
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Markovnikov-selective double hydrosilylation of challenging terminal aryl alkynes under cobalt and iron catalysis†
Łukasz
Banach
 *a,
Daria
Brykczyńska
b,
Adam
Gorczyński
b,
Bożena
Wyrzykiewicz
b,
Maciej
Skrodzki
ab and
Piotr
Pawluć
*ab
*a,
Daria
Brykczyńska
b,
Adam
Gorczyński
b,
Bożena
Wyrzykiewicz
b,
Maciej
Skrodzki
ab and
Piotr
Pawluć
*ab
aCentre for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego St. 10, 61-614 Poznań, Poland. E-mail: lukasz.banach@amu.edu.pl; piotr.pawluc@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego St. 8, 61-614 Poznań, Poland
First published on 24th November 2022
Abstract
Geminal bis(silanes) are unique compounds with interesting properties. The most straightforward way to access them is double hydrosilylation of alkynes, which was established only recently. Previous articles about transition metal-catalysed double hydrosilylation show that terminal aryl alkynes are a challenge. We report on cobalt(II) and iron(III) complexes with the easy-to-synthesise N,N,N-tridentate hydrazone ligand being active precatalysts in Markovnikov-selective double hydrosilylation of terminal aryl alkynes. The influence of the hydrazone ligand structure and the potential role of the sodium triethylborohydride activator were studied. Sets of geminal bis(silanes) with two identical or different silyl groups were synthesised, showing the applicability of the reported method.
Organosilanes are valuable compounds with vast applications, including in sophisticated fundamental research,1 organic synthesis,2 materials science,3 biotechnology4 and medicinal chemistry.5Gem-bis(silanes) are gaining particularly increasing attention due to their unique reactivity6 and structural properties.7 However, access to these compounds and further development of their chemistry was hampered for a long time due to the scarcity of synthetic methods for placing two silyl groups on one carbon atom.8Gem-bis(silanes) that could be obtained with these protocols8 had no Si–H bonds within the bis(silyl) moiety and the lack thereof could have been holding back further developments.9 Currently, strenuous efforts are underway to improve the field of gem-bis(silane) synthesis, focusing on Si–H containing products.
Catalytic hydrosilylation stands out as a very straightforward option to access organosilanes with perfect atom-economy.10–12 Due to reactivity and selectivity issues with hydrosilylation of vinylsilanes, access to gem-bis(silanes) through double hydrosilylation of alkynes is a challenge, and at first, such processes were observed only as side reactions.13 However, several research groups have recently reported successful double hydrosilylation of alkynes leading to gem-bis(silanes) (Scheme 1). Zhu's team utilised an iron(II) complex with a phenanthroline derived ligand that catalysed double anti-Markovnikov hydrosilylation of terminal aliphatic alkynes (Scheme 1a).14 Concomitantly, Lu's group published their results describing chiral cobalt(II) complexes with either imidazoline-iminopyridine or oxazoline-iminopyridine ligands that were capable of catalysing the transformation of terminal aliphatic alkynes (Scheme 1b and c).15,16 Notably, these chiral cobalt(II) precatalysts, when used in sequential one-pot hydrosilylation with two different silanes, led to optically pure products. Li, Cui and co-workers further expanded the scope of double hydrosilylation of alkynes by employing a lanthanum(III) bis(amido)ate complex which enabled that process on internal aryl–alkyl and silyl–alkyl alkynes (Scheme 1e).17 However, terminal aryl alkynes still remained problematic as they underwent only monohydrosilylation. Most recently, Li and team have addressed this issue by using B(C6F5)3 as the catalyst allowing sequential anti-Markovnikov double hydrosilylation of terminal aryl alkynes, starting with tertiary silanes and followed by primary silanes (Scheme 1d).18 We would like to offer a further narrowing of the gap regarding transformation of terminal aryl alkynes into gem-bis(silanes) by presenting the results of our research on Markovnikov-selective cobalt- and iron-catalysed double hydrosilylation.
