
The cubic structure of Li3As stabilized by substitution – Li8TtAs4 (Tt = Si, Ge) and Li14TtAs6 (Tt = Si, Ge, Sn) and their lithium ion conductivity†
Martin
Schmid
,
Florian
Wegner
,
Claudia
De Giorgi
,
Florian
Pielnhofer
 and
Arno
Pfitzner
*
and
Arno
Pfitzner
*
Institut für Anorganische Chemie, Universität Regensburg, Universitätsstrasse 31, 93053 Regensburg, Germany. E-mail: Arno.Pfitzner@chemie.uni-regensburg.de
First published on 23rd April 2024
Abstract
The new lithium arsenidotetrelates Li8SiAs4, Li8GeAs4, Li14SiAs6, Li14GeAs6 and Li14SnAs6 were synthesized via ball milling and structurally characterized by Rietveld analysis of X-ray powder diffraction data. The aliovalent substitution of lithium in hexagonal Li3As by introducing a tetravalent tetrel cation stabilizes cubic structures for Li8TtAs4 (Tt = Si, Ge) in the space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) and for the lithium richer compound Li14TtAs6 (Tt = Si, Ge, Sn) in the higher symmetrical space group Fmm (no. 225). Thermal properties of the arsenidotetrelates were investigated via high temperature powder diffraction and differential thermal analysis revealing a decomposition process of the lithium richer arsenidotetrelate (Li14TtAs6 → Li8TtAs4 + 2Li3As) into the lithium poorer arsenidotetrelates and lithium arsenide at moderate temperatures. Impedance spectroscopy shows moderate to good lithium ion conductivity for the lithium arsenidotetrelates.
and for the lithium richer compound Li14TtAs6 (Tt = Si, Ge, Sn) in the higher symmetrical space group Fmm (no. 225). Thermal properties of the arsenidotetrelates were investigated via high temperature powder diffraction and differential thermal analysis revealing a decomposition process of the lithium richer arsenidotetrelate (Li14TtAs6 → Li8TtAs4 + 2Li3As) into the lithium poorer arsenidotetrelates and lithium arsenide at moderate temperatures. Impedance spectroscopy shows moderate to good lithium ion conductivity for the lithium arsenidotetrelates.
Introduction
Lithium phosphidotetrelates have become an interesting and intensely discussed family of compounds over the last years, because of their interesting structural diversity and high ionic conductivity.1–3 First mentioned in the 1950s by Juza, the nitridosilicates, phosphidosilicates and arsenidosilicates received little attention.4,5 In 2016, the compounds Li2SiP2 and LiSi2P3 were synthesized and characterized independently by Johrendt et al. and by Fässler et al. who also discovered Li8SiP4 in the same year.3,6 Exchanging silicon by germanium leads to Li8GeP4 which crystallizes in two modifications, one of them is isostructural to Li8SiP4.7 In 2019 the group of Fässler reported the hitherto most lithium-rich phosphidosilicate Li14SiP6 and its high lithium conductivity up to 1 mS cm−1 at room temperature.1 Replacing silicon for germanium or tin subsequently led to the discovery of Li14GeP6 and Li14SnP6 in 2020.8 Arsenidosilicates, the corresponding heavier homologues to the phosphidosilicates, are known since the 1950s, but were only scarcely mentioned since then. Kovnir et al. synthesized the ternary compound Li1−xSn2+xAs2 (0.2 < x < 0.4) via solid-state reaction of the elements and Li0.9Ge2.9As3.1 and Li3Si7As8 by electrochemical lithiation of the tetrel arsenides GeAs and SiAs.9,10 One year later the same group reported on the silicon compounds Li2SiAs2 featuring adamantane-like Si4As10 units and LiSi3As6 build up by corner-sharing SiAs4-tetrahedra.11 In 2022 we could proof ion conductivity for Li3As.12 DFT modelling and conductivity experiments on Li3P and Li3As show significant similarities between the two compounds. Recently our group presented the lithium arsenidotrielates Li3AlAs2, Li3GaAs2 and Li3InAs2 showing, that we can exchange phosphorus by arsenic not just in binary, but also in ternary compounds.13 With the new compounds Li8TtAs4 (Tt = Si, Ge) and Li14TtAs6 (Tt = Si, Ge, Sn) discussed here we expand the family of arsenidotetrelates to higher lithium content showing similar structural features and properties like the phosphide-based compounds. These ternary phases can be understood as an aliovalent substitution of four lithium cations in Li3As by one tetrel cation (Li12As4 → Li8Tt4+As4, Tt = Si, Ge; Li18As6 → Li14Tt4+As6, Tt = Si, Ge, Sn). The formal aliovalent substitution of a tetrel cation in hexagonal Li3As forces the ternary phase into a cubic crystal system. Both crystal structures of Li8TtAs4 and Li14TtAs6 can be derived from the antifluorite structure type or alternatively can be described as a deficient Li3Bi structure type. The formal substitution of Li+ ions in Li3As by a highly charged cation resulting in a cubic structure was first described for transition metals. The systems Li–M–Pn (M = Ti, Zr, Hf; Pn = P, As, Sb, Bi) investigated by Adam and Schuster show the formation of ternary cubic phases stabilized by the substitution of lithium with four valent titanium, zirconium or hafnium. Monconduit et al. describe the same stabilizing effect for the Nb4+-cation substituting four lithium cations in Li3As leading to a cubic antifluorite-type ternary phase (Li12As4 → Li7M5+As4).14,15 The new phases in the systems Li–Tt–As (Tt = Si, Ge, Sn) show that this substitution also works for tetrels, i.e., main group elements. The crystal structures of the new arsenidotetrelates were determined by Rietveld refinement of X-ray powder diffraction (XRPD) data. High temperature XRPD and differential thermal analysis (DTA) reveal structural changes for different thermal conditions. Further, impedance spectroscopy proves good lithium ion mobility.Results and discussion
Synthesis and crystal structure determination
The synthesis of the compounds under discussion was performed via ball milling of stoichiometric amounts of the elements and subsequent annealing of the pre-reacted mixture at elevated temperatures. Samples with the composition Li8TtAs4 (Tt = Si, Ge) were taken out of the furnace and cooled in air, while ampoules containing the samples Li14TtAs6 (Tt = Si, Ge, Sn) had to be quenched faster in water to ensure phase pure samples. The X-ray powder diffraction patterns of the stoichiometric mixtures 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:6 show intense reflections which can be assigned to the product already after ball milling (Fig. S1–S3†). Li3As and unreacted tetrel are present in small amounts as side phases (broadening of reflections is due to micro crystallinity after ball milling). In case of Si, increasing numbers of ball mill cycles lead to lower intensities of Li3As. Therefore, the completion of the reaction between the elements in the milling process depends on the duration (higher energy input over time) of the ball mill experiment. For Li14GeAs6 only unreacted Ge is present after ball milling. Performing additional milling cycles showed no improvement, leading to the conclusion that the ball milled mixture needs subsequent thermal treatment for total conversion into the ternary phase. The reaction mixture with Sn shows no side phase after ball milling.
