
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase separation of p53 precedes aggregation and is affected by oncogenic mutations and ligands†
Elaine C.
Petronilho‡
a,
Murilo M.
Pedrote‡
a,
Mayra A.
Marques
 a,
Yulli M.
Passos
b,
Michelle F.
Mota
a,
Benjamin
Jakobus
c,
Gileno dos Santos de
Sousa
a,
Filipe
Pereira da Costa
a,
Adriani L.
Felix
a,
Giulia D. S.
Ferretti
a,
Fernando P.
Almeida
d,
Yraima
Cordeiro
b,
Tuane C. R. G.
Vieira
a,
Guilherme A. P.
de Oliveira
*a and
Jerson L.
Silva
*a
a,
Yulli M.
Passos
b,
Michelle F.
Mota
a,
Benjamin
Jakobus
c,
Gileno dos Santos de
Sousa
a,
Filipe
Pereira da Costa
a,
Adriani L.
Felix
a,
Giulia D. S.
Ferretti
a,
Fernando P.
Almeida
d,
Yraima
Cordeiro
b,
Tuane C. R. G.
Vieira
a,
Guilherme A. P.
de Oliveira
*a and
Jerson L.
Silva
*a
aInstitute of Medical Biochemistry Leopoldo de Meis, National Institute of Science and Technology for Structural Biology and Bioimaging, National Center of Nuclear Magnetic Resonance Jiri Jonas, Federal University of Rio de Janeiro, Rio de Janeiro RJ 21941-902, Brazil. E-mail: gaugusto@bioqmed.ufrj.br; jerson@bioqmed.ufrj.br
bFaculty of Pharmacy, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
cModal Informática Ltda, Almeida Godinho, 19, 304, Rio de Janeiro RJ 22741-140, Brazil
dNational Center for Structural Biology and Bioimaging (CENABIO), Federal University of Rio de Janeiro, Rio de Janeiro RJ 21941-902, Brazil
First published on 26th April 2021
Abstract
Mutant p53 tends to form aggregates with amyloid properties, especially amyloid oligomers inside the nucleus, which are believed to cause oncogenic gain-of-function (GoF). The mechanism of the formation of the aggregates in the nucleus remains uncertain. The present study demonstrated that the DNA-binding domain of p53 (p53C) underwent phase separation (PS) on the pathway to aggregation under various conditions. p53C phase separated in the presence of the crowding agent polyethylene glycol (PEG). Similarly, mutant p53C (M237I and R249S) underwent PS; however, the process evolved to a solid-like phase transition faster than that in the case of wild-type p53C. The data obtained by microscopy of live cells indicated that transfection of mutant full-length p53 into the cells tended to result in PS and phase transition (PT) in the nuclear compartments, which are likely the cause of the GoF effects. Fluorescence recovery after photobleaching (FRAP) experiments revealed liquid characteristics of the condensates in the nucleus. Mutant p53 tended to undergo gel- and solid-like phase transitions in the nucleus and in nuclear bodies demonstrated by slow and incomplete recovery of fluorescence after photobleaching. Polyanions, such as heparin and RNA, were able to modulate PS and PT in vitro. Heparin apparently stabilized the condensates in a gel-like state, and RNA apparently induced a solid-like state of the protein even in the absence of PEG. Conditions that destabilize p53C into a molten globule conformation also produced liquid droplets in the absence of crowding. The disordered transactivation domain (TAD) modulated both phase separation and amyloid aggregation. In summary, our data provide mechanistic insight into the formation of p53 condensates and conditions that may result in the formation of aggregated structures, such as mutant amyloid oligomers, in cancer. The pathway of mutant p53 from liquid droplets to gel-like and solid-like (amyloid) species may be a suitable target for anticancer therapy.
Introduction
Many proteins undergo aggregation, leading to severe human diseases, such as Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis.1 However, neurodegenerative diseases are not the only conditions affected by protein aggregation. Cancer has been recently shown to be affected by aggregation, particularly by the aggregation of mutant variants of the tumor suppressor protein p53, which are present in more than 50% of malignant tumors.2–4 In general, effective therapeutic strategies for all these diseases require the prevention of these aggregation processes.4–7 The aggregation of large amorphous or amyloid fibrils terminates a pathway and includes the formation of intermediate oligomeric species and protofibrils. Prior to the formation of more stable aggregate species, several proteins involved in neurodegenerative diseases tend to undergo phase separation and form biomolecular condensates usually by transiting from liquid-like to gel-like and solid-like states.8–11 This transition is particularly applicable for several nuclear proteins that associate with RNAs and DNA to generate membraneless organelles, such as the nucleolus.8–10,12Malignant mutations of p53 are mainly located in the DNA-binding domain (DBD) (p53C), leading to misfolding and aggregation of the mutant isoforms.5,7 Aggregation of p53C results in a mixture of amorphous, oligomeric and fibrillar amyloid species revealed by staining with thioflavin-T (ThT) and thioflavin-S (ThS), electron microscopy and X-ray diffraction.13–15 The aggregation of mutant p53 was suggested to contribute to loss-of-function (LoF) and gain-of-function (GoF) of p53 in cancer.4,5 However, the misfolding routes that lead to aggregation are unknown. Similar to other amyloid proteins, liquid–liquid phase separation (LLPS) can be an important step in p53 aggregation. Polyanions can modulate protein phase separation (PS),12 and both DNA and RNA were shown to modulate p53 aggregation.16,17 LLPS of p53 was suggested to be related to the cellular functions of the protein.18 Additionally, liquid condensates of p53 were suggested to serve as preassembled precursors that facilitate fibril assembly.19 However, additional investigation is needed to determine whether PS of oncogenic mutants is related to cancer.
The present study aimed to determine whether the formation of aggregates of p53 is preceded by PS. p53 is highly active in the nucleolus and in other membraneless nuclear compartments, which are themselves phase-separated organelles. p53 is only transiently expressed at higher concentrations under stress conditions; however, mutant p53 is known to accumulate, generating species that are larger than tetramers and demonstrate amyloid features, which are believed to have GoF effects. In vitro analyses performed in the present study demonstrated phase separation of various p53 constructs using polyethylene glycol (PEG) to simulate a crowding environment and variable temperature to stimulate aggregation. The data showed that before aggregation, p53 underwent PS depending on the protein concentration and temperature. Under the same conditions, p53 mutants (R249S and M237I) underwent PS faster and evolved more rapidly into solid-like aggregates compared with the corresponding properties of the wild-type protein. Additional live cell analysis demonstrated the formation of biomolecular condensates of mutant p53 in the nuclear bodies. Fluorescence recovery after photobleaching (FRAP) experiments were performed in cell and in vitro to evaluate the dynamics of p53 condensates. Full recovery of the fluorescence signal indicated liquid characteristics of the condensates in the nuclei containing wild-type (wt) p53. Incomplete recovery of the fluorescence signal after photobleaching indicated that mutant p53 tended to undergo an solid-like phase transition (SLPT) in the nuclear bodies. The results of in vitro FRAP experiments indicated that the properties of the condensates of wt p53C corresponded to a gel-like state, and the condensates of the M237I mutant p53 underwent a faster PT to a solid-like state. The intrinsically disordered transactivation domain (TAD) is known to delay p53C aggregation20 and was shown to influence phase separation. Additionally, the modulatory effects of biological polyanions have been tested, especially the effects of heparin. A low concentration of guanidinium, which was previously shown to induce a molten globule conformation of p53,21 induced the formation of liquid droplets even in the absence of a crowding agent. The process of phase separation of p53 and its transition to the aggregates described in the present study is likely to be related to the oncogenic activity of p53 and may be an important therapeutic target.
Results
Aggregation of the mutant forms of the core domain of p53
Accumulating evidence indicates that p53 is predisposed to aggregation in vitro13,14,22 and in tumor cells and tissues.13,15,23,24 In general, mutant p53 proteins have higher aggregation propensity than that of wild type protein.13,14 However, the main question is how monomer or tetramers of mutant p53 evolve to higher order oligomers and aggregates resulting in an oncogenic GoF. Phase transitions to solid-like amorphous and amyloid-like states of mutant p53 are a promising target for the development of novel therapeutic strategies against cancer. Oligomers and aggregates were detected in tumor cells by antibodies that recognize amyloid oligomers15,24 and by ThS fluorescence25 or in live cells by fluorescence fluctuation spectroscopy.26 We decided to use wt and two mutants of p53, R249S and M237I (Fig. 1), to determine whether the aggregation is preceded by phase separation. R249S is a mutation highly represented in several types of cancer, especially hepatocellular carcinoma. M237I mutation has been detected in glioblastomas and is usually associated with resistance to temozolomide. Fig. 1a shows various constructs used in the present study.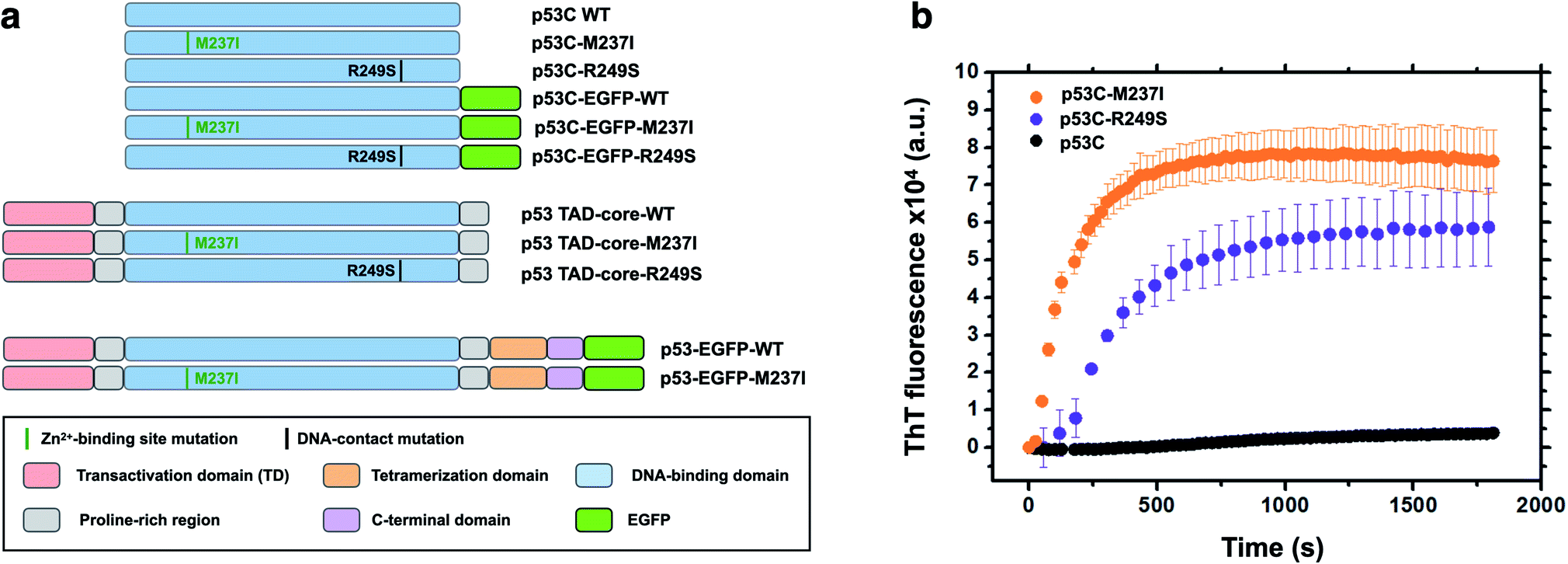 | ||
| Fig. 1 Scheme of the protein constructs and amyloid-like characteristics of mutant p53C in vitro. (a) Scheme of the constructs used in the present study. P53C corresponds to the DNA-binding domain; P53 TAD-core corresponds to the transactivation domain (TAD) and the DNA-binding domain. P53 corresponds to the full-length version of p53. EGFP, enhanced green fluorescent protein. M237I and R249S correspond to the mutations in the Zn2+-binding site and DNA contact site, respectively. Domains are color-coded. (b) Line plot of fluorescence of thioflavin T (ThT) as a function of time for wt, M237I, and R249S p53C constructs. The data are shown as the mean ± s.e.m. of n = 3 independent experiments. | ||
Fig. 1b shows in vitro aggregation kinetics for wt, R249S and M237I p53C forms. In vitro, the R249S and M237I mutant p53C constructs aggregated considerably faster than wt p53C according to an increase in fluorescence of ThT, which is a dye generally used to detect amyloid aggregation. We decided to use these two mutants for the in vitro and in cell studies.
