
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven photooxidation of alcohols using surface-doped graphitic carbon nitride†
Wuyuan
Zhang
 *a,
Anna
Bariotaki
b,
Ioulia
Smonou
b and
Frank
Hollmann
*a
*a,
Anna
Bariotaki
b,
Ioulia
Smonou
b and
Frank
Hollmann
*a
aDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629HZ Delft, The Netherlands. E-mail: W.Zhang-1@tudelft.nl; F.Hollmann@tudelft.nl
bDepartment of Chemistry, University of Crete, Heraklion-Voutes 71003, Crete, Greece
First published on 3rd April 2017
Abstract
Carbon-nanodot-doped g-C3N4 is used as a photocatalyst to promote the aerobic oxidation of alcohols and oxyfunctionalisation of activated hydrocarbons. A critical E-factor analysis of the current reaction system reveals its limitations en route to environmentally acceptable oxidation procedures.
In recent years, graphitic carbon nitride (g-C3N4) has received substantial interest as a photocatalyst for metal-free, visible-light promoted reactions.1 It exhibits a graphite-like, layered structure wherein tris-triazine units are connected through C–N-bonds forming a two-dimensional layer. g-C3N4 can be synthesized via various methods such as pyrolysis of urea or other nitrogen-rich precursors or layer exfoliation of bulk materials.2
Pure g-C3N4, however, is a rather poor photocatalyst, mainly due to the fast recombination of photoexcited, charge-separated states. Therefore, one focus of research lies in the improvement of its photocatalytic properties by modulating the potential of g-C3N4's conducting- and valence bands.1 Particularly doping of g-C3N4 with other elements such as Y,3 Fe,4 Pt,5 Au/Pd6,7 K, Ag,8,9 C10 or carbon-nanodots11 and many more has proven to be an efficient handle to modulate its properties. Also, doping with carbon-nanodots appears promising to increase the quantum efficiency of photocatalytic processes.
Interestingly, g-C3N4 is mostly considered as a photocatalyst for (sun)-light driven water splitting, remediation of organic pollutants and catalytic CO2 reduction.1 Applications for preparative organic synthesis are comparably few. For example, Goettmann et al. reported g-C3N4 catalysed Friedel–Crafts acylation.12 More recently, photocatalytic acetalisation of aldehydes and ketones,13 and hydrazine-driven reductions of alkenes and alkynes were reported using g-C3N4.14,15 Selective oxidations especially of benzylic C–H-bonds have been reported using mesoporous g-C3N4 together with N–OH-cocatalysts,16–19 or using transition metal doped g-C3N4.14,20–22 Also the oxidative coupling of amines has been reported.23
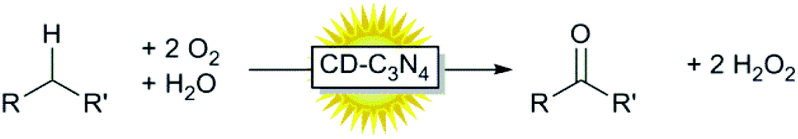
However, to the best of our knowledge, carbon-nanodot doped g-C3N4 has so far not been evaluated as a catalyst for photocatalytic oxidation reactions. Therefore, we set out to evaluate carbon-nanodot-doped g-C3N4 (CD-C3N4) as a visible-light-driven photocatalyst for the aerobic oxidation of alcohols (Scheme 1).