:1:6 show intense reflections which can be assigned to the product already after ball milling (Fig. S1–S3†). Li3As and unreacted tetrel are present in small amounts as side phases (broadening of reflections is due to micro crystallinity after ball milling). In case of Si, increasing numbers of ball mill cycles lead to lower intensities of Li3As. Therefore, the completion of the reaction between the elements in the milling process depends on the duration (higher energy input over time) of the ball mill experiment. For Li14GeAs6 only unreacted Ge is present after ball milling. Performing additional milling cycles showed no improvement, leading to the conclusion that the ball milled mixture needs subsequent thermal treatment for total conversion into the ternary phase. The reaction mixture with Sn shows no side phase after ball milling.
Li8SiAs4 and Li8GeAs4 are isotypic and crystalize in the cubic space group Pa (no. 218) with the lattice parameters 12.096(1) Å and 12.170(1) Å, respectively (Fig. 1 and S4†). Crystallographic data of all compounds are displayed in the ESI (Table S1†) together with displacement parameters (Table S2†) and atomic coordinates (Table S3†). All crystal structures were refined from powder diffraction data by Rietveld analysis using the corresponding isotypic phosphide structures as starting models (Fig. 3).3,7 The structures can be derived from the antifluorite structure type (Fig. 4) leading to a 2 × 2 × 2 supercell (Z = 8) which is built up by a cubic close packed arrangement of As atoms. The formation of covalent bonds between arsenic and the tetrel atom form isolated [TtAs4]8−-tetrahedra, which distort the ideal arrangement of the cubic close packed As atoms. To balance the negative charge these [TtAs4]8− tetrahedra are surrounded by 7 + 1 Li+ cations occupying all other tetrahedral voids (Li/Ge tetrahedra occupancy 7:1) and 25% of the octahedral voids. The [SiAs4]8−-tetrahedra in Li8SiAs4 are slightly distorted with interatomic distances of d(Si–As) = 2.371 Å (1×), 2.398 Å (1×) and 2.399 Å (3×). These distances are comparable to related compounds like Na5SiAs3 (2.351–2.428 Å),16 KSi3As3 (2.323–2.414 Å)17 and SiAs (2.364–2.414 Å).18 The [GeAs4]8−-tetrahedra of the isotypic compound Li8GeAs4 have interatomic distances d(Ge–As) of 2.468 Å (3×) and 2.471 Å (1×) similar to related compounds as Na5GeAs3 (2.433–2.520 Å),19 KGe3As3 (2.414–2.509 Å)20 and GeAs (2.449–2.488 Å).21 All coordination polyhedra are shown in the ESI (Fig. S5 and S6†).
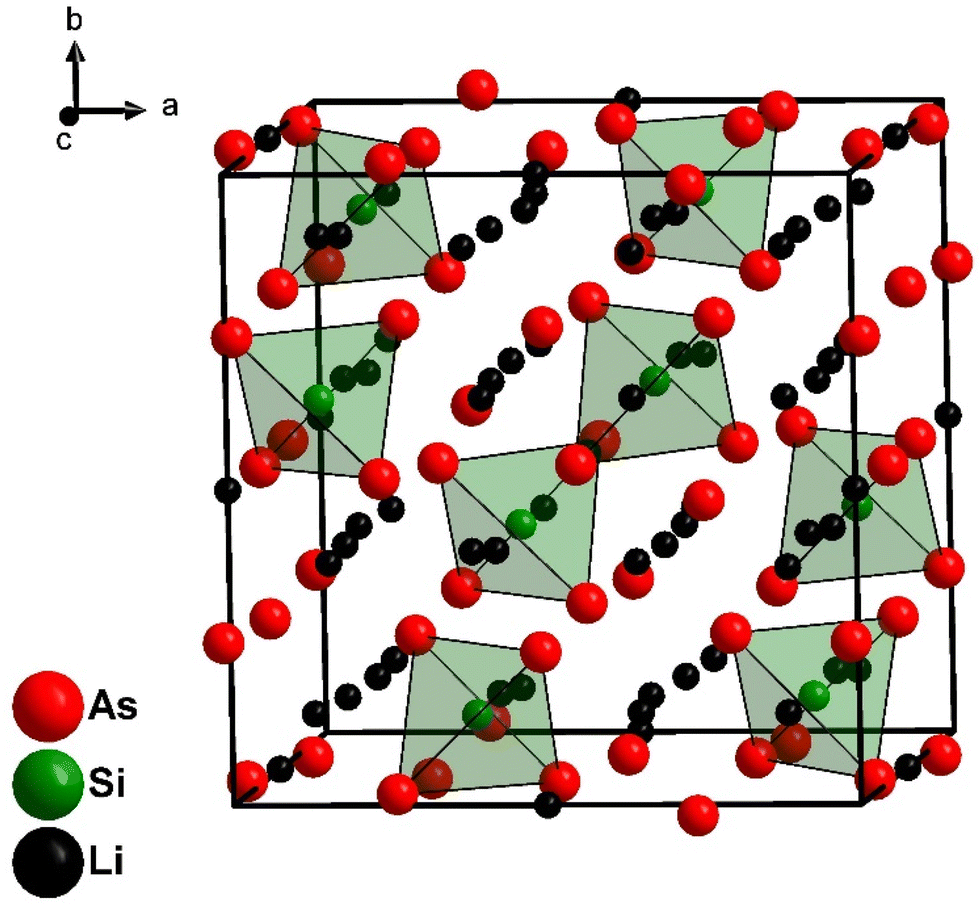 | ||
| Fig. 1 Cubic crystal structure of Li8SiAs4 with [SiAs4]8−-tetrahedra coloured in green. | ||
The lithium richer arsenidotetrelates Li14TtAs6 (Tt = Si, Ge, Sn) crystalize with even higher symmetry in space group Fmm (no. 225) with lattice parameters of a = 6.131(1) Å (Li14SiAs6), 6.143(1) Å (Li14GeAs6) and 6.193(1) Å (Li14SnAs6). XRPD patterns with Rietveld analysis are displayed in Fig. 3 for the Si compound and the corresponding Ge and Sn diffraction patterns are shown in the ESI (Fig. S8 and S9†). For Rietveld refinements the isotypic phosphidotetrelates were used as initial structure models with As localized on the P positions.18 The crystal structure can be described as a face-centered cubic lattice of arsenic atoms where all tetrahedral voids are occupied by a mixed site of Li1 and the corresponding tetrel with a ratio of tetrel/lithium 1:11 (Fig. 2). A second Li position (Li2) occupies all octahedral voids with a SOF of 0.5. A detailed discussion of the Rietveld refinement is given in the ESI.† Due to the highly symmetric crystal structure several interatomic distances are identical like the distance of Tt/Li1–Tt/Li1 and Li2–As1. Selected interatomic distances are listed in Table 1. Arsenidotetrelates and phosphidotetrelates show similar behaviour with respect to the increase of lattice parameter and interatomic distances. The lattice parameters of the arsenidotetrelates increase from Si to Ge by 0.20% and from Ge to Sn by 0.81%. A similar increase is observed for the phosphidotetrelates: from Si to Ge by 0.29% and from Ge to Sn by 1.02%. Fässler et al. assign the smaller increase of lattice parameters from Li14SiP6 to Li14GeP6 compared to Li14GeP6 to Li14SnP6 to the fact, that Ge could compensate the larger atomic radius of Ge compared to Si by its higher electronegativity.8 The overall slightly larger lattice parameters and interatomic distances of the arsenidotetrelates compared to their corresponding phosphidotetrelates can be explained with the larger atomic radius of arsenic compared to phosphorus. It must be noted that to the best of our knowledge no reliable values for the ionic radii of P3− and As3− can be found in textbooks or scientific literature.
