateau and A∞ the absorbance for the final plateau), was plotted against temperature. In this way, a sigmoidal curve was obtained according to eqn (3).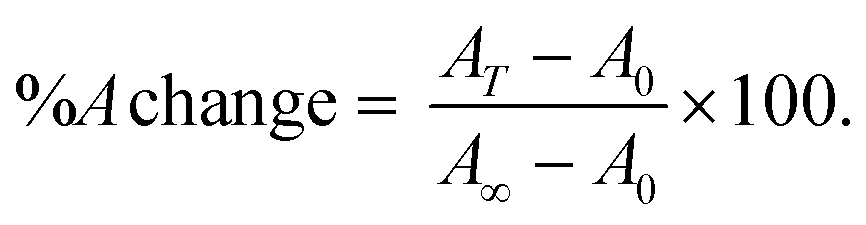 | (3) |
The melting temperature (Tm) was derived as the maximum of the first derivative of the sigmoidal curve. The concentration of the polynucleotide was around 20 μM and the metal complex was placed in an equal amount for the first experiment. If the stabilisation was too high, lower complex concentrations could be investigated. All the experiments were performed by modulating the optimal ionic strength conditions so that the melting temperature of the polynucleotide alone was around 50–60 °C. These conditions allowed the appreciation of a possible enhancement in Tm. The relevant reference melting temperatures are summarised in Table S9.†
Viscosity
The viscometer used was a semi-micro type “Cannon-Ubbelohde” capillary viscometer. To carry out the measurement, 3.0 mL of buffer solution (0.1 M NaCl, 2.5 mM NaCac, pH = 7.0) volume was taken with a glass pipette and inserted into the viscosimeter, which reached the lower tank. The viscometer was placed in a water bath at a constant temperature of 25.0 ± 0.1 °C and left to rest for 15 min to allow the solution to reach the desired temperature. The solution was sucked up with a small rubber pump until the liquid almost reached the exit. After that, DNA was added with a Hamilton micro-syringe connected to a Mitutoyo micrometric screw (the same as the spectroscopic titrations) to have a CDNA = 118 μM and the solution was mixed by pushing the liquid up and down. Each addition of the tested compound was performed the same way as the addition of DNA, like a titration (no incubation time). The flow time was measured with a digital stopwatch. The operation was carried out for the solvent used (measuring tsolv), for the solvent + DNA (tDNA) mixture and for different solvent + DNA + molecule mixtures (tsample). The flow times were measured at least six times and the average flow time was considered. After each use, a washing cycle was carried out, as follows: water–acetone–water–ethanol–N2. The relative viscosity of the polynucleotide was calculated as follows (eqn (4)):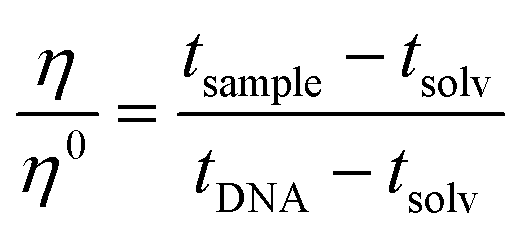 | (4) |
The relative viscosity is related to the polynucleotide elongation by L/L0 = (η/η0)1/3, where L is the length of the bound polynucleotide and L0 is the length of the free one.