Concentration- and temperature-induced demixing of the p53 core domain
To determine whether p53C undergoes demixing, we analyzed the effect of increasing concentrations of p53C-EGFP by differential interference contrast (DIC) and fluorescence microscopy (Fig. 2a). Demixing of wt and R249S hotspot mutant of p53C was concentration-dependent (Fig. 2a, ESI Fig. S1a and b†). Demixing of wt p53C evolved into liquid-like spheroids, and R249S acquired aggregation-like morphology (Fig. 2b and c). Phase separation resulting in the formation of liquid-like droplets promptly occurred in the presence of the molecular crowding agent PEG and demixing concentrations of wt p53C (Fig. 2d).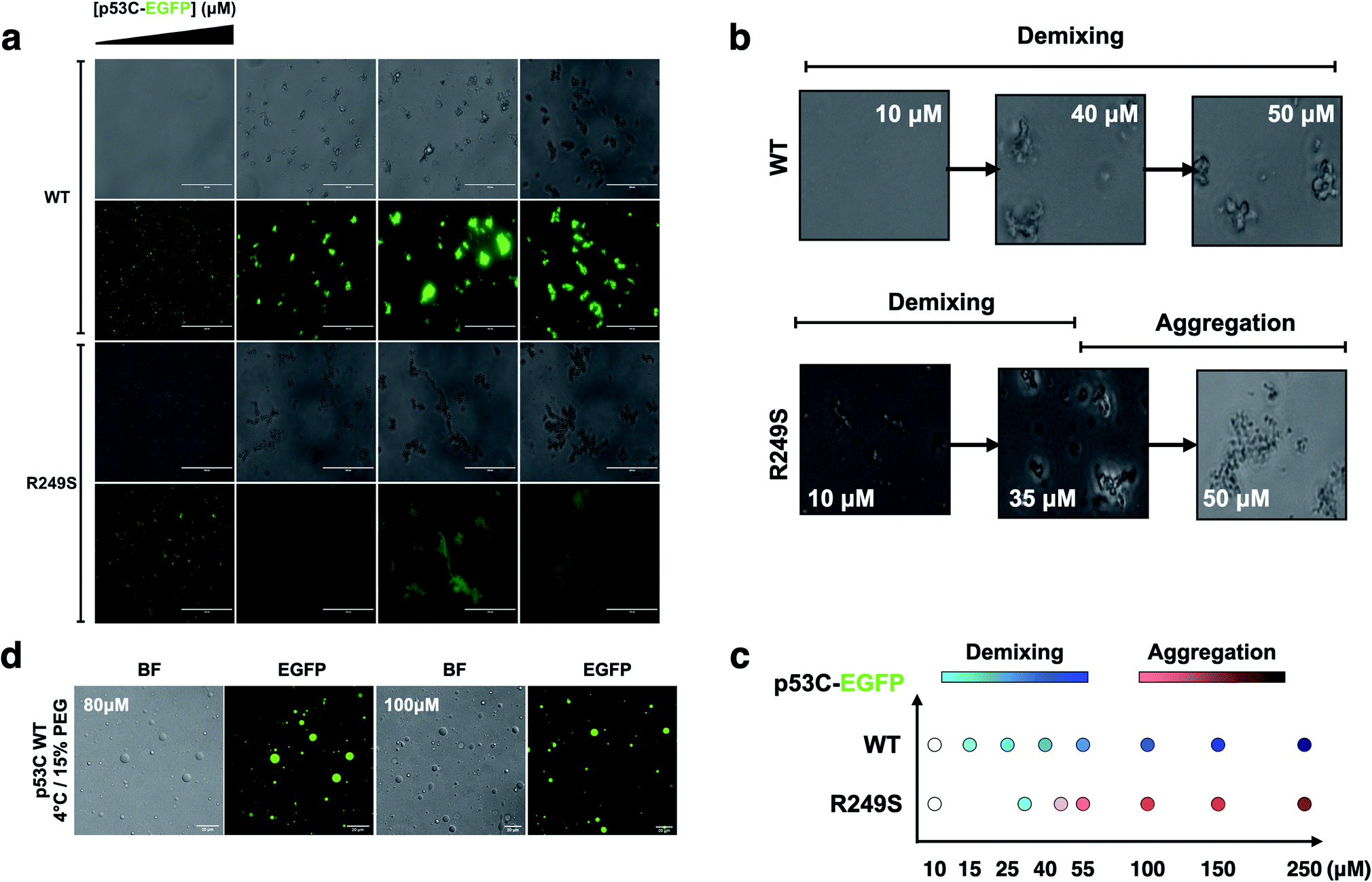 | ||
| Fig. 2 The DNA-binding domain of p53 undergoes phase separation. (a) Image panel showing bright-field (1st and 3rd row) and fluorescent channel (2nd and 4th row) images obtained at increasing concentrations (10, 55, 140 and 250 μM) of wt and R249S p53C-EGFP in the presence of 15% PEG at 4 °C (scale bar, 50 μm) (incubation time = 3 min). (b) Confocal images showing that the concentration influences the demixing of liquid-like spheroids of wt p53C (HIS-p53C) (10, 40 and 50 μM) and aggregation-like spheroids of the R249S mutant (10, 35 and 50 μM) at 4 °C. (c) Schematic representation of demixing concentrations (10, 15, 25, 40, 55, 100, 150, and 250 μM) of the wt and R249S mutant proteins. (d) Droplets of various concentrations (80 μM and 100 μM) of wt p53C in the presence of 15% w/v PEG at 4 °C detected by DIC microscopy (bright-field) and EGFP; scale bars, 20 μm. | ||
We next examined the influence of temperature on p53C PS in the presence of PEG. DIC images were acquired at various time points (from 15 to 60 min) at indicated temperature (Fig. 3). The results showed that wt p53C phase separated into the droplets within 15 min at 4 °C and evolved into spheroids after 60 min (Fig. 3a and b). At 15 °C, a mixture of the droplets and spheroids was detected during the timeframe of the analysis; however, at 37 °C, the reaction diverged rapidly into aggregate-like structures (Fig. 3a–c, ESI Fig. S2a and S3†).
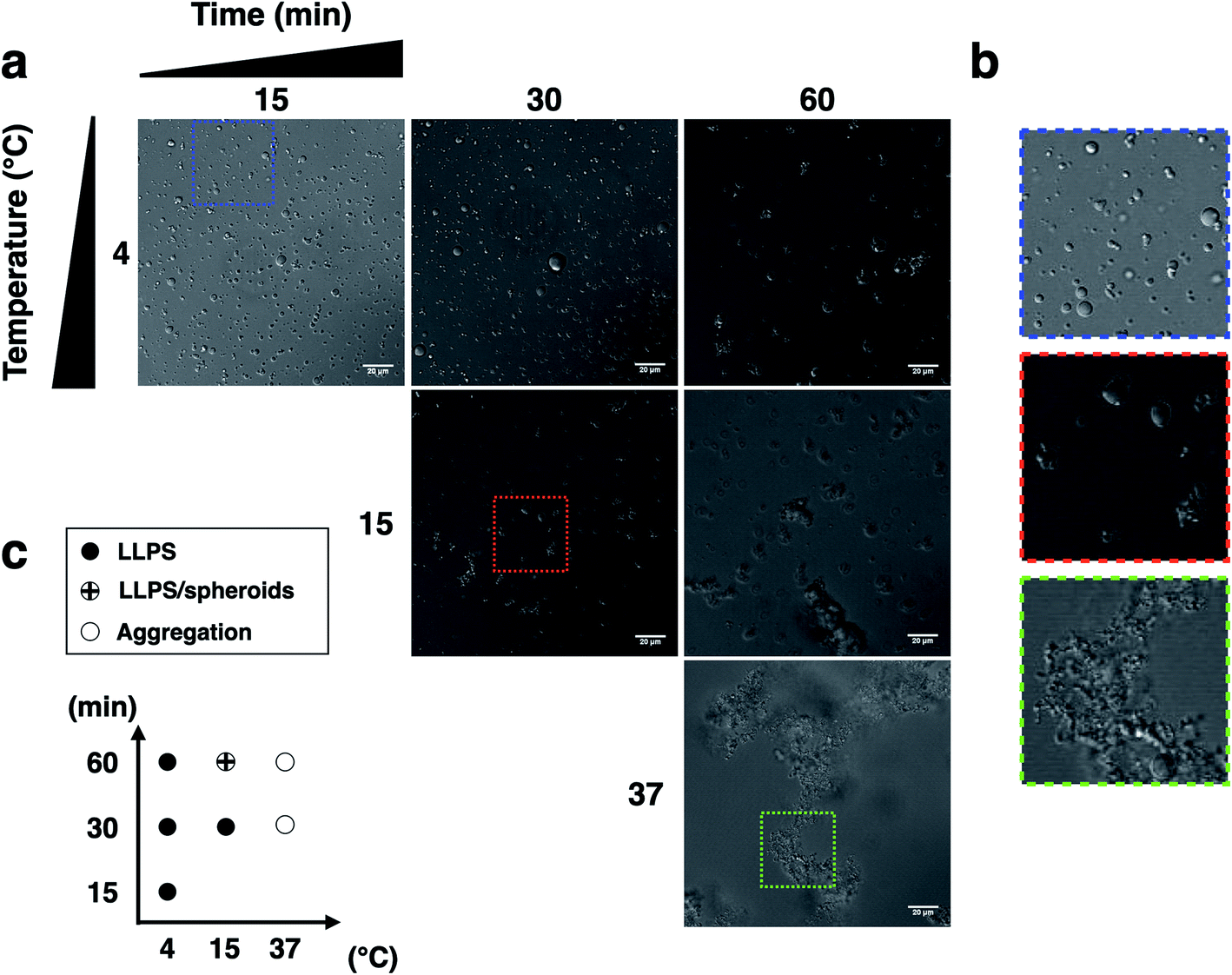 | ||
| Fig. 3 Liquid droplet characteristics of wt p53C (100 μM) at various temperatures and times. (a) and (b) Phase separation (PS) of p53C in the presence of a molecular crowding agent (15% w/v PEG-4000). No phase separation is observed in the absence of PEG or in the absence of p53 protein. (c) Diagram of p53C LLPS temperature (4, 15 and 37 °C) versus time (15, 30 and 60 min), showing the morphology of droplets, spheroids and aggregates under various conditions. | ||
The distribution of the droplet diameter after 15 and 30 min at 4 °C was evaluated by an automatic procedure (ESI Fig. S2b†). After 60 min, the results of distribution analysis were unreliable because the droplets evolved into pleomorphic spheroids (ESI Fig. S2c†). We detected a total of 5683 droplets, with a median diameter of 2.99 μm and a mean diameter of 4.16 μm with a standard deviation of 3.73 μm. The total number of the droplets formed under each condition and their diameters are shown in ESI Table S1† (see Methods).