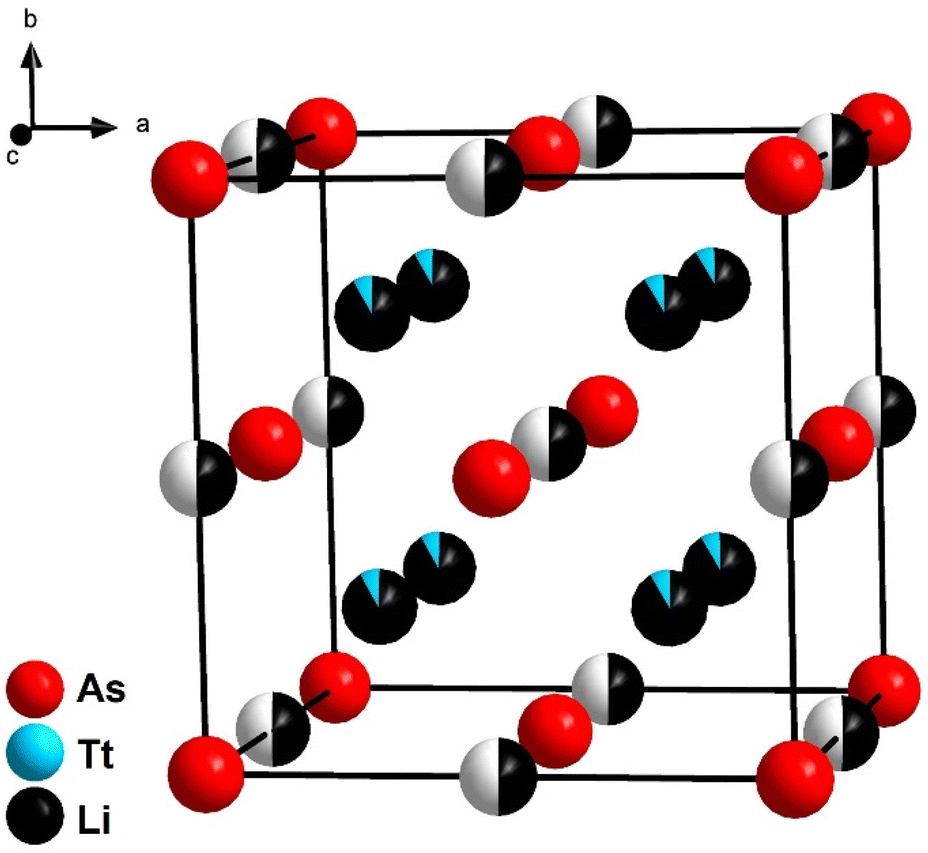 | ||
| Fig. 2 Cubic crystal structure of Li14TtAs6 (Tt = Si, Ge, Sn). Tetrahedral voids are occupied by lithium and tetrel cation in a ratio of 11:1 and the octahedral voids are half occupied by lithium. | ||
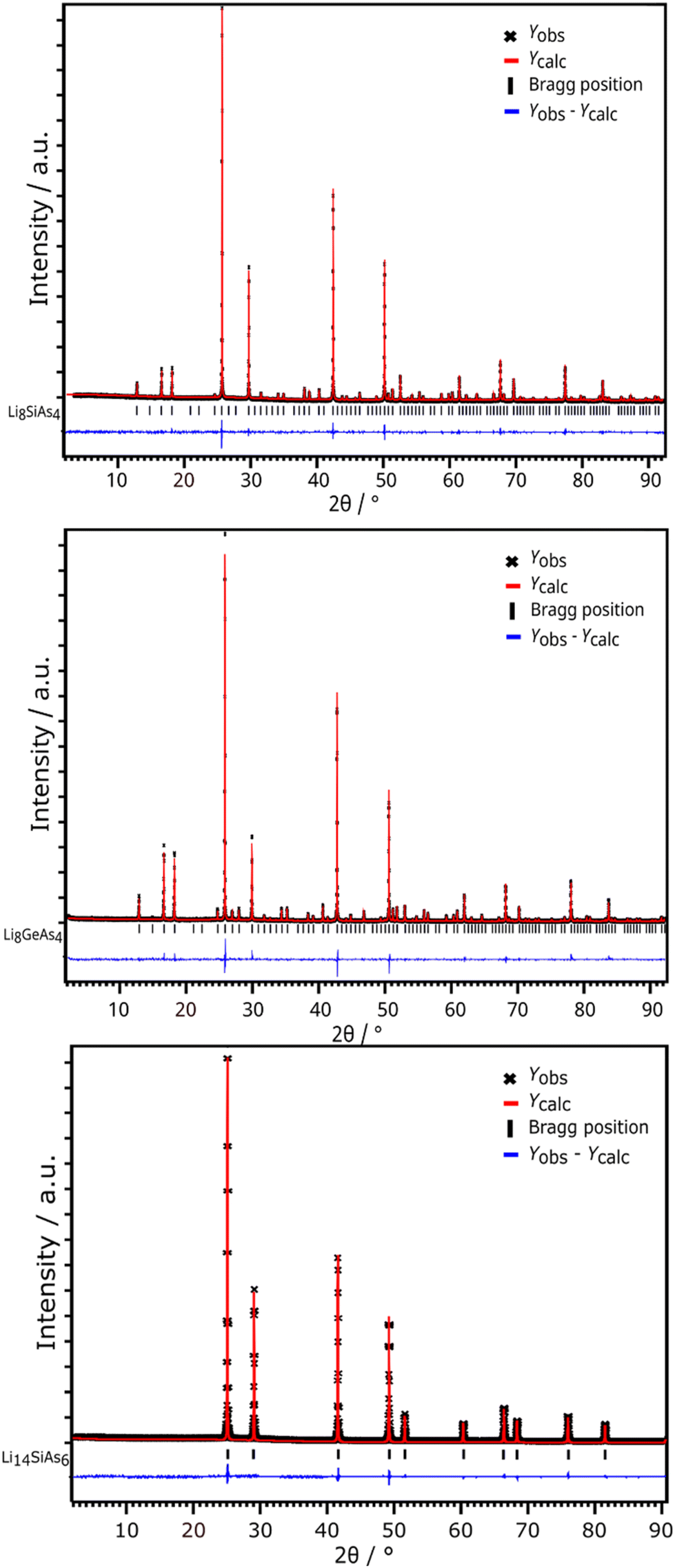 | ||
| Fig. 3 X-ray powder diffraction data with Rietveld analyses of Li8SiAs4 (top), Li8GeAs4 (middle) and Li14SiAs6 (bottom). Observed intensities Yobs are displayed with black crosses, the red line indicates the calculated intensities Ycalc, Bragg positions are marked with black lines and the difference plot (Yobs − Ycalc) is drawn in blue. | ||
| Formula | Tt/Li1–Li2/Å | As1–As1/Å | Li2–As1/Å |
|---|---|---|---|
| Li14SiAs6 | 2.654(1) | 4.335(1) | 3.065(1) |
| Li14GeAs6 | 2.660(1) | 4.343(1) | 3.071(1) |
| Li14SnAs6 | 2.681(1) | 4.379(1) | 3.096(1) |
The crystal structures of Li8TtAs4 (Tt = Si, Ge) with space group Pa (no. 218) and the crystal structures of Li14TtAs6 (Tt = Si, Ge, Sn) with space group Fmm (no. 225) are closely related and can be both derived from the antifluorite structure type (stuffed type) or alternatively from the Li3Bi structure type (defect type). The main structural difference between the lithium poorer and lithium richer arsenidotetrelates is that the lower amount of tetrel atoms in Li14TtAs6 (1 of 21 atoms) is evenly distributed over all tetrahedral voids creating mixed occupancy with lithium whereas the higher amount of tetrel atoms in Li8TtAs4 (1 of 13 atoms) is distributed in an ordered way occupying 1/8 of all tetrahedral voids by 100%. The structural relationship between Li8TtAs4 and Li14TtAs6 and the relation to the antifluorite structure type (or alternatively to the Li3Bi structure type) are visualized in Fig. 4. The ordering of tetrel cations in Li8TtAs4 reduces the space group symmetry from Fmm in Li14TtAs6 to Pa (Fmm → Pmm → Fmc → Fm → Pa). A detailed description of these symmetry relations is given by Fässler et al.7
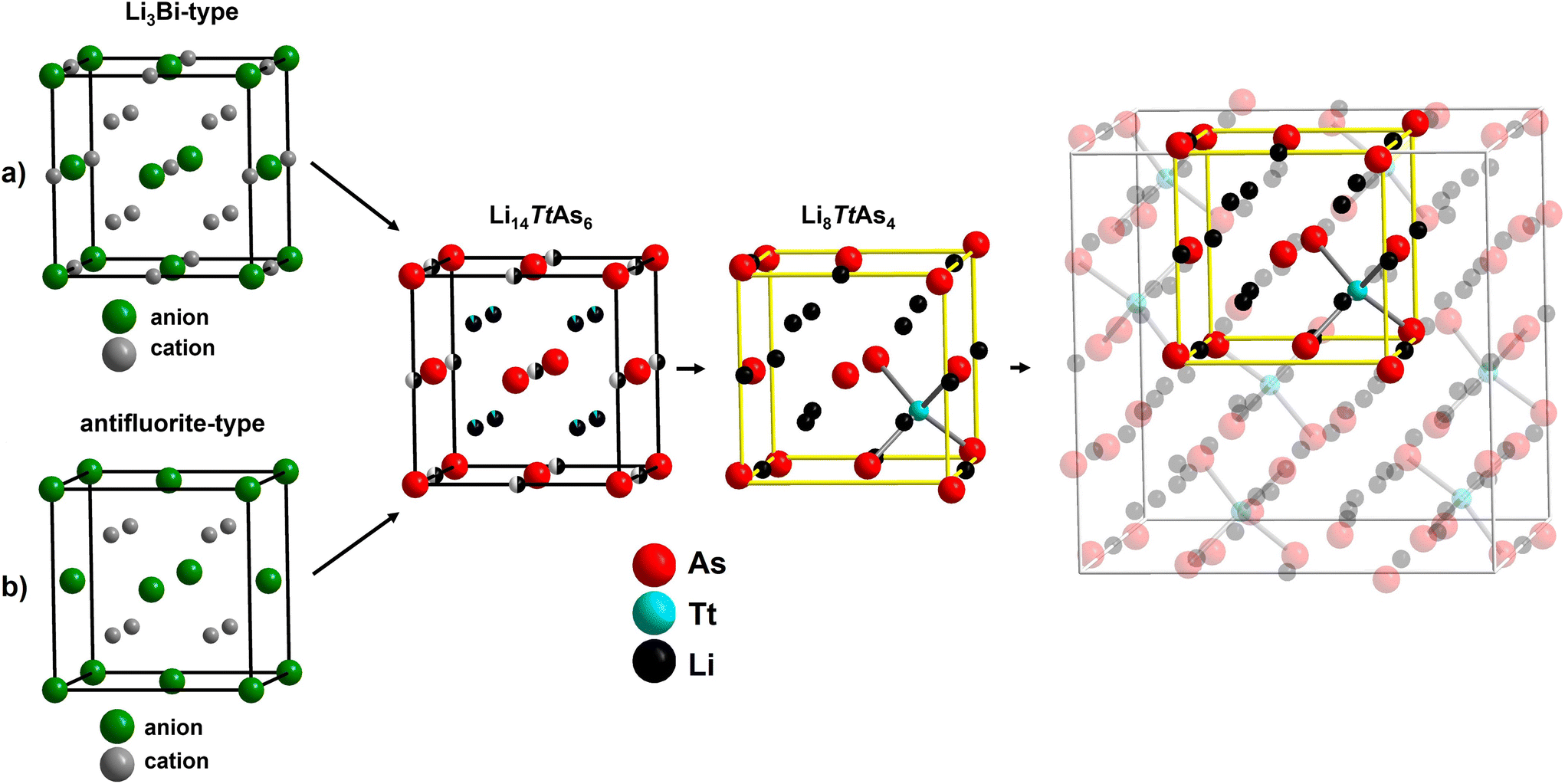 | ||
| Fig. 4 Representation of the structural relation of the compounds Li8TtAs4 and Li14TtAs6 to the antifluorite structure type and the Li3Bi structure type. (a) Li3Bi structure type, (b) antifluorite structure type, middle: structure of Li14TtAs6 which can be derived from the Li3Bi structure type by removing half of the occupation from the octahedral voids forming a half-occupied lithium position and adding a mixed position of lithium and tetrel cation in the tetrahedral voids (deficient Li3Bi structure). Second, the structure of