 | ||
| Scheme 1 Photocatalytic aerobic oxidation using carbon nanodot-doped g-C3N4 (CD-C3N4) as a photocatalyst. | ||
For the synthesis of g-C3N4 we followed the procedure by Tang and coworkers24 due to the more porous structure of the material and the resulting higher activity (due to the increased surface area). In short, calcination of urea at 600 °C for 4 h gave the desired mesoporous g-C3N4 as confirmed by TEM imaging and X-ray diffraction (Fig. S1 and S2†). Next, carbon nanodots were synthesized via thermal decomposition of sucrose.25 The latter were deposited on the g-C3N4 surface via thermal treatment of both materials.11 The XRD pattern of the such-obtained composite material did not change significantly compared to the starting material (g-C3N4) most probably due to the amorphous character of the carbon nanodots adsorbed. The UV/Vis spectrum showed the characteristic increase in absorption at wavelengths below 600 nm (Fig. S3†), and the BET measurement revealed a surface area of 105 m2 g−1 (Fig. S8†).
Having both catalysts at hand, we next compared their catalytic activity in the oxidation of benzyl alcohol to benzaldehyde as a model reaction (Fig. 1). Due to the volatility of benzaldehyde and the poor water solubility of the benzyl alcohol starting material we used a two-liquid phase approach employing benzyl alcohol as the second organic phase (phase ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 organic:aqueous).
:7 organic:aqueous).
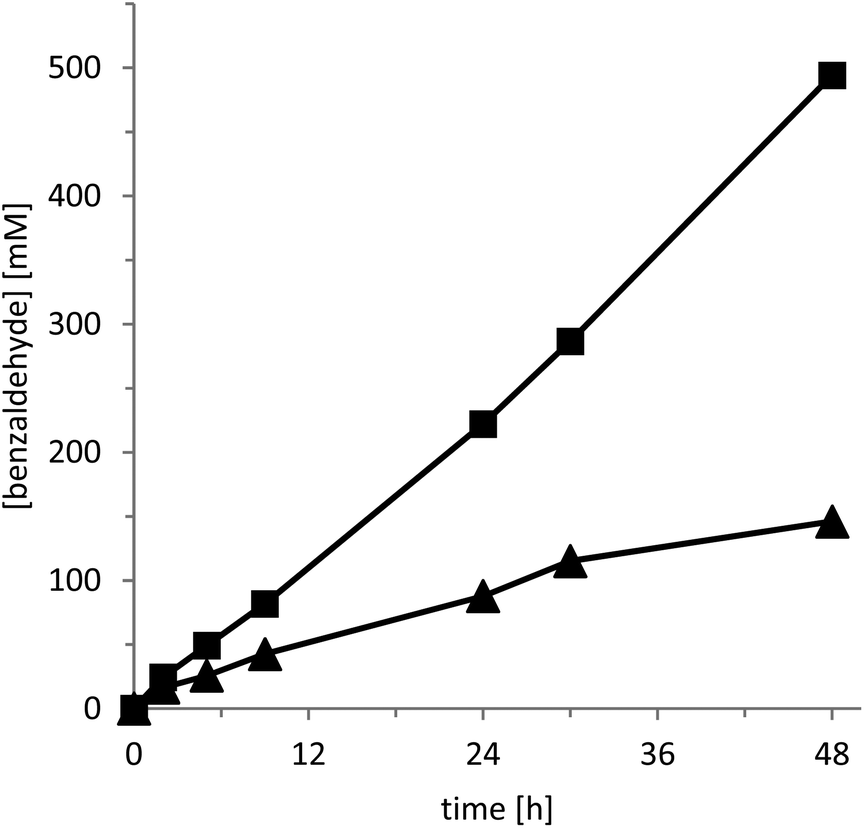 | ||
| Fig. 1 Photocatalytic oxidation of benzyl alcohol to benzaldehyde using g-C3N4 (▲) and CD-C3N4 (■) as photocatalysts. Conditions: 5 g L−1 of photocatalyst, two phase reaction: 700 μL of water + 300 μL of benzyl alcohol, 30 °C and oxygen atmosphere under visible light illumination using setup 1 (λ > 400 nm). | ||
As shown in Fig. 1, CD-C3N4 excelled over g-C3N4 both in terms of activity and robustness. Not only was the initial product formation rate roughly two times higher but also the long term-stability of the reaction: the reaction rate with g-C3N4 levelled off significantly after several hours whereas with CD-C3N4 linear product accumulation was observed for at least 48 h. Overall, with CD-C3N4 more than 500 mM of product accumulated corresponding to a product to catalyst ratio of more than 4:1 (g g−1), under the non-optimized conditions.
It is worth mentioning here, that in the absence of either the photocatalyst or a light source, no noticeable conversion of the starting material was observed. Also, hydrogen peroxide as a by-product was observable in trace amounts only throughout the experiments. This observation is in line with previous findings that CD-C3N4 is also an efficient H2O2 decomposition catalyst.11