The effect of transactivation domain on phase separation and transition of p53
We explored the influence of intrinsically disordered regions (IDRs) in p53 by monitoring PS and solid-like phase transition (SLPT) events of wt and two cancer-related mutant proteins. We selected the M237I mutation located in the Zn2+-binding site and the R249S mutation within the DNA-binding site. We used p53C and a larger construct comprising the TAD and both proline-rich segments (p53 TAD-core, Fig. 1a) to determine the influence of p53 IDRs. Two different temperature cycles were used to shift the reaction to the targeted species (droplets, oligomers and amyloids) (Fig. 4a), and the formed species were imaged. Negative staining transmission electron microscopy (TEM) demonstrated the difference in SLPT events between p53C wt (Fig. 4b and c) and p53 TAD-core wt (Fig. 4d and e). p53C wt and p53C M237I showed negligible aggregation after thawing (TH) in contrast to p53C R249S (Fig. 4b). However, p53C M237I demonstrated an SLPT to small oligomers (Fig. 4b). The presence of strongly contrasting materials after the temperature cycle (TC) indicated that all p53C constructs were severely aggregated most likely as amorphous precipitates (Fig. 4c). Interestingly, the pathways of the TAD-core of M237I and wt p53 were diverging after thawing or temperature cycle, respectively (Fig. 4d and e). The species with weak contrast and sphere-like morphology were detected at low magnification (Fig. 4d and e), which was possibly related to PS events detected by DIC microscopy. However, numerous small oligomer-like species detected at higher magnification appeared to form the building blocks of larger droplets (Fig. 4f). PS is a macroscopic physical property, and condensation into oligomers is at the detection limit of a microscopic/mesoscopic event, as predicted by Weber in the early 1990s.27,28 The droplets formed by TAD-core of wt p53 were smaller than the droplets formed by TAD-core of M237I p53 and were characterized by a narrow diameter distribution (Fig. 4g).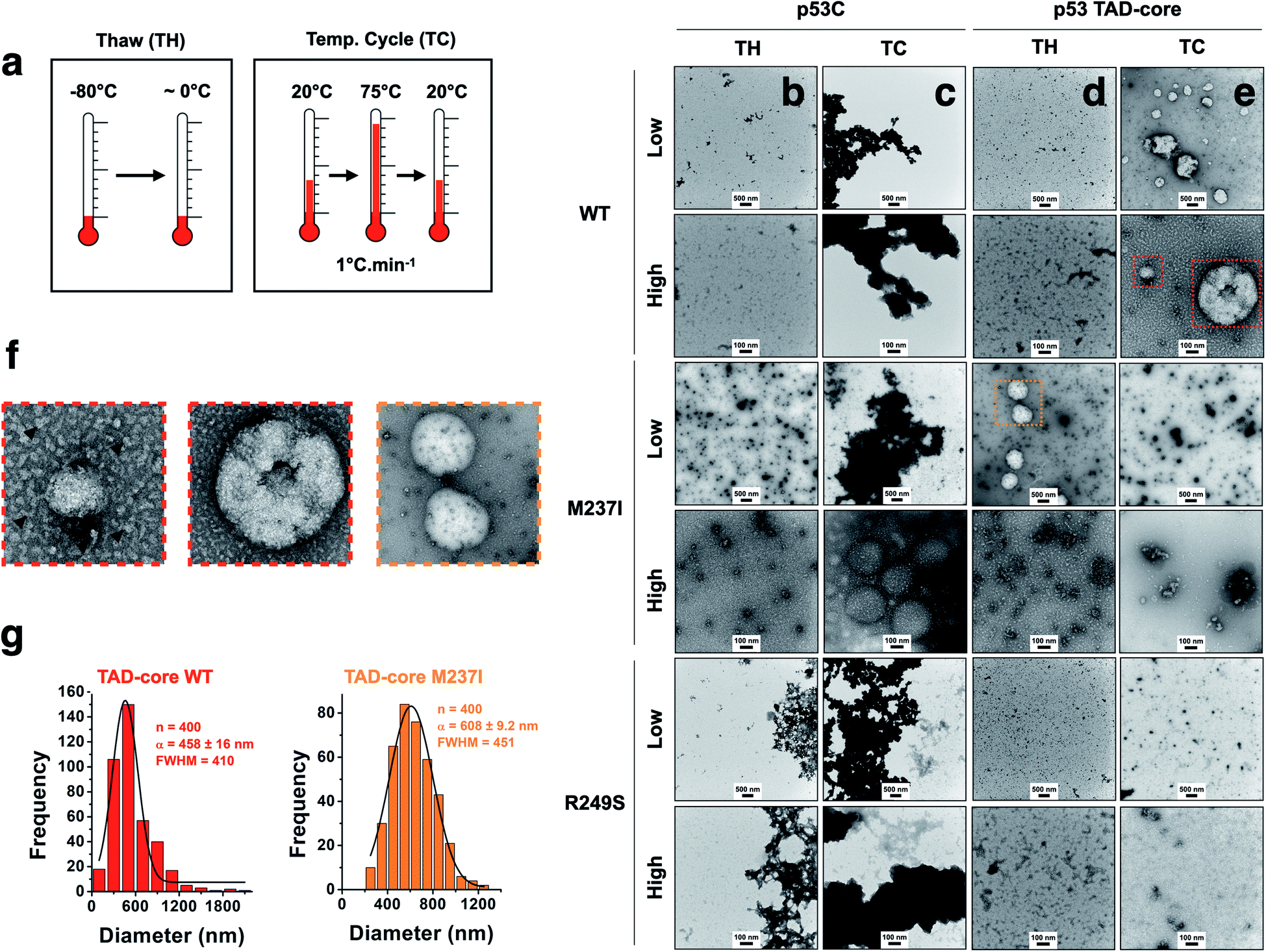 | ||
| Fig. 4 Influence of intrinsically disordered regions of p53 on the aggregation process. (a) Scheme showing two tested temperature schemes: Thawing (TW) and increase and decrease in temperature (TC). (b–e) Transmission electron micrographs showing the behavior of the wt, M237I and R249S proteins within the context of p53C and p53 TAD-core with respect to solid-like phase transition events at 4 °C (scale bars, 100 and 500 nm). Low and high labels refer to low (scale bar = 500 nm) and high (scale bar = 100 nm) magnification images. (f) Enlarged transmission electron micrographs showing small oligomer-like species (arrowheads). (g) Histogram (bar plots) showing the diameter distribution of droplets of wt and M237I within the context of the p53 TAD-core protein. The data were fitted to a Gaussian distribution (black lines) and show the mean (α) ± s.e.m. of n = 400 measured droplets. FWHM corresponds to full width at half maximum. | ||
p53 condensates and solid-like phase transition in the nucleus
To evaluate whether p53 forms condensates in the cells associated with regions enriched with nucleic acids, we performed the transfection of p53 wt-EGFP, M237I-EGFP and EGFP into H1299 cells. The data of Fig. 5a and b showed that both wt and M237I proteins were dispersed into the nucleus; however, the M237I protein was more likely to be associated with the nuclear compartments, including the nucleolus, which is an immiscible compartment full of nucleic acid molecules (Fig. 5a, b and ESI Fig. S4a–d†). | ||
| Fig. 5 Evidence of p53 LLPS in a cell-based model. (a) Collection of confocal images showing the fluorescence and bright-field (BF) channels. EGFP-fused proteins were used as reporters to determine the localization of wt and M237I full-length p53 after the transfection of H1299 cells (scale bars, 5 μm). (b) Zoomed-in images of cropped areas in ‘a’ showing the nuclear body compartments (nucleolar regions) containing wt (red arrows) and M237I p53 (blue arrows). (c) DIC images of wt and M237I full-length p53 showing fluorescence recovery after photobleaching of the nuclei and nucleolar condensates. Red circle shows bleached area. White arrow shows nucleolar p53 before bleaching (scale bars, 20 μm). (d and e) Line plots showing fluorescence recovery of (d) wt and the M237I p53 and (e) M237I p53 in two different cellular compartments, the nucleus and nucleolus. The data are shown as the mean ± s.d. of n = 5 measurements. | ||
PS may be reversible or can proceed to SLPT events.9,29 Fluorescence recovery after photobleaching (FRAP) is a suitable assay to evaluate the molecular dynamics of the phase-separated liquid droplets.30,31 Fluorescence recovery is fast if the phase-separated droplets are liquid-like. In contrast, fluorescence recovery is slow and may not recover completely in the case of the gel-like and solid-like droplets.
FRAP experiments were performed to evaluate the dynamics of p53 condensates. Comparison of the recovery kinetics of p53 wt-EGFP and M237I-EGFP condensates is shown in Fig. 5c–e. The recovery of M237I dispersed in the nucleoplasm was slightly slower than that of p53 wt (Fig. 5d). However, full recovery of fluorescence indicated that p53 condensates were in a liquid-like state in the nucleoplasm. However, a very slow recovery indicated that the droplets trapped in the nuclear compartments behaved more like a gel or a solid state (Fig. 5e), especially in the case of the M237I mutant of p53. These results indicated that mutant p53 underwent SPLT. Full recovery of fluorescence demonstrated liquid characteristics of the condensates of wt p53 in the nucleus. Mutant p53 tended to undergo an SLPT, especially in the nuclear bodies, as shown by slow and incomplete recovery of fluorescence after photobleaching.
Gel-like states and solid-like phase transition (SLPT) detected by FRAP
FRAP was used to evaluate the dynamics of wt and M237I p53C in vitro. The data of Fig. 6 indicated that the wt p53C droplets only partially recovered the fluorescence signal in contrast to the conditions in the nucleus, indicating that the DBD domain tended to evolve into a gel-like state (Fig. 6a and b). Mutant p53C was characterized by a typical solid-like state dynamics with essentially undetectable fluorescence recovery in contrast to the wild type isoform that partially recovered the signal (Fig. 6a and b). The influence of the EGFP-tagging of the protein on the aggregation of M237I p53C was assessed by measuring soluble fractions after temperature-induced aggregation over time (see Methods). Untagged M237I p53C had a slightly greater tendency to aggregate compared to that of EGFP-tagged protein at all three tested protein concentrations (Fig. 6c).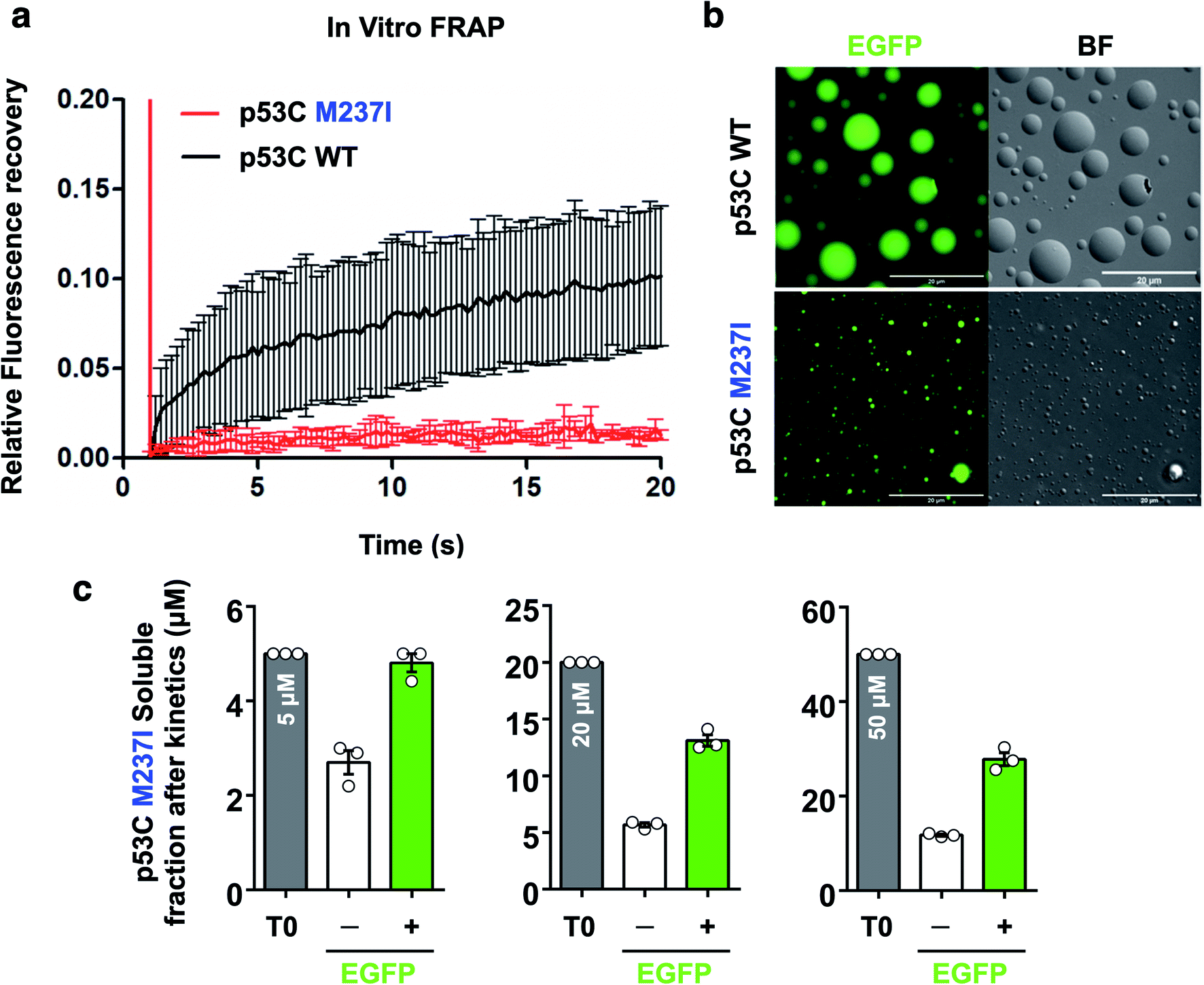 | ||
| Fig. 6 Fluorescence recovery after photobleaching of p53C in vitro. (a) Line plot showing fluorescence recovery of wt and M237I p53C in the presence of 15% PEG. The data are shown as the mean ± s.d. of n = 5 droplets. (b) Images showing the fluorescence and the bright-field (BF) channels of spherical droplets of wt and M237I p53C at 4 °C (time, 0 s; scale bars, 20 μm). (c) Scatter plots representing the soluble fractions after the kinetic assay of M237I p53C in the absence (−) or in the presence (+) of the EGFP-tagged protein. T0 corresponds to the monomer concentration used before kinetic assays. The data are shown as the mean ± s.e.m. of n = 3 independent experiments using the same protein batch. | ||
Effects of polyanions on phase separation and phase transition of p53
The nucleus contains a very high concentration on negative polyelectrolytes, such as RNA and DNA. Nuclear environment plays an important role in PS of nuclear proteins.12,29,32 We have reported that various RNAs influence the aggregation of p53C and full-length p53.17,29,32 High stoichiometric RNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :p53C ratios inhibited aggregation, and low stoichiometric ratios caused aggregation.17 We used dihodrofolate reductase (DHFR) RNA that has been previously shown to affect p53C aggregation.17 DHFR RNA at low stoichiometry induced aggregation of p53C under the tested conditions (100 μM p53C) (Fig. 7a).