Li14TtAs6 can be understood as a filled antifluorite structure, where the octahedral voids are half filled with lithium and tetrahedral voids are occupied with the mixed position. In the structure of Li8TtAs4 the mixed positions in the tetrahedral voids are replaced by an ordered arrangement of the tetrel cation occupying one of the eight octahedral voids in the yellow cell. The formation of ordered [TtAs4]8−-tetrahedra leads to a distortion in the ccp of the arsenide anions. The other seven tetrahedral voids are fully occupied by lithium and the lithium occupancy in the octahedral voids is reduced. Right: unit Cell of Li8TtAs4. The yellow cell (former equivalent of the unit cell of Li14TtAs6) is highlighted to underline the structural relation of both structures. The unit cell of Li8TtAs4 can be understood as a 2 × 2 × 2 supercell of eight yellow cells and an additional origin shift of (½, 0, 0). | ||
Thermal analysis
Quenching is crucial for phase pure samples of Li14TtAs6 (Tt = Si, Ge, Sn) indicating the stability of this cubic phase only at high temperatures. This is proven by high temperature powder diffraction data of previously quenched samples up to 305 °C. A decomposition process is most pronounced for Li14GeAs6 (Fig. 5) by the reformation of Li3As and Li8GeAs4 starting at 125 °C (Fig. S13†). The diffraction pattern of the Si compound shows weak intensity reflections at a temperature of 305 °C belonging to Li8SiAs4 (Fig. S10 and S12†) and the Sn compound decomposes at a temperature of 285 °C to Li3As and additional reflections which could possibly be assigned to yet unknown Li8SnAs4 (Fig. S11†). Reasonable diffraction data for Li8SnAs4 suitable for structural characterization are not yet accessible.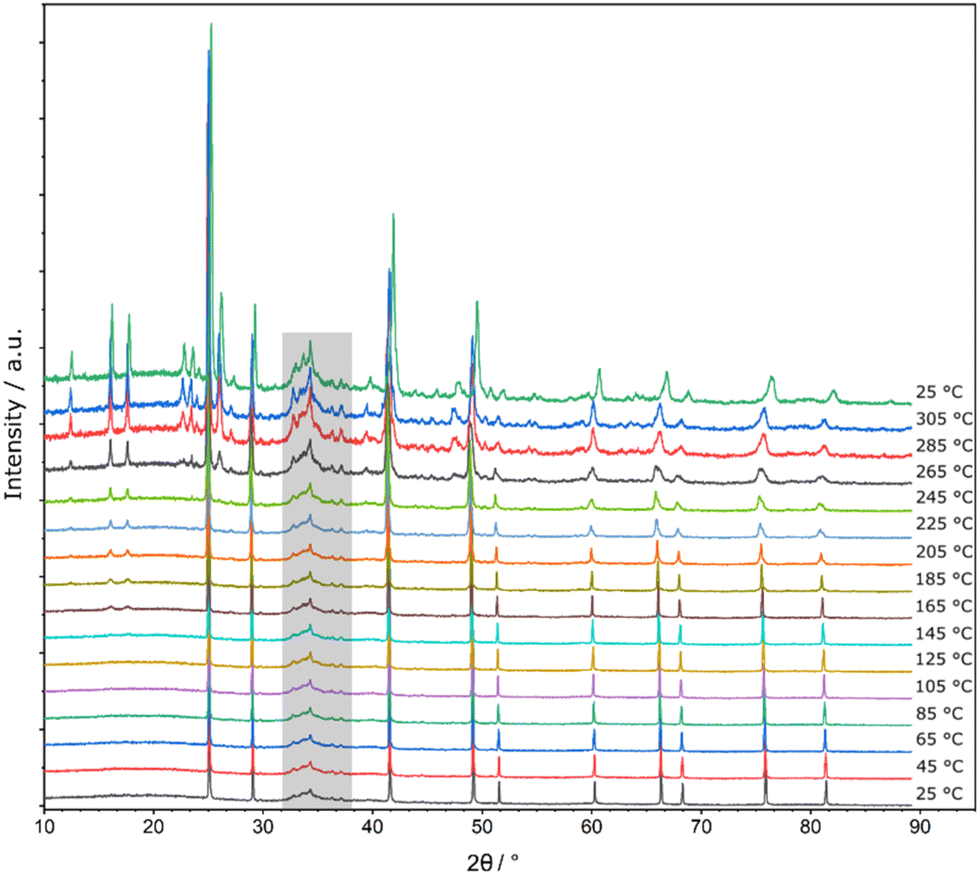 | ||
| Fig. 5 High temperature powder diffraction data (25 °C → 305 °C → 25 °C) of Li14GeAs6 in steps of 20 °C. Additional reflexes are emerging at a temperature of 165 °C which can be assigned to Li3As and Li8GeAs4. Please note that artifacts of the heating device are present in a 2θ range of 32°–38° (grey background). | ||
For temperatures above 305 °C differential thermal analysis (DTA) (Fig. S14–S16†) was performed showing significant and clear endothermal effects for Li14SiAs6 at an onset point of 489 °C (Li14GeAs6 at 464 °C, and Li14SnAs6 at 533 °C) in the heating cycles and exothermal effects in the cooling cycles at onset points of 473 °C and 442 °C for Li14SiAs6 (Li14GeAs6 at 480 °C and Li14SnAs6 at 534 °C and 465 °C). We assume a behaviour similar to the recently