The rate of the oxidation reaction exhibited a saturation-type dependency on both the catalyst concentration (Fig. 2) and the intensity of the light source applied (Fig. 3).
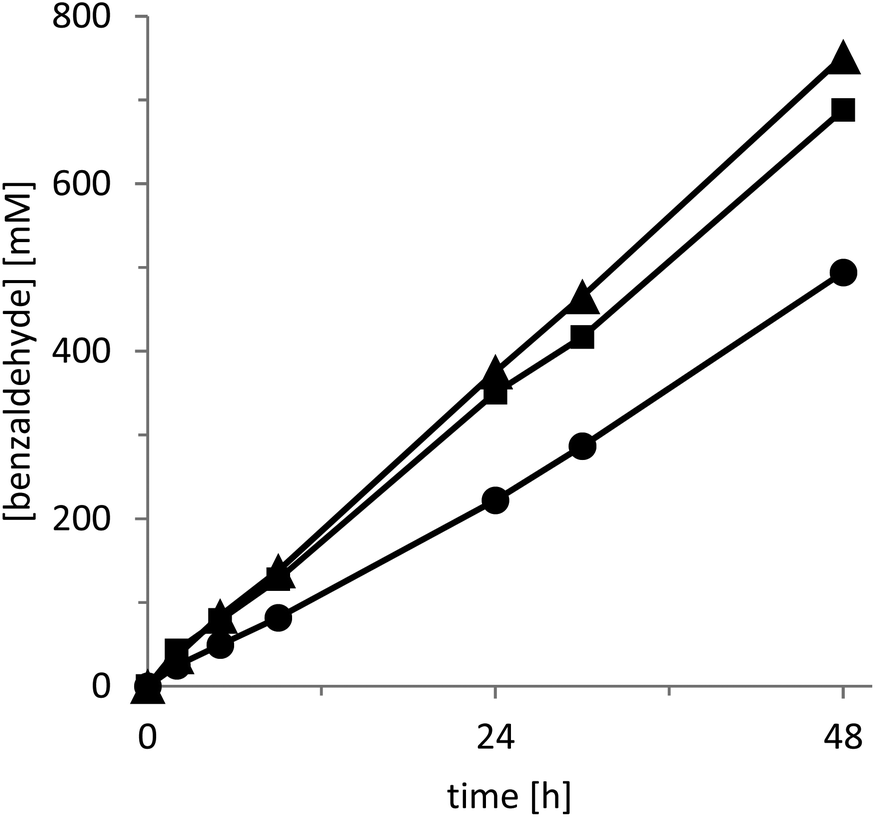 | ||
| Fig. 2 Influence of the catalyst loading on the rate of the photocatalytic oxidation of benzyl alcohol. [CD-C3N4] = 5(●), 10 (■), 25 (▲) g L−1. Conditions: Two phase reaction with 700 μL of water + 300 μL of benzyl alcohol, 30 °C and oxygen atmosphere under visible light illumination using setup 1 (λ > 400 nm). | ||
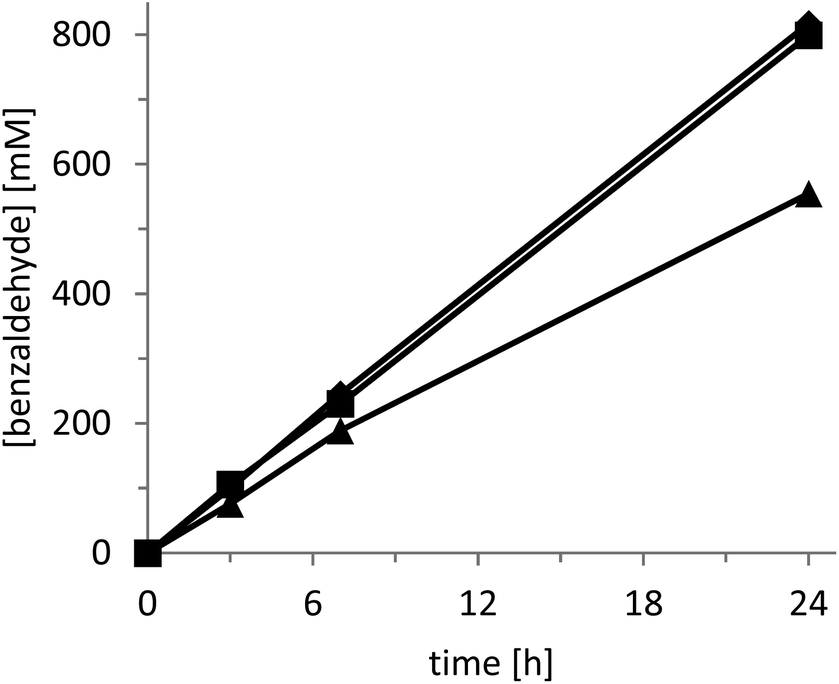 | ||
| Fig. 3 Influence of the light intensity on the rate of the photocatalytic oxidation of benzyl alcohol. Light intensity of 79 (▲), 197 (■), 341 (◆) W cm−2. Conditions: 5 g L−1 of photocatalyst, two phase reaction with 700 μL of water + 300 μL of benzyl alcohol, 30 °C and oxygen atmosphere under visible light illumination using setup 2 (λ > 400 nm). | ||
In the case of increasing catalyst concentrations, we suspect the decreasing transparency of the reaction mixture to account for this observation. The converging reaction rate at increasing light intensities may well be attributed to oxygen diffusion becoming overall rate-limiting. It should be mentioned here that for the latter experiments we utilised a specialized light-setup to control the light intensity (setup 2, Fig. S5†). Despite the much higher product formation rate attainable with this system (Fig. 3) we decided to perform the following experiment using a cheap white-light bulb in order to enable simple reproduction by others (setup 1, Fig. S4†). Nevertheless, the productivities shown in Fig. 3 (using a simple light source) of more than 0.2 gproduct g−1catalyst h−1 demonstrate the preparative potential of the photochemical alcohol oxidation system.
We investigated the recyclability of CD-C3N4 by performing benzyl alcohol oxidation reactions followed by filtration, washing and re-loading with reaction medium (Fig. S6†). As a result CD-C3N4 could be recycled at least 5 times. From linear regression of the initial rates, a catalyst deactivation of less than 4% per cycle was estimated.
Encouraged by these results we further explored the product scope of the reaction system (Table 1).
|
|
||
|---|---|---|
| Product | Producta [mM] | Rate [g g−1 h−1] |
| Reaction conditions: 5 g L−1 of photocatalyst, two phase reaction with 700 μL of water + 300 μL of alcohol, 30 °C and oxygen atmosphere under visible light illumination using setup 1 (λ > 400 nm) for 24 h.a Product concentration in the aqueous phase. | ||
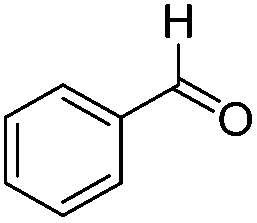
|
223 | 0.059 |
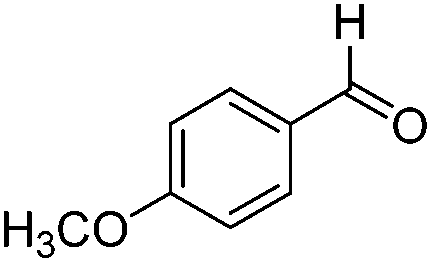
|
60.1 | 0.020 |

