
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Damming an electronic energy reservoir: ion-regulated electronic energy shuttling in a [2]rotaxane†
Shilin
Yu
 ad,
Arkady
Kupryakov
b,
James E. M.
Lewis
ce,
Vicente
Martí-Centelles
a,
Stephen M.
Goldup
*c,
Jean-Luc
Pozzo
a,
Gediminas
Jonusauskas
b and
Nathan D.
McClenaghan
*a
ad,
Arkady
Kupryakov
b,
James E. M.
Lewis
ce,
Vicente
Martí-Centelles
a,
Stephen M.
Goldup
*c,
Jean-Luc
Pozzo
a,
Gediminas
Jonusauskas
b and
Nathan D.
McClenaghan
*a
aInstitut des Sciences Moléculaires, University of Bordeaux/CNRS, Talence, France. E-mail: nathan.mcclenaghan@u-bordeaux.fr
bLaboratoire Ondes et Matière d'Aquitaine, University of Bordeaux/CNRS, Talence, France
cSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: s.goldup@soton.ac.uk
dDepartment of Chemistry, University of Jyvaskyla, 40014, Jyväskylä, Finland
eDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 82 Wood Lane, London W12 0BZ, UK
First published on 4th June 2021
Abstract
We demonstrate the first example of bidirectional reversible electronic energy transfer (REET) between the mechanically bonded components of a rotaxane. Our prototypical system was designed such that photoexcitation of a chromophore in the axle results in temporary storage of electronic energy in a quasi-isoenergetic “reservoir” chromophore in the macrocycle. Over time, the emissive state of the axle is repopulated from this reservoir, resulting in long-lived, delayed luminescence. Importantly, we show that cation binding in the cavity formed by the mechanical bond perturbs the axle chromophore energy levels, modulating the REET process, and ultimately providing a luminescence read-out of cation binding. Modulation of REET processes represents an unexplored mechanism in luminescent molecular sensor development.
Introduction
Luminescent rotaxanes1 in which the mechanical bond2 between a linear axle and an encircling macrocycle alters the properties of a luminophore in either component have been developed for a range of purposes, including monitoring the relative co-conformation of the sub-units,3 sensing of guests bound within the cavity of the rotaxane,4 and improving the stability5 and emission properties6 of the luminophore itself. Light has also been successfully used to promote processes in interlocked structures such as topological transformations7 dethreading/threading,8 and shuttling.9 The mechanical bond has also been used to study or optimize photophysical processes that rely on electronic energy10 (e.g. quenching, FRET), or electron transfer11 between molecular sub-units by taking advantage of the fact that, although there is no covalent bond between the components of a rotaxane, they are unable to separate and their relative motions are highly circumscribed. Thus, through-bond processes are explicitly prevented, as no such bond exists, and although the chromophores are able to move relative to one another in defined ways thanks to the flexibility of the interlocked structure, they are prevented from diffusing apart.In the current work, we sought to study reversible electronic energy transfer (REET) between matched chromophores within a rotaxane framework, as represented in Fig. 1. Such REET processes have previously been observed between covalently-linked chromophores12 but equivalent processes have not been observed between independent molecules in solution, at least in part due to the strong distance dependence of the REET phenomenon, and triplet–triplet energy transfer in general. The rotaxane scaffold serves as a suitable platform to study this relatively rare photochemical behaviour. Furthermore, we show that binding of a cation into the cavity formed by the mechanical bond modulates the REET process and thus the luminescence response of the system. To our knowledge, this is the first report of a chemical stimulus modulating REET, suggesting a new luminescence lifetime-based reporting mode for the development of molecular chemosensors.
