
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N- to C-sulfonyl photoisomerisation of dihydropyridinones: a synthetic and mechanistic study†
Pei-Pei
Yeh
,
James E.
Taylor
 ,
Daniel G.
Stark
,
David S. B.
Daniels
,
Charlene
Fallan
,
John C.
Walton
and
Andrew D.
Smith
*
,
Daniel G.
Stark
,
David S. B.
Daniels
,
Charlene
Fallan
,
John C.
Walton
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 16th October 2017
Abstract
The scope and limitations of a photoinitiated N- to C-sulfonyl migration process within a range of dihydropyridinones is assessed. This sulfonyl transfer proceeds without erosion of either diastereo- or enantiocontrol, and is general across a range of N-sulfonyl substituents (SO2R; R = Ph, 4-MeC6H4, 4-MeOC6H4, 4-NO2C6H4, Me, Et) as well as C(3)-(aryl, heteroaryl, alkyl and alkenyl) and C(4)-(aryl and ester) substitution. Crossover reactions indicate an intermolecular step is operative within the formal migration process, although no crossover from C-sulfonyl products was observed. EPR studies indicate the intermediacy of a sulfonyl radical and a mechanism is proposed based upon these observations.
Introduction
The sulfonamide is a ubiquitous functional group that is widely employed in synthetic, analytical, and medicinal chemistry.1 The N-sulfonyl functionality is typically regarded as a versatile and stable protecting group for nitrogen and is commonly used to moderate the nucleophilicity of nitrogen functional groups in synthesis.2 Sulfonamides are usually easy to install, inert to a range of experimental conditions, yet can be reductively deprotected by various methods including the use of either dissolving metals3 or treatment with SmI2.4 Alternatively, sulfonamides can be labile through the cleavage of their N–S bond that can be promoted by either photolysis5 or electrochemical means.6Photochemical irradiation of N-sulfonyl anilines can also promote Fries-type rearrangements, giving products resulting from N- to C-sulfonyl transfer.7 Such photochemical Fries rearrangements often result in a mixture of regioisomeric products and/or fully deprotected anilines. For example, irradiation of N-sulfonyl aniline 1 at 254 nm leads to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of para- and ortho-substituted N- to C-sulfonyl transfer products 2 and 3 (Scheme 1a).7e The reaction regioselectivity can be switched to favour the ortho-substituted product by performing photolysis in the solid phase after encapsulation in β-cyclodextrin.7d Related photochemical N- to C-sulfonyl transfer processes have also been observed for N-sulfonyl carbazoles8 and for intramolecular rearrangements of benzothiadiazine9a and sultam derivatives.9b
:1 mixture of para- and ortho-substituted N- to C-sulfonyl transfer products 2 and 3 (Scheme 1a).7e The reaction regioselectivity can be switched to favour the ortho-substituted product by performing photolysis in the solid phase after encapsulation in β-cyclodextrin.7d Related photochemical N- to C-sulfonyl transfer processes have also been observed for N-sulfonyl carbazoles8 and for intramolecular rearrangements of benzothiadiazine9a and sultam derivatives.9b
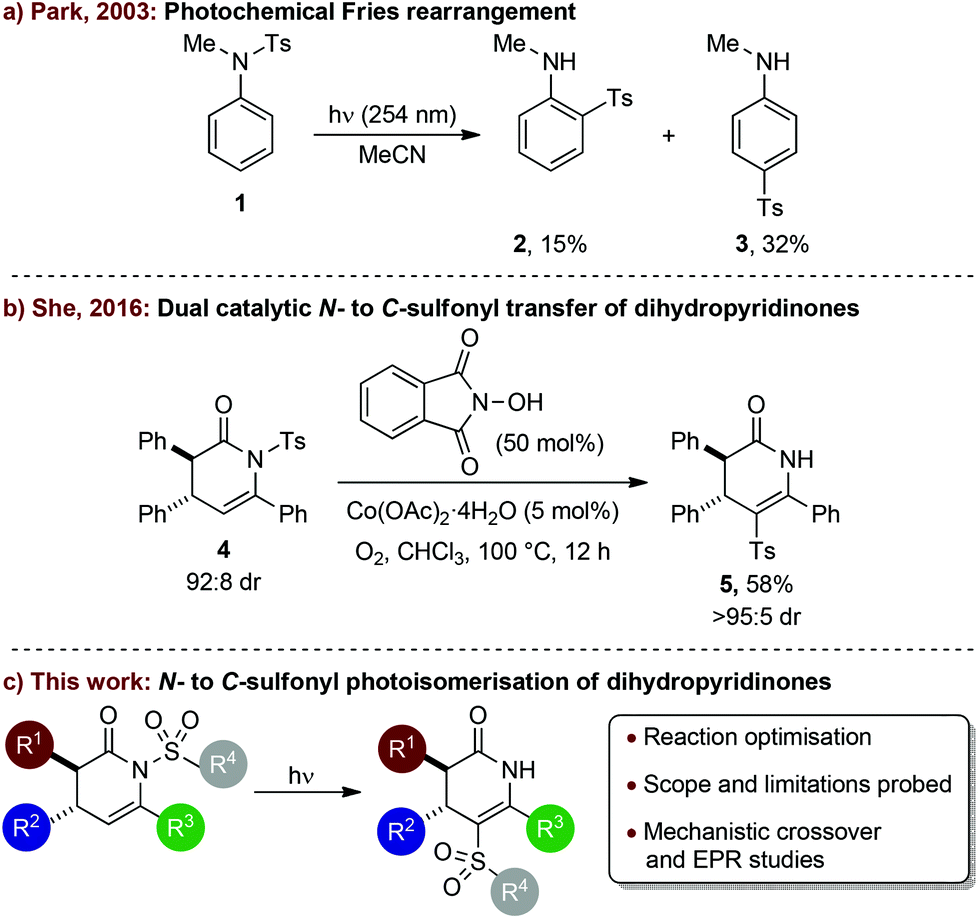 | ||
| Scheme 1 Literature examples of N- to C-sulfonyl transfer. | ||
To date, photochemical N- to C-sulfonyl transfer10 has mostly been explored for migration onto aryl rings, with only a few reports of sulfonyl transfer onto substituted alkenes. Henning and co-workers reported that N-tosyl β-aminovinyl phenyl ketones rearrange into the corresponding α-tosyl β-aminovinyl phenyl ketones upon photochemical irradiation.11 We have previously observed an interesting N- to C-sulfonyl transfer from a substituted N-tosyl dihydropyridinone to selectively give a C-tosyl-dihydropyridinone that occurred upon either prolonged standing, heating or photoirradiation.12 She and co-workers subsequently reported that related dihydropyridinones such as 4 also undergo N- to C-sulfonyl transfer into 5 in the presence of catalytic N-hydroxyphthalimide and cobalt(II) acetate under an oxygen atmosphere at high temperature (Scheme 1b).14 However, to date there have been no detailed investigations into the photochemical N- to C-sulfonyl transfer onto the alkene within substituted dihydropyridinone derivatives.
In this manuscript, the photochemical N- to C-sulfonyl migration of stereodefined substituted dihydropyridinone derivatives is optimised to give the corresponding rearranged products in high yields without compromising stereointegrity. The scope and limitations are explored through variation of the sulfonyl group and the dihydropyridinone substituents (Scheme 1c). Mechanistic work, including crossover experiments and EPR studies, give insight to the potential reaction mechanism of this transformation.
Results and discussion
Reaction optimisation
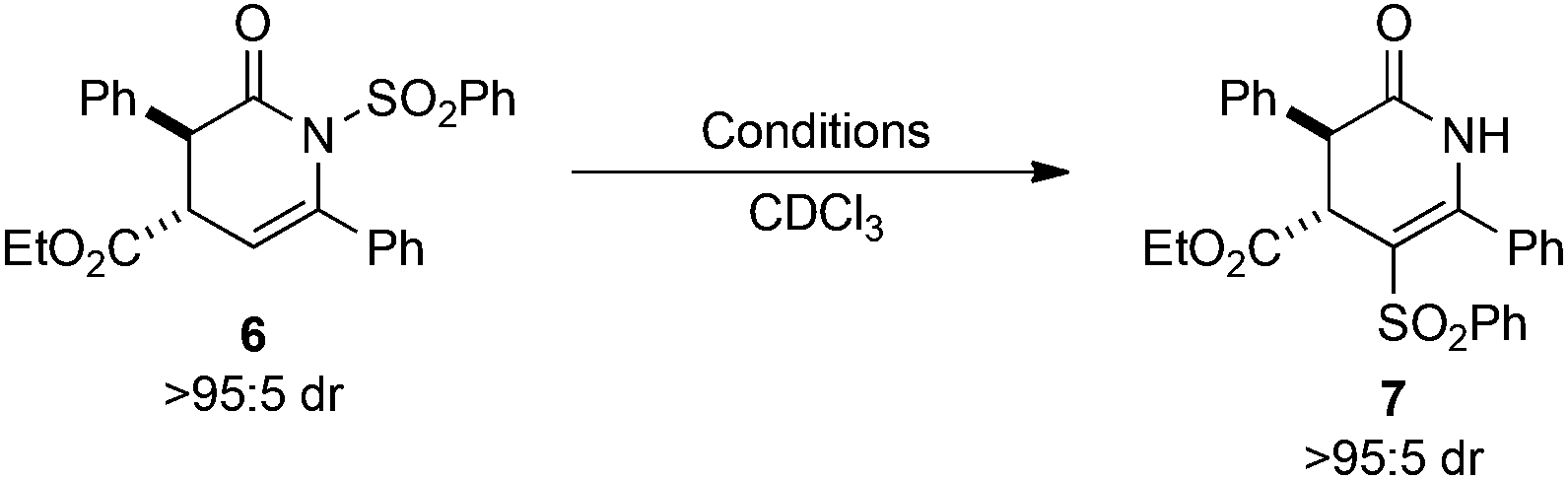
Initial studies assessed the influence of a range of reaction conditions capable of mediating the N- to C-sulfonyl transfer process using the transformation of dihydropyridinone 6 into isomeric 7 as a model. Initial experiments used (rac)-6 (>95:5 dr) dissolved in CDCl3 in an NMR tube wrapped in aluminium foil to eliminate ambient UV exposure. Monitoring the sample by 1H NMR spectroscopy at rt showed 49% conversion into 7 after 40 h, with no further conversion observed up to 112 h (Table 1, entries 1–3). Thermal mediation of the process was examined by heating a solution of 6 in CHCl3 at 50 °C for 16 h, giving 50% conversion into 7 as determined by 1H NMR spectroscopy (Table 1, entry 4). Notably, using CDCl3 pre-treated with K2CO3 as the reaction solvent gave no conversion into the desired product at either rt or 50 °C, indicating some dependence on reaction pH (Table 1, entries 5 and 6). Further studies used photoirradiation to facilitate the N- to C-sulfonyl transfer process. Irradiation of 6 using a broad-spectrum UV lamp showed a promising 58% conversion into 7 (Table 1, entry 7), although a number of unidentified decomposition products were also observed in the crude reaction mixture by 1H NMR spectroscopic analysis. However, the use of a 365 nm light source15 led exclusively to C-sulfonyl product 7, which was isolated in 91% yield as a single diastereoisomer without noticeable decomposition or side-reactions observed.