 | ||
| Scheme 1 Double hydrosilylation of alkynes without (d) or in the presence of bidentate (a, e) and tridentate (b, c, f) ligand. | ||
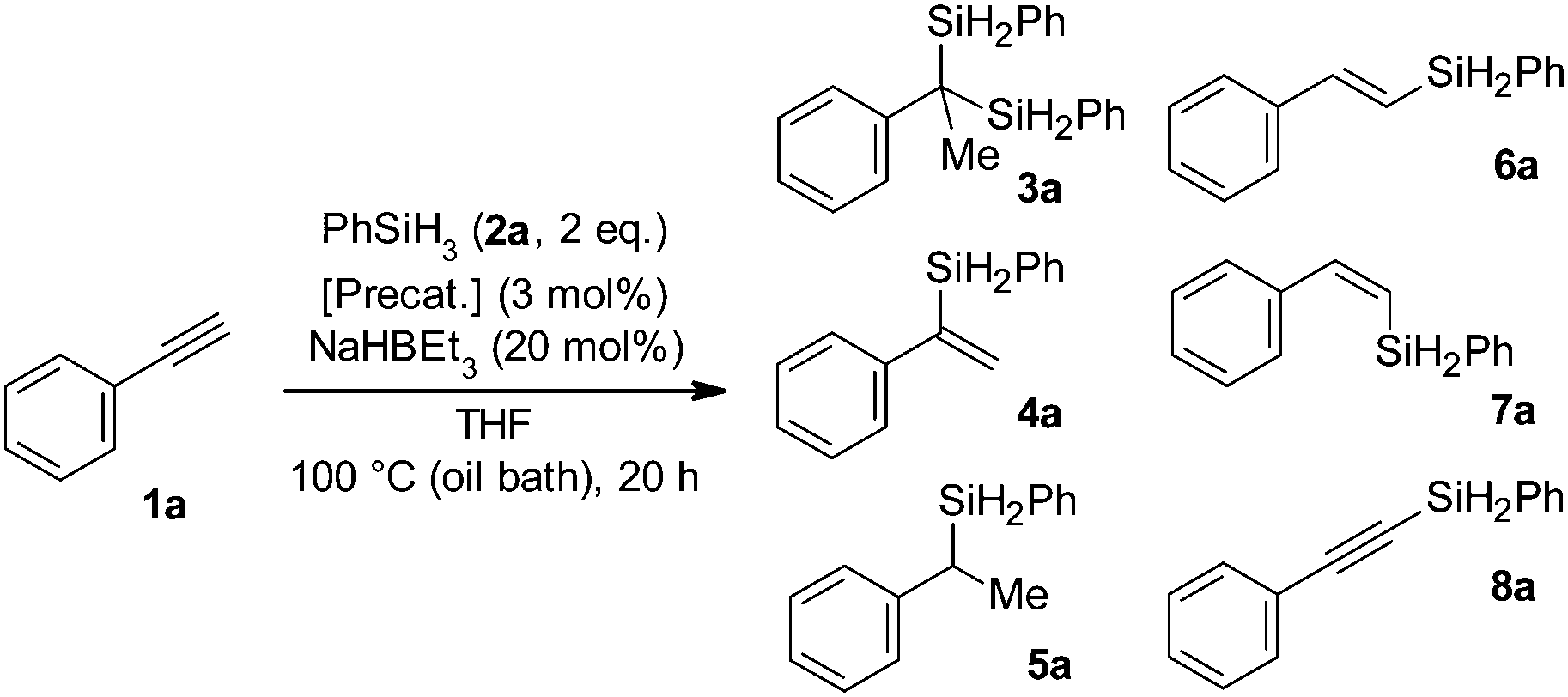
N,N,N-Tridentate hydrazone ligands, which can be easily and efficiently prepared in various structures through modular synthesis,19 served well in our studies on cobalt-catalysed hydrosilylation of alkenes20 and alkynes.21 Thus, we also decided to utilize this system for iron complexes (Chart 1) and verify their potential catalytic activity in alkyne hydrosilylation reactions. Initial tests were conducted in THF on a range of alkynes using phenylsilane with the Fe-L1 precatalyst and the NaHBEt3 activator. In the case of the phenylacetylene substrate we obtained an intriguing result – among a multitude of GC-MS signals from different products, one stood out due to a much higher retention time and an unusually high m/z ratio of 318 for M+ ions. We isolated the compound responsible for this signal and identified it as a product of Markovnikov-oriented double hydrosilylation of phenylacetylene (3a). This result prompted us to focus our efforts on iron-catalysed double hydrosilylation of terminal aryl alkynes, especially given the fact that phenylacetylene was repeatedly reported as a very challenging substrate in this type of transformation.14–17 Initial reaction optimisation studies focusing on solvent and activator selection allowed us to determine that THF and NaHBEt3 work the best in these regards; however, an NMR yield of only 28% was obtained (Table 1, entry 1; please see the ESI† for reaction optimisation details). Control experiments substituting Fe-L1 for either FeCl3 or L1 ligand confirmed that Fe-L1 is necessary for formation of 3a. We then tested related iron complexes which were different from Fe-L1 in the single aspect of the hydrazone ligand structure. First we exchanged one heterocycle on each side of L1 for another heterocyclic structure with only one nitrogen atom (2-pyridyl or 2-quinoyl), as in