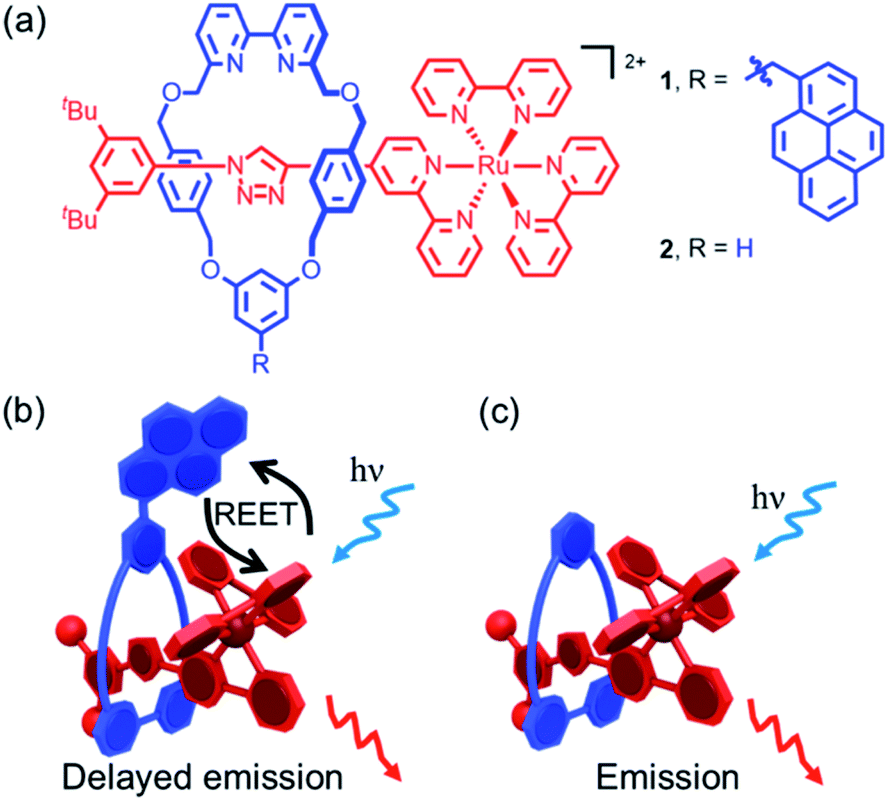 | ||
| Fig. 1 (a) Rotaxanes 1 and 2. (b) Cartoon of the REET process and consequent delayed luminescence in bichromophoric [2]rotaxane 1; (c) cartoon of the luminescent process in simple pyrene-free rotaxane 2. | ||
Results and discussion
The prerequisites for interchromophore REET, following photoexcitation, are that their lowest-lying energy levels are quasi-isoenergetic (within a few hundred cm−1) and that the kinetics for energy transfer should be faster than other major deexcitation processes.12,13 These conditions can be satisfied using combinations of transition metal complexes (microsecond excited-state lifetimes) and organic chromophores (millisecond excited-state triplet lifetimes), which allows ample time for bidirectional energy transfer. Thus, prototype bichromophoric rotaxane 1 (Fig. 1a) was designed with a pyrene-like chromophore on the ring component and a Ru(bpy)32+-based chromophore (bpy = 2,2′-bipyridine) as a stopper of the axle component.14 Rotaxane 1 was prepared using an active template15 Cu-mediated azide–alkyne cycloaddition (AT-CuAAC)16 reaction between the corresponding pyrene-functionalized bipyridine-containing macrocycle (Scheme S1†),17 an acetylene functionalized Ru(bpy)32+ complex, and an aryl azide (Scheme S2†). Analogue 2, which lacks the pyrene chromophore, was prepared as a reference compound (Scheme S2†).The electronic absorption spectrum of 1 (Fig. S1†) contains the distinct absorption bands of both the pyrene and Ru(bpy)32+ chromophores, indicating minimal ground-state electronic interaction. The MLCT absorption bands in the visible spectral region in 1 and 2 are identical, as is the red room temperature MLCT-based emission (λem. max. = 620 nm, λex = 450 nm, Φ = 0.11 for both) in degassed CH3CN (Fig. S2†). A small energy difference (ΔE = 515 cm−1) between the lowest-lying triplets of the two chromophores in rotaxane 1, as required for REET, was estimated as the difference between the highest energy MLCT and pyrene features in 77 K phosphorescence spectra (Fig. S3†).
Despite similar emission spectra, enhanced O2 sensitivity was observed for 1 compared with 2 (Φdeoxy/Φoxy = 18.3 vs. Φdeoxy/Φoxy = 5.0). This is consistent with enhanced quenching by dissolved O2 in the case of 1 due to a longer excited-state lifetime, as confirmed by luminescence decay values of τ = 9.75 μs vs. τ = 1.2 μs for 1 and 2, respectively (Fig. 2). Analysis of luminescence decays on shorter timescales at 300 K (Fig. S4†) revealed an additional short decay component (τ ≈ 15 ns) in the case of 1 but not 2, which corresponds to the time required for energy partitioning between the chromophores to give a thermally-equilibrated excited system, whose deexcitation corresponds to the slow component.