|
|
||||
|---|---|---|---|---|
| Entry | UV-source | Temp. (°C) | Time (h) | Conversionb (%) |
| a Reactions performed on a 0.05 mmol scale in an NMR tube. b Determined by 1H NMR spectroscopy using 1-methyl naphthalene as an internal standard. c CDCl3 pre-treated with K2CO3. d Broad spectrum UV lamp. e 365 nm wavelength irradiation. f Isolated yield following column chromatography. | ||||
| 1 | Off | rt | 16 | 37 |
| 2 | Off | rt | 40 | 49 |
| 3 | Off | rt | 112 | 50 |
| 4 | Off | 50 | 16 | 53 |
| 5c | Off | rt | 16 | — |
| 6c | Off | 50 | 16 | — |
| 7 | Ond | 30 | 16 | 58 |
| 8 | One | 30 | 16 | 94 (91)f |
Next, the effect of the solvent and overall reaction concentration on the photochemical migration process was examined (Table 2). Reactions were performed in round bottomed flasks, with the internal temperature within the UV light box chamber kept constant at 30 °C. During our investigations, it was found that optimal reaction reproducibility was obtained using degassed solvents (30 min Ar sparge) and glassware that was cleaned with aqueous KOH (1 M) prior to use. Irradiation of (rac)-6 in MeOH resulted in a low 9% conversion into isomerised product 7 (Table 2, entry 1). Enantiomerically pure 6 (>95:5 dr, 99:1 er) underwent photochemical N- to C-sulfonyl transfer in both EtOAc and THF, giving 7 in 48% and 68% yields, respectively, with no erosion in dr or er detected in either case (Table 2, entries 2 and 3). Chlorinated solvents were optimal, with reaction in either CH2Cl2 or CHCl3 giving product 7 in 84% and 71% yield, respectively (Table 2, entries 4 and 5). Changing the reaction concentration led to further improvements (Table 2, entries 6–8), with 7 obtained in excellent 95% yield when using either CH2Cl2 or CHCl3 at 0.1 M, again with no erosion in stereointegrity observed. Therefore, CH2Cl2 (0.1 M) was chosen as the solvent for subsequent investigations into the scope and limitations of this process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solventb | Conc. (M) | Yieldc (%) | erd | dre |
|
a Reactions preformed on a 0.1 mmol scale in round bottomed flask, cleaned in aq. KOH prior to use.
b Solvents degassed by sparging with Ar for 30 min prior to use.
c Isolated yield following column chromatography.
d Determined by HPLC analysis.
e dr determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
f Reaction using (rac)-6 (>95:5 dr).
g Conversion determined by 1H NMR spectroscopy using 1-methyl naphthalene as an internal standard.
|
|||||
| 1f | MeOH | 0.4 | (9)g | N/A | >95:5 |
| 2 | EtOAc | 0.4 | 48 | 99:1 |
>95:5 |
| 3 | THF | 0.4 | 68 | 99:1 |
>95:5 |
| 4 | CH2Cl2 | 0.4 | 84 | 99:1 |
>95:5 |
| 5 | CHCl3 | 0.4 | 71 | 99:1 |
>95:5 |
| 6 | CH2Cl2 | 0.2 | 87 | 99:1 |
>95:5 |
| 7 | CH2Cl2 | 0.1 | 95 | 99:1 |
>95:5 |
| 8 | CHCl3 | 0.1 | 95 | 99:1 |
>95:5 |
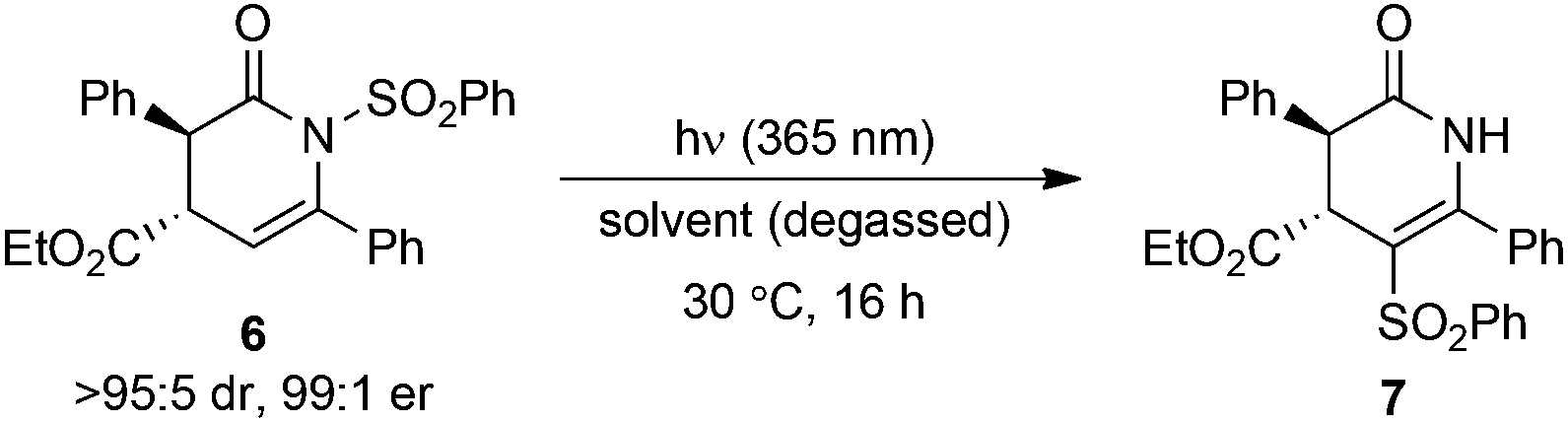
Substrate scope
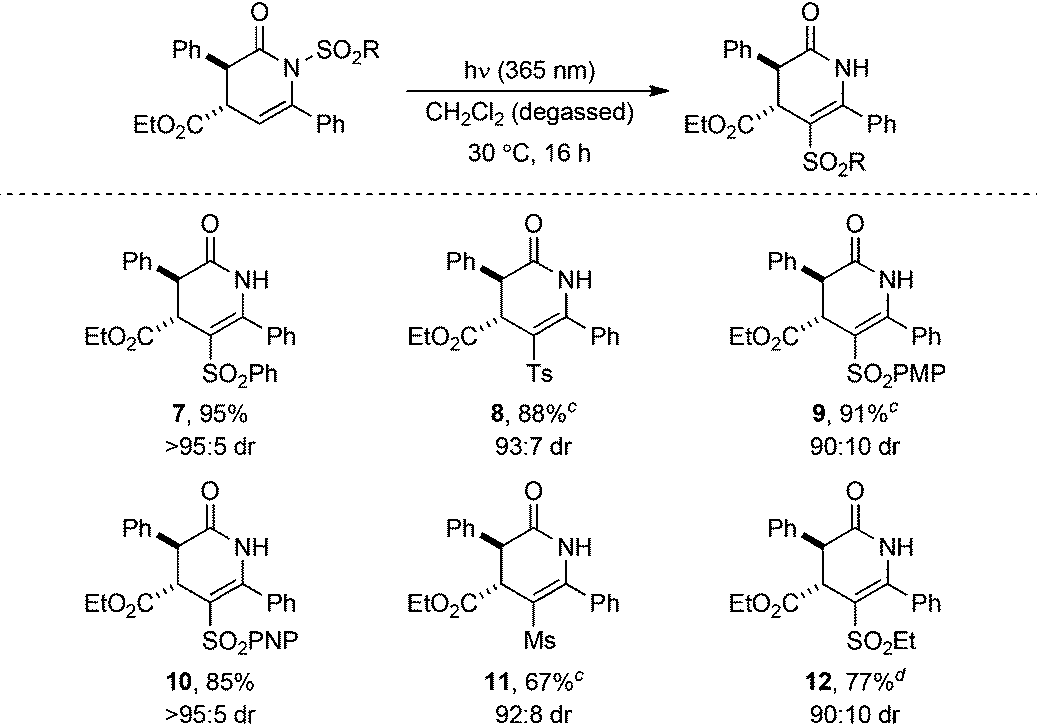
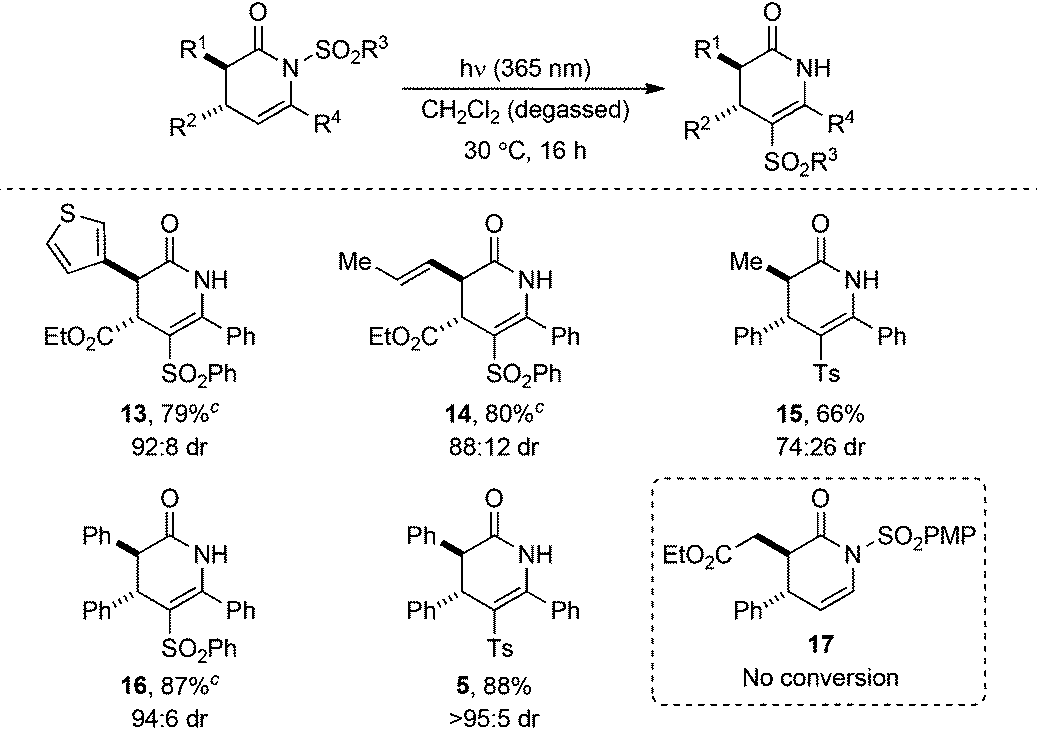
The generality of the photoisomerisation process was first investigated through variation of the sulfonyl substituent. A range of racemic N-sulfonyl dihydropyridinones was prepared following established isothiourea-catalysed methods12,16 in 90:10 to >95:5 dr and subjected to irradiation under the previously optimised conditions (Table 3). In all cases, no change in dr from the N-sulfonyl starting material (90:10 to >95:5 dr) to C-sulfonyl product (90:10 to >95:5 dr) was observed upon photoisomerisation, although chromatographic purification gave some products as single diastereoisomers. Incorporation of electron-rich aryl units within the sulfonyl group, 4-MeC6H4SO2 (Ts) and 4-MeOC6H4SO2 (PMP), gave isomerised products 8 and 9 in excellent 88% and 91% yields, respectively. Similarly, an electron-deficient N-sulfonyl substituent, 4-NO2C6H4SO2 (PNP), provided 10 in 85% yield. Changing the N-sulfonyl substituent from aryl to alkyl did not affect the photoisomerisation, with C-mesyl and C-ethanesulfonyl substituted dihydropyridinones 11 and 12 also obtained in good yields.