Agarose gel electrophoresis
Agarose gel electrophoresis of pUC18 was performed after overnight incubation of the plasmid (6.52 μM, base pairs) at 37.0 °C in the presence of different increasing concentrations of the tested metal complex or cisplatin in buffer (0.1 M NaCl, 2.5 mM NaCac, pH = 7.0). A vehicle-treated pUC18 sample was included with the maximum DMSO concentration used in the electrophoresis experiment. After 24 h of incubation at 37.0 °C, loading buffer (4 μL) was added to each sample (20 μL) before being loaded onto 1% agarose gel. Electrophoresis was run at 6.5 V cm−1 for 130 min. After the run, the gel was stained with 1 μg mL−1 solution of ethidium bromide in TAE 1× (buffer solution containing a mixture of Tris base, acetic acid and EDTA) for 1 h. Finally, the gel was visualized by exposure to UV light (312 nm) using a Gel Doc XR + Imaging System (Bio-Rad).FRET melting
The FRET melting assay was performed in a real-time polymerase chain reaction (7500 Fast Real-Time PCR, Applied Biosystems) with the oligonucleotides described in Table S10.† Here, the melting of the oligonucleotide was determined by the increase in the fluorescence of FAM with an increase in temperature. The sigmoidal fit of the normalised FAM fluorescence against temperature provided the melting temperature as the inflexion point of the curve. DMSO and meso-tetrakis(N-methyl-4-pyridyl)porphyrin (TMPyP4) were used in the vehicle and positive control experiments, respectively. Stock solutions of the oligonucleotides in 10 mM KCl, 90 mM LiCl, and 10 mM LiCac were heated to 92 °C, kept at that temperature for 6 min, and cooled by immediately immersing them in ice to perform annealing just before the experiment. 12 × 8-well optical tube strips were filled with 25 μL each sample (20 μL oligonucleotide + 5 μL metal complex), providing a final oligonucleotide concentration of 0.2 μM, with the metal complex concentration of 0.2, 1.0, 2.0 and 4.0 μM (CD/CP = 1, 5, 10, and 20), and for the positive control TMPyP4, a concentration of 2.0 μM (CD/CP = 10), respectively. The samples were heated from 25 °C to 95 °C at a rate of 1 °C min−1. The FAM fluorescence intensity (λexc = 492 nm, λem = 516 nm) was recorded at 0.4 °C intervals. The experiments were performed in 10 mM KCl, 90 mM LiCl, and 10 mM LiCac with no incubation. The difference between the mid-transition temperature of the oligonucleotide with and without the drug (ΔTm) was calculated.In silico studies. PCM calculations
The initial structures of the Ag(I) and Au(I) complexes were optimized in water as an implicit solvent at the ωb97XD/Def2TZVP level of theory, as suggested in previous benchmark works.63,64 The polarizable continuum model (PCM) was used to account for long-range solvation effects.65–67 All calculations were carried out with the Gaussian16 software.Target set up
The target DNA G-quadruplex structures (G4s), i.e., Tel-23, CTA-22, and c-myc, were obtained from the Protein Data Bank (PDB) with PDB IDs 2JSM,682KM3,69 and 1XAV,70 respectively. In the case of CTA-22 and Tel-23, two K+ atoms were added between the tetrads. Each structure was solvated with a truncated octahedron TIP4P-Ew water box,71 as suggested in a recent work conducted on a G-quadruplex system.72 The parm-BSC1 force field was used for the G4 structures.73 Then, the solvated system was electrically neutralized with 0.10 M KCl and subjected to two minimization cycles of 5000 steps each: 2500 steps using steepest descent and 2500 steps using conjugate gradient.74 In the first cycle, we restrained the G4 heavy atoms with a 10 kcal mol−1 Å−2 harmonic potential. Finally, the entire system was minimized without constraints. A 1 ns-long heating to 300 K in an NVT ensemble was followed, restraining the movement of the G4 heavy atoms with a 10 kcal mol−1 Å−2 harmonic potential. To ensure the slow release of the restraints, we performed three different 2 ns-long NPT equilibration runs by gradually decreasing the restraints to 1 kcal mol−1 Å−2. In the first run, constraints were applied to the heavy atoms, whereas in the remaining two, only to the G4 backbone. The production run was carried out without any restraint for 1.5 μs in the NPT ensemble. All simulations were performed applying the particle mesh Ewald (PME) truncation method (with a short-range cut-off of 10 Å), an integration step of 2 fs, the SHAKE algorithm, a Langevin thermostat with a friction coefficient of 1 ps−1, and the Monte Carlo thermostat for NPT simulations. All molecular dynamics simulations were run with AMBER18.Molecular docking
Given that we did not know “a priori” the binding site, we performed blind docking with the AutoDock v4.2.2 program suite.75 In particular, a search grid was centred on the Cartesian coordinates of the centre of mass of the G4 structure and defined by a box large enough to include the whole receptor macromolecule (120 × 120 × 120 points in x, y, and z directions with a spacing of 0.375 Å). The ligand molecules were treated as flexible during docking to allow for structural changes, while the G4 target was kept frozen in its original conformation throughout the docking procedure. The search was carried out with a Lamarckian genetic algorithm: a total of 200 docking runs for each target–ligand complex were carried out with a maximum of 25000000 energy evaluations. All other parameters were set to default values.