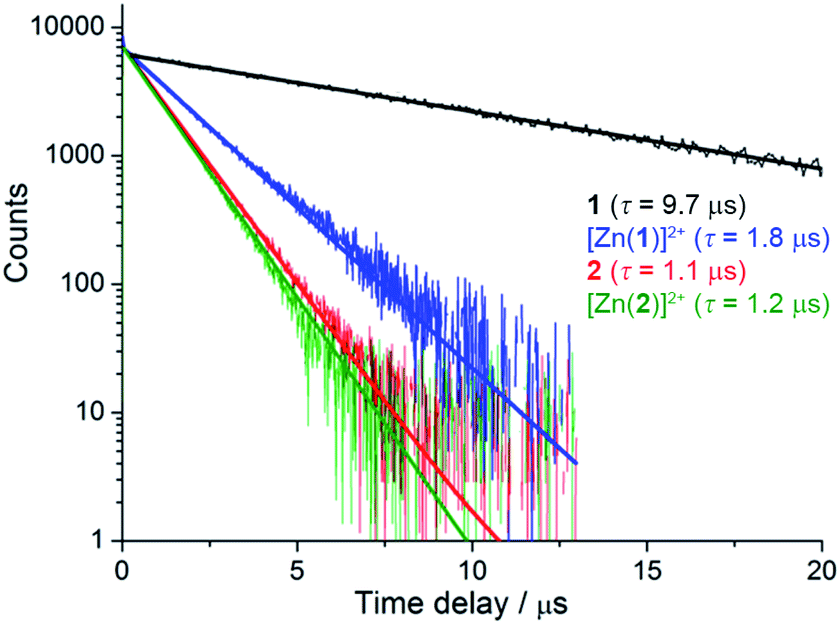 | ||
| Fig. 2 Luminescence decays of rotaxanes 1, [Zn(1)]2+, 2 and [Zn(2)]2+ in degassed acetonitrile (λex = 450 nm, λem = 620 nm). | ||
Unambiguous evidence for the formation of an excited-state equilibrium as a result of REET between ring and stopper was provided by transient absorption spectroscopy (Fig. 3). Selective excitation in the MLCT band of the Ru(bpy)32+ chromophore revealed an initial negative ground state-bleaching band (and positive signature at 370 nm) that evolves synchronously with the growth of a positive band (410 nm) ascribed to the pyrene Tn ← T1 transient with a time constant similar to that of the prompt luminescence decay component (Fig. S5† for kinetic analysis). This corresponds to the real-time observation of energy redistribution between the rotaxane ring and axle chromophores, with a rate that is the sum of forward (kf) and back (kb) interchromophore energy transfer rates.13
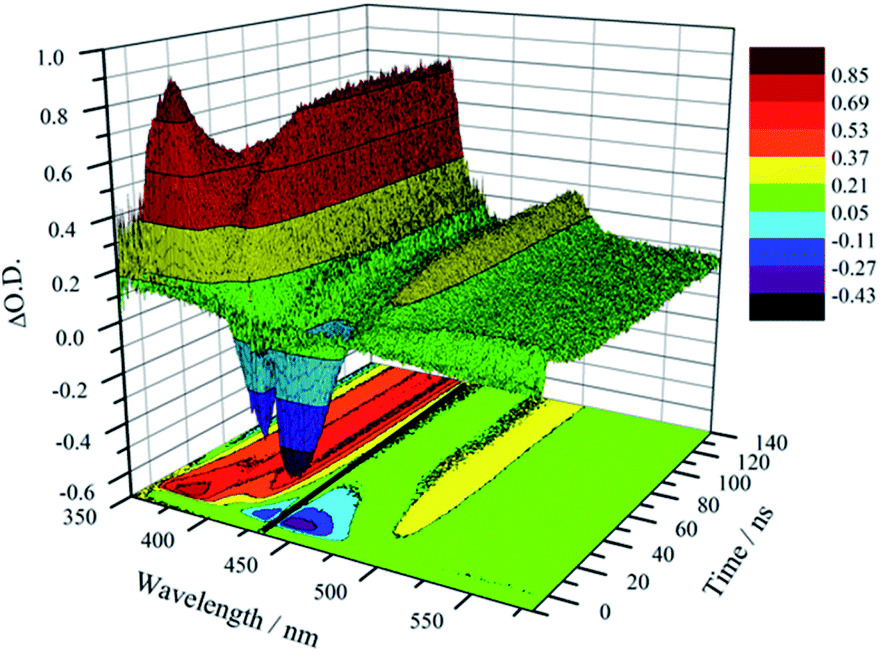 | ||
| Fig. 3 Transient absorption map showing excitation energy redistributing between pyrene and Ru(bpy)32+ in 1 in CH3CN (λex = 450 nm) at 300 K. Note: artefact at 442 nm is due to notch filter blocking parasite laser excitation. | ||
After the initial energy redistribution between ring and axle chromophores, the concomitant decay of the transient signals associated with pyrene triplet and MLCT transient unambiguously confirmed that a dynamic excited-state equilibrium had been established. The dynamic behaviour of excited rotaxane 1, which is governed by interplay of axle and ring components, was mathematically modelled and described by a set of differential equations (ESI†) and may further be rationalised by the equation below. Here α and (1 − α) correspond to the relative populations of the excited ring and axle chromophores at equilibrium, respectively. Using this notation, α/(1− α) equates to the excited-state equilibrium constant, Keq.