the case of Fe-L2 and Fe-L3. The yields of 3a for Fe-L2 and Fe-L3 were similar between them (6% and 4%, respectively), but significantly lower than that for Fe-L1 (Table 1, entries 2 and 3). Some possible explanations for such results are that the NH groups at the heterocyclic units of the ligand are somehow important in the catalytic process and that a binding geometry of a five-membered heterocycle to the iron centre serves better than a six-membered system in the catalytically active species. In order to verify these potential explanations we have synthesised Fe-L4 with all the NH positions that are present in Fe-L1 transformed into NMe moieties; thus the binding geometry of the ligand should stay the same as that in Fe-L1 and the role of NH could be verified. Tests with Fe-L4 gave a negligible yield of 3a (Table 1, entry 4), thus confirming the practical significance of NH groups. Changing the orientation of the imidazolyl ring from 2-imidazolyl in Fe-L1 to 4-imidazolyl in Fe-L5 decreased the yield of 3a to only 5% (Table 1, entry 5). Interestingly, a decrease in the loading of Fe-L1 from 3 mol% to 1 mol% resulted in an increase of the yield of 3a to 43% (Table 1, entry 6), but further decrease in the Fe-L1 loading to 0.5 mol% was not helpful (see the ESI† for details). As we anticipated that 4a was an intermediate on the way to 3a we decided to also employ the Co-L1 complex in order to help in the first hydrosilylation of phenylacetylene. At first this seemed to be beneficial as the yield of 3a improved to 60% (Table 1, entry 7). However, a control experiment using Co-L1 without Fe-L1 gave a 63% yield of 3a (Table 1, entry 8). This showed that the cobalt complex is a better precatalyst than its iron analogue. Interestingly, Co-L4 was drastically worse than Co-L1 as 3a was not produced when using this complex (Table 1, entry 9). Thus, a negative effect of introducing methyl groups on nitrogen atoms in heterocyclic systems of the ligand was also present in the case of cobalt complexes.