Next, the C(3), C(4), and C(6) substituents around the dihydropyridinone core were varied to further assess the scope of the N- to C-sulfonyl transfer. A range of racemic dihydropyridinones (74:26 to >95:5 dr) containing a variety of aryl, heteroaryl, alkenyl and alkyl substituents was prepared using either NHC or isothiourea-catalysed organocatalytic methodologies.16–18 The dihydropyridinones were then subjected to photoirradiation under the previously optimised conditions (Table 4). Again, in all examples where photoisomerisation occurred, no change in dr from N-sulfonyl starting material (74:26 to >95:5 dr) to C-sulfonyl product (74:26 to >95:5 dr) was observed, although purification led to some products being isolated as single diastereoisomers. Heteroaromatic substituents positioned at C(3) were well tolerated, with 3-thienyl dihydropyridinone 13 obtained in 79% yield. Photolysis of a C(3)-alkenyl substituted dihydropyridinone gave 14 in excellent 80% yield and C(3) alkyl groups were also well tolerated, with product 15 obtained in 78% yield. Altering the C(4) substituent from an ethyl ester to phenyl showed no loss in efficiency of photoisomerisation, with phenylsulfonyl- and tosyl dihydropyridinones 16 and 5 obtained in 87% and 88% yield, respectively. Notably, irradiation of 17 containing a C(6)-H substituent under the standard conditions gave no isomerisation, with only starting material returned.
Mechanistic studies
To probe the possible mechanistic pathway of the UV-mediated N- to C-sulfonyl transfer process a crossover reaction was performed. A 46:54 mixture of dihydropyridinones 4 and 6 was irradiated at 365 nm in CDCl3, with the reaction conversion and product distribution monitored over a total of 18 h using 1H NMR spectroscopy (Table 5). After 45 min irradiation the reaction had proceeded to a total of 38% conversion, with an approximately even distribution of all four possible isomerisation products observed (Table 5, entry 3). The reaction conversion increased over time, but the product distribution did not vary significantly. After 18 h, the photoisomerisation had proceeded to full conversion, giving C-sulfonyl products 5, 7, 8 and 16 in a final ratio of 23:27:25:25 (Table 5, entry 5). Throughout the reaction, potential N-sulfonyl crossover products 18 and 19 arising from N- to N-sulfonyl transfer were not observed.
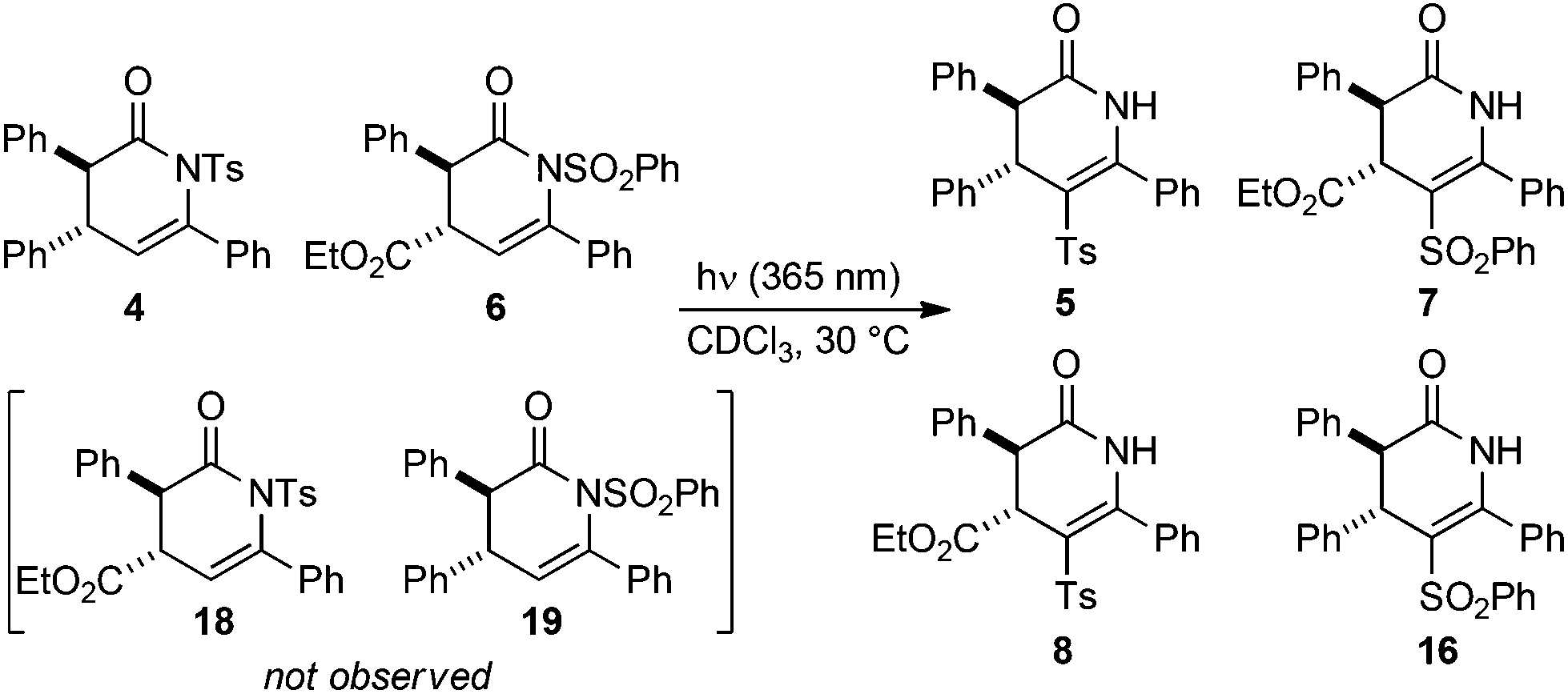
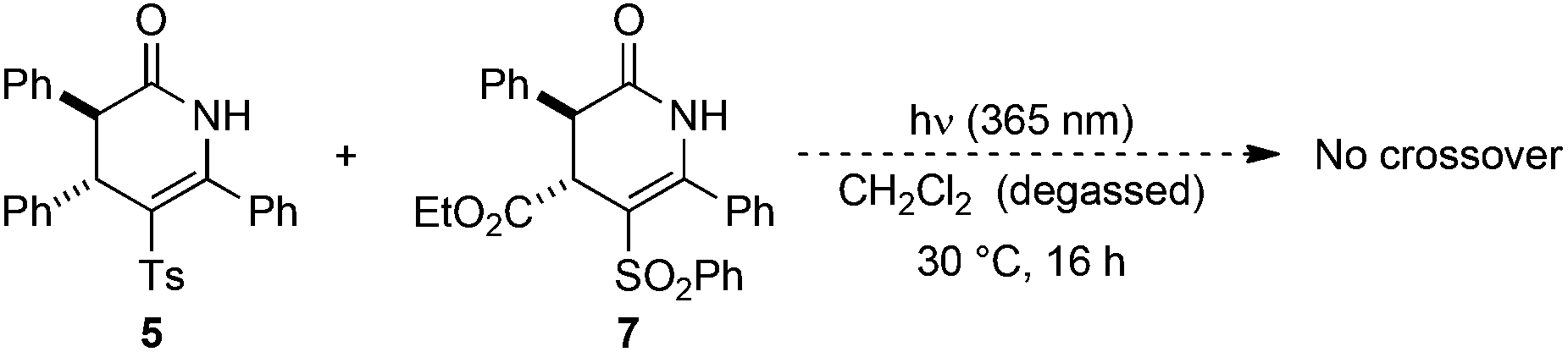
Next, the stability of the C-sulfonyl products to photoisomerisation was examined. Irradiation of a 50:50 mixture of C-sulfonyl dihydropyridinones 5 and 7 under the standard conditions gave no crossover, with starting materials returned (Scheme 2). The observation of significant crossover upon photoisomerisation of N-sulfonyl dihydropyridinones, coupled with no observed N- to N-sulfonyl transfer within the starting materials or C- to C-sulfonyl transfer in the products suggests the reaction proceeds via N–S bond cleavage followed by intermolecular reaction at carbon.