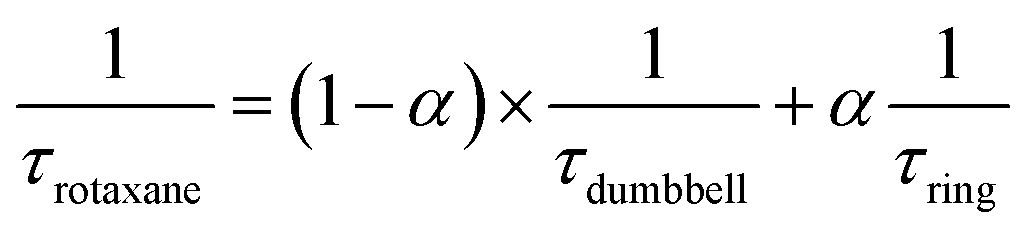
Analysis of variable temperature spectroscopic data (see ESI†) afforded a triplet–triplet energy gap of 475 cm−1, similar to the value obtained from the low temperature phosphorescence data (515 cm−1) and the population of each excited state at equilibrium as a function of temperature, which was also determined through kinetic analysis. At equilibrium at 298 K, the excitation energy is predominantly localized on the ring pyrene unit (81%) compared with the inorganic stopper (19%), giving a Keq of ca. 4. Furthermore, the rate of the back transfer (from ring to stopper) and forward (stopper to ring) transfer was calculated (7.07 × 107 s−1vs. 5.36 × 108 s−1, respectively).
Having demonstrated that REET occurs efficiently in rotaxane 1, we turned our attention to how this process might be modulated using the properties of the interlocked structure. Rotaxanes with cavities similar to those of 1 and 2 have been shown to bind first row transition metal ions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in a mechanically chelating fashion involving the bipyridine unit and the triazole.18 As the triazole unit of 1 is directly conjugated to the Ru(bpy)32+ chromophore, coordination of an ion such as Zn2+ is expected to stabilize the MLCT state by rendering the ligand more electron deficient. Even a slight modification of the 3MLCT energy level with respect to that of the pyrene, which is largely insensitive to the medium, is expected to perturb and potentially interrupt the REET process with an accompanying effect on luminescence lifetime. Thus, we embarked on a proof-of-principle demonstration of luminescent lifetime-based sensing through analyte-mediated perturbation of REET, an unexplored approach that may have advantages for time-gated detection and sensor concentration-independent measurements.
:1 stoichiometry in a mechanically chelating fashion involving the bipyridine unit and the triazole.18 As the triazole unit of 1 is directly conjugated to the Ru(bpy)32+ chromophore, coordination of an ion such as Zn2+ is expected to stabilize the MLCT state by rendering the ligand more electron deficient. Even a slight modification of the 3MLCT energy level with respect to that of the pyrene, which is largely insensitive to the medium, is expected to perturb and potentially interrupt the REET process with an accompanying effect on luminescence lifetime. Thus, we embarked on a proof-of-principle demonstration of luminescent lifetime-based sensing through analyte-mediated perturbation of REET, an unexplored approach that may have advantages for time-gated detection and sensor concentration-independent measurements.
1H-NMR analysis (Fig. S7†) of rotaxane 1 in the absence or presence of Zn2+ revealed differences consistent with binding of the metal ion in the rotaxane cavity; several resonances were seen to shift, notably the triazole C–H and the central proton of the resorcinol moiety of the macrocycle. Spectrophotometric titrations of rotaxane 1 with Zn(ClO4)2·6H2O (Fig. S8†) were carried out to further investigate ion binding and obtain binding constants. On adding Zn2+, the MLCT absorption bands (400–500 nm) became slightly less intense and broader. Several isosbestic points (477 nm, 374 nm and 325 nm) along with the Job plot (Fig. S9†) are consistent with a 1:1 stoichiometry for Zn2+ binding by 1. Unsurprisingly, binding constants of the same order of magnitude were obtained for rotaxanes 1 and 2 (7.14 ± 2.6 × 106 M−1 and 1.15 ± 0.2 × 106 M−1, respectively), as they integrate structurally similar binding cavities (Fig. S10†).
More striking Zn2+-induced changes were observed in the luminescence spectra. The luminescence band maximum of 1 (CH3CN, λex = 475 nm, air-equilibrated) shifts from 626 nm to 649 nm (Δω = 705 cm−1) on adding Zn2+ and the emission intensity increases (Φ([Zn(1)]2+)/Φ(1) = 5, Fig. 4). In the case of 2, Zn2+ coordination gives rise to a similar shift in the emission maxima, but with a smaller luminescence enhancement (Φ([Zn(2)]2+)/Φ(2) = 1.5, Fig. S10†). The enhanced luminescence efficiency of rotaxane 1 in the presence of Zn2+ is consistent with an ion-induced disruption of energy shuttling between the axle and macrocycle chromophores as, if this process is rendered less efficient, the lifetime of the emission is shortened and quenching by ambient oxygen is reduced, resulting in luminescence turn “on”. Luminescence decay analysis supports this hypothesis as the delayed luminescence lifetime components of rotaxanes 1 and 2 in degassed solution in the presence of 5 eq. Zn2+ are, respectively, 1.8 μs and 1.2 μs versus 9.75 μs and 1.1 μs for 1 and 2 alone (Fig. 2).