|
73.0 | 0.023 |

|
40.2 | 0.012 |

|
12.7/1.9a | 0.003/0.001a |
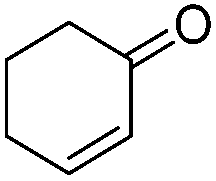
|
228.1/41a | 0.055/0.023a |

|
701.2 | 0.193 |
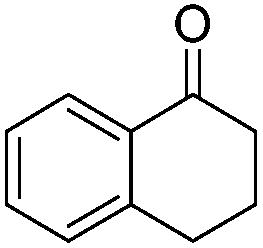
|
108.4 | 0.040 |
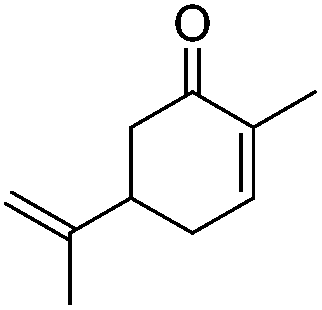
|
247.3 | 0.093 |
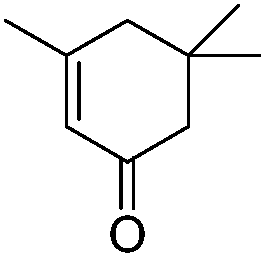
|
252.4 | 0.087 |