reported homologous phosphides, for which the following interpretation was suggested.18 The endothermal effects can be explained by a reformation process of Li3As and Li8TtAs4 to Li14TtAs6 after gradual decomposition of Li14TtAs6 during the previous annealing period. The initial decomposition which is detected in the high temperature powder data must therefore be too slow to be detectable by DTA. While cooling the metastable phase Li14TtAs6 from high temperatures the samples again undergo decomposition to Li3As and Li8TtAs4 indicated by the exothermal effects. Powder diffraction data of the Si compound after DTA (Fig. S11†) shows a mixture of Li14SiAs6, Li8SiAs4 and Li3As proofing the decomposition of the lithium rich arsenidotetrelate after the cooling cycle (Li14SiAs6 → Li8SiAs4 + 2Li3As). The described decomposition and reformation processes are also observed for the lithium phosphidotetrelates Li14TtP6 (Tt = Si, Ge, Sn) which decompose to Li8TtP4 (Tt = Si, Ge, Sn) and Li3P upon decreasing temperatures and therefore confirm the strong similarity between the arsenidotetrelates and phosphidotetrelates not only in structural aspects but also in the thermal properties.1,8
Impedance spectroscopy
Lithium ion mobility was measured using ac-impedance spectroscopy with ion blocking electrodes (indium foil) in a temperature range of 50 °C to 100 °C. Measurements at higher temperatures were not investigated, because of the limited thermal stability of the lithium rich arsenidotetrelates. Conductivities of the title compounds were obtained from Nyquist plots (Fig. S17 and S18†) showing semi-circles with low frequency tails, typical for lithium ion conductors. The temperature dependent ionic conductivities are displayed in the Arrhenius plot shown in Fig. 6. The lithium poor arsenidotetrelates Li8SiAs4 and Li8GeAs4 show specific ion conductivities of σspec(50 °C) = 1.9 × 10−5 S cm−1 (Ea = 0.39 eV) and σspec(50 °C) = 3.0 × 10−5 S cm−1 (Ea = 0.40 eV). In the system Li–Si–As only Li2SiAs2 is characterized in terms of ion conductivity with a specific ion conductivity of σspec(25 °C) = 8 × 10−8 S cm−1.11 Thus, Li8SiAs4 exceeds Li2SiAs2 in lithium conductivity by three orders of magnitude. The homologous lithium phosphidotetrelate Li8SiP4 shows similar ion conducting behaviour with a specific ion conductivity of σspec(75 °C) = 1.2(2) × 10−5 S cm−1 and the corresponding Ge containing compound with a specific ion conductivity of σspec(25 °C) = 1.8 × 10−5 S cm−1 shows also similar values like Li8GeAs4.3,7 The lithium rich