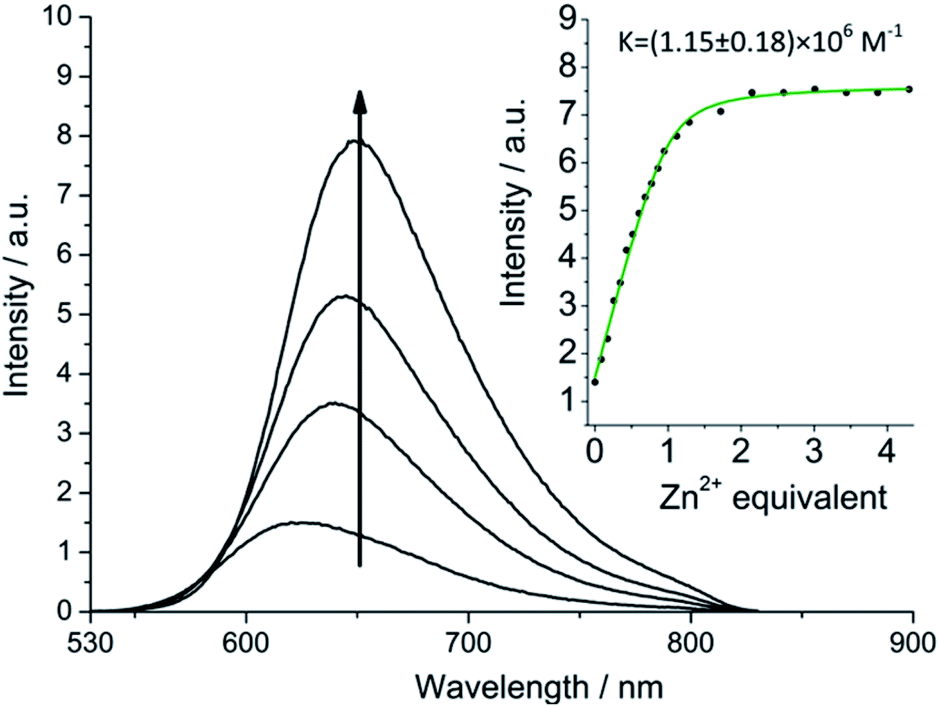 | ||
| Fig. 4 Luminescence spectra (λex = 475 nm) of 1 on adding Zn(ClO4)2 in air equilibrated acetonitrile showing ion-regulated oxygen sensitivity (equivalents of Zn2+ in order of increasing emission intensity at 650 nm: 0, 0.4, 0.8, 4.5). Inset: changing emission intensity (λ = 640 nm) on adding Zn2+. | ||
The transient absorption map of excited [Zn(1)]2+ shows several qualitatively similar features to that of 1 (see Fig. S11 and kinetic analysis in Fig. S12†). However, after the initial relaxation (τ = 13 ns), a larger degree of the excitation energy is localized on the axle chromophore (67% versus 19% for 1). This demonstrates that the excited-state equilibrium is significantly perturbed by ion binding, which is reflected in a luminescence lifetime modulation and increased emission intensity in oxygenated solution because of decreased O2 sensitivity. Indeed, binding of Zn2+ stabilizes/lowers the 3MLCT state energy by ca. 700 cm−1, while the pyrene triplet states are unaltered, thus decreasing the proportion of energy that is temporarily stored on the pyrene group. Fig. 5 summarises the ensemble of pertinent low-lying states, relative energies and rates of ring-axle energy transfer processes in 1 compared with [Zn(1)]2+.