 | ||
| Scheme 2 Crossover of C-sulfonyl dihydropyridinones. | ||
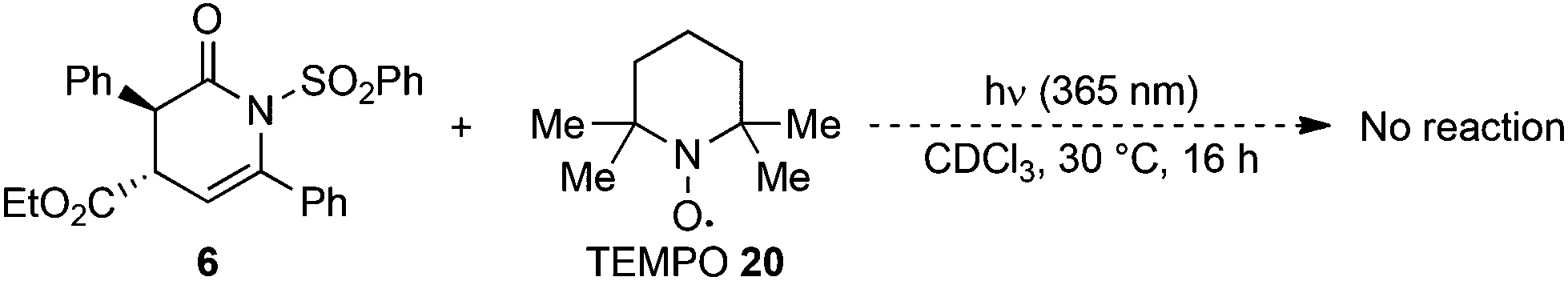
A final control experiment was conducted by irradiating 6 in the presence of an equivalent of TEMPO 20 as an additive (Scheme 3). This led to full suppression of the N- to C-sulfonyl transfer with only starting material returned, which is consistent with the photoisomerisation process proceeding via a radical reaction mechanism.
 | ||
| Scheme 3 Reaction in the presence of TEMPO 20. | ||
EPR spectroscopic study
A plausible initiation of the reaction mechanism involves direct homolysis of the N–S bond on UV photolysis at 365 nm wavelength. The UV/vis absorption spectrum of dihydropyridinone 6 showed absorption maxima at 261.8 nm and 366.8 nm.18 Strong absorption of the incident 365 nm radiation would therefore be expected and hence fission of the N–S bond is plausible. In an attempt to confirm the intermediacy of radicals 21 and 22 generated in this way, a sample of N-phenylsulfonyl dihydropyridinone 6 was examined by EPR spectroscopy (Scheme 4). Dihydropyridinone 6 (10 mg, 0.024 mmol) was dispersed in t-BuPh (0.5 mL) and the suspension sonicated. A 0.2 mL sample was transferred to a quartz tube (0.4 mm dia.) and de-aerated by bubbling nitrogen for ca. 15 min. This tube was placed in the resonant cavity of the EPR spectrometer and irradiated with the full spectrum from a 500 Watt high pressure Hg arc. Continuous-wave (CW) EPR spectra were scanned repeatedly at ambient temperature (295 K). Spectra were obtained after different photolysis times and were computer simulated using the NIEHS software package WinSim2002 (Fig. 1). A large central broad feature, most likely due to radicals trapped and immobilised within the solid particles in the dispersion, was removed by digital filtration. A best fit of spectrum (R = 0.900) was obtained with the following parameters: major radical: 77%, g-factor = 2.0044, a(2H) = 1.2, a(1H) = 0.6, a(2H) = 0.4 G. Minor radical: 23%, g-factor = 2.0090, a(3H) = 2.5 G. | ||
| Scheme 4 EPR study of the photoisomerisation of dihydropyridinone 6. | ||
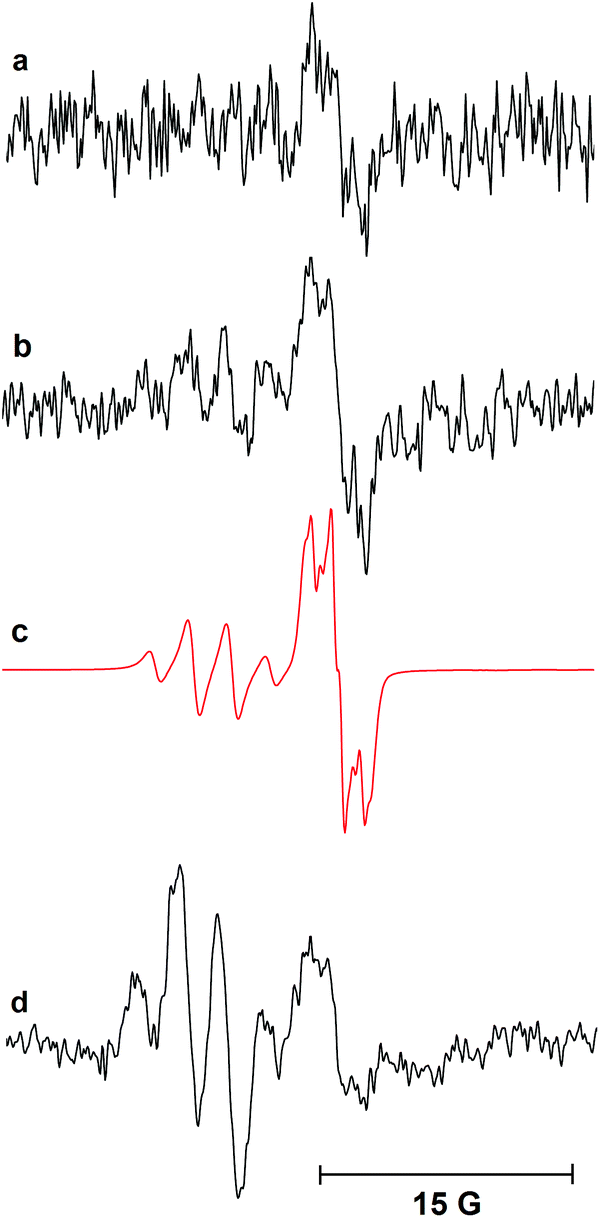 | ||
| Fig. 1 EPR spectra during continuous photolysis of a solution of dihydropyridinone 6 in t-BuPh at 295 K. (a) 1 scan after 1 min UV irradiation. (b) 40 scans after 15 min UV irradiation. (c) Computed simulation. (d) 70 scans after 60 min UV irradiation. | ||
The experimental g-factor and the hyperfine splitting (hfs) of the initial species in Fig. 1a and the major component in Fig. 1b are very close to the published parameters for the 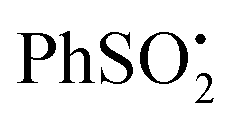 radical 22 (g = 2.0045, a(2Hm) = 1.13 G, a(1H)p = 0.52 G, a(2Ho) = 0.33 (in toluene)).19 This is positive evidence in support of the formation of the phenyl sulfonyl radical 22 during the photolysis. Furthermore, the EPR signal was isotropic which agrees with this radical becoming detached from the precursor dihydropyridinone and freely tumbling in the solution.
radical 22 (g = 2.0045, a(2Hm) = 1.13 G, a(1H)p = 0.52 G, a(2Ho) = 0.33 (in toluene)).19 This is positive evidence in support of the formation of the phenyl sulfonyl radical 22 during the photolysis. Furthermore, the EPR signal was isotropic which agrees with this radical becoming detached from the precursor dihydropyridinone and freely tumbling in the solution.
As photolysis was continued, a second radical appeared and dominated the spectrum after 60 min. That this was not the partner radical 21 was clear from comparison of its hfs with those of related radicals. While the observed radical has essentially the structure of an 1-aza-allyl radical, its hfs differ markedly from those published for this species.20 The g-factor (2.0090) of the second radical is unusually large, indicating that it was not centred on either C or N. Reference to the literature suggested it was actually the phenyl sulfinyl radical [PhS˙O]. The published EPR parameters for this radical in toluene are g = 2.0091, a(2Ho) = 2.4, a(Hm) = 2.4, a(2Hp) = 0.7 G.21 The small hfs from the para-H-atom was not resolved in our spectrum, but otherwise the correspondence was close. Evidently some degradation/reduction took place on prolonged photolysis such that PhS˙O was generated from either 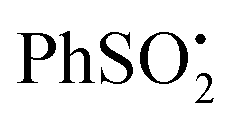 22, reactant 6, or product 7.
22, reactant 6, or product 7.
A similar EPR experiment was carried out with the N-ethylsulfonyl dihydropyridinone precursor to product 12. On photolysis at 290 K in the EPR cavity an isotropic spectrum with the following parameters was observed: g = 2.0048, a(2H) = 1.7, a(3H) = 2.2 G.18 Comparison with literature data indicated this was the expected 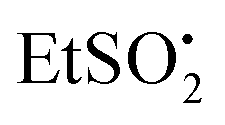 radical.22 The two EPR studies therefore provided positive evidence that both aryl sulfonyl and alkyl sulfonyl radicals are readily produced on UV irradiation of the corresponding N-substituted dihydropyridinones. In each case, the partner dihydropyridinonyl radical 21 would have a complex spectrum of at least 24 lines (probably more from delocalisation into one or more aromatic rings). As such, it is likely that it was not detected because individual lines would be below the observed signal-to-noise level (Fig. 1).
radical.22 The two EPR studies therefore provided positive evidence that both aryl sulfonyl and alkyl sulfonyl radicals are readily produced on UV irradiation of the corresponding N-substituted dihydropyridinones. In each case, the partner dihydropyridinonyl radical 21 would have a complex spectrum of at least 24 lines (probably more from delocalisation into one or more aromatic rings). As such, it is likely that it was not detected because individual lines would be below the observed signal-to-noise level (Fig. 1).