arsenidotetrelates show specific ion conductivities of σspec(50 °C) = 4.6 × 10−4 S cm−1 (Ea = 0.28 eV), σspec(50 °C) = 6.6 × 10−4 S cm−1 (Ea = 0.34 eV) and σspec(50 °C) = 3.9 × 10−4 S cm−1 (Ea = 0.40 eV) for Li14SiAs6, Li14GeAs6, and Li14SnAs6, respectively. These specific ion conductivities in the range of 10−4 S cm−1 at 50 °C are quite high and only about one order of magnitude lower than for the corresponding phosphides (σ ∼ 1 × 10−3 S cm−1 at room temperature1,7). Conductivity measurements originating from different experimental setups can differ for several orders of magnitude. Therefore, a direct comparison of the conductivity values of the phosphides and the arsenides under discussion is difficult.22 Nevertheless, in all conductivity measurements we observed lower specific lithium ion conductivities for the arsenides. Overall, impedance spectroscopy shows that Li8SiAs4 and Li8GeAs4 exhibit lower lithium ion mobility than the Li14TtAs6 (Tt = Si, Ge, Sn) compounds which could be explained by the higher mole fraction of lithium, since Li8SiAs4 and Li8GeAs4 contain 61.5% lithium whereas Li14SiAs6 and Li14GeAs6 contain 66.6% lithium.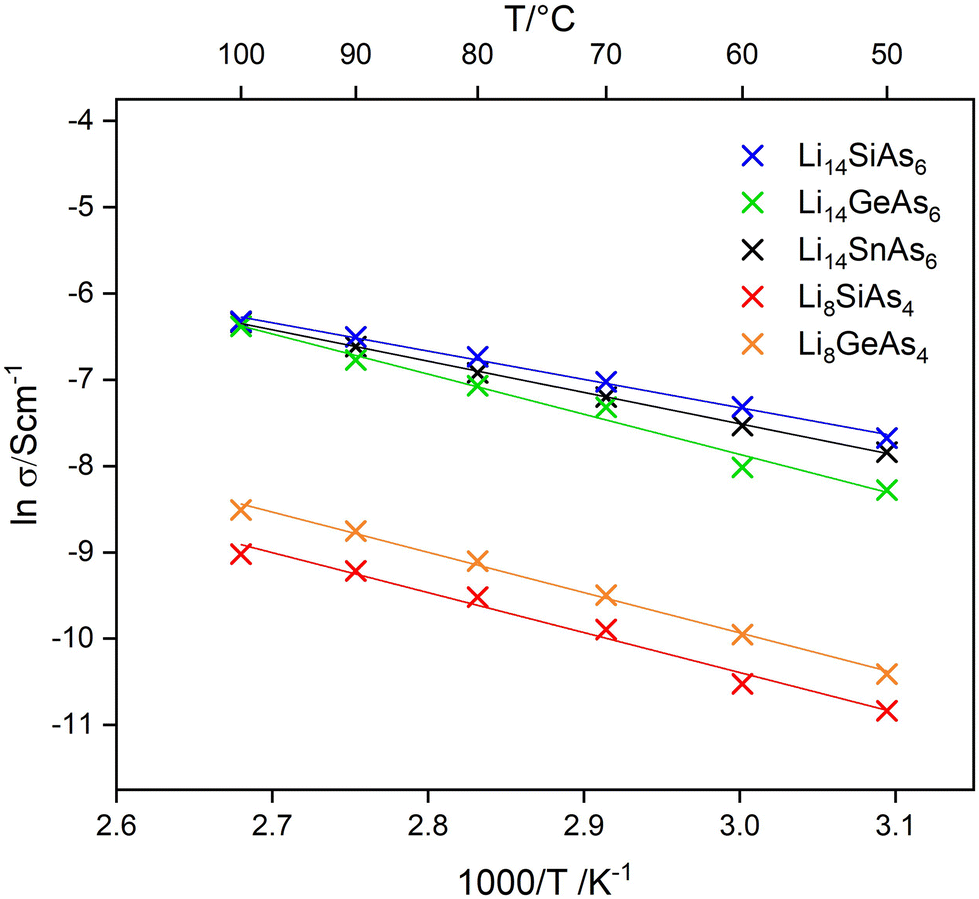 | ||
| Fig. 6 Arrhenius plots of the ion conductivities of Li8SiAs4, Li8GeAs4, Li14SiAs6, Li14GeAs6 and Li14SnAs6. It becomes obvious that the conductivity is strongly increasing with the Li content of the materials. | ||
Conclusions
We report on new members of the family of lithium arsenidotetrelates. Li8SiAs4, Li8GeAs4, Li14SiAs6, Li14GeAs6, and Li14SnAs6 can be synthesized via ball milling and phase pure samples of the compounds are obtained after annealing at high temperatures. The crystal structure (space group Pa (no. 218)) of the lithium poorer arsenidotetrelates Li8SiAs4 and Li8GeAs4 is built up by a distorted ccp of arsenide ions. The tetrel cation occupies 1/8 of all tetrahedral voids by 100% and lithium cations occupy the remaining tetrahedral voids and partly some octahedral voids. Both crystal structures feature slightly distorted [TtAs4]8−-tetrahedra enclosed by lithium atoms. The compounds Li14TtAs6 (Tt = Si, Ge, Sn) crystallize in a higher symmetric space group Fmm (no. 225). In contrast to the lithium poorer compounds all tetrahedral voids are occupied with a mixture of lithium and the tetrel cation in a ratio of 1:11. The higher tetrel content leads to an occupation of 50% of the octahedral voids by lithium. The structural relation between Li8TtAs4 and Li14TtAs6 can be understood looking at the corresponding unit cells in detail.