:p53C ratios inhibited aggregation, and low stoichiometric ratios caused aggregation.17 We used dihodrofolate reductase (DHFR) RNA that has been previously shown to affect p53C aggregation.17 DHFR RNA at low stoichiometry induced aggregation of p53C under the tested conditions (100 μM p53C) (Fig. 7a).
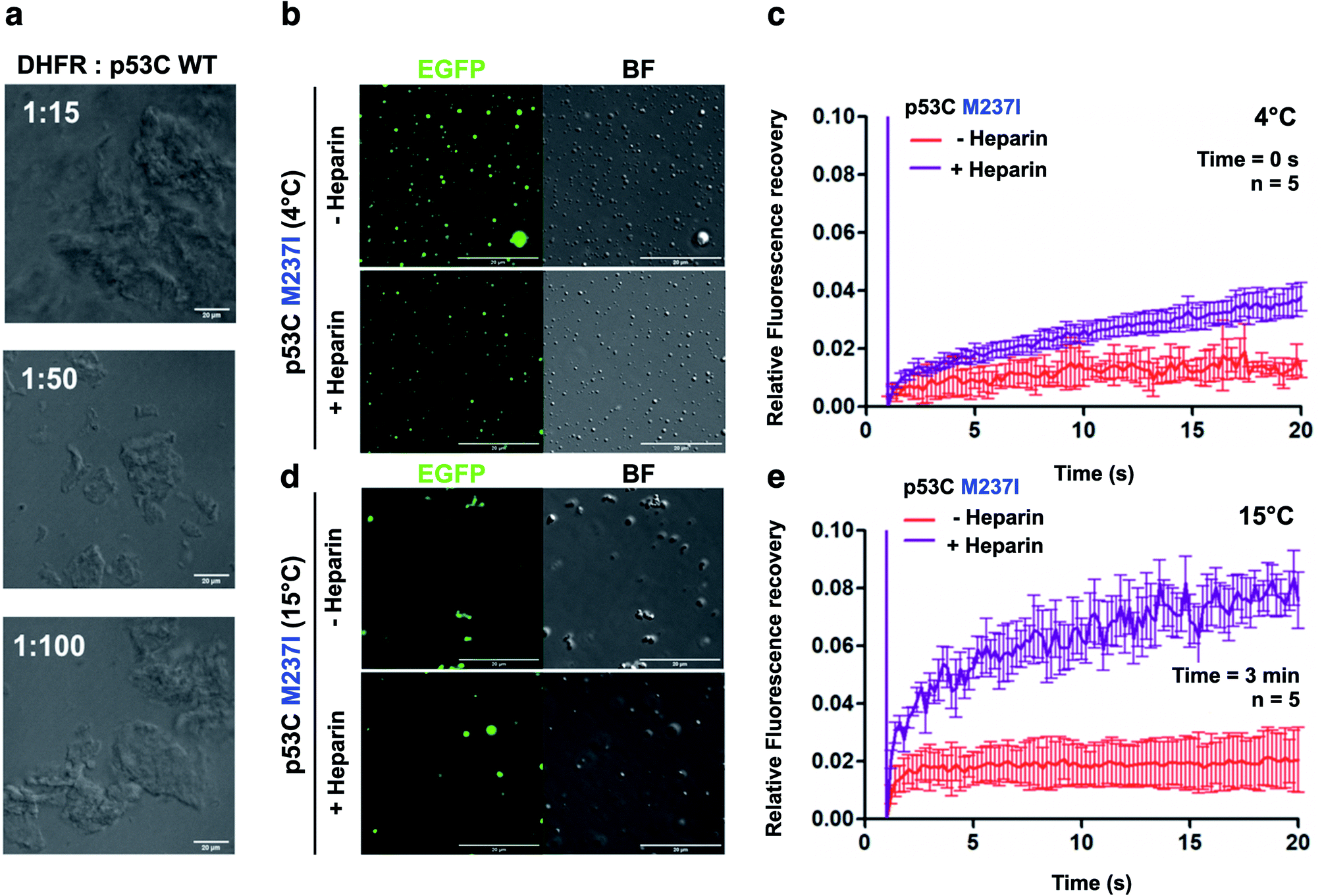 | ||
| Fig. 7 Polyanions modulate p53C phase separation and transition in vitro. (a) Bright field images at various stoichiometries of DHFR:p53C wt. Scale bar, 20 μm. DHFR corresponds to dihydrofolate reductase. (b and d) Images showing the bright field (BF) and the fluorescence channels of p53C M237I in the absence (−) and in the presence (+) of heparin at (b) 4 °C and (d) 15 °C. Scale bar, 20 μm. (c and e) Line plots showing fluorescence recovery of p53C M237I in the absence (−) and in the presence (+) of heparin at (c) 4 °C and (e) 15 °C. The data are shown as the mean ± s.d. of n = 5 FRAP measurements. Time corresponds to incubation time at indicated temperature before PEG addition. | ||
We also performed DIC and FRAP analyses to investigate the effect of heparin, which is a highly negatively charged molecule, on p53C PS. M237I p53C formed small droplets in the presence of PEG and PEG with heparin at 4 °C (Fig. 7b). FRAP recovery was low under both conditions and was higher in the presence of heparin (Fig. 7c). Low FRAP recovery and an aggregated morphology were demonstrated by analysis of the samples of M237I p53C after incubation for 3 min at 15 °C (Fig. 7d, e and ESI Fig. S5†), and these results were consistent with the greater tendency of this mutant for aggregation. Addition of heparin after 3 min resulted in the appearance of the droplet structures and an increase in FRAP recovery (Fig. 7d and e), suggesting that the presence of a polyanion was able to modulate the transition between the solid and gel protein states. The effect of heparin was also observed in the case of wt p53C; however, this effect was less pronounced and was manifested only as changes in the droplets size (ESI Fig. S6†).
SLPT was also detected by binding of Congo Red (CR) (Fig. 8 and ESI Fig. S7†).CR is a dye usually used to identify amyloids in the cells and tissues. CR can be used to identify both gel-like and solid-like amyloid states in the phase-separated materials.33,34 CR fluorescence was detected only after the addition of PEG to both M237I p53C and wt p53C, showing the formation of aggregated structures and suggesting a solid state (Fig. 8a and b). The signal was observed at 4 and 15 °C (Fig. 8c and d). CR binding was independent of its concentration (Fig. 8c, insets). Addition of heparin to the aggregates after CR addition resulted in the reversal of the morphology to a droplet form, which retained CR binding, suggesting the transition to a gel-like state (Fig. 8c). Addition of heparin before the addition of CR blocked the transition to an aggregated morphology (Fig. 8d).
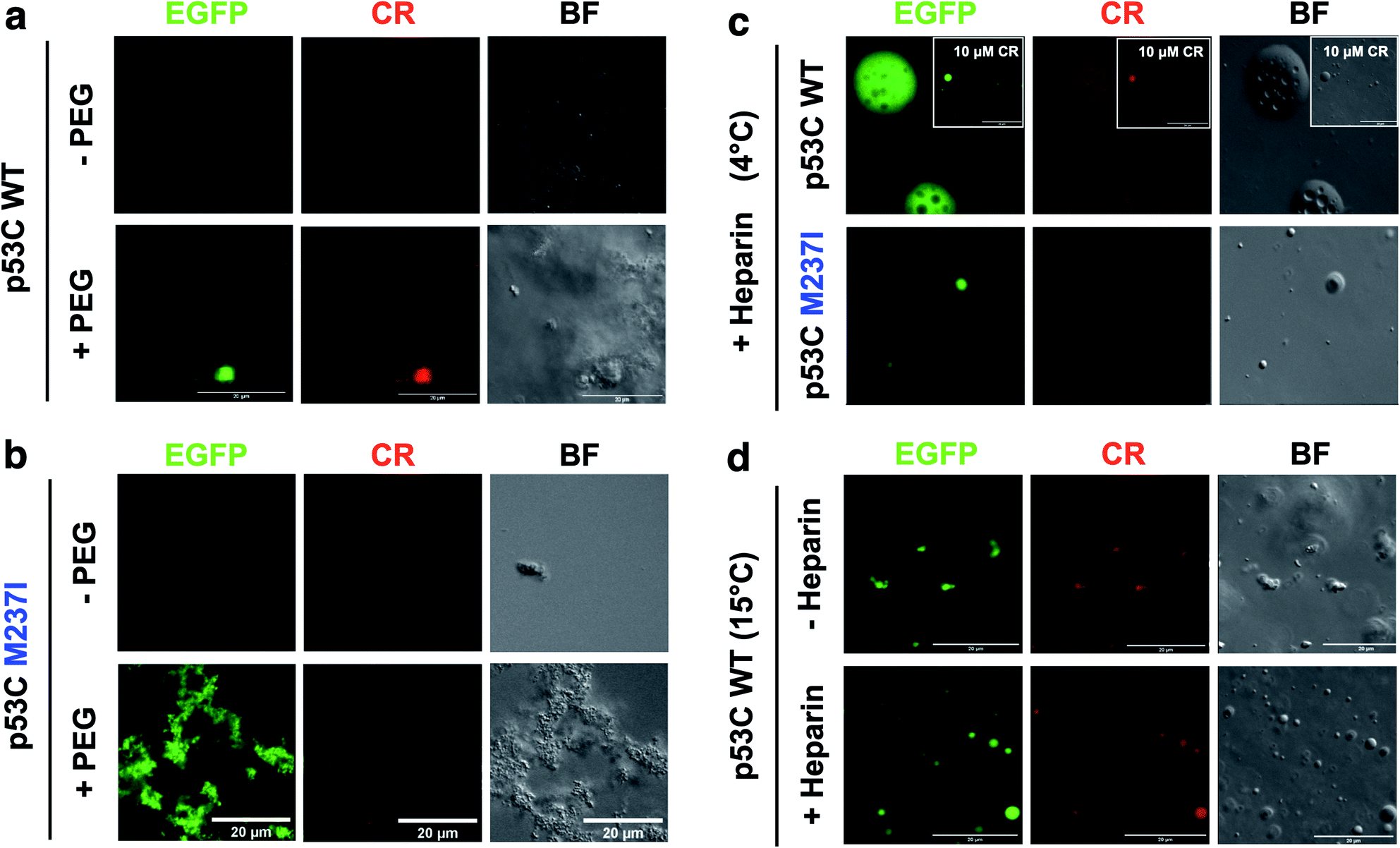 | ||
| Fig. 8 Heparin-induced p53C phase separation based on Congo Red binding under crowded conditions. (a and b) Fluorescence channels of EGFP and Congo Red (CR) (100 μM) and the bright field (BF) images of (a) wt and (b) M237I p53C in the absence (−) and in the presence (+) of PEG. (c) Fluorescent channels of EGFP and Congo Red (CR) and the bright field (BF) images of wt and M237I p53C at 4 °C after the addition of heparin. The inset shows the result obtained in the presence of 10 μM CR. (d) Fluorescence channels of EGFP and Congo Red (CR) and the bright field (BF) images of WT p53C at 15 °C in the absence (−) or in the presence (+) of heparin. Heparin was added before CR. | ||
These results suggested that p53 rapidly evolved to a gel-like state in vitro in the presence of a molecular crowding agent, transforming to a solid-like state via an amyloid route. Anionic ligands interfered with this aggregation route: RNA accelerated the transition to a solid state, and heparin stabilized a gel state.