Proposed mechanism
The crossover and EPR mechanistic investigations allow the reaction mechanism depicted in Scheme 5 to be proposed. Photolysis of N-sulfonyl dihydropyridinone 6 promotes homolysis of the N–S bond to give the corresponding phenyl sulfonyl radical 22 (observed by EPR) and dihydropyridinonyl radical 21. This homolytic N–S bond fission is analogous to the first step proposed for the photo-Fries rearrangement of N-sulfonyl anilines.7 Intermolecular addition of 22 to dihydropyridinone 6 generates radical 23, which can undergo β-scission to generate imine 24 and another phenyl sulfonyl radical 22. Enhanced resonance stabilisation of the intermediate benzyl radical 23 rationalises the importance of the C(6) aryl substituent for the observed reactivity and the absence of product from irradiation of 17 containing a C(6)-H substituent Finally, tautomerisation of 24 generates C-sulfonyl dihydropyridinone product 7. Possible termination processes include recombination of 21 and 22 through either N- or C- to regenerate starting material 6 or form product 7, respectively. Termination through homodimerization or recombination of radicals 21 or 23 is also possible, although these are likely to be minor processes given the high yields obtained and no products from such reactions could be identified in the crude reaction mixtures.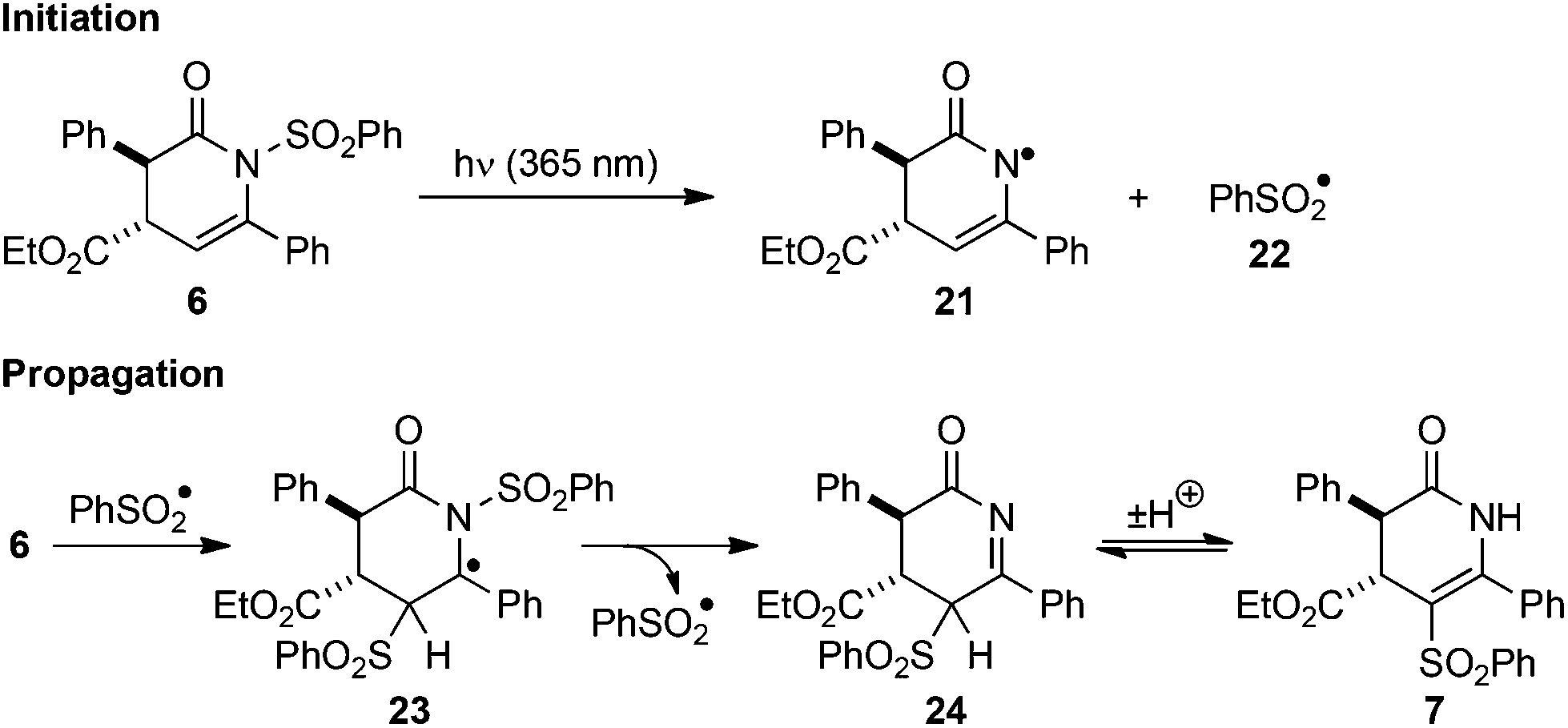 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
A UV-light induced N- to C-sulfonyl transfer process of N-sulfonyl dihydropyridinones has been studied. Various N-sulfonyl and C-substituents around the dihydropyridinone ring are tolerated, leading to the formation of the corresponding C-sulfonyl products in high yields with no erosion in stereointegrity observed. Mechanistic studies indicate an intermolecular radical chain process is operative, with a key phenyl sulfonyl radical observed by EPR spectroscopy.Experimental
General information
Reactions involving moisture sensitive reagents were carried out in flame-dried glassware under an inert atmosphere (N2 or Ar) using standard vacuum line techniques. Anhydrous solvents (CH2Cl2, THF and PhMe) were obtained after passing through an alumina column (Mbraun SPS-800). Petrol is defined as petroleum ether 40–60 °C. All other solvents and commercial reagents were used as received without further purification unless otherwise stated.Room temperature (rt) refers to 20–25 °C. A temperature of 0 °C was obtained using an ice/water bath. Reactions involving heating were performed using DrySyn blocks and a contact thermocouple.
Analytical thin layer chromatography was performed on pre-coated aluminium plates (Kieselgel 60 F254 silica) and visualisation was achieved using ultraviolet light (254 nm) and/or staining with aqueous KMnO4 solution, followed by heating. Column chromatography was performed in glass columns fitted with porosity 3 sintered discs over Kieselgel 60 silica using the solvent system stated.
Melting points were recorded on an Electrothermal 9100 melting point apparatus, (dec) refers to decomposition.
HPLC analyses were obtained on a Shimadzu HPLC consisting of a DGU-20A5 degassing unit, LC-20AT liquid chromatography pump, SIL-20AHT autosampler, CMB-20A communications bus module, SPD-M20A diode array detector and a CTO-20A column oven. Separation was achieved using a CHIRALPAK IA column using the method stated, with traces compared with authentic racemic spectra.
Infrared spectra were recorded on a Shimadzu IRAffinity-1 Fourier transform IR spectrophotometer fitted with a Specac Quest ATR accessory (diamond puck). Spectra were recorded of either thin films or solids, with characteristic absorption wavenumbers (νmax) reported in cm−1.
1H and 13C{1H} spectra were acquired on either a Bruker AV400 with a BBFO probe (1H 400 MHz; 13C{1H} 101 MHz), a Bruker AVII 400 with a BBFO probe (1H 400 MHz; 13C{1H} 101 MHz), or a Bruker AVIII-HD 500 with a SmartProbe BBFO+ probe (1H 500 MHz, 13C{1H} 126 MHz) in the deuterated solvent stated. All chemical shifts are quoted in parts per million (ppm) relative to the residual solvent peak. All coupling constants, J, are quoted in Hz. Multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and multiples thereof. The abbreviation Ar denotes aromatic and br denotes broad.
Mass spectrometry (m/z) data were acquired by either electrospray ionisation (ESI) or nanospray ionisation (NSI) at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
General procedure for N- to C-sulfonyl photoisomerisation
All glassware was cleaned in a KOH/i-PrOH bath, rinsed with deionised water followed by acetone and subsequently dried prior to use. Solvents were degassed prior to use by sparging with a baloon of argon for at least 15 min. A solution of the appropriate dihydropyridinone in degassed CH2Cl2 (0.1 M) was irradiated with UV light at 365 nm (internal temperature within the UV chamber recorded as 30 °C)15 for 16 h. The solvent was removed under reduced pressure and the crude reaction mixture purified by column chromatography in the solvent system stated.:5 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (174.6 mg, 95%, >95:5 dr) as a white solid. Mp 144–145 °C; HPLC analysis, Chiralpak IA (hexane/i-PrOH 70:30, 1.0 mL min−1, 254 nm, 30 °C) tR (3R,4R) 11.4 min, tR (3S,4S) 13.7 min, 99:1 er; νmax (ATR, cm−1) 3267, 2962, 2926, 1730, 1627, 1446, 1261, 1143, 1089, 1022, 802; 1H NMR (500 MHz, CDCl3) δH: 1.38 (3H, t, J 7.1, CH2CH3), 4.28–4.39 (3H, m, C(3)H and CH2CH3), 4.52 (1H, d, J 1.0, C(4)H), 6.82 (2H, d, J 7.7, ArH), 6.99–7.03 (3H, m, ArH), 7.14 (1H, br s, NH), 7.27–7.34 (6H, m, ArH), 7.37–7.41 (4H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.3 (CH2CH3), 47.3 (C(3)), 48.0 (C(4)), 62.5 (CH2CH3), 114.8 (C(5)), 127.1 (ArCH), 127.6 (ArCH), 128.3 (ArCH), 128.8 (ArCH), 129.4 (ArCH), 130.7 (ArCH), 131.9 (C(3)ArC(1)), 132.5 (ArCH), 135.6 (SO2ArC(1)), 141.1 (C(6)ArC(1)), 147.1 (C(6)), 168.4 (C(2)O), 170.5 (CO2Et); HRMS (ESI+) C26H24NO5S [M + H]+ found 462.1372 requires 472.1370 (+0.5 ppm).
:7 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (41.9 mg, 88%, >95:5 dr) as a white solid. Mp 169–170 °C; νmax (film/cm−1) 3240, 2924, 1726, 1691, 1631, 1452, 1348, 1145, 1087, 1014, 813; 1H NMR (500 MHz, CDCl3) δH: 1.38 (3H, t, J 7.1, CH2CH3), 2.30 (3H, s, ArCH3), 4.23–4.40 (3H, m, C(3)H and CH2CH3), 4.47 (1H, d, J 1.5, C(4)H), 6.76 (2H, d, J 8.2 SO2Ar(3,5)H), 6.83 (2H, d, J 8.2 SO2Ar(2,6)H), 7.17 (2H, m, ArH), 7.26–7.34 (5H, m, ArH), 7.36 (3H, m, ArH), 7.41 (1H, t, J 7.5, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.3 (CH2CH3), 21.6 (SO2Ar(4)CH3), 47.3 (C(3)), 49.2 (C(4)), 62.4 (CH2CH3), 115.6 (C(5)), 127.1 (ArCH), 127.6 (ArCH), 128.1 (ArCH), 128.4 (ArCH), 128.9 (ArCH), 129.1 (ArC), 129.3 (ArCH), 130.5 (ArC), 132.0 (ArC), 135.7 (ArC), 138.1 (ArC), 143.3 (ArC), 146.8 (C(6)), 168.6 (C(2)O), 170.5 (CO2Et); HRMS (ESI+) C27H26NO5S [M + H]+ found 476.1524, requires 476.1526 (−0.5 ppm).