 | ||
| Chart 1 Cobalt and iron complexes used in this study. | ||
|
|
|||
|---|---|---|---|
| Entrya | [Precat.] | Yield of 3ab (%) |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a:5a:6a:7a:8ab :4a:5a:6a:7a:8ab |
| a Reaction conditions: 1a (0.25 mmol), 2a (0.50 mmol), precatalyst (3 mol%), NaHBEt3 (20 mol%) in THF (0.5 mL) in an argon-filled Schlenk bomb flask immersed in an oil bath (100 °C) for 20 h. b Determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. c 1 mol% of precatalyst and 7 mol% of NaHBEt3, reaction time of 12 h. d 1 mol% of each precatalyst (Fe-L1 in two portions) and a total of 10 mol% of NaHBEt3. | |||
| 1 | Fe-L1 | 28 | 28:2:2:9:6:0 |
| 2 | Fe-L2 | 6 | 6:2:3:10:1:4 |
| 3 | Fe-L3 | 4 | 4:2:3:16:19:0 |
| 4 | Fe-L4 | 2 | 2:1:2:11:0:0 |
| 5 | Fe-L5 | 5 | 5:2:4:10:5:20 |
| 6c | Fe-L1 | 43 | 43:1:3:7:2:0 |
| 7d | Fe-L1 + Co-L1 | 60 | 60:3:8:1:0:0 |
| 8c | Co-L1 | 63 | 63:1:4:1:0:0 |
| 9c | Co-L4 | 0 | 0:1:1:4:0:0 |
Based on our results obtained for different cobalt and iron complexes we concluded that NaHBEt3 might be involved in the modification of the ligand structure. We hypothesised that NaHBEt3 not only serves to reduce the metal centre and clear it of chlorine atoms, but also deprotonates the NH groups present in the heterocyclic units of the ligand and binds to nitrogen atoms – that is why Fe-L1 and Fe-L4, as well as Co-L1 and Co-L4 have significantly different activities.22 In order to verify our hypothesis we conducted a series of 1H and 11B NMR experiments (see the ESI† for details). First we added NaHBEt3 to L1 in THF-d8 and the 1H NMR results showed swift deprotonation of NH units. After addition of 3 eq. of NaHBEt3 to L1 its 11B NMR spectrum showed a clear singlet at −0.13 ppm which we assign to the BEt3 bonded to the nitrogen atom.23 Further NMR experiments were carried out on Fe-L1 and Co-L1 complexes. Due to their paramagnetic nature their 1H NMR spectra are illegible for the most part. However, after addition of 1 eq. of NaHBEt3 to Fe-L1 a broad and irregular signal in the range of −3.25 ppm to −4.86 ppm appeared which we tentatively assign to Fe–H species. This signal seemed to migrate slightly as more NaHBEt3 was added. Similarly, addition of NaHBEt3 to Co-L1 gave rise to a broad singlet at −12.05 ppm which can be a manifestation of Co-H species.24 After addition of 7 eq. of NaHBEt3 to Fe-L1 or Co-L1, their respective 11B NMR spectra showed two signals at 1.19 ppm and −0.41 ppm which we interpret as resulting from the BEt3 units bonded to the nitrogen atoms from deprotonated NH groups in the L1 ligand.23
When investigating the alkyne substrate scope in the reaction with phenylsilane, we focused on Co-L1 and started with phenylacetylene derivatives with additional substituents on the aromatic ring (Scheme 2). When compared to an unsubstituted system, the methyl group in the meta or para position essentially did not impact the yield of the gem-bis(silyl) product. A somewhat stronger influence was exerted by the methyl substituent in the ortho position as the product 3d was isolated with a 40% yield. This is clearly due to the steric effect, which overall is not as strong as it could be expected, given the bulkiness of the quaternary centre being formed in the final product. Other substituents, like –Ph, –OMe, –N(Me)2 and –F, were also tolerated and the respective products were obtained in moderate yields. We also encountered phenylacetylene derivatives that did not yield the expected gem-bis(silyl) products. These had substituents that have the ability to coordinate to the metal centre and/or can be the subject of hydride attack either in reduction, deprotonation or substitution processes (–CN, –CO2Et, –NH2, and –Br). Further studies revealed that an extended aromatic system worked well in double hydrosilylation (3j). We additionally tested a couple of heteroaromatic substrates. While 3-ethynylpyridine did not give the desired product, 2-ethynylthiophene gave a modest 37% yield of 3k. Terminal alkyl alkynes did not undergo double hydrosilylation. Among internal alkynes only aryl–alkyl 1-phenyl-1-propyne yielded 18% of respective gem-bis(silane) (see the ESI† for details). Investigations on the silane substrate scope showed that our protocol can be applied to primary alkylsilanes, as n-hexylsilane also served to obtain 3l. Interestingly, 3l was contaminated with a homologous compound 3l′ with 14 g mol−1 lower mass. Based on a set of NMR analyses (1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC; see the ESI† for details) we identified 3l′ as an analogue of 3l devoid of the methyl group bonded to the benzylic carbon atom. Formation of 3l′ suggests the occurrence of an intriguing side reaction that is probably facilitated by the electron donating nature of alkylsilyl group(s). Secondary silanes (diethylsilane and diphenylsilane) or tertiary silanes (dimethylphenylsilane, triethylsilane and triphenylsilane) when tested on phenylacetylene did not work to obtain respective gem-bis(silanes).