Conclusions
A new biscarbene complex of Ag(I) was synthesised and characterised. Also, some tests were performed on its Au(I) counterpart to evidence the role of the metal centre. The higher cytotoxic activity of the metal complexes compared to cisplatin can possibly be correlated with their cellular uptake and lipophilicity. Alternatively, the differences in the cytotoxicity between the two complexes were not correlated with their cellular uptake, indicating the importance of a different mode of action depending on the metal centre. Both [Ag(BIA)2]+ and [Au(BIA)2]+ could overcome the cisplatin resistance in ovarian cancer cells but the silver complex showed better results with a lower resistance factor and higher selectivity for cancer cells.[Ag(BIA)2]+ showed high equilibrium constants (higher affinity with respect to gold) for protein and the non-canonical structures of DNA (see Table S11†), with the latter being possible targets for this drug. Interestingly, a non-common and strong binding towards an i-motif was observed here for Ag(I)–NHCs. The same occurred for all parallel (c-myc), anti-parallel (CTA-22) and hybrid (Tel-23) G4 topologies, with different features. The dramatic changes in the anthracene spectroscopic profiles upon oligonucleotides binding indicated that these moieties are strongly involved in the reaction. This is true for both G4s and the i-motif. No similar discussion is possible for the wavelengths correlated with the ligand–metal coordination, given that they overlap too much with the DNA absorbance range.76
The computational data for G4s agreed with the experimental data. The stacking interaction with the thymine residue on the loops was found to be the preferred binding mode for c-myc (the lowest value among the three G4s), while for Tel-23, groove binding was the most stable pose. In the case of CTA-22, the intermediate situation was found, in accordance with the melting data. Actually, the binding energies found for these metal-G4 complexes were estimated; thus, molecular dynamics simulations are necessary to better describe the ligand–receptor interactions.
Provided that the results are different for the different biosubstrates and considering the output of the theoretical calculations, the anthracenes seem to play a major role by interacting with the loops and the grooves of the oligos. Indeed, simple stacking over the G-tetrads or C-dyads would likely be much less tunable.77 This seems to be the case where oppositely, the compatibility between the geometrical constraints for both metal complexes and oligo grooves/loops plays a major role.78 In this frame, it should be noted that the metal centre and the presence of two anthracenes will be fundamental to drive some peculiar geometrical arrangement.
The few experiments on [Au(BIA)2]+ showed an interaction with double-strand DNA of low interest given that aggregation on DNA may be the major binding mode. Only FRET experiments gave positive results, where a strong interaction with mitochondrial G4 (Fmt9438T) was found. The computational data demonstrated a slightly different geometry compared to its silver analogue and a different preferential binding mode with c-myc emerged. As a gold compound, the affinity for cysteine residues of the proteins was preserved, showing the formation of a kinetic adduct at short incubation times, with two gold complexes in an Au⋯S⋯Au coordination geometry, with the loss of one of the two carbene ligands.
It is worth noting that the complexes studied in our previous work,26 with an ethyl carbon chain on the imidazole, instead of a butyl one, demonstrated different behaviour from two different points of view. Firstly, the silver biscarbene could not bind the G4 structure, validating our hypothesis that a longer carbon chain could enhance the affinity for this non-canonical DNA structure. Secondly, the binding with the serum albumin could not form covalent binding with the protein, and the driving force of the whole interaction was found to be based on electrostatics (related to lower hydrophobicity), while in this study, the silver biscarbene covalently binds the Cys34 residue with the loss of one carbene ligand.
Notably, this is the first example of a silver carbene that can bind G4s and i-motif structures. This can put into play new mechanisms of action that can improve the efficiency of anticancer drugs.
Author contributions
Conceptualisation: F.B., E.G., G.S., A.P., T.B.; data curation: F.B., E.G., G.S. and D.C.; formal analysis: F.B, E.G., G.S., D.C., M.S., A.D. and L.G.; funding acquisition: F.B, E.G., G.S., A.P. and T.B.; investigation: F.B, E.G., G.S., D.C., M.S., A.D. and L.G.; methodology: D.C., N.B., B.G., A.P and T.B.; supervision: D.C., N.B., B.G., A.P. and T.B.; writing – original draft: F.B., E.G. and G.S.; writing – review and editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FB, EG and GS thank the DSCM and DCCI of the University of Pisa for financial support with the interdisciplinary project PRIMULE (PRomoting Innovative and MULtidisciplinary Experiments at DSCM). This contribution is part of the work from COST Action CA18202, NECTAR Network for Equilibria and Chemical Thermodynamics Advanced Research, supported by COST (European Cooperation in Science and Technology). AP gratefully acknowledges funding by the University of Pisa under the “PRA–Progetti di Ricerca di Ateneo” (Institutional Research Grants), project no. PRA_2022–2023_12 “New challenges of transition metal and lanthanide complexes in the perspective of green chemistry”.References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed.