Formation of the droplets of p53 in the molten globule state
Crowding conditions support the formation of the condensates by providing a compact network for protein interactions.8,29 p53 droplets were dissolved in 1,6-hexanediol, which is known to disturb weak hydrophobic interactions involved in the PS process (ESI Fig. S8†).p53C has a flexible structure, which enables multivalent interactions. Hence, we aimed to determine whether the conditions that are known to increase the flexibility leading to a molten globule conformation of p53 increase the chance of the formation of liquid droplets. Under the conditions that induce mild denaturation by hydrostatic pressure, p53C undergoes denaturation and irreversible aggregation into amyloid structures.22,35 This phenomenon was confirmed to occur at 37 °C, and mutant p53C was shown to be more prone to aggregation, which is apparently related to oncogenic GoF.13,16 Sub-denaturing conditions in the presence of guanidinium hydrochloride (Gnd–HCl) (0.5–0.8 M) result in the transition of p53C to a molten globule state, followed by aggregation depending on the protein concentration.21,36,37 Thus, we determined whether these sub-denaturing conditions trigger PS of wt p53C. The data of DIC microscopy indicated the formation of liquid droplets at 0.5 and 0.8 M Gnd–HCl, with greater formation at the latter concentration (Fig. 9a).
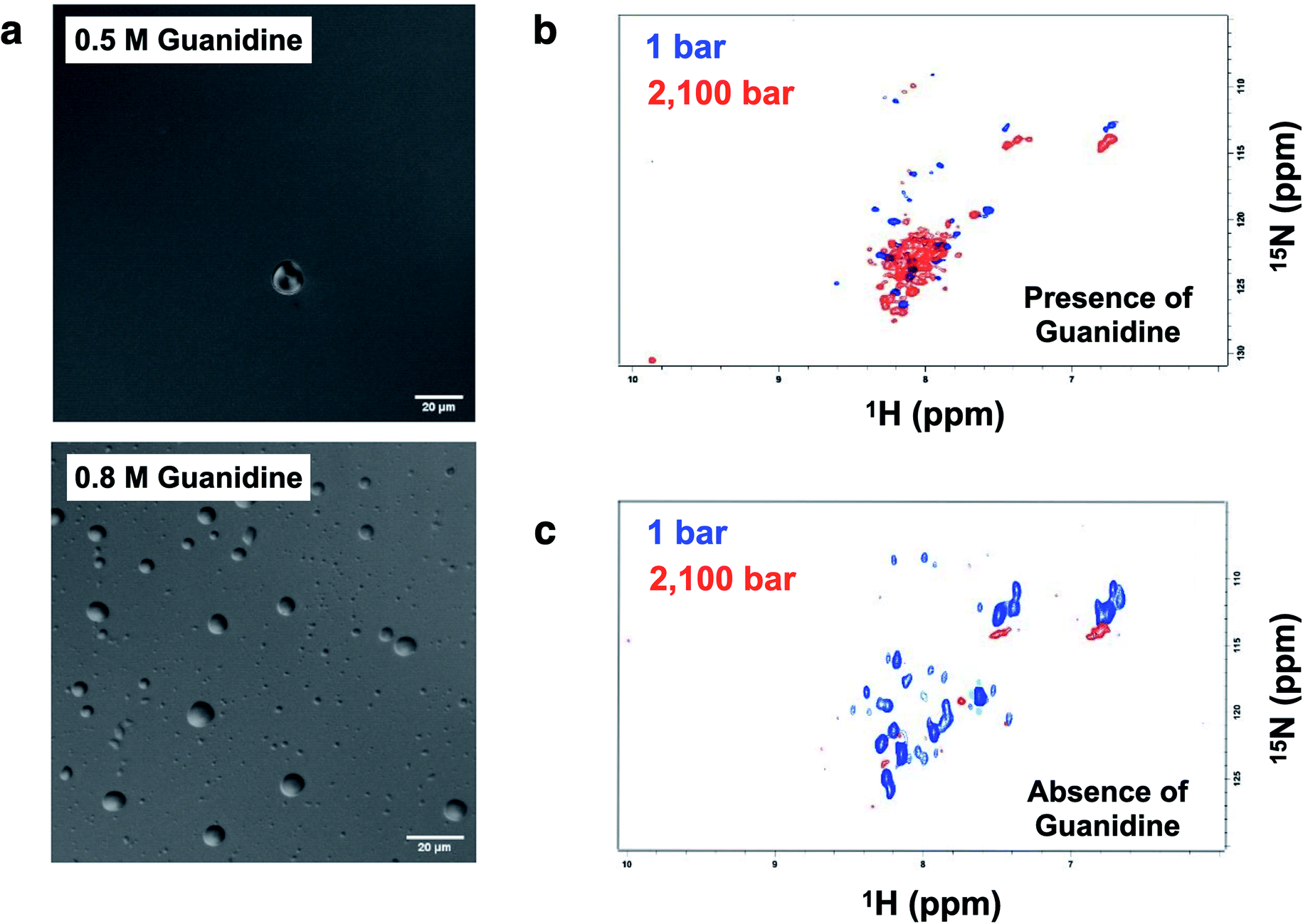 | ||
| Fig. 9 Formation of droplets induced by low concentrations of Gnd–HCl and high concentrations of p53. (a) DIC images of p53C (100 μM) in the presence of 0.5 M and 0.8 M guanidinium hydrochloride (Gnd–HCl) at 4 °C (scale bars, 20 μm). (b and c) 1H–15N TROSY NMR spectra of p53C (500 μM) in the presence and in the absence of 0.5 M Gnd–HCl, respectively. | ||
Hydrostatic pressure nuclear magnetic resonance (HP-NMR) at 1 bar and 2100 bar was used to evaluate a p53C wt sample in the presence of 0.5 M Gnd–HCl (Fig. 9b and c). The intensity of the NMR signal was decreased in the presence of 0.5 M Gnd–HCl (Fig. 9b), which can be attributed to phase separation and aggregation because the data of Fig. 9a suggested the presence of liquid droplets under these conditions. An increase in the pressure in this sample resulted in an increase in the intensity of the signals with low dispersion (Fig. 9b), suggesting that PS of p53C wt can be reversed by pressure variations, inducing a soluble state and most likely molten globule conformation. In contrast, in the absence of Gnd–HCl, an increase in the pressure to 2100 bar essentially abolished the NMR signal, indicating complete and irreversible aggregation of the sample (Fig. 9c).
Discussion
Cancer is a very complex disease, and multiple factors contribute to its pathophysiology.2,3 Tumor cells multiply faster than normal cells, elude tumor suppressor activity, induce angiogenesis and are able to invade various tissues, forming metastases. PS has been proposed to be a key event in oncogenicity.8,29,38,39 A recent study linked mutations in the tumor suppressor SPOP (speckle-type BTB/POZ protein) to PS defects.40 SPOP mutations were shown to disrupt substrate interactions that influence PS of SPOP and localization of the protein to membraneless organelles. The oncogenic effect results from cellular accumulation of oncogenic SPOP substrate proteins, which subsequently promote cell growth.40 Anticancer compounds can also partition into various nuclear condensates.41 Thus, these drugs encounter their targets in the nuclear condensates containing transcription factors and proteins involved in RNA processing. The disturbance of biomolecular condensates in the nucleus was linked to oncogenesis,39 and understanding of these processes is expected to provide new therapeutic tools.p53 is the second tumor suppressor protein related to cancer in which a mutation can disturb PS, likely leading to PT to amyloid aggregates with GoF effects. In general, a condensate is formed by the scaffold and client macromolecules.38,42 Scaffolds induce the formation of the condensates, and the client proteins subsequently partition into the condensates generated by the scaffolds. In physiological sites in the nucleus, especially in the membraneless compartments, RNA and proteins probably cooperate in the formation of the condensates of p53C, especially nucleolar proteins that have been shown to contribute to the multilayered structure of the nucleolus.43 Indeed, we detected the formation of the droplets by mutant p53 inside the nuclear compartments, including the nucleolus (Fig. 5). In addition to be involved in diseases, both phase separation and amyloids have important roles in the functional cells of various living organisms.1,11
Understanding of PS of p53 is important because of the role of this protein in cancer.44–46 More than half of malignant tumors have mutations in p53, which are generally associated with worse prognosis. Until a few years ago, it was a general belief that mutant p53 is very difficult to target, despite its presence in half of the malignancies. If new therapies are not developed, malignancies harboring mutations of the TP53 gene will kill more than half a billion people currently alive.2,5,47 Oncology was suggested as the best pharmaceutical application of the studies on PS.8,38,47 A compound that affects the PS and progression to amyloid structures of an oncogenic mutant of p53 is expected to suppress the corresponding GoF effects.
Why does p53 phase separate?
p53 orchestrates an important response to various attacks on the cell and deserves its name as “the guardian of the genome”. Most of the targets of p53 are located in the nucleus and depend on p53 binding to the recognition sites in DNA. Modular structure of p53 provides for multiplicity of functions and modulations. The role of p53 as the guardian of the genome relies on complex interactions in the nucleus and nuclear compartments, such as the nucleolus.2,5,26,48,49 The nucleolus has a complex multilayer of at least three distinct liquid phases that are highly dependent on the constitutive RNAs and proteins.43 Similar to the nucleolar proteins NPM1 and FIB1, p53 PS appears to be modulated by RNA and polyanions (Fig. 7 and 8).Our recent publication reported visualization of the M237I mutant p53-EGFP as an oligomer that is larger than tetramers in the nuclei of live cells.26 Number and brightness analysis of the data of fluorescence fluctuation spectroscopy were used to compare the oligomerization status of wt and M237I p53. We detected mostly monomers and dimers of wt p53 in the cytosol and nucleus, whereas abundant tetramers of M237I p53 were detected in the cytosol, and larger oligomers were densely distributed within the nuclei of live cells.26
The results of the present study showed that higher loads of M237I p53 result in better detection of the formation of a liquid droplet state. On the other hand, we have previously demonstrated that amyloid structures are abundant in the nuclei of glioblastoma cells expressing the M237I mutant.26 FRAP studies revealed that the condensates of M237I p53 in the nuclear membraneless compartments, including the nucleolus, are characterized by dynamics indicative of a gel- or solid-like state, whereas the condensates in the nucleoplasm appear to be in a more liquid state (Fig. 5). This coexistence of liquid droplets and solid-state aggregates of mutant p53 should determine the oncogenic fate of the cells. Additionally, the in vitro data of the present study indicated that PS precedes PT into amyloid aggregates. Physiologically, PS of p53 is expected to be transient, especially within the nuclear condensates (Fig. 10).