 | ||
| Scheme 2 Substrate scope studies on double hydrosilylation of terminal aryl alkynes. Reaction conditions: 1 (0.50 mmol), 2 (1.00 mmol), Co-L1 (1 mol%), NaHBEt3 (7 mol%) in THF (1.0 mL) in an argon-filled Schlenk bomb flask immersed in an oil bath (100 °C) for 3 h. The reported yields are for isolated products. a Product 3l contaminated with 25% of a homologue 3l′. The reported yield accounts for this contamination. | ||
Despite the fact that our procedure did not work for secondary and tertiary silanes we sought an intermediate solution. Knowing that Co-L1 works well in monohydrosilylation of alkynes with secondary and even tertiary silanes21a we decided to utilise this in the preparation of mixed gem-bis(silanes) with two different silyl groups. In order to do so, we changed the reaction conditions in such a way that in the first stage only 1 eq. of more substituted silane was used at lower temperature, while in the second stage a fresh portion of Co-L1 and NaHBEt3 was added with the second silane at higher temperature (Scheme 3). This approach allowed us to obtain a set of racemic mixed gem-bis(silanes).
 | ||
| Scheme 3 Synthesis of mixed geminal bis(silanes). The reported yields are for isolated products. Reaction conditions: 1a (0.50 mmol) and 2 (0.50 mmol) added to Co-L1 (0.5 mol%) activated by NaHBEt3 (2 mol%) in THF (0.5 mL) and heated up to 40–60 °C for 20 h, followed by addition of the Co-L1 (1 mol%) and NaHBEt3 (7 mol%) mixture in THF (0.5 mL) and 2′ (0.5 mmol). | ||
In summary, we have developed a method for Markovnikov-selective double hydrosilylation of terminal aryl alkynes using a cobalt(II) complex with the N,N,N-tridentate hydrazone ligand. A similar, yet lower, activity was also found for the analogous iron complex. Studies on various cobalt(II) and iron(III) complexes with different N,N,N-tridentate hydrazone ligands showed that even subtle changes in the ligand structure play an important role with respect to catalytic activity. NMR experiments revealed that the NaHBEt3 activator may not only react with the metal centre of the precatalyst, but also modify the ligand and thus influence the catalytic activity of the metal centre.
This work was supported by the National Science Centre, Poland (grant no. UMO-2016/23/B/ST5/00177). We would like to thank Ms Joanna Jałoszyńska, M.Sc., for the help in the synthesis of ligand L3, Ms Monika Szukowska, M.Sc., for conducting the SEM analyses and Dr Samuel Bennett for language editing of the manuscript. We would also like to thank the reviewers for their thoughtful comments and suggestions, which helped us to strengthen our manuscript. Ł. B. would like to thank Dr Rafał Januszewski and Dr Maciej Zaranek for their helpful discussions on hydrosilylation processes and related topics.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Chem. Rev., 2022, 122, 3996–4090 CrossRef CAS PubMed; (b) A. Fernandes, C. Laye, S. Pramanik, D. Palmeira, Ö. Ö. Pekel, S. Massip, M. Schmidtmann, T. Müller, F. Robert and Y. Landais, J. Am. Chem. Soc., 2020, 142, 564–572 CrossRef CAS PubMed; (c) M. J. Cowley, V. Huch, H. S. Rzepa and D. Scheschkewitz, Nat. Chem., 2013, 5, 876–879 CrossRef CAS PubMed; (d) S. Ishida, T. Iwamoto, C. Kabuto and M. A. Kira, Nature, 2003, 421, 725–727 CrossRef CAS PubMed; (e) T. Müller, Angew. Chem., Int. Ed., 2001, 40, 3033–3036 CrossRef.