:10 dr. Purification by column chromatography (CH2Cl2/Et2O, 95:5) gave the title compound (70.8 mg, 91%, >95:5 dr) as a white solid. Mp 196–198 °C; νmax (film/cm−1) 3232, 2978, 1722, 1703, 1633, 1593, 1496, 1454, 1267, 1141, 1091, 1016; 1H NMR (400 MHz, CDCl3) δH: 1.37 (3H, t, J 7.1, CH2CH3), 3.76 (3H, s, OCH3), 4.24–4.38 (3H, m, C(3)H and CH2CH3), 4.46 (1H, d, J 1.7, C(4)H), 6.48 (2H, d, J 9.0, SO2ArC(3,5)H), 6.79 (2H, d, J 9.0, SO2ArC(2,6)H), 7.12–7.20 (3H, m, NH and ArH), 7.28–7.32 (4H, m, ArH), 7.32–7.36 (3H, m, ArH), 7.35–7.42 (1H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.2 (CH2CH3), 47.3 (C(4)), 48.1 (C(3)), 55.7 (OCH3), 62.4 (CH2CH3), 113.5 (SO2ArC(3)), 115.2 (C(5)), 127.1 (ArCH), 128.1 (ArCH), 128.5 (ArCH), 129.1 (ArCH), 129.3 (ArCH), 129.7 (ArCH), 130.6 (ArCH), 132.1 (C(6)ArC(1)), 132.7 (SO2ArC(1)), 135.7 (C(3)ArC(1)), 146.7 (C(6)), 162.8 (SO2ArC(4)), 168.6 (C(2)O), 170.6 (CO2Et); HRMS (ESI+) C27H26NO6S [M + H]+ found 492.1472, requires 492.1475 (−0.7 ppm).
:5 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (43.1 mg, 85%, >95:5 dr) as a white solid. Mp 220–221 °C; νmax (film/cm−1) 3255, 2981, 1728, 1624, 15.27, 1448, 1348, 1303, 1145, 1089, 854; 1H NMR (400 MHz, CDCl3) δH: 1.40 (3H, t, J 7.1, CH2CH3), 4.29–4.43 (3H, m, C(3)H and CH2CH3), 4.52 (1H, d, J 1.8, C(4)H), 6.87 (2H, d, J 9.0, SO2ArC(2,6)H), 7.07–7.18 (2H, m, NH and ArH), 7.28–7.32 (5H, m, ArH), 7.37–7.47 (4H, m, ArH), 7.78 (2H, d, J 9.0, SO2ArC(3,5)H); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.2 (CH2CH3), 47.3 (C(4)), 47.6 (C(3)), 62.6 (CH2CH3), 113.7 (C(5)), 123.1 (SO2ArC(3)), 126.9 (ArCH), 128.3 (C(6)ArC(1)), 128.6 (SO2ArC(2)), 128.7 (ArCH), 129.4 (ArCH), 131.1 (ArCH), 135.3 (C(3)ArC(1)), 146.6 (SO2ArC(1)), 148.3 (C(6)), 149.5 (SO2ArC(4)), 168.2 (C(2)O), 170.1 (CO2Et); HRMS (ESI+) C27H26NO5S [M + NH4]+ found 524.1481, requires 524.1486 (−0.9 ppm).
:8 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (42.3 mg, 67%, >95:5 dr) as a white solid. Mp 86–87 °C; νmax (film/cm−1) 3250, 2926, 1726, 1701, 1629, 1448, 1300, 1132, 954; 1H NMR (500 MHz, CDCl3) δH: 1.34 (3H, t, J 7.1, CH2CH3), 2.49 (3H, s, SO2CH3), 4.24–4.33 (2H, m, CH2CH3), 4.35 (1H, s, C(3)H), 4.44 (1H, d, J 1.9, C(4)H), 7.32–7.36 (1H, m, ArH), 7.39–7.51 (9H, m, ArH), 7.53–7.58 (1H, br s, NH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.3 (CH2CH3), 44.0 (SO2CH3), 46.4 (C(4)), 47.7 (C(3)), 62.5 (CH2CH3), 113.8 (C(5)), 127.0 (ArCH), 128.4 (ArCH), 128.8 (ArCH), 128.9 (ArCH), 129.3 (ArCH), 131.1 (ArCH), 132.0 (C(6)ArC(1)), 135.2 (C(3)ArC(1)), 147.6 (C(6)), 169.0 (C(2)O), 170.6 (CO2Et); HRMS (ESI+) C27H26NO5S [M + H]+ found 400.1218, requires 400.1213 (+1.2 ppm).
:10 dr. Purification by column chromatography (hexane/EtOAc, 50:50) gave the title compound (20 mg, 77%, 90:10 dr) as a colourless oil. νmax (neat, cm−1) 3248, 1728 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1708 (CO), 1631, 1540, 1303 (C–N), 1130; 1H NMR (500 MHz, CDCl3) δH: 0.83 (3H, t, J 7.4, SO2CH2CH3), 1.35 (3H, t, J 7.1, OCH2CH3), 2.34 (1H, dd, J 14.0, 7.4, SO2CHAHB), 2.54 (1H, dd, J 14.0, 7.4, SO2CHAHB), 4.25–4.34 (2H, m, OCH2CH3), 4.36 (1H, br s, C(4)H), 4.38 (1H, d, J 1.8, C(3)H), 7.30–7.52 (11H, m, NH and ArH); 13C{1H} NMR (126 MHz, CDCl3) δC: 6.3 (SO2CH2CH3), 14.3 (OCH2CH3), 46.8 (C(4)H), 47.9 (C(3)H), 49.7 (SO2CH2), 62.5 (OCH2), 112.0 (C(5)), 127.1 (ArCH), 128.4 (ArCH), 128.8 (ArCH), 128.9 (ArCH), 129.4 (ArCH), 131.1 (ArCH), 132.1 (ArC), 135.2 (ArC), 147.4 (C(6)), 168.7 (C(2)O), 170.5 (CO2Et); HMRS (ESI+) C22H23NO5SNa [M + Na]+ found 436.1184, requires 436.1189 (−1.2 ppm).
:8 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (54.2 mg, 79%, >95:5 dr) as a white solid. Mp 152–153 °C; νmax (film/cm−1) 3265, 2980, 2360, 1730, 1701, 1627, 1446, 1301, 1145, 1089, 756; 1H NMR (400 MHz, CDCl3) δH: 1.35 (3H, t, J 7.1, CH2CH3), 4.21–4.35 (3H, m, C(3)H and CH2CH3), 4.58 (1H, d, J 1.7, C(4)H), 7.00–7.04 (2H, m, ArH), 7.05–7.14 (6H, m, NH and ArH), 7.21–7.27 (3H, m, ArH), 7.31–7.39 (3H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.3 (CH2CH3), 44.6 (C(3)), 46.0 (C(4)), 62.4 (CH2CH3), 114.7 (C(5)), 122.3 (C(3)ArCH), 126.9 (ArCH), 127.1 (ArCH), 127.6 (ArCH), 128.4 (ArCH), 128.6 (ArCH), 128.9 (ArCH), 130.5 (ArCH), 131.9 (C(3)ArC(1)), 132.6 (ArCH), 135.2 (SO2ArC(1)), 141.1 (C(6)ArC(1)), 147.0 (C(6)), 168.2 (C(2)O), 170.3 (CO2Et); HRMS (ESI+) C24H22NO5S2 [M + H]+ found 468.0931, requires 468.0934 (−0.6 ppm).
:12 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (48.0 mg, 80%, >95:5 dr) as a white solid. Mp 155–156 °C; νmax (film/cm−1) 3265, 2978, 1728, 1701, 1629, 1446, 1303, 1226, 1178, 1145, 1091, 1031, 964; 1H NMR (400 MHz, CDCl3) δH: 1.30 (3H, t, J 7.1, OCH2CH3), 1.68 (3H, dt, J 6.5, 1.4 CHCH3), 3.57–3.61 (1H, m, C(3)H), 4.09 (1H, d, J 1.8, C(4)H), 4.15–4.28 (1H, m, OCH2CH3), 5.32 (1H, ddq, J 15.3, 6.3, 1.6, C(3)CH), 5.74–5.84 (1H, m, CHCHCH3), 6.87 (1H, br s, NH), 7.15–7.20 (2H, m, ArH), 7.25–7.33 (4H, m, ArH), 7.39–7.46 (4H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.2 (CH2CH3), 18.3 (CHCH3), 45.5 (C(4)), 46.4 (C(3)), 62.2 (OCH2CH3), 113.9 (C(5)), 124.3 (C(3)CH), 127.9 (ArCH), 128.5 (2 × ArCH), 129.0 (ArCH), 130.6 (ArC and CHCHCH3), 132.2 (ArC), 132.8 (ArCH), 141.4 (ArC), 147.4 (C(6)), 168.7 (C(2)O), 170.4 (CO2Et); HRMS (ESI+) C23H24NO5S [M + H]+ found 426.1370, requires 426.1370 (+0.1 ppm).
:26 dr) in CH2Cl2 (1.2 mL) for 16 h gave the crude product in 74:26 dr. Purification by column chromatography (CH2Cl2/Et2O, 95:5) gave the title compound (31.6 mg, 66%, 66:34 dr) as a white solid. Mp 182–184 °C; νmax (film/cm−1) 3244, 2972, 1697, 1629, 1597, 1456, 1288, 1145, 1089, 813; 1H NMR (500 Hz, CDCl3) major diastereoisomer (selected) δH: 1.39 (3H, d, J 7.3, CHCH3), 2.31 (3H, s, ArCH3), 2.83 (1H, q, J 7.4, C(3)H), 4.24 (1H, d, J 1.0, C(4)H), 6.83 (1H, br s, NH), 6.96 (2H, d, J 8.1, SO2ArC(3,5)H), 7.03 (2H, d, J 8.3, SO2ArC(2,6)H); minor diastereoisomer (selected) δH: 1.06 (3H, d, J 6.9, CHCH3), 2.27 (3H, s, ArCH3), 3.19–3.25 (1H, m, C(3)H), 4.43 (1H, d, J 7.4, C(4)H), 6.75 (2H, d, J 8.3, SO2ArC(3,5)H), 6.83–6.85 (2H, m, SO2ArC(2,6)H), 6.89 (1H, br s, NH); both diastereoisomers δH: 7.18–7.49 (10H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) major diastereoisomer (selected) δC: 18.0 (CH3), 21.6 (SO2Ar(4)CH3), 43.9 (C(3)), 46.8 (C(4)), 118.3 (C(5)), 172.2 (C(2)O); minor diastereoisomer (selected) δC: 11.8 (CH3), 21.6 (SO2Ar(4)CH3), 40.3 (C(3)), 45.3 (C(4)), 122.1 (C(5)), both diastereoisomers δC: 127.1, 127.5, 127.7, 127.8, 127.9, 128.5, 128.6, 128.7, 128.8, 129.0, 129.0, 129.2, 130.2, 130.4, 132.7, 138.6, 140.1, 142.6, 143.6, 145.6; HRMS (ESI+) C25H24NO3S [M + H]+ found 418.1470, requires 418.1471 (−0.3 ppm).