The main difference between the crystal structures is that in Li8TtAs4 the tetrel cations occupy 1/8 of the tetrahedral voids by 100% in a well-ordered arrangement whereas in Li14TtAs6 the tetrel cations are statistically distributed over all tetrahedral voids and therefore forming a mixed occupied site in addition with lithium in a ratio of 1:11 (tetrel/lithium). The unit cell of Li8TtAs4 can be derived from a 2 × 2 × 2 supercell of Li14TtAs6 unit cells (additional origin shift of the Li8TtAs4 unit cell by (1/2, 0, 0) is necessary) considering the different tetrel cation arrangement in the tetrahedral voids, the varied lithium content and distribution, and the distortion of the ccp of As anions. Both crystal structures can be derived from the anti-fluorite structure type or alternatively from the Li3Bi structure type.
Considering the sum formulae of the arsenidotetrelates under discussion the ternary phases can also be described as an formal aliovalent substitution of lithium in Li3As by an tetrel cation (Li12As4 → Li8Tt4+As4, Tt = Si, Ge; Li18As6 → Li14Tt4+As6, Tt = Si, Ge, Sn) exchanging four lithium cations with one tetrel cation. The introduced tetravalent tetrel cation forces the former hexagonal structure of Li3As into the cubic ternary phases Li8TtAs4 and Li14TtAs6, depending on the synthesis route and exact composition.
From high temperature diffraction data and DTA, it can be concluded that the compounds Li14TtAs6 (Tt = Si, Ge, Sn) are only stable at high temperatures and therefore are only accessible via quenching samples from high temperatures to ambient conditions. At moderate temperatures a decomposition to lithium arsenide and Li8TtAs4 (Li14TtAs6 → Li8TtAs4 + 2Li3As) is observed. This decomposition shows that the mixed occupancy of a site by tetrel and lithium in the higher symmetrical structure is thermodynamically unstable at room temperature. It is replaced by an ordered distribution of the tetrel cations at lower temperatures in the thermodynamically stable structure of Li8TtAs4. Impedance spectroscopy reveals lithium ion mobility for all compounds under discussion with specific ion conductivities for Li8SiAs4 and Li8GeAs4 in the range of 10−5 S cm−1 showing one order of magnitude lower in conductivity compared to the lithium richer compounds.
Experimental
Warning: Since elemental arsenic and its compounds are highly toxic all manipulations should be executed with extreme caution. In addition, safety considerations on a high level are advised, because all arsenic containing samples can reach a high vapor pressure at elevated temperatures and lithium is able to damage quartz ampoules by corrosion phenomena.Since all synthesised compounds are highly sensitive against air and moisture all manipulations were carried out in a Glovebox (M Braun) with oxygen and moisture levels below 0.5 ppm.
Synthesis
All compounds were synthesized in a two-step synthesis. First a pre-reacted powder mixture was obtained by ball milling (FRITSCH Pulverisette 7 premium line, 25 mL zirconia grinding bowls, 10 zirconia grinding balls (∅ = 10 mm)) the elements lithium (Merck, 99%), silicon (Alfa Aesar, 99.9%), germanium (Chempur, 99.9%), tin (Chempur, 99.999%) and arsenic (Chempur, sublimed) in stoichiometric amounts (batch size: 2 g) for 12 milling cycles (24 cycles for Li14SiAs6). One milling cycle was 5 minutes of milling (rotation speed: 600 rpm) followed by 4 minutes of letting the sample equilibrate. Lithium was used in excess (up to 15%) to compensate for evaporation during the second step. The pre-reacted mixture obtained from ball milling was placed in a graphite container (∅ = 6 mm) that was closed with a lid. The loaded container was then placed in an evacuated silica tube. Phase pure samples of the compounds were obtained by heating the mixtures to the reaction temperature (Li8SiAs4 and Li8GeAs4: 650 °C, 12 h; Li14SiAs6, Li14GeAs6, Li14SnAs6: 700 °C, 24 h) followed by quenching the silica tube in water for Li14TtAs6 (Tt = Si, Ge, Sn). The silica tubes for Li8TtAs4 (Tt = Si, Ge) were removed from the furnace and cooled on air.Powder diffraction
The samples were ground in a mortar and filled in a glass capillary (∅ = 0.3 mm), which was sealed with glue in the glove box. The subsequent flame sealed capillary was mounted on a STOE STADI P diffractometer (Stoe & Cie) equipped with a Mythen 1K detector for data collection and a graphite furnace for high temperature measurements. CuKα1-radiation (λ = 1.540598 Å) was used in all measurements. Raw data were processed with the WinXPOW software package (Stoe & Cie), Rietveld refinements were performed with Jana2006.23,24DTA
Samples were ground in argon atmosphere and prepared in flame sealed quartz tubes (∅ = 2 mm). Measurements were carried out using a SETARAM TG–DTA 92.16.18.Impedance spectroscopy
Impedance measurements were carried out on a Zahner Zennium impedance analyzer coupled with an Eurotherm controlled furnace located in a Glovebox (M Braun) with oxygen and moisture levels below 0.5 ppm. The cold-pressed pelletized samples (∅ = 8 mm) were sandwiched between indium foil and contacted with platinum electrodes. Measurements were carried out in a temperature range of 50–100 °C in steps of 10 °C and a frequency range of 1 MHz to 100 mHz. The measurement of ionic conductivities at room temperature was not possible since our measuring cell is located in an argon filled glovebox and cannot be actively cooled. The software Zahner Analysis was used for raw data processing.25Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Free State of Bavaria and the University of Regensburg for excellent working conditions. Dedicated to Prof. H. D. Lutz on the occasion of his 90th birthday.References
- S. Strangmüller, H. Eickhoff, D. Müller, W. Klein, G. Raudaschl-Sieber, H. Kirchhain, C. Sedlmeier, V. Baran, A. Senyshyn, V. L. Deringer, L. van Wüllen, H. A. Gasteiger and T. F. Fässler, Fast Ionic Conductivity in the Most Lithium-Rich Phosphidosilicate Li14SiP6, J. Am. Chem. Soc., 2019, 141, 14200–14209 CrossRef PubMed
.