- (a) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS; (b) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (c) H. Ottosson and P. G. Steel, Chem. – Eur. J., 2006, 12, 1576–1585 CrossRef CAS PubMed.
- (a) Y. Zuo, Z. Gou, W. Quan and W. Lin, Coord. Chem. Rev., 2021, 438, 213887 CrossRef CAS; (b) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC; (c) S. Inagaki, S. Guan, Q. Yang, M. P. Kapoor and T. Shimada, Chem. Commun., 2008, 202–204 RSC.
- (a) S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed; (b) S. Hudson, J. Cooney, B. K. Hodnett and E. Magner, Chem. Mater., 2007, 19, 2049–2055 CrossRef CAS; (c) K. Szymańska, J. Bryjak, J. Mrowiec-Białoń and A. B. Jarzębski, Micropor. Mesopor. Mat., 2007, 99, 167–175 CrossRef; (d) C. Airoldi and O. A. C. Monteiro Jr., J. Appl. Polym. Sci., 2000, 77, 797–804 CrossRef CAS.
- (a) N. F. Lazareva and I. M. Lazarev, Russ. Chem. Bull., 2015, 64, 1221–1232 CrossRef CAS; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed; (c) J. Wang, C. Ma, Y. Wu, R. A. Lamb, L. H. Pinto and W. F. DeGrado, J. Am. Chem. Soc., 2011, 133, 13844–13847 CrossRef CAS PubMed.
- (a) Y. Chen, X. Song, L. Gao and Z. Song, Org. Lett., 2021, 23, 124–128 CrossRef CAS PubMed; (b) L. Gao, Y. Zhang and Z. Song, Synlett, 2013, 139–144 Search PubMed; (c) D. R. Williams, Á. I. Morales-Ramos and C. M. Williams, Org. Lett., 2006, 8, 4393–4396 CrossRef CAS PubMed.
- (a) Z. Liu, X. Lin, N. Yang, Z. Su, C. Hu, P. Xiao, Y. He and Z. Song, J. Am. Chem. Soc., 2016, 138, 1877–1883 CrossRef CAS PubMed; (b) Y. Wu, L. Li, H. Li, L. Gao, H. Xie, Z. Zhang, Z. Su, C. Hu and Z. Song, Org. Lett., 2014, 16, 1880–1883 CrossRef CAS PubMed.
- (a) H. Hazrati and M. Oestreich, Org. Lett., 2018, 20, 5367–5369 CrossRef CAS PubMed; (b) Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800–12803 CrossRef CAS PubMed; (c) Z. Song, Z. Lei, L. Gao, X. Wu and L. Li, Org. Lett., 2010, 12, 5298–5301 CrossRef CAS PubMed; (d) T. N. Mitchell and M. Schütze, Tetrahedron, 1999, 55, 1285–1294 CrossRef CAS; (e) R. J. P. Corriu, M. Granier and G. F. Lanneau, J. Organomet. Chem., 1998, 562, 79–88 CrossRef CAS; (f) R. Calas, J. Dunogues, C. Biran and N. Duffaut, J. Organomet. Chem., 1969, 20, P22–P24 CrossRef CAS.
- (a) H. Liang, L.-J. Wang, Y.-X. Ji, H. Wang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1839–1844 CrossRef CAS PubMed; (b) K. Kuciński, H. Stachowiak and G. Hreczycho, Inorg. Chem. Front., 2020, 7, 4190–4196 RSC; (c) S. Bähr, S. Brinkmann-Chen, M. Garcia-Borràs, J. M. Roberts, D. E. Katsoulis, K. N. Houk and F. H. Arnold, Angew. Chem., Int. Ed., 2020, 59, 15507–15511 CrossRef PubMed; (d) C. Bellini, V. Dorcet, J.-F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS PubMed; (e) S. Wu, X. Li, Z. Xiong, W. Xu, Y. Lu and H. Sun, Organometallics, 2013, 32, 3227–3237 CrossRef CAS; (f) X. Shang and Z.-Q. Liu, Org. Biomol. Chem., 2016, 14, 7829–7831 RSC; (g) R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 2347–2366 CrossRef CAS; (h) U. Schubert and C. Lorenz, Inorg. Chem., 1997, 36, 1258–1259 CrossRef CAS PubMed; (i) E. Lukevics and M. Dzintara, J. Organomet. Chem., 1985, 295, 265–315 CrossRef CAS.