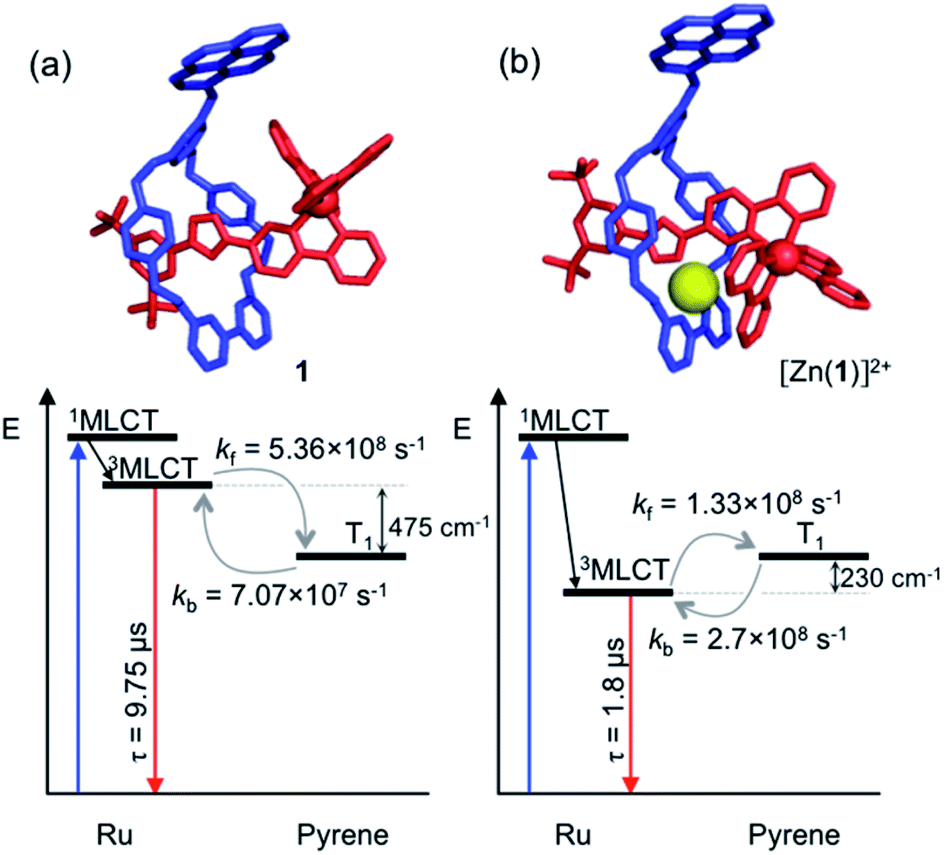 | ||
| Fig. 5 Jablonski–Perrin diagram representing pertinent low lying excited states and electronic energy transfer between 3MLCT and triplet pyrene of rotaxanes 1 (a) and [Zn(1)]2+ (b) at 300 K. Molecular modelling of rotaxane structures used Spartan'18 – MMFF force field.19 | ||
Finally, as might be anticipated from the above discussion, the analogous titration of 1 with Zn2+ in degassed solution is rather different from that obtained under oxygenated conditions (Fig. S13†). The steady-state emission intensity of 1 varies much less on adding Zn2+ (ratio 1:1.4) and an isoemissive point (616 nm) is observed. Thus, whereas under oxygenated conditions rotaxane 1 reports the binding of Zn2+ in a “switch on” manner, under deoxygenated conditions the rotaxane host facilitates ratiometric detection of Zn2+ by monitoring unchanging emission intensity (616 nm) versus significant MLCT changes at, for example, 645 nm, making Zn2+ detection largely rotaxane concentration independent.
Conclusions
We have reported the first example of reversible electronic energy transfer between chromophores that are not covalently linked by using the mechanical bond as a platform. Integrating energetically and kinetically matched chromophores onto the ring and axle units of a rotaxane is shown to offer a design principle to enable fast reversible intercomponent energy exchange to take place resulting in delayed luminescence and long luminescence lifetimes. The reversibility of the energy transfer process is shown to be highly efficient as identical luminescence quantum yields are measured in the bichromophoric rotaxane and pyrene-free variant, indicating no significant additional energy loss pathway in the current case. Furthermore, the high sensitivity of this energy shuttling to perturbation of the relative energies of the excited states involved in the REET process was used to develop a simple chemosensor with the potential for ratiometric detection of metal ions, an unexplored sensing mechanism. This rotaxane platform could further be adapted to integrate alternative kinetically and energetically matched dye pairs exhibiting REET, to regulate excitation and emission wavelength, luminescence lifetime range and modulate oxygen sensitivity. We are now exploring coupled ring and energy shuttling as a method for developing new approaches to lifetime-based chemosensing and photosensitizer development.Data availability
Principle data and characterisation is available in ESI.Author contributions
All authors contributed to manuscript preparation. Additionally:- SY: Synthesis, characterization, titrations; AK: Steady-state & time-resolved spectroscopy; JEML: Synthesis, analysis and supervision. VM-C: Experimental revisions & molecular modelling; GJ: Fast spectroscopy & data analysis; J-LP: Supervision & editing; SMG: Molecule design, analysis and supervision. NMcC: Concepts, photoactive molecule designs, supervision and data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Agence Nationale de la Recherche [grant number ANR-16-CE29-0011], China Scholarship Council (S. Y.), European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreements: no. 796612 (V. M.-C.) & no. 660731 (J. E. M. L.) and CNRS. SMG thanks the Royal Society for a Wolfson Research Fellowship.Notes and references
- For recent reviews see: (a) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC; (b) D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543 CrossRef CAS PubMed; (c) S. Yu, N. D. McClenaghan and J.-L. Pozzo, Photochem. Photobiol. Sci., 2019, 18, 2102 CrossRef CAS PubMed; (d) S. Corra, M. Curcio, M. Baroncini, S. Silvi and A. Credi, Adv. Mater., 2020, 32, 1906064 CrossRef CAS PubMed.