- T. M. F. Restle, S. Strangmüller, V. Baran, A. Senyshyn, H. Kirchhain, W. Klein, S. Merk, D. Müller, T. Kutsch, L. van Wüllen and T. F. Fässler, Super-Ionic Conductivity in ω-Li9TrP4 (Tr = Al, Ga, In) and Lithium Diffusion Pathways in Li9AlP4 Polymorphs, Adv. Funct. Mater., 2022, 32, 2112377 CrossRef CAS
- L. Toffoletti, H. Kirchhain, J. Landesfeind, W. Klein, L. van Wüllen, H. A. Gasteiger and T. F. Fässler, Lithium Ion Mobility in Lithium Phosphidosilicates: Crystal Structure, 7Li, 29Si, and 31P MAS NMR Spectroscopy, and Impedance Spectroscopy of Li8SiP4 and Li2SiP2, Chem. – Eur. J., 2016, 22, 17635–17645 CrossRef CAS PubMed
- R. Juza and W. Schulz, Ternäre Phosphide und Arsenide des Lithiums mit Elementen der 3. und 4. Gruppe, Z. Anorg. Allg. Chem., 1954, 275, 65–78 CrossRef CAS
- R. Juza, H. H. Weber and E. Meyer-Simon, Über ternäre Nitride und Oxonitride von Elementen der 4. Gruppe, Z. Anorg. Allg. Chem., 1953, 273, 48–64 CrossRef CAS
- A. Haffner, T. Bräuniger and D. Johrendt, Supertetrahedral Networks and Lithium-Ion Mobility in Li2SiP2 and LiSi2P3, Angew. Chem., Int. Ed., 2016, 55, 13585–13588 CrossRef CAS PubMed
- H. Eickhoff, S. Strangmüller, W. Klein, H. Kirchhain, C. Dietrich, W. G. Zeier, L. van Wüllen and T. F. Fässler, Lithium Phosphidogermanates α- and β-Li8GeP4 – A Novel Compound Class with Mixed Li+ Ionic and Electronic Conductivity, Chem. Mater., 2018, 30, 6440–6448 CrossRef CAS
- S. Strangmüller, H. Eickhoff, G. Raudaschl-Sieber, H. Kirchhain, C. Sedlmeier, L. van Wüllen, H. A. Gasteiger and T. F. Fässler, Modifying the Properties of Fast Lithium-Ion Conductors – The Lithium Phosphidotetrelates Li14SiP6 Li14GeP6 and Li14SnP6, Chem. Mater., 2020, 32, 6925–6934 CrossRef
- K. Lee, D. Kaseman, S. Sen, I. Hung, Z. Gan, B. Gerke, R. Pöttgen, M. Feygenson, J. Neuefeind, O. I. Lebedev and K. Kovnir, Intricate short-range ordering and strongly anisotropic transport properties of Li(1−x)Sn(2+x)As2, J. Am. Chem. Soc., 2015, 137, 3622–3630 CrossRef CAS PubMed
- J. Mark, M. P. Hanrahan, K. E. Woo, S. Lee, A. J. Rossini and K. Kovnir, Chemical and Electrochemical Lithiation of van der Waals Tetrel-Arsenides, Chem. – Eur. J., 2019, 25, 6392–6401 CrossRef CAS PubMed
- J. Mark, K. Lee, M. A. T. Marple, S. Lee, S. Sen and K. Kovnir, LiSi3As6 and Li2SiAs2 with flexible SiAs2 polyanions: synthesis, structure, bonding, and ionic conductivity, J. Mater. Chem. A, 2020, 8, 3322–3332 RSC
- F. Wegner, F. Kamm, F. Pielnhofer and A. Pfitzner, Li3As and Li3P revisited: DFT modelling on phase stability and ion conductivity, Z. Anorg. Allg. Chem., 2022, 648, e202100358 CrossRef CAS
- F. Wegner, F. Kamm, F. Pielnhofer and A. Pfitzner, Li3TrAs2 (Tr = Al, Ga, In) – Derivatives of the antifluorite type structure, conductivities and electronic structures, Z. Anorg. Allg. Chem., 2023, 649, e202200330 CrossRef CAS
- A. Adam and H.-U. Schuster, Ternäre intermetallische Phasen des Lithiums mit Elementen der 4. Neben- und 5. Hauptgruppe mit statistischer Metallverteilung im “Kationen”-Teilgitter, Z. Anorg. Allg. Chem., 1991, 597, 33–39 CrossRef CAS
- L. Monconduit, M. Tillard-Charbonnel and C. Belin, Transition Metal-Substituted Lithium Pnictogenide phases. Synthesis and Crystal Structure Determinations of Novel Phases in the Li–M–X Systems (M=V, Nb, Ta; X=P, As), J. Solid State Chem., 2001, 156, 37–43 CrossRef CAS
- B. Eisenmann, J. Klein and M. Somer, Crystal structure of decasodium di-μ-arsenidobis(diarsenidosilicate), Na10Si2As6, Z. Kristallogr. – Cryst. Mater., 1991, 197, 267–268 CAS
- W. M. Hurng, J. D. Corbett, S. L. Wang and R. A. Jacobson, A Zintl phase with a layered network structure, potassium silicon arsenide (KSi3As3), Inorg. Chem., 1987, 26, 2392–2395 CrossRef CAS
- T. Wadsten, M. Vikan, C. Krohn, Å. Nilsson, H. Theorell, R. Blinc, S. Paušak, L. Ehrenberg and J. Dumanović, The Crystal Structures of SiP2, SiAs2, and GeP, Acta Chem. Scand., 1967, 21, 593–594 CrossRef CAS
- B. Eisenmann, J. Klein and M. Somer, Crystal structure of decasodium di-μ-arsenidobis(diarsenidogermanate), Na10Ge2As6, Z. Kristallogr. – Cryst. Mater., 1991, 197, 265–266 CAS
- M. Khatun, S. S. Stoyko and A. Mar, Ternary arsenides ATt3As3 (A=K, Rb; Tt=Ge, Sn) with layered structures, J. Solid State Chem., 2016, 238, 229–235 CrossRef CAS
- K. Lee, S. Kamali, T. Ericsson, M. Bellard and K. Kovnir, GeAs: Highly Anisotropic van der Waals Thermoelectric Material, Chem. Mater., 2016, 28, 2776–2785 CrossRef CAS
- S. Ohno, T. Bernges, J. Buchheim, M. Duchardt, A.-K. Hatz, M. A. Kraft, H. Kwak, A. L. Santhosha, Z. Liu, N. Minafra, F. Tsuji, A. Sakuda, R. Schlem, S. Xiong, Z. Zhang, P. Adelhelm, H. Chen, A. Hayashi, Y. S. Jung, B. V. Lotsch, B. Roling, N. M. Vargas-Barbosa and W. G. Zeier, How Certain Are the Reported Ionic Conductivities of Thiophosphate-Based Solid Electrolytes? An Interlaboratory Study, ACS Energy Lett., 2020, 5, 910–915 CrossRef CAS
-
STOE-WinXPOW, Vol. Version 3.0.2.5, STOE & Cie GmbH, Darmstadt, 2011 Search PubMed
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS
-
Zahner-Meßtechnik GmbH & Co. KG, Vol. Version Z.3.03, Thales-Flink, Kronach Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332047–2332051. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00664j |
| This journal is © The Royal Society of Chemistry 2024 |