- (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (b) S. Pawlenko, Organosilicon Chemistry, De Gruyter, 1986, pp. 17–108 CrossRef.
- (a) L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed; (b) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC; (c) B. Marciniec, Comprehensive handbook on hydrosilylation, Elsevier, 2013, pp. 8–159 Search PubMed.
- (a) D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 983–1010 CAS; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 853–887 CrossRef CAS; (c) A. Ramani, B. Desai, M. Patel and T. Naveen, Asian J. Org. Chem., 2022, e202200047 CAS.
- (a) C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef CAS PubMed; (b) A. Nurseiit, J. Janabel, K. A. Gudun, A. Kassymbek, M. Segizbayev, T. M. Seilkhanov and A. Y. Khalimon, ChemCatChem, 2019, 11, 790–798 CrossRef CAS; (c) M. Zhang, Y. Ji, Z. Zhang and C. Zhang, Org. Lett., 2022, 24, 2756–2761 CrossRef CAS PubMed.
- M.-Y. Hu, J. Lian, W. Sun, T.-Z. Qiao and S.-F. Zhu, J. Am. Chem. Soc., 2019, 141, 4579–4583 CrossRef CAS PubMed.
- Z. Cheng, S. Xing, J. Guo, B. Cheng, L.-F. Hu, X.-H. Zhang and Z. Lu, Chin. J. Chem., 2019, 37, 457–461 CrossRef CAS.
- J. Guo, H. Wang, S. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881–895 CAS.
- W. Chen, H. Song, J. Li and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 2365–2369 CrossRef CAS PubMed.
- G. Wang, X. Su, L. Gao, X. Liu, G. Li and S. Li, Chem. Sci., 2021, 12, 10883–10892 RSC.
- A. Bocian, A. Szymańska, D. Brykczyńska, M. Kubicki, M. Wałęsa-Chorab, G. N. Roviello, M. A. Fik-Jaskółka, A. Gorczyński and V. Patroniak, Molecules, 2019, 24, 3173 CrossRef CAS PubMed.
- (a) A. Bocian, M. Skrodzki, M. Kubicki, A. Gorczyński, P. Pawluć and V. Patroniak, Appl. Catal., A, 2020, 602, 117665 CrossRef CAS; (b) A. Gorczyński, M. Zaranek, S. Witomska, A. Bocian, A. R. Stefankiewicz, M. Kubicki, V. Patroniak and P. Pawluć, Catal. Commun., 2016, 78, 71–74 CrossRef.
- (a) M. Skrodzki, V. Patroniak and P. Pawluć, Org. Lett., 2021, 23, 663–667 CrossRef CAS PubMed; (b) M. Skrodzki, V. O. Garrido, A. G. Csáky and P. Pawluć, J. Catal., 2022, 411, 116–121 CrossRef CAS.
- (a) Z. Chen, H. W. Schmalle, T. Fox, O. Blacque and H. Berke, J. Organomet. Chem., 2007, 692, 4875–4885 CrossRef CAS; (b) W. Zou, L. Gao, J. Cao, Z. Li, G. Li, G. Wang and S. Li, Chem. – Eur. J., 2022, 28, e202104004 CrossRef CAS PubMed.
- H. Nöth and B. Wrackmeyer, in Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer-Verlag, 1978, pp. 90, 115 and 317 Search PubMed.
- H. Stachowiak, K. Kuciński, F. Kallmeier, R. Kempe and G. Hreczycho, Chem. – Eur. J., 2022, 28, e202103629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04015h |
| This journal is © The Royal Society of Chemistry 2022 |