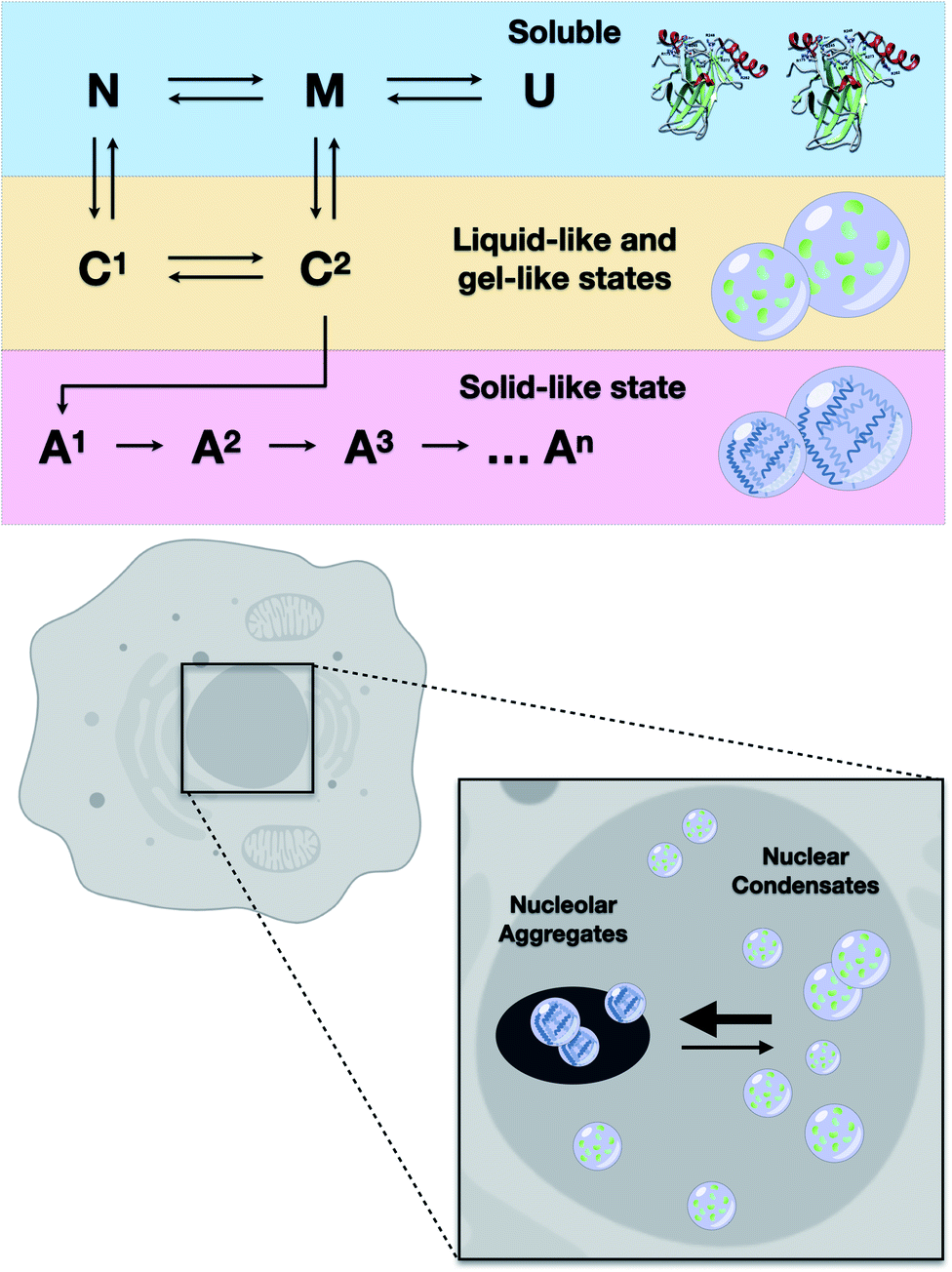 | ||
| Fig. 10 Scheme showing the results of in vitro and cell-based experiments. Upper panel: transitions from soluble protein to protein condensates (C1 and C2) likely involve native-like (N) or molten-globule states (M). U stands for the unfolded state. Condensates progressively evolve to several types of heterogeneous aggregates (A1, A2, A3, An). Lower panel: mutant p53 undergoes liquid-like and gel-like phase separation inside the nucleus and solid-like phase transition within the nucleolar compartments. | ||
Indeed, a recent report by Kilic and co-workers50 demonstrated that p53 is enriched within optoDroplets of p53-binding protein 1 (53BP1). Conditions that affected the phase separation of 53BP1 impaired the 53BP1-dependent induction of p53 and decreased the expression of p53 target genes. Thus, wt p53 is expected to be a client component of 53BP1 droplets integrated with localized DNA damage recognition sites.
Targeting the pathway LLPS → amyloid aggregation
The ability to identify and intercept the precursor states of p53 aggregation provides a unique approach for tracking the aggregation of p53 and testing the efficacy of the treatments that aim to stabilize the native form of p53.5,7,46,51–53 Several therapeutic strategies can be used to search for the compounds that can reactivate mutant p53.5 Our group has demonstrated that PRIMA-1 acts by inhibiting the aggregation of mutant p53.6 We showed how PRIMA-1 releases amyloid aggregates of mutant p53 and thereby decreases the dominant negative (DN) and GoF effects, indicating that aggregation of mutant p53 is an excellent target for the development of new drugs for cancer treatment. Other approaches to counteract p53 aggregation include a designed peptide that can penetrate into the cells and inhibit amyloid aggregation of mutant p53,52 resveratrol, which inhibits the aggregation of p53 mutants in the tumor cells and in animal models,53 and a bifunctional ligand with high activity that prevents p53 aggregation in the tumor cell lines.54These studies showed that the aggregates of mutant p53 can be suppressed by various molecules. However, it is unclear which step of the aggregation pathway is influenced. We have previously shown that sub-denaturing concentrations of Gnd–HCl induce the formation of native-like molten globule states that were highly prone to amyloid aggregation.21 We proposed that these precursor states are promising therapeutic targets. The present study demonstrated that these states initially induced the formation of liquid droplets that preceded the formation of the solid amyloid aggregates. The droplets of p53C formed under these conditions (low Gnd–HCl) were highly sensitive to pressure (Fig. 9). On the other hand, amyloid aggregates of p53 were not sensitive to pressure. The scheme in Fig. 10 shows how p53 transitions between the soluble, condensate and aggregated states. In the nucleus, the transition from a liquid-like to a gel-like and eventually to a solid-like state is expected to be influenced by ligands (such as RNA and DNA) and mutations (Fig. 10). Highly flexible structure and multiple binding of p53 to other nuclear components, such as RNAs and other proteins, can regulate the structural transitions. Mutant p53 is expected to rapidly transition to an aggregated state, forming amyloid oligomers that appear to have GoF effects.4,13,15,20,23,25,55–57
A relevant question is whether p53 forms biomolecular condensates or is a component of a large nuclear body. In addition to binding DNA and RNA,16,17,58 p53 can noncovalently or covalently bind poly(ADP-ribose) (PAR).59–61 PARylated p53 is not exported to the cytoplasm.60 On the other hand, PAR is known as the key participant in the formation of nuclear condensates. 53BP1 is an important component of DNA double-strand break (DSB) signaling and repair in mammalian cells. 53BP1 was recently shown to undergo phase separation, resulting in a scaffold with p53 as a client molecule.50 The results of the present study indicated that RNA and heparin modulated phase separation and the solid-like transition of p53 and demonstrated an intricate role of cofactors in the aggregation of mutant p53. RNA is highly enriched in the nucleus and nuclear bodies and is thus expected to play a key role in the PS and SLPT events. In vitro experiments of the present study used only one RNA molecules (an mRNA) to clearly show the acceleration of PT similar to the effects reported for several proteins involved in neurodegenerative diseases. On the other hand, heparin appears to have a distinct effect, interfering with the protein transition state from solid to gel and leading to a more dynamic state (Fig. 7 and 8). Recently, sulfated glycosaminoglycans (GAGs) were shown to mediate the prion-like aggregation behavior of p53 by specifically acting on the GAG-dependent cellular uptake of p53 aggregates.62 Low molecular weight heparin was proposed to modulate mammalian PrP prion conversion.63 The anticancer effect of heparin may be related to both inhibition of the prion effect and modulation of the phase transition.
Some molecules have been described to modify the condensates inside the tumor cells although they did not influence aggregation.64 A study described small molecule compounds that induce the dissolution of the structural condensates of mutant p53 and stabilize the condensates of DNA-binding mutant p53.64 Interestingly, transient p53 overexpression in yeast induced the formation of p53 prion aggregates; however, liquid-like p53 condensates were generated in response to stress and disappeared after stress removal.65
The present study demonstrated that mutant p53 tends to form condensates inside the nucleus. Other domains of p53 apparently modulated PS and SLPT of the protein. We have previously reported that the TAD decreases the amyloid aggregation of p53C.20 The modulatory role of the TAD in both PS and SLPT was clearly demonstrated by the data shown in Fig. 4. The droplets formed by TAD-core of M237I p53 were larger than those formed by TAD-core of wt p53. This difference may reflect the findings of fluorescence microscopy, which indicated that M237I has a high tendency to coalesce into the droplets in the nuclear subcompartments. This phenomenon may directly influence the GoF effect of this mutation in glioblastoma.26
In summary, our results clearly indicated how the phase separation of p53 and its oncogenic mutant may drive the protein into a physiological action (reversible pathway) or a phase transition into solid states (less reversible), such as amyloid oligomers and larger aggregates that contribute to oncogenic GoF.
Methods
Site-directed mutagenesis
Point mutations were introduced into pET15b p53C (Addgene #24866) and p53 TAD-core (Addgene #24864) with synthetic complementary oligonucleotides (Eurofins Genomics LLC) using a QuickChange Lightning site-directed mutagenesis kit (Agilent Technologies, USA, #210519) according to the manufacturer's instructions. DNA Sanger sequencing was performed by GENEWIZ (South Plainfield, NJ). The following oligonucleotides were used: M237I – 5′-caccatccactacaactacatatgtaacagttcctgcat-3′ and 5′-atgcaggaactgttacatatgtagttgtagtggatggtg-3′; R249S – 5′-gcatgaaccggagccccatcctcacca-3′ and 5′-tggtgaggatggggctccggttcatgc-3′. P53C-EGFP wt, M237I, and R249S were inserted into pET24a by Genscript. The vectors used in the present study were prepared and expressed as described previously.26Protein purification
The soluble fractions of the cell lysate were centrifuged and purified on an SP-Sepharose cation exchange column (Pharmacia) eluted with a NaCl gradient (0–1 M), followed by size exclusion chromatography on a Superdex 75 10/300 GL column (GE Life Sciences, #17-5174-01). Protein concentration was measured spectrophotometrically at 595 nm by the Bradford method. Elution of the proteins was monitored by a high-performance liquid chromatography (HPLC) system, and the purity was determined by 12.5% SDS-PAGE. The purified p53 proteins were concentrated and stored in liquid nitrogen in the presence of 5% glycerol. The constructs p53 M237I-EGFP, p53 wt-EGFP and p53 R249S were isolated on a Ni-NTA Superflow resin column (Qiagen, #30450) equilibrated in buffer A (50 mM Tris–Cl, 150 mM NaCl, and 2.5 mM TCEP, pH 7.4) using an Äkta Prime system at a flow rate of approximately 0.8–1 mL min−1 p53 was eluted using a linear gradient of 5–100% of buffer B (buffer A plus 0.5 M imidazole). The protein concentration was determined at 280 nm using an extinction coefficient of 17420 M−1 cm−1.