:6 dr. Recrystallisation (Et2O) gave the title compound (40.2 mg, 87%, >95:5 dr) as a white solid. Mp 249–250 °C; νmax (film/cm−1) 3209, 2897, 1695, 1672, 1494, 1315, 1145, 1089, 800; 1H NMR (500 MHz, CDCl3) δH: 4.12 (1H, s, C(3)H), 4.87 (1H, s, C(4)H), 6.72 (2H, d, J 7.4, ArH), 6.96–6.99 (2H, m, ArH), 7.15 (1H, br s, NH), 7.18–7.34 (6H, m, ArH), 7.38–7.48 (10H, m, ArH); 13C{1H} NMR (126 MHz, CDCl3) δC: 47.3 (C(4)), 53.9 (C(3)), 119.2 (C(5)), 127.1 (ArCH), 127.3 (ArCH), 127.5 (ArCH), 127.9 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.6 (ArCH), 129.4 (ArCH), 129.5 (ArCH), 130.5 (ArCH), 132.3 (SO2ArC(1)), 132.4 (ArCH), 137.2 (C(3)ArC(1)), 139.7 (C(4)ArC(1)), 141.3 (C(6)ArC(1)), 146.2 (C(6)), 168.9 (C(2)O); HRMS (ESI+) C29H24NO3S [M + H]+ found 466.1468, requires 466.1471 (−0.7 ppm).
:5 dr. Recrystallisation (Et2O) gave the title compound (42.4 mg, 88%, >95:5 dr) as a white solid. Mp 232–234 °C {lit.12 238–240 °C}; 1H NMR (500 MHz, CDCl3) δH: 2.29 (3H, s, CH3), 4.10 (1H, s, C(3)H), 4.82 (1H, s, C(4)H), 6.63 (2H, d, J 8.3, SO2ArC(3)H), 6.77 (2H, d, J 8.0, SO2ArC(2)H), 7.10 (1H, br s, NH), 7.24–7.51 (15H, m, ArH). Data in accordance with the literature.12
O), 1708 (CO), 1631, 1540, 1303 (C–N), 1130; 1H NMR (500 MHz, CDCl3) δH: 0.83 (3H, t, J 7.4, SO2CH2CH3), 1.35 (3H, t, J 7.1, OCH2CH3), 2.34 (1H, dd, J 14.0, 7.4, SO2CHAHB), 2.54 (1H, dd, J 14.0, 7.4, SO2CHAHB), 4.25–4.34 (2H, m, OCH2CH3), 4.36 (1H, br s, C(4)H), 4.38 (1H, d, J 1.8, C(3)H), 7.30–7.52 (11H, m, NH and ArH); 13C{1H} NMR (126 MHz, CDCl3) δC: 6.3 (SO2CH2CH3), 14.3 (OCH2CH3), 46.8 (C(4)H), 47.9 (C(3)H), 49.7 (SO2CH2), 62.5 (OCH2), 112.0 (C(5)), 127.1 (ArCH), 128.4 (ArCH), 128.8 (ArCH), 128.9 (ArCH), 129.4 (ArCH), 131.1 (ArCH), 132.1 (ArC), 135.2 (ArC), 147.4 (C(6)), 168.7 (C(2)O), 170.5 (CO2Et); HMRS (ESI+) C22H23NO5SNa [M + Na]+ found 436.1184, requires 436.1189 (−1.2 ppm).
:8 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (54.2 mg, 79%, >95:5 dr) as a white solid. Mp 152–153 °C; νmax (film/cm−1) 3265, 2980, 2360, 1730, 1701, 1627, 1446, 1301, 1145, 1089, 756; 1H NMR (400 MHz, CDCl3) δH: 1.35 (3H, t, J 7.1, CH2CH3), 4.21–4.35 (3H, m, C(3)H and CH2CH3), 4.58 (1H, d, J 1.7, C(4)H), 7.00–7.04 (2H, m, ArH), 7.05–7.14 (6H, m, NH and ArH), 7.21–7.27 (3H, m, ArH), 7.31–7.39 (3H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.3 (CH2CH3), 44.6 (C(3)), 46.0 (C(4)), 62.4 (CH2CH3), 114.7 (C(5)), 122.3 (C(3)ArCH), 126.9 (ArCH), 127.1 (ArCH), 127.6 (ArCH), 128.4 (ArCH), 128.6 (ArCH), 128.9 (ArCH), 130.5 (ArCH), 131.9 (C(3)ArC(1)), 132.6 (ArCH), 135.2 (SO2ArC(1)), 141.1 (C(6)ArC(1)), 147.0 (C(6)), 168.2 (C(2)O), 170.3 (CO2Et); HRMS (ESI+) C24H22NO5S2 [M + H]+ found 468.0931, requires 468.0934 (−0.6 ppm).
:12 dr. Purification by column chromatography (CH2Cl2/Et2O, 97:3) gave the title compound (48.0 mg, 80%, >95:5 dr) as a white solid. Mp 155–156 °C; νmax (film/cm−1) 3265, 2978, 1728, 1701, 1629, 1446, 1303, 1226, 1178, 1145, 1091, 1031, 964; 1H NMR (400 MHz, CDCl3) δH: 1.30 (3H, t, J 7.1, OCH2CH3), 1.68 (3H, dt, J 6.5, 1.4 CHCH3), 3.57–3.61 (1H, m, C(3)H), 4.09 (1H, d, J 1.8, C(4)H), 4.15–4.28 (1H, m, OCH2CH3), 5.32 (1H, ddq, J 15.3, 6.3, 1.6, C(3)CH), 5.74–5.84 (1H, m, CHCHCH3), 6.87 (1H, br s, NH), 7.15–7.20 (2H, m, ArH), 7.25–7.33 (4H, m, ArH), 7.39–7.46 (4H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) δC: 14.2 (CH2CH3), 18.3 (CHCH3), 45.5 (C(4)), 46.4 (C(3)), 62.2 (OCH2CH3), 113.9 (C(5)), 124.3 (C(3)CH), 127.9 (ArCH), 128.5 (2 × ArCH), 129.0 (ArCH), 130.6 (ArC and CHCHCH3), 132.2 (ArC), 132.8 (ArCH), 141.4 (ArC), 147.4 (C(6)), 168.7 (C(2)O), 170.4 (CO2Et); HRMS (ESI+) C23H24NO5S [M + H]+ found 426.1370, requires 426.1370 (+0.1 ppm).
:26 dr) in CH2Cl2 (1.2 mL) for 16 h gave the crude product in 74:26 dr. Purification by column chromatography (CH2Cl2/Et2O, 95:5) gave the title compound (31.6 mg, 66%, 66:34 dr) as a white solid. Mp 182–184 °C; νmax (film/cm−1) 3244, 2972, 1697, 1629, 1597, 1456, 1288, 1145, 1089, 813; 1H NMR (500 Hz, CDCl3) major diastereoisomer (selected) δH: 1.39 (3H, d, J 7.3, CHCH3), 2.31 (3H, s, ArCH3), 2.83 (1H, q, J 7.4, C(3)H), 4.24 (1H, d, J 1.0, C(4)H), 6.83 (1H, br s, NH), 6.96 (2H, d, J 8.1, SO2ArC(3,5)H), 7.03 (2H, d, J 8.3, SO2ArC(2,6)H); minor diastereoisomer (selected) δH: 1.06 (3H, d, J 6.9, CHCH3), 2.27 (3H, s, ArCH3), 3.19–3.25 (1H, m, C(3)H), 4.43 (1H, d, J 7.4, C(4)H), 6.75 (2H, d, J 8.3, SO2ArC(3,5)H), 6.83–6.85 (2H, m, SO2ArC(2,6)H), 6.89 (1H, br s, NH); both diastereoisomers δH: 7.18–7.49 (10H, m, ArH); 13C{1H} NMR (101 MHz, CDCl3) major diastereoisomer (selected) δC: 18.0 (CH3), 21.6 (SO2Ar(4)CH3), 43.9 (C(3)), 46.8 (C(4)), 118.3 (C(5)), 172.2 (C(2)O); minor diastereoisomer (selected) δC: 11.8 (CH3), 21.6 (SO2Ar(4)CH3), 40.3 (C(3)), 45.3 (C(4)), 122.1 (C(5)), both diastereoisomers δC: 127.1, 127.5, 127.7, 127.8, 127.9, 128.5, 128.6, 128.7, 128.8, 129.0, 129.0, 129.2, 130.2, 130.4, 132.7, 138.6, 140.1, 142.6, 143.6, 145.6; HRMS (ESI+) C25H24NO3S [M + H]+ found 418.1470, requires 418.1471 (−0.3 ppm).
:6 dr. Recrystallisation (Et2O) gave the title compound (40.2 mg, 87%, >95:5 dr) as a white solid. Mp 249–250 °C; νmax (film/cm−1) 3209, 2897, 1695, 1672, 1494, 1315, 1145, 1089, 800; 1H NMR (500 MHz, CDCl3) δH: 4.12 (1H, s, C(3)H), 4.87 (1H, s, C(4)H), 6.72 (2H, d, J 7.4, ArH), 6.96–6.99 (2H, m, ArH), 7.15 (1H, br s, NH), 7.18–7.34 (6H, m, ArH), 7.38–7.48 (10H, m, ArH); 13C{1H} NMR (126 MHz, CDCl3) δC: 47.3 (C(4)), 53.9 (C(3)), 119.2 (C(5)), 127.1 (ArCH), 127.3 (ArCH), 127.5 (ArCH), 127.9 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.6 (ArCH), 129.4 (ArCH), 129.5 (ArCH), 130.5 (ArCH), 132.3 (SO2ArC(1)), 132.4 (ArCH), 137.2 (C(3)ArC(1)), 139.7 (C(4)ArC(1)), 141.3 (C(6)ArC(1)), 146.2 (C(6)), 168.9 (C(2)O); HRMS (ESI+) C29H24NO3S [M + H]+ found 466.1468, requires 466.1471 (−0.7 ppm).