Especially allylic alcohols were converted at excellent rates and selectivities while benzylic alcohols were converted somewhat slower and non-activated alcohols such as cyclohexanol were rather sluggish substrates. This is roughly in-line with the general bond-dissociation energies of the C–H bonds oxidised. However, it also should be taken into account that the reactions reported in Table 1 have been obtained from two-liquid phase systems and that, depending on the partitioning coefficient of the starting material, the aqueous concentrations may vary very significantly thereby influencing the reaction kinetics.
The preparative applicability of the proposed photocatalytic oxidation was exemplarily demonstrated in the oxidation of carveol to carvone. Performing this reaction on a 6.8 mmol-scale (1.03 g) gave more than 95% conversion into the desired product (GC yield) and 0.773 g of isolated carvone (74.8% isolated yield) under non-optimised reaction- and DSP conditions.
An E-factor analysis26 of this reaction revealed the current limitations of this reaction setup from an environmental point-of-view (Table 2). The ‘classical’ E-factor (including the weighable compounds only) of the overall reaction is rather moderate (144) with solvents (used both for the reaction and for the extraction of the product) contributing over 95% to the total E-factor. Obviously, dichloromethane used in this reaction is not acceptable and will be substituted by more acceptable solvents in future studies.27 Also decreasing the contribution of water (e.g. by further increasing the concentration of the starting material) will be highly desirable. In fact, preliminary experiments using neat reagents (i.e. CD-C3N4 suspended in pure benzyl alcohol or cyclohexanol) showed an even faster product accumulation than in the biphasic system (Fig. S7†). Probably this is also to be attributed to a higher O2 solubility in these media than in aqueous systems. Another advantage of using neat reagents is that extraction can be omitted as physical methods to separate the product (e.g. distillation) are sufficient.
| Contributor | E-factor contribution [kg kg−1] |
|---|---|
| Reaction | |
| Water | 38.8 |
| CD-C3N4 | 0.26 |
| CO2 from a light source | 12.800 |
| DSP | |
| CH2Cl2 | 102.8 |
| MgSO4 | 1.9 |
However, the ‘hidden’ E-factor contributors demand more attention en route to an environmentally acceptable reaction system. Using setup 2 enabled us to quantify the power input (197 W for 90 h) and energy used for the illumination reaction (17.7 kWh). According to the European Energy Agency this corresponds to CO2 emission of approximately 9.9 kg CO228 and an E-factor contribution of 12.800 obviously ‘outshining’ the values discussed above. Of course the current setup has not been optimised for efficient utilisation of light and further geometric optimisation together with the increase of the reagent payload will certainly reduce this number to acceptable values. Also, provided the aspirational trend towards renewable energies continues, less CO2 emissions and thereby a reduced ‘CO2’-E-factor may be assumed. Furthermore, using sunlight will almost entirely eliminate this contribution.
Also, it should not be forgotten that the preparation of the photocatalyst (though exhibiting very low classical E-factors) is based on high-temperature calcination processes.
Overall, despite the potential of photochemical, aerobic oxidation we prefer to refrain from calling the current procedure green or environmentally benign.
Finally, we evaluated oxidation/oxyfunctionalisation of non-functionalized C–H bonds (Table 3). In general, the same trend in the reaction rate was observed here as well whereas the reaction rates were significantly lower than observed for the corresponding alcohols. This is in line with the higher C–H-bond dissociation energy of these non-functionalized C–H bonds. Furthermore, accumulation of the intermediate alcohol product did not occur (generally the alcohol product accounted for less than 25% of the final product) indicating that the initial C–H-bond oxidation is overall rate-limiting.
|
|
|||
|---|---|---|---|
| Product | Product [mM] | Selectivitya (%) | Rate [g g−1 h−1] |
| Reaction conditions: 5 g L−1 of photocatalyst, two phase reaction with 700 μL of water + 300 μL of alkane, 30 °C and oxygen atmosphere under visible light illumination using setup 2 (λ > 400 nm) for 24 h.a Selectivity = [aldehyde/ketone]/([alcohol] + [aldehyde/ketone])%.b Product concentration in the aqueous phase. | |||
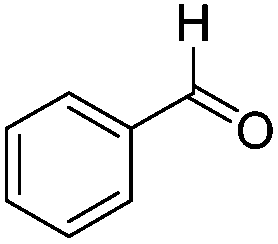
|
16.3 | 77 | 0.004 |

|
19.0 | 83.3 | 0.0065 |

|
3.0 | 86.2 | 0.001 |

|
12.6 | — | 0.0044 |

|
3.9 | — | 0.002 |
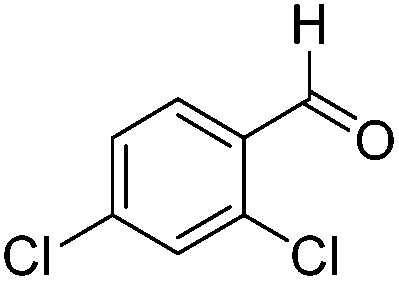
|
4.1 | — | 0.0018 |