- Selected reviews on the synthesis and properties of MIMs: (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260 CrossRef CAS PubMed; (b) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398 CrossRef CAS; (c) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 2016 CrossRef; (d) X. Hou, C. Ke and J. F. Stoddart, Chem. Soc. Rev., 2016, 45, 3766 RSC; (e) J. R. J. Maynard and S. M. Goldup, Chem, 2020, 6, 1914 CrossRef CAS.
- (a) W. Zhou, J. Li, X. He, C. Li, J. Lv, Y. Li, S. Wang, H. Liu and D. Zhu, Chem.–Eur. J., 2008, 14, 754 CrossRef CAS; (b) W.-K. Wang, Z.-Y. Xu, Y.-C. Zhang, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Chem. Commun., 2016, 52, 7490 RSC; (c) X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582 RSC; (d) Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584 CrossRef CAS PubMed; (e) A. H. G. David, R. Casares, J. M. Cuerva, A. G. Campana and V. Blanco, J. Am. Chem. Soc., 2019, 141, 18064 CrossRef CAS PubMed.
- Selected examples and reviews: (a) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC; (b) J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228 CrossRef CAS PubMed; (c) M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5310 CrossRef CAS PubMed; (d) J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584 CrossRef CAS PubMed; (e) M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315 CrossRef CAS PubMed; (f) K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052 RSC; (g) R. C. Knighton, S. Dapin and P. D. Beer, Chem.–Eur. J., 2020, 26, 5288 CrossRef CAS PubMed.
- Selected examples: (a) J. E. H. Buston, J. R. Young and H. L. Anderson, Chem. Commun., 2000, 905 RSC; (b) J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329 RSC; (c) J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025 CrossRef CAS; (d) H. Masai, J. Terao, S. Seki, S. Nakashima, M. Kiguchi, K. Okoshi, T. Fujihara and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 1742 CrossRef CAS; (e) H. Masai and J. Terao, Bull. Chem. Soc. Jpn., 2019, 92, 529 CrossRef CAS; (f) Z. Zhang, G. J. Tizzard, J. A. G. Williams and S. M. Goldup, Chem. Sci., 2020, 11, 1839 RSC.
- Selected examples: (a) M. Inouye, K. Hayashi, Y. Yonenaga, T. Itou, K. Fujimoto, T. A. Uchida, M. Iwamura and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 14392 CrossRef CAS; (b) X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef; (c) K. Hayashi, Y. Miyaoka, Y. Ohishi, T. A. Uchida, M. Iwamura, K. Nozaki and M. Inouye, Chem.–Eur. J., 2018, 24, 14613 CrossRef CAS; (d) A. Garci, Y. Beldjoudi, M. S. Kodaimati, J. E. Hornick, M. T. Nguyen, M. M. Cetin, C. L. Stern, I. Roy, E. A. Weiss and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 7956 CrossRef CAS PubMed.
- (a) A. Tron, H.-P. Jacquot de Rouville, A. Ducrot, J. H. R. Tucker, M. Baroncini, A. Credi and N. D. McClenaghan, Chem. Commun., 2015, 51, 2810 RSC; (b) A. Tron, P. J. Thornton, C. Lincheneau, J.-P. Desvergne, N. Spencer, J. H. R. Tucker and N. D. McClenaghan, J. Org. Chem., 2015, 80, 988 CrossRef CAS PubMed; (c) P. Wan, Y. Jiang, Y. Wang, Z. Wang and X. Zhang, Chem. Commun., 2008, 5710 RSC.
- (a) Q.-H. Guo, Y. Qiu, X. Kuang, J. Liang, Y. Feng, L. Zhang, Y. Jiao, D. Shen, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 14443 CrossRef CAS; (b) L. Scarpantonio, A. Tron, C. Destribats, P. Godard and N. D. McClenaghan, Chem. Commun., 2012, 48, 3981 RSC; (c) G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F.-H. Huang, J. Am. Chem. Soc., 2012, 134, 8711 CrossRef CAS.