- A. Citta, E. Schuh, F. Mohr, A. Folda, M. L. Massimino, A. Bindoli, A. Casini and M. P. Rigobello, Metallomics, 2013, 5, 1006 CrossRef CAS PubMed.
- W. Streciwilk, A. Terenzi, F. Lo Nardo, P. Prochnow, J. E. Bandow, B. K. Keppler and I. Ott, Eur. J. Inorg. Chem., 2018, 2018, 3104–3112 CrossRef CAS.
- C. K. Mirabelli, R. K. Johnson, M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS.
- Available at, https://classic.clinicaltrials.gov/ct2/results?term=auranofin&Search=Apply&age_v=&gndr=&type=&rslt=.
- M. Kupiec, R. Ziółkowski, L. Massai, L. Messori and K. Pawlak, J. Inorg. Biochem., 2019, 198, 110714 CrossRef CAS PubMed.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Sci. Rep., 2019, 9, 12335 CrossRef PubMed.
- M. P. Sullivan, H. U. Holtkamp and C. G. Hartinger, in Metallo-Drugs: Development and Action of Anticancer Agents, ed. A. Sigel, H. Sigel, E. Freisinger and R. K. O. Sigel, De Gruyter, 2018, pp. 351–386 Search PubMed.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- S. K. Raju, A. Karunakaran, S. Kumar, P. Sekar, M. Murugesan and M. Karthikeyan, Ger. J. Pharm. Biomater., 2022, 1, 06–28 CrossRef.
- S. B. Aher, P. N. Muskawar, K. Thenmozhi and P. R. Bhagat, Eur. J. Med. Chem., 2014, 81, 408–419 CrossRef CAS PubMed.
- N. Pandya, S. R. Bhagwat and A. Kumar, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1876, 188594 CrossRef CAS PubMed.
- V. Bernat and M. D. Disney, Neuron, 2015, 87, 28–46 CrossRef CAS PubMed.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. de Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS PubMed.
- S. Gama, I. Rodrigues, F. Mendes, I. C. Santos, E. Gabano, B. Klejevskaja, J. Gonzalez-Garcia, M. Ravera, R. Vilar and A. Paulo, J. Inorg. Biochem., 2016, 160, 275–286 CrossRef CAS PubMed.
- C. Nakanishi and H. Seimiya, Biochem. Biophys. Res. Commun., 2020, 531, 45–50 CrossRef CAS PubMed.
- T. Kench and R. Vilar, in Annual Reports in Medicinal Chemistry, Elsevier, 2020, vol. 54, pp. 485–515 Search PubMed.
- S. M. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. C. Gerner, K. G. Schmetterer, G. Del Favero, R. Bonsignore, M. Cichna-Markl, G. Koellensperger, A. Casini and C. Gerner, Chem. – Eur. J., 2020, 26, 15528–15537 CrossRef CAS PubMed.
- L. Martino, B. Pagano, I. Fotticchia, S. Neidle and C. Giancola, J. Phys. Chem. B, 2009, 113, 14779–14786 CrossRef CAS PubMed.
- P. Alberti, J. Ren, M. P. Teulade-Fichou, L. Guittat, J.-F. Riou, J. B. Chaires, C. Hélène, J.-P. Vigneron, J.-M. Lehn and J.-L. Mergny, J. Biomol. Struct. Dyn., 2001, 19, 505–513 CrossRef CAS PubMed.
- S. Shi, X. Geng, J. Zhao, T. Yao, C. Wang, D. Yang, L. Zheng and L. Ji, Biochimie, 2010, 92, 370–377 CrossRef CAS PubMed.
- S. Shi, J. Zhao, X. Geng, T. Yao, H. Huang, T. Liu, L. Zheng, Z. Li, D. Yang and L. Ji, Dalton Trans., 2010, 39, 2490 RSC.
- H. Xu, H. Zhang and X. Qu, J. Inorg. Biochem., 2006, 100, 1646–1652 CrossRef CAS PubMed.
- A. S. Tikhomirov, M. A. S. Abdelhamid, G. Y. Nadysev, G. V. Zatonsky, E. E. Bykov, P. J. Chueh, Z. A. E. Waller and A. E. Shchekotikhin, J. Nat. Prod., 2021, 84, 1617–1625 CrossRef CAS PubMed.