Differential interference contrast (DIC) and fluorescence microscopy
LLPS analyses of p53 were performed using DIC and fluorescence microscopy tests under various conditions, including variations in temperature, concentration and molecular crowding. The samples (20 μL) in buffer A (50 mM Tris–Cl, pH 7.4, 150 mM NaCl and 5 mM DTT) were loaded onto a glass slide and cover slip system prepared as described previously.66 The images were acquired using a Leica TCS-SPE confocal microscope with a 63×/1.4 oil objective and a 488 nm laser. For fluorescence analysis, the samples were excited at 405 nm, and fluorescence signals were acquired at 450–500 nm. The images were processed using Fiji (a distribution package of ImageJ software, USA).Transmission electron microscopy (TEM)
All samples were prepared and analyzed as described previously.26 Images were acquired using a Philips Tecnai microscope operated at 80 kV at 46000× magnification.
NMR spectroscopy
The 1H–15N-TROSY NMR spectra of wt p53C were obtained using an 800 MHz Bruker Advance III spectrometer (Jiri Jonas National Center of Nuclear Magnetic Resonance, Rio de Janeiro, Brazil) connected to a high hydrostatic pressure (HHP) system equipped with an Xtreme-60 syringe pump (Daedalus Innovations). The labeled protein was expressed according to Fersht and coworkers in the absence of zinc sulfate.67 The purification method was described previously. Protein concentration was 500 μM in 50 mM Tris–HCl, pH 7.4, containing 200 mM NaCl, 5 mM DTT, and 10% D2O. The protein was partially aggregated and then subjected to pressurization steps from 300 to 2100 bar using a zircon tube at 293 K. All spectra were processed using TopSpin version 3.2.Transfection assay
The lung carcinoma cell line H1299 purchased from the American Type Culture Collection (ATCC) was used for the transfection assays. The cells were transfected as described by Pedrote and coworkers.26 The plasmids (5 μg) containing full-length wt-p53 and mutant M237I fused to EGFP and the pEGFP-N1 vector (Clontech, Inc.) were used. The images were acquired by confocal microscopy using a super resolution Elyra LSM 710 confocal laser scanning microscope (Carl Zeiss, Inc.), Plan-Apochromat 63×/1.4 oil DICM27 objective, 256 × 256 pixel frame scan mode and 1.94 second scan time. A 488 nm laser was used, with a pinhole value of 52.5 or 0.8 μm. The gain has been adjusted for all images. A sequence of 100 images was acquired, including each cell compartment (e.g., the nucleus and cytoplasm) in 5–8 cells for each experimental condition.Immunofluorescence assay
H1299 cells were transfected with p53 M237I-EGFP. Then, the cells were washed three times with cold PBS and incubated with ice-cold methanol:acetone (1:1) for 20 min at 20 °C. The cells were labeled overnight with a mouse monoclonal anti-nucleophosmin 1 antibody (ThermoFisher) diluted to 2 μg mL−1 in 2% bovine serum albumin (BSA), 0.2% Tween 20, and 10% (v/v) glycerol in PBS. The cells were subsequently incubated with Alexa 647-conjugated goat anti-mouse (Life Technologies) secondary antibodies (1:2000) in the same buffer for 1 h at room temperature in the dark. The cells were washed twice with PBS, incubated with DAPI (Life Technologies) for 10 min at room temperature in the dark, and washed twice with PBS. Then, the cells were analyzed by confocal microscopy at 60000 magnification.
Automatic droplet detection
We automated the analysis of 62 microscopy images to determine (i) the number of formed liquid droplets and (ii) characteristics of these droplets. Specifically, we obtained the following measurements of the droplets: (1) median axis lengths, (2) mean axis lengths, (3) standard deviation and variance of the axis length and (4) high median and low median axis length. Use of software to extract the data allowed us to process a dataset, which was impossible to analyze manually in an accurate fashion (we discuss the dataset in section A: “The dataset” below). However, to give an indication of the size of the dataset, the microscopy images include over 5000 droplets. These data supported our hypothesis that p53 (p53C) underwent LLPS under the test conditions, forming liquid droplets in the presence of the crowding agent PEG.8 images using PEG 15% + 1 h, 4 °C.
6 images using PEG 15% + 1 h, 15 °C.
7 images using PEG 15% + 1 h, 37 °C.
15 images using PEG 15% + 15 min, 4 °C.
8 images using PEG 15% + 30 min, 15 °C.
6 images using PEG 15% + 30 min, 37 °C.
12 images using PEG 15% + 30 min, 4 °C.
Each image was 512 × 512 pixels, with a resolution of 72 pixels per inch.
Digital image processing is a field of computer science pertaining to the theory of the applications of computational systems to obtain and process the information present in the images. Image processing techniques have proven useful in addressing a range of problems. Successful applications, often in combination with other fields, have been implemented in diverse areas, ranging from vehicle tracking and traffic surveillance to computational pathology,68 biomedicine69 and chemistry. A specific example of the applications includes the use in the segmentation of abdominal CT images.70
We applied a commonly used image processing technique known as image labeling to extract the information pertaining to the formation of the droplets from 62 microscopy images. We wrote a Python program using the popular scikit-image library.71 Scikit-image consists of a collection of commonly used image processing and computer vision algorithms. The library has been extensively used in industry and in the scientific community and contains a large range of sample scripts, illustrating common solutions to extract information from various types of images. We based our information extraction software on one of these scripts, with minor modifications. The program works as follows:
For each image:
(S1) Compute the luminance of the (RGB) microscopy image.
(S2) Generate an elevation map using the Sobel filter.
(S3) Apply canny edge detection to the elevation map generated in step 2.
(S4) Apply the edge detector and fill any holes in the image.
(S5) Threshold the resulting luminance (grayscale) to produce a binary image (i.e., segment the image). We used the OTSU thresholding.
(S6) Remove artefacts and holes using binary closing.
(S7) Label the segmented image.
(S8) For each labeled image region with a major axis length greater than 20 μm, extract the region properties and use them to (i) derive the axis length in pixels and (ii) draw a red rectangle around the droplet.
(S9) Save each processed image (containing the marked droplets/regions).
The process is visually outlined in ESI Fig. S9.† Since each microscopy image contained a size indicator in μm, we can convert actual distance to and from pixels. In the images, 2.90 pixels equaled 1 μm. Outliers were detected based on annotations on the images (i.e., μm to pixel overlay) and were manually removed from the dataset. The complete source code is available in the ESI.†
Due to image artefacts, the analysis (i.e., number of droplets) may be slightly skewed. For example, ESI Fig. S3† shows the labeled and original images side by side. Comparison indicated that some smaller droplets, which are easily discerned by human eye, were not identified automatically due to slightly blurry edges. Thus, due to image artefacts, automatic analysis of 6 microscopy images in 15% PEG (1 h at 15 °C) was inconclusive, and the data had to be removed. Regardless of these minor inaccuracies and/or noise in the images, the data support our hypothesis that p53 (p53C) undergoes LLPS under the test conditions, forming liquid droplets in the presence of the crowding agent PEG.
Thioflavin T fluorescence
All experiments were performed and analyzed as described previously.26Measurements of soluble materials after kinetic assays
We measured the soluble fractions of p53C M237I and the corresponding EGFP-tagged construct after kinetic assays of the samples (100 μL) for 20 min at 37 °C and 600 rpm. The samples were centrifuged at 14000g for 10 min at 4 °C. The top part of the supernatant was used to avoid contamination with the aggregated material. The concentrations of the soluble material were measured based on absorbance at 280 nm and molar extinction coefficients of 17420 M−1 cm−1 (p53C-M237I) and 40060 M−1 cm−1 (EGFP-tagged) using a NanoDrop system (Thermo Scientific, Inc.).
Fluorescence recovery after photobleaching
FRAP experiments were performed using a Zeiss LSM 710 confocal laser scanning microscope with a Plan-Apochromat 63×/1.4 objective lens. Circular areas with diameter from 1 to 3 μm were bleached using 488 nm/100 mW argon ion laser. Fluorescence signal from the bleached area was measured over 200 frames, including 5 frames before and 195 frames after the bleaching; acquired images were 512 × 512 pixels, with 0.79 μs pixel dwell and 242 ms scan time. Fluorescence recovery data were evaluated using the FIJI ImageJ SimFRAP plugin in 3–5 replicates. The results were averaged using Graph Pad Prism.41Data availability
Not applicable.Author contributions
E. C. P., M. M. P., G. A. P. d. O., T. C. R. G. V. and J. L. S. designed the research; G. A. P. d. O., T. C. R. G. V. and J. L. S. conceived and coordinated the research. E. C. P., M. M. P., Y. M. P., G. S. S., M. F. M., F. P. C, M. A. M. and G. A. P. d. O. performed the research; J. L. S. and Y. C., contributed new reagents and analytic tools; E. C. P., M. M. P., T. C. R. G. V., J. L. S., and G. A. P. d. O. analyzed the data; E. C. P., M. M. P., B. J., G. A. P. d. O and J. L. S. prepared the figures; B. J. wrote the program for automatic droplet detection; and E. C. P. performed the NMR experiments. The manuscript was prepared by E. C. P., M. M. P., B. J., J. L. S. and G. A. P. d. O.Conflicts of interest
The authors declare that they have no conflicts of interest regarding the contents of this article.Abbreviations
The abbreviations used are as follows:| PEG | Polyethylene glycol |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| LLPS | Liquid–liquid phase separation |
| DIC | differential interference contrast |
| WT | Wild type |
| PS | Phase separation |
| SLPT | Solid-like phase transition |
| IDRs | Intrinsically disordered regions |
| TH | Thawed |
| TC | Temperature cycle |
| SPOP | Speckle-type BTB/POZ protein |
| N&B | Number and brightness |
| GoF | Gain of function |
| MG | Molten globule |
| PAR | Poly(ADP-ribose) |
| DN | dominant negative |
| 53BP1 | p53-binding protein 1 |
| DSB | Double-strand break |
| HHP | High hydrostatic pressure |
| Gnd–HCl | Guanidine hydrochloride |
| NMR | Nuclear magnetic resonance |
Acknowledgements
This research was supported by the Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro (FAPERJ) grants 210.008/2018 and 202840/2018, the National Council for Scientific and Technological Development (CNPq), and the National Institute of Science and Technology for Structural Biology and Bioimaging (INCT) grants 465395/2014-7 and 402321/2016-2 to J. L. S. and the Pew Charitable Trusts Foundation to G.A.P.d.O.References
- F. Chiti and C. M. Dobson, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed
.