- Selected examples: (a) F. G. Gatti, S. Leon, J. K. Wong, G. Bottari, A. Altieri, M. A. Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brouwer and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10 CrossRef CAS PubMed; (b) E. M. Perez, D. T. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210 CrossRef CAS PubMed; (c) Q. C. Wang, D. H. Qu, J. Ren, K. Chen and H. Tian, Angew. Chem., Int. Ed., 2004, 43, 2661 CrossRef CAS PubMed; (d) W. Zhou, D. Chen, J. Li, J. Xu, J. Lv, H. Liu and Y.-L. Li, Org. Lett., 2007, 9, 3929 CrossRef CAS PubMed; (e) B. Colasson, A. Credi and G. Ragazzon, Coord. Chem. Rev., 2016, 325, 125 CrossRef CAS; (f) A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R. Orenes, M. Alajarina and J. Berna, Chem. Sci., 2017, 8, 3775 RSC.
- Selected examples: (a) T. Ogoshi, D. Yamafuji, T. Yamagishi and A. M. Brouwer, Chem. Commun., 2013, 49, 5468 RSC; (b) S. Kirner, C. Henkel, D. M. Guldi, J. D. Megiatto and D. I. Schuster, Chem. Sci., 2015, 6, 7293 RSC; (c) L. D. Movsisyan, M. D. Peeks, G. M. Greetham, M. Towrie, A. L. Thompson, A. W. Parker and H. L. Anderson, J. Am. Chem. Soc., 2014, 136, 17996 CrossRef CAS PubMed; (d) H.-G. Fu, H.-Y. Zhang, H.-Y. Zhang and Y. Liu, Chem. Commun., 2019, 55, 13462 RSC; (e) J. Pruchyathamkorn, W. J. Kendrick, A. T. Frawley, A. Mattioni, F. Caycedo-Soler, S. F. Huelga, M. B. Plenio and H. L. Anderson, Angew. Chem., Int. Ed., 2020, 59, 16455 CrossRef CAS PubMed; (f) P. Rajamalli, F. Rizzi, W. Li, M. A. Jinks, A. K. Gupta, B. A. Laidlaw, I. D. W. Samuel, T. J. Penfold, S. M. Goldup and E. Zysman-Colman, Angew. Chem. Int. Ed., 2021, 60, 12066 CrossRef CAS PubMed.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece and N. Spencer, Chem.–Eur. J., 2000, 6, 3558 CrossRef CAS; (b) A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291(5511), 2124 CrossRef CAS PubMed; (c) B. Colasson, A. Credi and B. Ventura, Chem.–Eur. J., 2020, 26, 534 CrossRef CAS PubMed; (d) T. Kumpulainen, M. R. Panman, B. H. Bakker, M. Hilbers, S. Woutersen and A. M. Brouwer, J. Am. Chem. Soc., 2019, 141, 19118 CrossRef CAS PubMed; (e) F. Jia, H. V. Schroder, L.-P. Yang, C. von Essen, S. Sobottka, B. Sarkar, K. Rissanen, W. Jiang and C. A. Schalley, J. Am. Chem. Soc., 2020, 142, 3306 CrossRef CAS PubMed.
- (a) S. A. Denisov, S. Yu, G. Jonusauskas, J.-L. Pozzo and N. D. McClenaghan, ChemPhysChem, 2016, 17, 1794 CrossRef CAS PubMed; (b) X. Y. Wang, A. Del Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5, 55 CrossRef CAS.
- W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1992, 96, 2917 CrossRef CAS.
- For examples of other matched pairs see: (a) D. T. Yonemoto, C. M. Papa, C. Mongin and F. N. Castellano, J. Am. Chem. Soc., 2020, 142, 10883 CrossRef CAS PubMed; (b) S. Denisov, Y. Cudré, P. Verwilst, G. Jonusauskas, M. Marín-Suárez, J. Fernandez-Sanchez, E. Baranoff and N. D. McClenaghan, Inorg. Chem., 2014, 53, 2677 CrossRef CAS PubMed.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- Selected examples: (a) V. Aucagne, K. D. Hanni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186 CrossRef CAS PubMed; (b) H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151 CrossRef CAS PubMed; (c) A. Noor, W. K. C. Lo, S. C. Moratti and J. D. Crowley, Chem. Commun., 2014, 50, 7044 RSC; (d) M. J. Langton, Y. Xiong and P. D. Beer, Chem.–Eur. J., 2015, 21, 18910 CrossRef CAS PubMed.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154 RSC.
- Selected examples: (a) A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205 CrossRef CAS; (b) D. A. Leigh, P. J. Lusby, A. M. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 44, 4557 CrossRef CAS PubMed; (c) M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879 CrossRef CAS PubMed.
- P. E. Deppmeier, B. J. Driessen, A. J. Hehre, W. J. Hehre, T. S. Johnson, J. A. Ohlinger and S. Klunzinger, Spartan 18, build 1.3.0, Wavefunction Inc., Feb 19 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compounds characterization, optical spectroscopic experiments. See DOI: 10.1039/d1sc02225c |
| This journal is © The Royal Society of Chemistry 2021 |