|
7.6 | 76.9 | 10.0023 |

|
18.1 | 87.4 | 0.0067 |

|
30.3 + 37.7 | — | 0.008 + 0.010 |
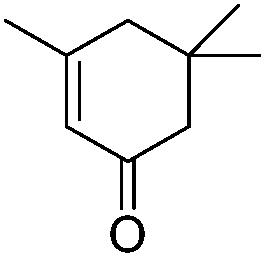
|
28.14 | 0.011 | |
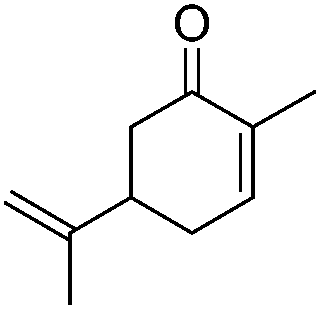
|
21.25 | 0.008 | |
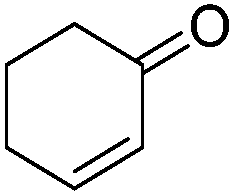
|
10.3/9.1b | 0.002/0.005b | |

|
3.0 | 0.0007 | |
Conclusions
With the current contribution we demonstrate that simple metal-free CD-C3N4 is a very suitable and recyclable photocatalyst for the oxidation of primary and secondary alcohols to the corresponding aldehydes and ketones. Furthermore, also extension of this concept to the corresponding alkanes appears feasible, albeit at reduced efficiencies.Ongoing mechanistic studies will reveal a more detailed understanding of the reaction and put the basis for optimised catalysts and reaction setups en route to truly practical catalysts.
The critical E-factor analysis of the current reaction setup will guide our further studies en route to truly environmentally acceptable oxidation processes.
Acknowledgements
Financial support by the European Research Council (ERC Consolidator Grant No. 648026) is gratefully acknowledged. The authors thank Ben Norder (Delft University of Technology) for XRD and Dr Wiel H. Evers (Delft University of Technology) for TEM measurements.Notes and references
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- Y. Wang, Y. Li, X. Bai, Q. Cai, C. Liu, Y. Zuo, S. Kang and L. Cui, Catal. Commun., 2016, 84, 179–182 CrossRef CAS.
- Q. Wang, A. Chen, X. Wang, J. Zhang, J. Yang and X. a. Li, J. Mol. Catal. A: Chem., 2016, 420, 159–166 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- C. Han, L. Wu, L. Ge, Y. Li and Z. Zhao, Carbon, 2015, 92, 31–40 CrossRef CAS.
- T. Xiong, W. Cen, Y. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS.
- Y. Fu, T. Huang, L. Zhang, J. Zhu and X. Wang, Nanoscale, 2015, 7, 13723–13733 RSC.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- Y. Wang, X. Bai, H. Qin, F. Wang, Y. Li, X. Li, S. Kang, Y. Zuo and L. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 17212–17219 CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- M. Abdullah Khan, I. F. Teixeira, M. M. J. Li, Y. Koito and S. C. E. Tsang, Chem. Commun., 2016, 52, 2772–2775 RSC.
- S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4, 1094–1098 CrossRef CAS.
- R. B. N. Baig, S. Verma, R. S. Varma and M. N. Nadagouda, ACS Sustainable Chem. Eng., 2016, 4, 1661–1664 CrossRef CAS.
- P. Zhang, J. Deng, J. Mao, H. Li and Y. Wang, Chin. J. Catal., 2015, 36, 1580–1586 CrossRef CAS.
- F. Z. Su, S. C. Mathew, G. Lipner, X. Z. Fu, M. Antonietti, S. Blechert and X. C. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- B. Long, Z. Ding and X. Wang, ChemSusChem, 2013, 6, 2074–2078 CrossRef CAS PubMed.
- P. Zhang, Y. Wang, J. Yao, C. Wang, C. Yan, M. Antonietti and H. Li, Adv. Synth. Catal., 2011, 353, 1447–1451 CrossRef CAS.
- H. Han, G. Ding, T. Wu, D. Yang, T. Jiang and B. Han, Molecules, 2015, 20, 12686 CrossRef CAS PubMed.
- Z. Ding, X. Chen, M. Antonietti and X. Wang, ChemSusChem, 2011, 4, 274–281 CAS.
- S. Verma, R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4, 2333–2336 CrossRef CAS.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- http://www.eea.europa.eu/ (accessed on 19.02.2017).
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedures and additional analytic material. See DOI: 10.1039/c7gc00539c |
| This journal is © The Royal Society of Chemistry 2017 |