:5 dr. Recrystallisation (Et2O) gave the title compound (42.4 mg, 88%, >95:5 dr) as a white solid. Mp 232–234 °C {lit.12 238–240 °C}; 1H NMR (500 MHz, CDCl3) δH: 2.29 (3H, s, CH3), 4.10 (1H, s, C(3)H), 4.82 (1H, s, C(4)H), 6.63 (2H, d, J 8.3, SO2ArC(3)H), 6.77 (2H, d, J 8.0, SO2ArC(2)H), 7.10 (1H, br s, NH), 7.24–7.51 (15H, m, ArH). Data in accordance with the literature.12
EPR spectroscopy
EPR spectra were obtained at 9.5 GHz with 100 kHz modulation employing a Bruker EMX 10/12 spectrometer fitted with a rectangular ER4122 SP resonant cavity. Stock suspensions of each substrate (10 mg) in tert-butylbenzene (0.5 mL) were prepared and sonicated. An aliquot (0.2 mL) was placed in a 4 mm o.d. quartz tube and de-aerated by bubbling nitrogen for 15 min. Photolysis in the resonant cavity was by unfiltered light from a 500 W super pressure mercury arc lamp. Sample temperature was maintained at 295 K by means of the Bruker VT unit. In all cases where spectra were obtained they were digitally filtered and hfs were assigned with the aid of the Bruker WinEPR and NIEHS WinSim2002 software packages. The majority of EPR spectra were recorded at a frequency of 9.48 GHz, 4.0 mW power, 1.0 Gpp modulation intensity and gain of ca. 106.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013) ERC Grant Agreement no. 279850 (CF and JET) and EPSRC grant number EP/J018139/1 (DSBD) for funding. ADS thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- For a recent review, see: Y. Chen, Synthesis, 2016, 2483–2522 CrossRef CAS and references cited within.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, 4th edn, 2007 Search PubMed.
- For selected examples, see: (a) S. Searles and S. Nukina, Chem. Rev., 1959, 59, 1077–1103 CrossRef CAS; (b) J. Kovacs and U. R. Ghatak, J. Org. Chem., 1966, 31, 119–121 CrossRef CAS; (c) S. Ji, L. B. Gortler, A. Waring, A. J. Battisti, S. Bank, W. D. Closson and P. A. Wriede, J. Am. Chem. Soc., 1967, 89, 5311–5312 CrossRef CAS; (d) E. H. Gold and E. Babad, J. Org. Chem., 1972, 37, 2208–2210 CrossRef CAS; (e) B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017–1018 RSC; (f) S. C. Bergmeier and P. P. Seth, Tetrahedron Lett., 1999, 40, 6181–6184 CrossRef CAS; (g) P. Nandi, M. Y. Redko, K. Petersen, J. L. Dye, M. Lefenfeld, P. F. Vogt and J. E. Jackson, Org. Lett., 2008, 10, 5441–5444 CrossRef CAS PubMed; (h) N. Shohji, T. Kawaji and S. Okamoto, Org. Lett., 2011, 13, 2626–2629 CrossRef CAS PubMed.
- For selected examples, see: (a) E. Vedejs and S. Lin, J. Org. Chem., 1994, 59, 1602–1603 CrossRef CAS; (b) H. Knowles, A. F. Parsons and R. M. Pettifer, Synlett, 1997, 271–272 CrossRef CAS; (c) H. S. Knowles, A. F. Parsons, R. M. Pettifer and S. Rickling, Tetrahedron, 2000, 56, 979–988 CrossRef CAS; (d) Z. Moussa and D. Romo, Synlett, 2006, 3294–3298 CAS; (e) T. Ankner and G. Hilmersson, Org. Lett., 2009, 11, 503–506 CrossRef CAS PubMed.
- For selected examples, see: (a) L. D'Souza and R. A. Day, Science, 1968, 160, 882–883 Search PubMed; (b) J. A. Pincock and A. Jurgens, Tetrahedron Lett., 1979, 20, 1029–1030 CrossRef; (c) T. Hamada, A. Nishida, Y. Matsumoto and O. Yonemitsu, J. Am. Chem. Soc., 1980, 102, 3978–3980 CrossRef CAS; (d) T. Hamada, A. Nishida and O. Yonemitsu, J. Am. Chem. Soc., 1986, 108, 140–145 CrossRef CAS; (e) Q. Liu, Z. Liu, Y.-L. Zhou, W. Zhang, L. Yang, Z.-L. Liu and W. Yu, Synlett, 2005, 2510–2512 CrossRef CAS; (f) J. E. T. Corrie and G. Papageorgiou, J. Chem. Soc., Perkin Trans. 1, 1996, 1583–1592 RSC; (g) J. Xuan, B.-J. Li, Z.-J. Feng, G.-D. Sun, H.-H. Ma, Z.-W. Yuan, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. – Asian J., 2013, 8, 1090–1094 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Lebouc, P. Martigny, R. Carlier and J. Simonet, Tetrahedron, 1985, 41, 1251–1258 CrossRef CAS; (b) E. R. Civitello and H. Rapoport, J. Org. Chem., 1992, 57, 834–840 CrossRef CAS; (c) B. Nyasse, L. Grehn, H. L. S. Maia, L. S. Monteiro and U. Ragnarsson, J. Org. Chem., 1999, 64, 7135–7139 CrossRef CAS; (d) V. Coeffard, C. Thobie-Gautier, I. Beaudet, E. Le Grognec and J.-P. Quintard, Eur. J. Org. Chem., 2008, 383–391 CrossRef CAS; (e) H. Senboku, K. Nakahara, T. Fukuhara and S. Hara, Tetrahedron Lett., 2010, 51, 435–438 CrossRef CAS; (f) P. Viaud, V. Coeffard, C. Thobie-Gautier, I. Beaudet, N. Galland, J.-P. Quintard and E. Le Grognec, Org. Lett., 2012, 14, 942–945 CrossRef CAS PubMed.
- (a) H. Nozaki, T. Okada, R. Noyori and M. Kawanisi, Tetrahedron, 1966, 22, 2177–2180 CrossRef CAS; (b) B. Weiss, H. Dürr and H. J. Haas, Angew. Chem., Int. Ed. Engl., 1980, 19, 648–650 CrossRef CAS PubMed; (c) M. Z. A. Badr, M. M. Aly and A. M. Fahmy, J. Org. Chem., 1981, 46, 4784–4787 CrossRef CAS; (d) K. Pitchumani, M. C. Durai Manickam and C. Srinivasan, Tetrahedron Lett., 1991, 32, 2975–2978 CrossRef CAS; (e) K. K. Park, J. J. Lee and J. Ryu, Tetrahedron, 2003, 59, 7651–7659 CrossRef CAS; (f) X. Hong, J. M. Mejía-Oneto, S. France and A. Padwa, Tetrahedron Lett., 2006, 47, 2409–2412 CrossRef CAS; (g) E. Torti, S. Protti, D. Merli, D. Dondi and M. Fagnoni, Chem. – Eur. J., 2016, 22, 16998–17005 CrossRef CAS PubMed.
- A. Chakrabarti, G. K. Biswas and D. P. Chakraborty, Tetrahedron, 1989, 45, 5059–5064 CrossRef CAS.
- (a) C. V. Kumar, K. R. Gopidas, K. Bhattacharyya, P. K. Das and M. V. George, J. Org. Chem., 1986, 51, 1967–1972 CrossRef CAS; (b) L. A. Paquette, R. D. Dura and I. Modolo, J. Org. Chem., 2009, 74, 1982–1987 CrossRef CAS PubMed.
- Alternative methods to promote N- to C-sulfonyl transfer include, lithium anions: (a) D. Hellwinkel and R. Lenz, Chem. Ber., 1985, 118, 66–85 CrossRef CAS. Electron-beams: (b) J. Kato, Y. Maekawa and M. Yoshida, Chem. Lett., 2005, 34, 266–267 CrossRef CAS; (c) J. Kato, H. Kakehata, Y. Maekawa and T. Yamashita, Chem. Commun., 2006, 4498–4500 RSC. Lewis acids: (d) B. Prasad, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, G. S. Deora, P. Misra and M. Pal, Chem. Commun., 2012, 48, 10434–10436 RSC. Lewis bases: (e) Y. Zhu, W.-T. Lu, H.-C. Sun and Z.-P. Zhan, Org. Lett., 2013, 15, 4146–4149 CrossRef CAS PubMed. Thermolysis: (f) J. Wang, K.-I. Son and J. Xu, Monatsh. Chem., 2016, 147, 1637–1649 CrossRef CAS.
- H.-G. Henning, M. Amin and P. Wessig, J. Prakt. Chem./Chem.-Ztg., 1993, 335, 42–46 CrossRef CAS.
- C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 CrossRef CAS PubMed.
- The research data underpinning this research can be found at: DOI:10.17630/55734a8c-cebb-4a38-9175-c9288c68ec83 .
- R. Han, L. He, L. Liu, X. Xie and X. She, Chem. – Asian J., 2016, 11, 193–197 CrossRef CAS PubMed.
- Photoisomerisation was performed using a UV light box (365 nm) built in-house. See the ESI† for pictures of the reaction set-up.
- P.-P. Yeh, D. S. B. Daniels, C. Fallan, E. Gould, C. Simal, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2015, 13, 2177–2191 CAS.
- (a) A. T. Davies, J. E. Taylor, J. Douglas, C. J. Collett, L. C. Morrill, C. Fallan, A. M. Z. Slawin, G. Churchill and A. D. Smith, J. Org. Chem., 2013, 78, 9243–9257 CrossRef CAS PubMed; (b) J. R. Struble, J. Kaeobamrung and J. W. Bode, Org. Lett., 2008, 10, 957–960 CrossRef CAS PubMed.
- See the ESI† for details.
- C. Chatgilialoglu, B. C. Gilbert and R. O. C. Norman, J. Chem. Soc., Perkin Trans. 2, 1979, 770–775 RSC.
- L. Maclnnes and J. C. Walton, J. Chem. Soc., Chem. Commun., 1986, 1604–1605 RSC.
- B. C. Gilbert, B. Gill and M. D. Sexton, J. Chem. Soc., Chem. Commun., 1978, 78–79 RSC.
- C. Chatgilialoglu, B. C. Gilbert and R. O. C. Norman, J. Chem. Soc., Perkin Trans. 2, 1980, 1429–1436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and spectra. Research data available.13 See DOI: 10.1039/c7ob01699a |
| This journal is © The Royal Society of Chemistry 2017 |