- W. A. Freed-Pastor and C. Prives, Mutant p53: one name, many proteins, Genes Dev., 2012, 26, 1268–1286 CrossRef CAS PubMed
- P. A. Muller and K. H. Vousden, p53 mutations in cancer, Nat. Cell Biol., 2013, 15, 2–8 CrossRef CAS
- J. L. Silva, C. V. De Moura Gallo, D. C. Costa and L. P. Rangel, Prion-like aggregation of mutant p53 in cancer, Trends Biochem. Sci., 2014, 39, 260–267 CrossRef CAS
- J. L. Silva, E. A. Cino, I. N. Soares, V. F. Ferreira and G. A. P. de Oliveira, Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer, Acc. Chem. Res., 2018, 51, 181–190 CrossRef CAS PubMed
- L. P. Rangel,
et al., p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells, J. Biol. Chem., 2019, 294, 3670–3682 CrossRef CAS PubMed
- A. Navalkar,
et al., Prion-like p53 Amyloids in Cancer, Biochemistry, 2020, 59, 146–155 CrossRef CAS PubMed
- S. Alberti and D. Dormann, Liquid-Liquid Phase Separation in Disease, Annu. Rev. Genet., 2019, 53, 171–194 CrossRef CAS PubMed
- T. Murakami,
et al., ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function, Neuron, 2015, 88, 678–690 CrossRef CAS PubMed
- E. Gomes and J. Shorter, The molecular language of membraneless organelles, J. Biol. Chem., 2019, 294, 7115–7127 CrossRef CAS PubMed
- S. Boeynaems,
et al., Protein Phase Separation: A New Phase in Cell Biology, Trends Cell Biol., 2018, 28, 420–435 CrossRef CAS
- S. Maharana,
et al., RNA buffers the phase separation behavior of prion-like RNA binding proteins, Science, 2018, 360, 918–921 CrossRef CAS
- A. P. Ano Bom,
et al., Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer, J. Biol. Chem., 2012, 287, 28152–28162 CrossRef CAS
- R. Wilcken, G. Wang, F. M. Boeckler and A. R. Fersht, Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13584–13589 CrossRef CAS PubMed
- S. Ghosh,
et al., p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis, Cell Death Differ., 2017, 24, 1784–1798 CrossRef CAS
- D. Ishimaru,
et al., Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation, Biochemistry, 2009, 48, 6126–6135 CrossRef CAS PubMed
- P. S. Kovachev,
et al., Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain, J. Biol. Chem., 2017, 292, 9345–9357 CrossRef CAS PubMed
- K. Kamagata,
et al., Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains, Sci. Rep., 2020, 10, 580 CrossRef CAS PubMed
- M. S. Safari,
et al., Anomalous Dense Liquid Condensates Host the Nucleation of Tumor Suppressor p53 Fibrils, iScience, 2019, 12, 342–355 CrossRef CAS PubMed
- N. Melo Dos Santos,
et al., Loss of the p53 transactivation domain results in high amyloid aggregation of the Delta40p53 isoform in endometrial carcinoma cells, J. Biol. Chem., 2019, 294, 9430–9439 CrossRef PubMed
- M. M. Pedrote,
et al., Aggregation-primed molten globule conformers of the p53 core domain provide potential tools for studying p53C aggregation in cancer, J. Biol. Chem., 2018, 293, 11374–11387 CrossRef CAS PubMed
- D. Ishimaru, L. R. Andrade, L. S. P. Teixeira, P. A. Quesado, L. M. Maiolino, P. M. Lopez, Y. Cordeiro, L. T. Costa, W. M. Heckl, G. Weissmüller, D. Foguel and J. L. Silva, Fibrillar aggregates of the tumor suppressor p53 core domain, Biochemistry, 2003, 42, 9022–9027 CrossRef CAS
- J. Xu,
et al., Gain of function of mutant p53 by coaggregation with multiple tumor suppressors, Nat. Chem. Biol., 2011, 7, 285–295 CrossRef CAS PubMed
- C. B. Levy,
et al., Co-localization of mutant p53 and amyloid-like protein aggregates
in breast tumors, Int. J. Biochem. Cell Biol., 2011, 43, 60–64 CrossRef CAS PubMed
- C. A. Lasagna-Reeves,
et al., Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity, Biochem. Biophys. Res. Commun., 2013, 430, 963–968 CrossRef CAS PubMed
- M. M. Pedrote,
et al., Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers, iScience, 2020, 23, 100820 CrossRef CAS
- G. Weber, Thermodynamics of the association and the pressure dissociation of oligomeric proteins, J. Phys. Chem., 1993, 97, 7108–7115 CrossRef CAS
- J. L. Silva, Y. Cordeiro and D. Foguel, Protein folding and aggregation: two sides of the same coin in the condensation of proteins revealed by pressure studies, Biochim. Biophys. Acta, 2006, 1764, 443–451 CrossRef CAS
- G. A. P. de Oliveira, Y. Cordeiro, J. L. Silva and T. Vieira, Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases, Adv. Protein Chem. Struct. Biol., 2019, 118, 289–331 Search PubMed
- D. Milovanovic, Y. Wu, X. Bian and P. De Camilli, A liquid phase of synapsin and lipid vesicles, Science, 2018, 361, 604–607 CrossRef CAS PubMed
- T. M. Franzmann,
et al., Phase separation of a yeast prion protein promotes cellular fitness, Science, 2018, 359, 1–8 CrossRef
- Y. Cordeiro, T. Vieira, P. S. Kovachev, S. Sanyal and J. L. Silva, Modulation of p53 and prion protein aggregation by RNA, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 933–940 CrossRef CAS PubMed
- W. M. Babinchak,
et al., Small molecules as potent biphasic modulators of protein liquid-liquid phase separation, Nat. Commun., 2020, 11, 5574 CrossRef CAS PubMed
- H. Tange,
et al., Liquid-liquid phase separation of full-length prion protein initiates conformational conversion in vitro, J. Biol. Chem., 2021, 100367, DOI:10.1016/j.jbc.2021.100367
- D. Ishimaru,
et al., Conversion of Wild-type p53 Core Domain into a Conformation that Mimics a Hot-spot Mutant, J. Mol. Biol., 2003, 333, 443–451 CrossRef CAS PubMed
- D. Ishimaru,
et al., Reversible aggregation plays a crucial role on the folding landscape of p53 core domain, Biophys. J., 2004, 87, 2691–2700 CrossRef CAS PubMed
- A. P. Bom,
et al., The p53 core domain is a molten globule at low pH: functional implications of a partially unfolded structure, J. Biol. Chem., 2010, 285, 2857–2866 CrossRef PubMed
- D. C. Costa,
et al., Aggregation and Prion-Like Properties of Misfolded Tumor Suppressors: Is Cancer a Prion Disease?, Cold Spring Harbor Perspect. Biol., 2016, 8, a023614 CrossRef PubMed
- A. Boija, I. A. Klein and R. A. Young, Biomolecular Condensates and Cancer, Cancer Cell, 2021, 39, 174–192 CrossRef CAS PubMed
- J. J. Bouchard,
et al., Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments, Mol. Cell, 2018, 72, 19–36 CrossRef CAS PubMed
- I. A. Klein,
et al., Partitioning of cancer therapeutics in nuclear condensates, Science, 2020, 368, 1386–1392 CrossRef CAS PubMed
- S. F. Banani,
et al., Compositional Control of Phase-Separated Cellular Bodies, Cell, 2016, 166, 651–663 CrossRef CAS PubMed
- M. Feric,
et al., Coexisting Liquid Phases Underlie Nucleolar Subcompartments, Cell, 2016, 165, 1686–1697 CrossRef CAS PubMed
- J. L. Silva,
et al., Ligand binding and hydration in protein misfolding: insights from studies of prion and p53 tumor suppressor proteins, Acc. Chem. Res., 2010, 43, 271–279 CrossRef CAS PubMed
- O. Laptenko and C. Prives, p53: master of life, death, and the epigenome, Genes Dev., 2017, 31, 955–956 CrossRef CAS PubMed
- J. J. Miller, C. Gaiddon and T. Storr, A balancing act: using small molecules for therapeutic intervention of the p53 pathway in cancer, Chem. Soc. Rev., 2020, 49, 6995–7014 RSC
- A. Mullard, Biomolecular condensates pique drug discovery curiosity, Nat. Rev. Drug Discovery, 2019, 18, 324–326 Search PubMed
- A. C. Joerger and A. R. Fersht, The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches, Annu. Rev. Biochem., 2016, 85, 375–404 CrossRef CAS PubMed
- J. L. Silva, L. P. Rangel, D. C. Costa, Y. Cordeiro and C. V. De Moura Gallo, Expanding the prion concept to cancer biology: dominant-negative effect of aggregates of mutant p53 tumour suppressor, Biosci. Rep., 2013, 33, e00054 CrossRef PubMed
- S. Kilic,
et al., Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments, EMBO J., 2019, 38, e101379 CrossRef PubMed
- G. Wang and A. R. Fersht, Multisite aggregation of p53 and implications for drug rescue, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2634–E2643 CrossRef CAS PubMed
- A. Soragni,
et al., A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas, Cancer Cell, 2016, 29, 90–103 CrossRef CAS PubMed
- D. C. Ferraz da Costa,
et al., Resveratrol prevents p53 aggregation in vitro and in breast cancer cells, Oncotarget, 2018, 9, 29112–29122 CrossRef PubMed
- J. J. Miller,
et al., Bifunctional ligand design for modulating mutant p53 aggregation in cancer, Chem. Sci., 2019, 10, 10802–10814 RSC
- W. Freed-Pastor and C. Prives, Targeting mutant p53 through the mevalonate pathway, Nat. Cell Biol., 2016, 18, 1122–1124 CrossRef CAS PubMed
- E. A. Cino, I. N. Soares, M. M. Pedrote, G. A. de Oliveira and J. L. Silva, Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds, Sci. Rep., 2016, 6, 32535 CrossRef CAS PubMed
- I. Lima,
et al., Biophysical characterization of p53 core domain aggregates, Biochem. J., 2020, 477, 111–120 CrossRef CAS PubMed
- J. L. Silva and Y. Cordeiro, The "Jekyll and Hyde" Actions of Nucleic Acids on the Prion-like Aggregation of Proteins, J. Biol. Chem., 2016, 291, 15482–15490 CrossRef CAS PubMed
- A. Kruger,
et al., Interactions of p53 with poly(ADP-ribose) and DNA induce distinct changes in protein structure as revealed by ATR-FTIR spectroscopy, Nucleic Acids Res., 2019, 47, 4843–4858 CrossRef PubMed
- M. Kanai,
et al., Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation, Nat. Cell Biol., 2007, 9, 1175–1183 CrossRef CAS PubMed
- A. K. L. Leung, Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation, Trends Cell Biol., 2020, 30, 370–383 CrossRef CAS PubMed
- N. Iwahashi,
et al., Sulfated glycosaminoglycans mediate prion-like behavior of p53 aggregates, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33225–33234 CrossRef CAS PubMed
- T. C. Vieira, Y. Cordeiro, B. Caughey and J. L. Silva, Heparin binding confers prion stability and impairs its aggregation, FASEB J., 2014, 28, 2667–2676 CrossRef CAS PubMed
- C. Lemos,
et al., Identification of Small Molecules that Modulate Mutant p53 Condensation, iScience, 2020, 23, 101517 CrossRef CAS PubMed
- S. K. Park, S. Park, C. Pentek and S. W. Liebman, Tumor suppressor protein p53 expressed in yeast can remain diffuse, form a prion, or form unstable liquid-like droplets, iScience, 2021, 24, 102000 CrossRef CAS PubMed
- C. O. Matos,
et al., Liquid-liquid phase separation and fibrillation of the prion protein modulated by a high-affinity DNA aptamer, FASEB J., 2020, 34, 365–385 CrossRef CAS PubMed
- K. B. Wong,
et al., Hot-spot mutants of p53 core domain evince characteristic local structural changes, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8438–8442 CrossRef CAS PubMed
- T. J. Fuchs and J. M. Buhmann, Computational pathology: challenges and promises for tissue analysis, Comput Med Imaging Graph, 2011, 35, 515–530 CrossRef PubMed
- F. Kanavati,
et al., Weakly-supervised learning for lung carcinoma classification using deep learning, Sci. Rep., 2020, 10, 9297 CrossRef CAS PubMed
- G. B. Goh, C. Siegel, A. Vishnu, N. O. Hodas, N. Baker, arXiv preprint arXiv:1706.06689, 2017.
- https://scikit-image.org/, visited 10/07/2020.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01739j |
| ‡ Elaine C. Petronilho and Murilo M. Pedrote are equally first authors. |
| This journal is © The Royal Society of Chemistry 2021 |