- F. Binacchi, F. Guarra, D. Cirri, T. Marzo, A. Pratesi, L. Messori, C. Gabbiani and T. Biver, Molecules, 2020, 25, 5446 CrossRef CAS PubMed.
- M. G. Fabbrini, D. Cirri, A. Pratesi, L. Ciofi, T. Marzo, A. Guerri, S. Nistri, A. Dell'Accio, T. Gamberi, M. Severi, A. Bencini and L. Messori, ChemMedChem, 2019, 14, 182–188 CrossRef CAS PubMed.
- F. Macii and T. Biver, J. Inorg. Biochem., 2021, 216, 111305 CrossRef CAS PubMed.
- M. H. Rahman, T. Maruyama, T. Okada, K. Yamasaki and M. Otagiri, Biochem. Pharmacol., 1993, 46, 1721–1731 CrossRef CAS PubMed.
- S. Awasthi and N. T. Saraswathi, RSC Adv., 2016, 6, 90739–90753 RSC.
- I. Rombouts, B. Lagrain, K. A. Scherf, M. A. Lambrecht, P. Koehler and J. A. Delcour, Sci. Rep., 2015, 5, 12210 CrossRef CAS PubMed.
- S. Sen, M. W. Perrin, A. C. Sedgwick, V. M. Lynch, J. L. Sessler and J. F. Arambula, Chem. Sci., 2021, 12, 7547–7553 RSC.
- A. A. Isab, C. F. Shaw, J. D. Hoeschele and J. Locke, Inorg. Chem., 1988, 27, 3588–3592 CrossRef CAS.
- M. T. Coffer, C. F. Shaw, M. K. Eidsness, J. W. Watkins and R. C. Elder, Inorg. Chem., 1986, 25, 333–339 CrossRef.
- A. Pratesi, D. Cirri, L. Ciofi and L. Messori, Inorg. Chem., 2018, 57, 10507–10510 CrossRef CAS PubMed.
- A. Pratesi, D. Cirri, D. Fregona, G. Ferraro, A. Giorgio, A. Merlino and L. Messori, Inorg. Chem., 2019, 58, 10616–10619 CrossRef CAS PubMed.
- C. Zoppi, L. Massai, D. Cirri, C. Gabbiani, A. Pratesi and L. Messori, Inorg. Chim. Acta, 2021, 520, 120297 CrossRef CAS.
- D. Cirri, L. Massai, C. Giacomelli, M. L. Trincavelli, A. Guerri, C. Gabbiani, L. Messori and A. Pratesi, Dalton Trans., 2022, 51, 13527–13539 RSC.
- A. Giorgio and A. Merlino, Coord. Chem. Rev., 2020, 407, 213175 CrossRef CAS.
- J. Xiao and C. F. Shaw, Inorg. Chem., 1992, 31, 3706–3710 CrossRef CAS.
- C. Bazzicalupi, A. Bencini, A. Bianchi, T. Biver, A. Boggioni, S. Bonacchi, A. Danesi, C. Giorgi, P. Gratteri, A. M. Ingraín, F. Secco, C. Sissi, B. Valtancoli and M. Venturini, Chem. – Eur. J., 2008, 14, 184–196 CrossRef CAS PubMed.
- G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660–672 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- J. D. McGhee and P. H. Von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS PubMed.
- N. Busto, J. Valladolid, C. Aliende, F. A. Jalón, B. R. Manzano, A. M. Rodríguez, J. F. Gaspar, C. Martins, T. Biver, G. Espino, J. M. Leal and B. García, Chem. – Asian J., 2012, 7, 788–801 CrossRef CAS PubMed.
- T. Biver, F. Secco, M. R. Tinè, M. Venturini, A. Bencini, A. Bianchi and C. Giorgi, J. Inorg. Biochem., 2004, 98, 1531–1538 CrossRef CAS PubMed.
- B. K. Paul and N. Guchhait, J. Phys. Chem. B, 2011, 115, 11938–11949 CrossRef CAS PubMed.
- S. Ramakrishnan and M. Palaniandavar, J. Chem. Sci., 2005, 117, 179–186 CrossRef CAS.
- L. Wang, G. Zhang, J. Pan, C. Xiong and D. Gong, J. Photochem. Photobiol., B, 2014, 141, 253–261 CrossRef CAS PubMed.
- A. R. Rubio, R. González, N. Busto, M. Vaquero, A. L. Iglesias, F. A. Jalón, G. Espino, A. M. Rodríguez, B. García and B. R. Manzano, Pharmaceutics, 2021, 13, 1540 CrossRef CAS PubMed.
- J. Amato, G. Miglietta, R. Morigi, N. Iaccarino, A. Locatelli, A. Leoni, E. Novellino, B. Pagano, G. Capranico and A. Randazzo, J. Med. Chem., 2020, 63, 3090–3103 CrossRef CAS PubMed.
- S. Mohammadi, Z. Narimani, M. Ashouri, R. Firouzi and M. H. Karimi-Jafari, Sci. Rep., 2022, 12, 410 CrossRef CAS PubMed.
- R. E. Amaro, J. Baudry, J. Chodera, Ö. Demir, J. A. McCammon, Y. Miao and J. C. Smith, Biophys. J., 2018, 114, 2271–2278 CrossRef CAS PubMed.
- W. Evangelista Falcon, S. R. Ellingson, J. C. Smith and J. Baudry, J. Phys. Chem. B, 2019, 123, 5189–5195 CrossRef CAS PubMed.
- P. Murat, Y. Singh and E. Defrancq, Chem. Soc. Rev., 2011, 40, 5293 RSC.
- H. Mach, C. R. Middaugh and R. V. Lewis, Anal. Biochem., 1992, 200, 74–80 CrossRef CAS PubMed.
- G. Felsenfeld and S. Z. Hirschman, J. Mol. Biol., 1965, 13, 407–427 CrossRef CAS PubMed.
- T. Biver, F. Secco and M. Venturini, Arch. Biochem. Biophys., 2005, 437, 215–223 CrossRef CAS PubMed.
- A. R. Rubio, N. Busto, J. M. Leal and B. García, RSC Adv., 2016, 6, 101142–101152 RSC.
- X. Long and M. D. Stone, PLoS One, 2013, 8, e83420 CrossRef PubMed.
- D. Cirri, A. Geri, L. Massai, M. Mannelli, T. Gamberi, F. Magherini, M. Becatti, C. Gabbiani, A. Pratesi and L. Messori, Molecules, 2023, 28, 1050 CrossRef CAS PubMed.
- A. Menconi, T. Marzo, L. Massai, A. Pratesi, M. Severi, G. Petroni, L. Antonuzzo, L. Messori, S. Pillozzi and D. Cirri, BioMetals, 2021, 34, 867–879 CrossRef CAS PubMed.
- G. Ciancaleoni, S. Rampino, D. Zuccaccia, F. Tarantelli, P. Belanzoni and L. Belpassi, J. Chem. Theory Comput., 2014, 10, 1021–1034 CrossRef CAS PubMed.
- Y. Minenkov, Å. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526 RSC.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. T. Phan, V. Kuryavyi, K. N. Luu and D. J. Patel, Nucleic Acids Res., 2007, 35, 6517–6525 CrossRef CAS PubMed.
- K. W. Lim, P. Alberti, A. Guédin, L. Lacroix, J.-F. Riou, N. J. Royle, J.-L. Mergny and A. T. Phan, Nucleic Acids Res., 2009, 37, 6239–6248 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Biochemistry, 2005, 44, 2048–2058 CrossRef CAS PubMed.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS PubMed.
- M. Castelli, F. Doria, M. Freccero, G. Colombo and E. Moroni, J. Chem. Theory Comput., 2022, 18, 4515–4528 CrossRef CAS PubMed.
- I. Ivani, P. D. Dans, A. Noy, A. Pérez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goñi, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpí, C. González, M. Vendruscolo, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Methods, 2016, 13, 55–58 CrossRef CAS PubMed.
- J. D. Schmit, N. L. Kariyawasam, V. Needham and P. E. Smith, J. Chem. Theory Comput., 2018, 14, 1823–1827 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- L. Messori, L. Marchetti, L. Massai, F. Scaletti, A. Guerri, I. Landini, S. Nobili, G. Perrone, E. Mini, P. Leoni, M. Pasquali and C. Gabbiani, Inorg. Chem., 2014, 53, 2396–2403 CrossRef CAS PubMed.
- T. Santos, G. F. Salgado, E. J. Cabrita and C. Cruz, Pharmaceuticals, 2021, 14, 769 CrossRef CAS PubMed.
- A. Arora and S. Maiti, J. Phys. Chem. B, 2008, 112, 8151–8159 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, stability and solubility in aqueous solution, cellular tests, BSA interaction, DNA/RNA interaction, G-quadruplexes, experimental details, K summary table. See DOI: https://doi.org/10.1039/d4dt00851k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |