DOI:
10.1039/C5DT04073F
(Paper)
Dalton Trans., 2016,
45, 2261-2272
3H-1,3-Azaphospholo[4,5-b]pyridines – novel heterocyclic P,N-bridging or hybrid ligands: synthesis and first d8-transition metal complexes†
Received
16th October 2015
, Accepted 24th November 2015
First published on 25th November 2015
Abstract
The first 3H-1,3-azaphospholo-pyridines 2a–c were synthesized as racemic mixtures in modest to medium yield by the reaction of N-(2-chloropyrid-3-yl)-trimethylacetimidoyl chloride 1 with RPLi2 (R = Ph, n-Bu, i-Bu), generated from RPH2 and BuLi in THF at −70 °C, and studied with respect to their suitability as ligands (L) in transition metal complexes. Reactions of 2a with group 6 metal(0) pentacarbonyls led to P-coordinated LM(CO)5 complexes 3a–5a (Cr, Mo, W) and the reaction of 2c with (norbornadiene)Mo(CO)4 surprisingly to 4c. [Rh(1,5-COD)Cl]2 and 2a,b, in metal/ligand ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, furnished LRh(1,5-COD)Cl complexes 6a,b with P-coordination, 6b accompanied by a minor contamination by the bis-coordinated L[Rh(COD)Cl]2 complex 7b. Reactions of 2a,b with [(allyl)PdCl]2 proceeded in THF with dismutation of N-coordinated (allyl)PdCl and formed with 2a a labile crude product [(2a){(allyl)PdCl}1.2(PdCl2)0.8]·C4H8O, with the composition close to L[Pd(allyl)Cl]PdCl2 THF (8a·THF), which converted during crystallization to 9a, whereas 2b directly formed the N,N′-PdCl2-bridged bis[LPd(allyl)chloride] complex 9b. Conversion of 2b with equimolar amounts of Pd(CH3CN)2Cl2 in THF, or Na2PdCl4 in methanol, gave rise to the dimeric P,N-bridging complex 10b. Crystal structure analyses of 6a (rac), 9b·2CDCl3 (meso), 10b·4.5THF and 10b·2D6-acetone (rac) provided detailed structural information. 10b, but more efficiently complexes formed in situ from 2a,b and Pd2(DBA)3 or Pd(OAc)2, catalysed the arylamination of 2-bromopyridine with 2,4,6-trimethylaniline.
:1, furnished LRh(1,5-COD)Cl complexes 6a,b with P-coordination, 6b accompanied by a minor contamination by the bis-coordinated L[Rh(COD)Cl]2 complex 7b. Reactions of 2a,b with [(allyl)PdCl]2 proceeded in THF with dismutation of N-coordinated (allyl)PdCl and formed with 2a a labile crude product [(2a){(allyl)PdCl}1.2(PdCl2)0.8]·C4H8O, with the composition close to L[Pd(allyl)Cl]PdCl2 THF (8a·THF), which converted during crystallization to 9a, whereas 2b directly formed the N,N′-PdCl2-bridged bis[LPd(allyl)chloride] complex 9b. Conversion of 2b with equimolar amounts of Pd(CH3CN)2Cl2 in THF, or Na2PdCl4 in methanol, gave rise to the dimeric P,N-bridging complex 10b. Crystal structure analyses of 6a (rac), 9b·2CDCl3 (meso), 10b·4.5THF and 10b·2D6-acetone (rac) provided detailed structural information. 10b, but more efficiently complexes formed in situ from 2a,b and Pd2(DBA)3 or Pd(OAc)2, catalysed the arylamination of 2-bromopyridine with 2,4,6-trimethylaniline.
Introduction
Various types of pyridylphosphines are known and have been applied as hybrid or hemilabile ligands in a large number of mono-, di- and polynuclear transition metal complexes and in a variety of transition-metal-catalysed organic transformations.1 Even four-membered P,N-chelate complexes can be formed with 2-PR2 derivatives2 if the rotation of this group around the C–P bond is not hindered by a substituent in 3-position of the pyridine ring. Pyrido[b]-anellated phospholes or phosphinines, or partially saturated derivatives thereof with the phosphino group fixed in a ring system, are, to the best of our knowledge, still unknown except for a single 4-aza-dibenzophosphole and a η1P-AuCl complex of the N-methylated ligand.3 In connection with our investigations of pyrido[b]-anellated 1H-1,3-azaphospholes,4 we were interested in establishing the consequences for the coordination behaviour if the phosphorus is fixed within a cyclic structure, fused with the pyridine ring. Since the dicoordinated phosphorus of the 1H-1,3-azaphospholes is a weak donor and has formed isolable transition metal complexes so far only with M0(CO)n fragments (M = Cr, Mo, W; n = 5, rarely 4 and 3)4,5 or, as shown for the related 1H-1,3-benzazaphospholes, with electron-rich d10 coinage metal compounds or HgCl2,6 the first pyrido[b]-anellated 3H-1,3-azaphospholes were synthesized and tested with respect to their reactions with some transition metal compounds and as ligands in a Pd-catalysed C–N cross-coupling reaction.
Results and discussion
Ligands and LM(CO)5 complexes
For the synthesis of the novel P,N-ligands we exploited the increased reactivity of 2-chloropyridines to electrophilic substitution compared to chlorobenzenes. The precursor N-(2-chloropyrid-3-yl)-imidoyl chloride 1, accessible by refluxing N-(2-chloropyrid-3-yl)-pivalamide with PCl5 in toluene, was found to react at −70 to 20 °C with RPLi2 species (R = Ph, n-Bu, i-Bu), freshly prepared from the corresponding primary phosphines and two equivalents of nBuLi in THF at −70 °C, to form the P-substituted 1,3-azaphospholo[4,5-b]pyridines 2a–c (Scheme 1). These compounds were isolated in low to moderate yields (21–59%) as oily racemic mixtures of 3R- and 3S-enantiomers. Compound 2a solidified on storage at room temperature. The non-aromatic phosphole-type heterocycles are more sensitive to air and to decomposition by acidic OH-groups on silica gel than their aromatic 1H-1,3-isomers,4b but vacuum distillation provided the compounds in sufficient purity for reactivity studies towards transition metal compounds.
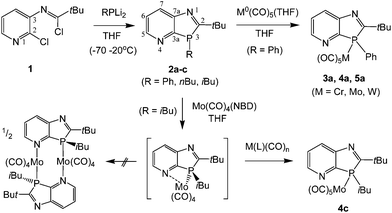 |
| | Scheme 1 Synthesis of 3H-1,3-azaphospholo[4,5-b]pyridines 2a–c and metal(0) pentacarbonyl complexes 3a–5a and 4c. | |
Treatment of 2a with M(CO)5(THF) (M = Cr, Mo, W) in THF, prepared in situ by UV-irradiation of the corresponding M(CO)6 solution, furnished at r.t. the carbonyl complexes 3a–5a as amorphous solids in high (Cr, W) to moderate (Mo) yield. The weak CO bands at 2068, 2076 and 2075 cm−1 (A1 mode, trans-CO stretching) of 3a–5a are similar or equal to the wave numbers published for the corresponding Ph3PM(CO)5 complexes (2065, 2074 and 2075 cm−1) whereas the strong CO bands of 4a and 5a at 1944 and 1936 cm−1 (in 3a superimposed by the absorption of Cr(CO)6 trace impurity) are bathochromically shifted relative to the E-bands (unsymmetric stretching of four coplanar CO ligands) of the respective Ph3PM(CO)5 complexes (1950 and 1942 cm−1) and comparable to those of (phenyldialkylphosphine)M(CO)5 complexes (1942 and 1937 cm−1).7 The one-bond 31P–183W coupling constant of 5a (1JPW = 225 Hz), known to correlate in a linear fashion with the CO(E-mode) stretching vibrations7a and the total electronegativity of the P-substituents,8 even adopts a value between that of PhBu2PW(CO)5 and Bu3PW(CO)5 (1JPW = 235 and 200 Hz).7a The higher donor strengths indicated by the E- than by the A1-mode bands might be attributable to interactions between the cis-CO ligands and the delocalized π-system of the phosphole-type ligand, which are lacking for the perpendicularly bound trans-CO ligand. The 31P coordination chemical shifts of 3a–5a, Δδ = 20.1, 36.8 and 60.0 ppm, are found at the lower end of the Δδ-ranges of the M(PR3)(CO)5 complexes of chromium, molybdenum and tungsten.7a The 13C NMR signals of C2 and C3a, in α-position to phosphorus, are slightly downfield-shifted by coordination of the metals at phosphorus, with a concomitant decrease of 1JPC of C2 from 35–38 in 2a–c to 3–6 Hz (or broad singlets) in 3a–5a but a strong increase of 1JPC of C3a from 17–20 to ca. 60–72 Hz. Coordination of M(CO)5 at nitrogen was not observed, even if excess or two equivalents of M(CO)5(THF) were used; this simply caused contamination by M(CO)6. The lack of additional N-coordination is revealed by the 13C NMR spectra, in which a second set of cis-M(CO)4 and trans-M(CO) signals, clearly recognizable in the spectra of the better soluble complexes 3a and 4c, is absent. The absence of N-coordination is further supported by minimal downfield coordination chemical shifts of C5 in 3a–5a and 4c (Δδ = 1.0–1.5 ppm), whereas in pyridine–W(CO)5 complexes the Δδ values are larger, amounting to 4.9–6.3 ppm.9 Attempts to prepare a mono- or dimeric N,P-bridging complex by the reaction of 2c (as ligand L) with an equimolar amount of Mo(NBD)(CO)4 (NBD = norbornadiene) failed. Instead, the Mo(κ1P-L)Mo(CO)5 complex 4c was isolated in fair yield (34%). It was unambiguously identified by its 13C and 31P NMR data (Δδ31P4c–2c = 40.8 ppm). Whether this is attributable to the higher stability of 4c compared to a dimeric [Mo(κ2P,N-L)(CO)4]2 complex, or to weak intramolecular interactions with the N-lone pair or π-density at the pyridine N-atom in a [Mo(L)(CO)4] monomer, promoting dismutation reactions, was not investigated during this study.
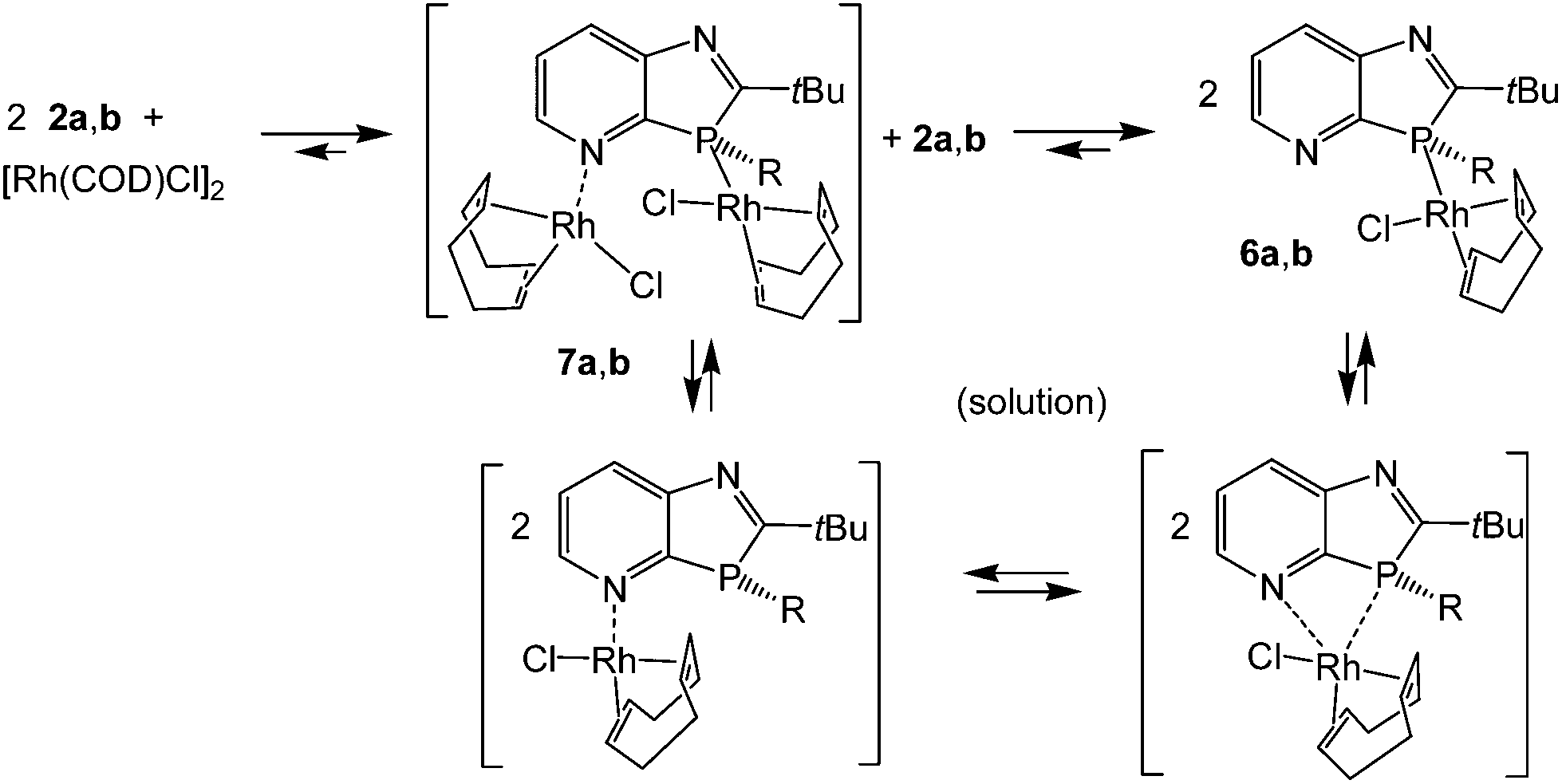
LRh(COD)Cl complexes
Reaction of [Rh(COD)Cl]2 in THF solution with 2a,b in a 1:2 molar ratio led to cleavage of the weak μ2-chloro bridging bond (Scheme 2). The higher coordination strength at rhodium(I) of the σ3P donor compared to the imino-N-atoms, known from early measurements of reaction enthalpies of [Rh(COD)Cl]2 with Ph3P and pyridine10 and from the P-coordination in (2-PyPPh2)Rh(COD)Cl,11 led to the expected coordination at phosphorus. The racemic complexes 6a,b were obtained in high yields as orange-yellow powders, and 6a also as single crystals, providing detailed structural information. The triclinic unit cell (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), contains one (S)- (Fig. 1) and (R)-enantiomer. The rhodium center is coordinated by the pyridoazaphosphole ligand through the phosphorus atom, and exhibits a distorted square-planar geometry. The Rh–P bond length is shorter than in the complexes (triarylphosphine)Rh(COD)Cl (2.297–2.3607(14) Å)12a and in (P-tert-butyl-2-trimethylsilyl-1,3-benzazaphospholine)RhCOD)Cl (2.3354(5) Å),13 thus implying a somewhat stronger coordination. The chloride ligand and the center of the C25–C26 double bond are arranged cis to phosphorus while the C21–C22 double bond is positioned cis to chloride. The angles of the C
), contains one (S)- (Fig. 1) and (R)-enantiomer. The rhodium center is coordinated by the pyridoazaphosphole ligand through the phosphorus atom, and exhibits a distorted square-planar geometry. The Rh–P bond length is shorter than in the complexes (triarylphosphine)Rh(COD)Cl (2.297–2.3607(14) Å)12a and in (P-tert-butyl-2-trimethylsilyl-1,3-benzazaphospholine)RhCOD)Cl (2.3354(5) Å),13 thus implying a somewhat stronger coordination. The chloride ligand and the center of the C25–C26 double bond are arranged cis to phosphorus while the C21–C22 double bond is positioned cis to chloride. The angles of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C centers to the Cl- and P-atoms in the trans-position are effectively linear (177.5, 176.7°) and the Rh-(CC) distance trans to phosphorus is 0.09 Å shorter than the Rh-(CC) trans to chloride, presumably because of back bonding (trans-influence). Because the C3A–P3–C2 angle in the five-membered ring (87.53(4)°) is smaller than the ideal tetrahedral angle, the opposite angle Rh–P3–C11 at the distorted tetrahedral phosphorus is widened (122.16(3)°). The two imino N-atoms are far away from rhodium, both intramolecularly and also in the packing (see the ESI†), where the 3R- and 3S-enantiomers each form homochiral chains with weak intermolecular contacts (H6⋯Cl 2.79 and H7⋯Cl 2.89 Å) between the chlorine atom of the Rh(COD)Cl group and H6 and H7 of the pyridine ring of the neighbouring molecule. The chains are linked by inversion via the contact H19B⋯Cl, 2.85 Å. The intramolecular contact H20A⋯Cl, 2.74 Å, may also be a stabilizing factor. Both these latter contacts involve tert-butyl hydrogens.
C centers to the Cl- and P-atoms in the trans-position are effectively linear (177.5, 176.7°) and the Rh-(CC) distance trans to phosphorus is 0.09 Å shorter than the Rh-(CC) trans to chloride, presumably because of back bonding (trans-influence). Because the C3A–P3–C2 angle in the five-membered ring (87.53(4)°) is smaller than the ideal tetrahedral angle, the opposite angle Rh–P3–C11 at the distorted tetrahedral phosphorus is widened (122.16(3)°). The two imino N-atoms are far away from rhodium, both intramolecularly and also in the packing (see the ESI†), where the 3R- and 3S-enantiomers each form homochiral chains with weak intermolecular contacts (H6⋯Cl 2.79 and H7⋯Cl 2.89 Å) between the chlorine atom of the Rh(COD)Cl group and H6 and H7 of the pyridine ring of the neighbouring molecule. The chains are linked by inversion via the contact H19B⋯Cl, 2.85 Å. The intramolecular contact H20A⋯Cl, 2.74 Å, may also be a stabilizing factor. Both these latter contacts involve tert-butyl hydrogens.
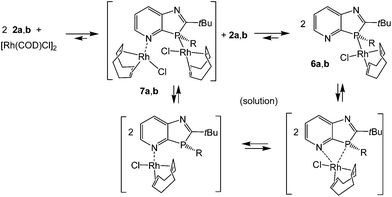 |
| | Scheme 2 Formation of (1,3-azaphospholo[4,5-b]pyridine)Rh(COD)Cl complexes 6a,b; possible N,P-bis-Rh(COD)Cl intermediates 7a,b and equilibrium species in solution. | |
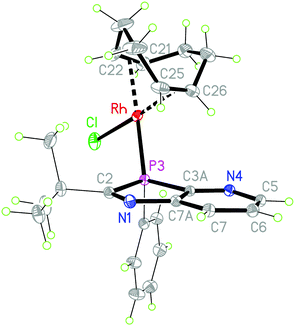 |
| | Fig. 1 Molecular structure of 6a the (3S)-enantiomer is depicted; ellipsoids with 50% probability. Selected bond lengths (Å) and angles (°): Rh–P3 2.2855(3), Rh–Cl 2.3547(3), Rh–C25 2.1274(11), Rh–C26 2.1364(11), Rh–C21 2.2085(11), Rh–C22 2.2176(11), Rh–(C21C22) 2.103, Rh–(C25C26) 2.013, C2–P3 1.8733(10), P3–C3A 1.8144(10), N1–C2 1.2902(13), N1–C7A 1.4159(12); P3–Rh–Cl 90.624(10), C3A–P3–C2 87.53(4), C11–P3–Rh 122.16(3), N1–C2–P3 113.53(7). | |
The solution NMR spectra in CDCl3 confirmed the formation of complexes 6a and 6b in terms of the slightly reduced downfield shifts of 13C2 and 13C3a, the characteristic changes of the 1JPC coupling constants as mentioned above for 3a–5a, and, in particular, by the downfield shifts of the phosphorus resonance (Δδ ≈ 29 ppm). The 31P doublets of 6a and 6b appear at δ = 27.2 and 28.2 ppm, respectively, and thus lie within the signal range of (2-PyPPh2)Rh(COD)Cl and (triarylphosphine)Rh(COD)Cl complexes.11,12 The one-bond 31P–103Rh-coupling constants, 1JPRh ≈ 143 (6a) and 141.7 Hz (6b), indicative of the sum of electronegativities of the ligands at Rh(I),13 were smaller by ca. 10 Hz than in the aforementioned complexes11,12 and also smaller than in the aryldialkylphosphine-ligated (P-tert-butyl-1,3-benzazaphospholine)Rh(COD)Cl (1JPRh = 150.7–151.9 Hz)14 and (2-PyPiPr2)Rh(COD)Cl complexes (1JPRh = 144.7 Hz).15 For the solution of 6a in CDCl3, besides the strong phosphorus doublet, a small but very broad singlet was observed at δ31P = −1.1 ppm (integral ratios 87–83 to 13–17%), close to that of the free ligand (δ31P = −1.9 ppm), which may be caused by small equilibrium amounts of N-coordinated species and/or 2a, formed by ligand dissociation in solution. Such processes are known for the sterically hindered (o-Tol3P)Rh(COD)Cl12a and double exchange reactions of (Ph3P)Rh(COD)Cl with (p-MeOC6H4)3P-ligated LRh(CO)(acac).16 The analogous dynamic behavior of 6a is further confirmed by rather broad aryl signals in the proton and 13C NMR spectra and very broad signals for the COD protons and 13C nuclei. An indication of involvement of pyridine nitrogen may be provided by the lack of equally intense CH proton signals for 6a in the range 3.1–3.6 and 5.2–5.7 ppm, typical of (triarylphosphine)Rh(COD)Cl complexes,11,12 and by the occurrence of a very broad signal at δ = 4.24 ppm, close to the averaged CH proton signal of (2-PyPPh2)Rh(COD)Cl (δ = 4.42 ppm).11,12 An averaged 13C signal for CH at δ = 85.8 ppm, close to that in (2-PyPPh2)Rh(COD)Cl at δ = 88 ppm and absent in (triarylphosphine)Rh(COD)Cl complexes, constitutes additional evidence in this direction. Thus, it can be assumed that the interactions with the pyridine N-atom lead in analogy to (2-PyPPh2)Rh(COD)Cl to trigonal bipyramidal intermediates that allow rapid pseudorotation with the interchange of axial and equatorial positions and thus of the CH nuclei in the trans- and cis-positions to phosphorus or chloride.11 Coordination at the pyridine nitrogen within the ring plane and at the phosphorus of 6a outside the ring plane disfavors intramolecular N- and P-coordination compared to pyridylphosphines, where the PR2 group may rotate around the P–C2-bond to a suitable position and even allow P,N-chelate formation. Weak interactions with π-electrons of pyridine nitrogen, however, and thus an intramolecular Rh(COD)Cl migration, may not a priori be excluded, though an intermolecular mechanism of two molecules 6avia an intermediate P,N-bis-Rh(COD)Cl complex 7a might be a more suitable pathway. Small equilibrium amounts of 2a might also cause similar (or further) line broadening by rapid ligand exchange reactions, long known for (Ph3P)Rh(COD)Cl in the presence of Ph3P.12 Whether the imino group of the five-membered ring is also involved in solution reactions is not clear. Since the behavior of 6a is certainly even more complicated than that of triaryl- or pyridylphosphinorhodium complexes, a closer investigation of the dynamic behavior is a case for specialists in this field.
The solution NMR spectra of the P-butyl-1,3-azaphospholo[4,5-b]pyridine)Rh(COD)Cl complex 6b differ from those of 6a. The proton and 13C NMR signals of the olefinic CH nuclei trans and cis to P or Cl, respectively, are not averaged out but appear in a similar region as in (Ph3P)Rh(COD)Cl11 or (2-PyPiPr2)Rh(COD)Cl) at low temperatures (≤−30 °C).15 This, together with the only marginal changes in VT 31P NMR spectra of 6b in the temperature range of −56 to +40 °C, means that the ligand exchange reactions are considerably slower than in 6a and in (2-pyridyldialkylphosphine)Rh(COD)Cl complexes. This may be attributed to the higher complex stability of P-alkyl- compared to P-aryl-azaphospholopyridine–Rh(I) complexes and to the rigidity of the bicyclic azaphospholopyridine ligands. A small sharp 31P doublet signal at δ = 17.2 ppm (1JPRh = 151.1 Hz), ca. 5% by 31P-integration, may be attributed, in accordance with the CHN values and a cationic fragment peak for [7b-Cl]+ in the high resolution mass spectrum, to a small amount of contamination by 7b. N-Coordinated (pyridyl)Rh(COD)Cl complexes are known17 and thus, in addition to phosphorus of 2b, the N-atom may also undergo coordination to the transition metal.
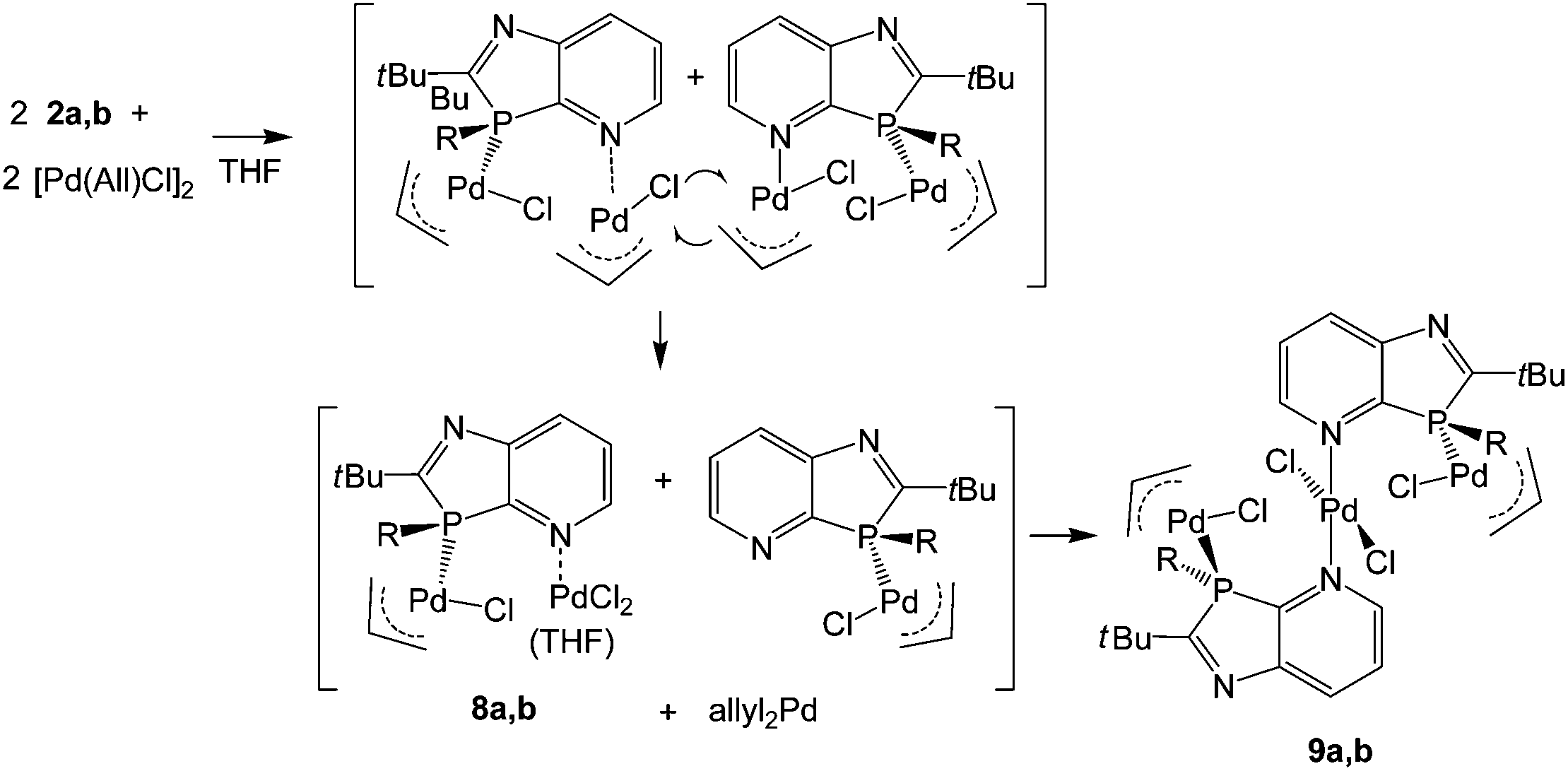
LPd(allyl)Cl and (LPdCl2)2 complexes
With the intention of ensuring complete conversion of the ligands to complexes with a 1:1 ligand/metal ratio, reactions of 2a and 2b were performed with [η3-(allyl)PdCl]2 in a molar ratio 2:1.5. This led in THF to orange-yellow products, soluble in THF and sparingly soluble in hexane. CHN analyses of the yellow powder, obtained from 2a after the extraction of hexane-soluble components, were roughly consistent with a composition [(2a){(allyl)PdCl}1.2(PdCl2)0.8]·C4H8O (close to 8a·C4H8O); those of the product formed with 2b corresponded to 9b with a composition [(2b)2{(allyl)PdCl}2(PdCl2)]. Crystals were first obtained from a solution of the crude product formed from 2a in THF by overlayering with hexane. These were studied by XRD and identified as complex 9a, crystallised with THF, showing that 2a reacts more slowly, but analogously to 2b to form complexes of type 9. Severe disorder of the allyl group and unexpected peaks, possibly caused by twinning, prevented satisfactory refinement. However, single crystals were then grown by slow concentration of an NMR sample of crude 9b in CDCl3, allowing a full and satisfactory refinement of the XRD data and unambiguous identification as 9b·2CDCl3 (Fig. 2).
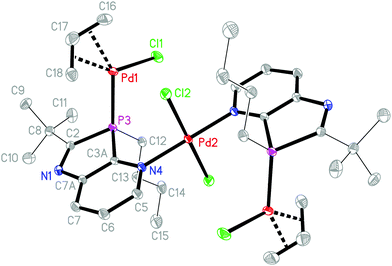 |
| | Fig. 2 Crystal structure of 9b·2CDCl3 (hydrogens and solvent omitted for clarity). Ellipsoids represent 50% probability levels. Selected bond lengths (Å) and angles (°): Pd1–P3 2.2685(5), Pd1–Cl1 2.3542(5), Pd1–C18 2.125(2), Pd1–C17 2.157(2), Pd1–C16 2.197(2), C16–C17 1.384(4), C17–C18 1.396(4), Pd2–N4 2.0121(15), Pd2–Cl2 2.2976(5), C2–P3 1.8506(19), P3–C3A 1.8027(18), C2–N1 1.288(2), N1–C7A 1.417(2) Å; C16–Pd1–Cl1 100.35(7), C18–Pd1–Cl1 167.66(7), C16–Pd1–P3 165.00(8), C18–Pd1–P3 97.89(7)°. | |
The crystal structure analysis of 9b·2CDCl3 revealed an inversion-symmetric molecule in which a central PdCl2 unit connects two (η3-allyl)(κ1P-azaphospholopyridine)palladium chloride fragments by κ1N-coordination within the ring plane; as a consequence of symmetry, the mutual orientation of the two pyridoazaphosphole rings is trans with anti-orientation, and the configuration at phosphorus is R in one and S is in the other ring. The planes of the pyridine ring and the almost perfectly square planar N2Pd2Cl2 moiety are arranged perpendicularly (interplanar angle 89.9°). The atoms Pd1, P3, Cl1, C16 and C17 are coplanar (mean deviation 0.02 Å); the angles P3–Pd1–C16 and Cl1–Pd1–C18 are roughly linear (165.00(8), 167.66(7)°). Similarly to the Rh(COD) complex 6a, the Pd1–C16 bond length trans to phosphorus is shorter by 0.072 Å than the Pd1–C18 bond length trans to chloride. Despite this back-bonding effect, C17–C18 is still shorter than C16–C17, which suggests a slightly increased weight of a resonance structure with the C–Pd σ-bond trans to P and the (CC)–Pd π-bond trans to the Cl atom. The P3–Pd1–Cl1 angle (94.182(19)°) is close to the ideal 90°, but the bite angle C18–Pd1–C16 (67.44(10)°) of the allyl group is substantially smaller and causes significant deviations of C16 and C18 from true cis- and trans-positions to Cl and P. The phosphorus atom is a distorted tetrahedral with a narrow C2–P3–C3A angle (87.36(8)°), enforced by the five-membered azaphosphole ring, and a concomitantly increased C12–P3–Pd1 angle (121.33(6)°). The palladium Pd1 is coordinated 2.00 Å above the mean ligand plane (average deviation 0.03 Å), and the n-butyl group is directed towards the opposite side, with C12 lying 1.56 Å out of the plane. In the crystal packing the molecules are connected to form ribbons parallel to [110] by weak interactions of 2.89, 2.76 Å between chlorine Cl1 of the Pd(allyl)Cl groups and H6 and H7 of the pyridine rings of the neighbouring molecules, in a fashion similar to that observed for 6a (an intramolecular contact H5⋯Cl1, 2.71 Å across the inversion centre, is also observed). In addition, short N⋯D and Cl⋯Cl contacts of 2.53 and 3.49 Å respectively are observed between CDCl3 and the molecules of 9b (Fig. 3).
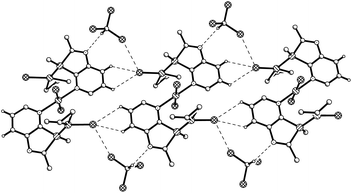 |
| | Fig. 3 Packing of 9b·2CDCl3 in the crystal (n- and tert-butyl groups indicated by their α-C atoms). Dashed lines represent weak C–H⋯Cl and C–D⋯N hydrogen bonds or Cl⋯Cl contacts. | |
The surprising coordination of PdCl2 as well as (allyl)PdCl may be explained by primary formation of P- and N-bis(allylPdCl) azaphospholopyridine complexes, formed by the association of the P,N-ligand with the weakly chloro-bridged (allylPdCl) dimer and subsequent dismutation of the less stable N-coordinated (allyl)PdCl fragment. Since N-pyridyl-Pd(allyl)Cl complexes18 have not been reported to convert to N-pyridyl-PdCl2 complexes, activation of this process is assumed to be connected with the proximity of the P-coordinated (allyl)PdCl group. A possible scenario might be a chloro-bridging interaction, leading to a more labile pentacoordinated Pd(allyl)Cl group that could undergo allyl-chloride exchange reaction with a second molecule. This would lead to 8a, which together with THF and a small residual amount of the primary product would be consistent with the composition of the crude product formed from 2a and, after combination with the intermediate LRh(allyl)Cl, with the formation of 9a,b (Scheme 3). The atomic balance of the chloro atoms in 9a,b shows that the reaction actually proceeded in a ligand/metal ratio of 1:2. The assumption of an allyl-Cl exchange implies the formation of (allyl)2Pd, removed by washing the products with hexanes on work-up. A small shoulder (δ = 58 ppm) at the upfield end of the allyl 13CH2 resonance (δ = 62.8 ppm) in the 13C solution NMR spectrum of [(2a){(allyl)PdCl}1.2(PdCl2)0.8]·C4H8O, close to the upfield CH2 signal of diallylpalladium (δ = 54.6 in D8-toluene19), might be a hint at this species, formed by dismutation of the residual (2a)(allylPdCl)2 complex of the crude product. The corresponding CH2-signals of N- and P-(allyl)PdCl compounds absorb at lower field (δ = 61–63 ppm)18,20 and are not responsible for this shoulder.
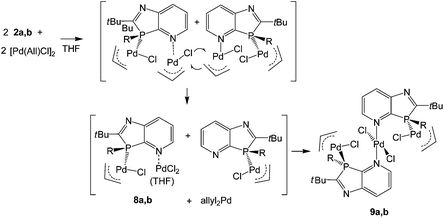 |
| | Scheme 3 Reaction of 2a,b with [η3-(allyl)PdCl]2via assumed intermediates 8a,b to 9a,b. | |
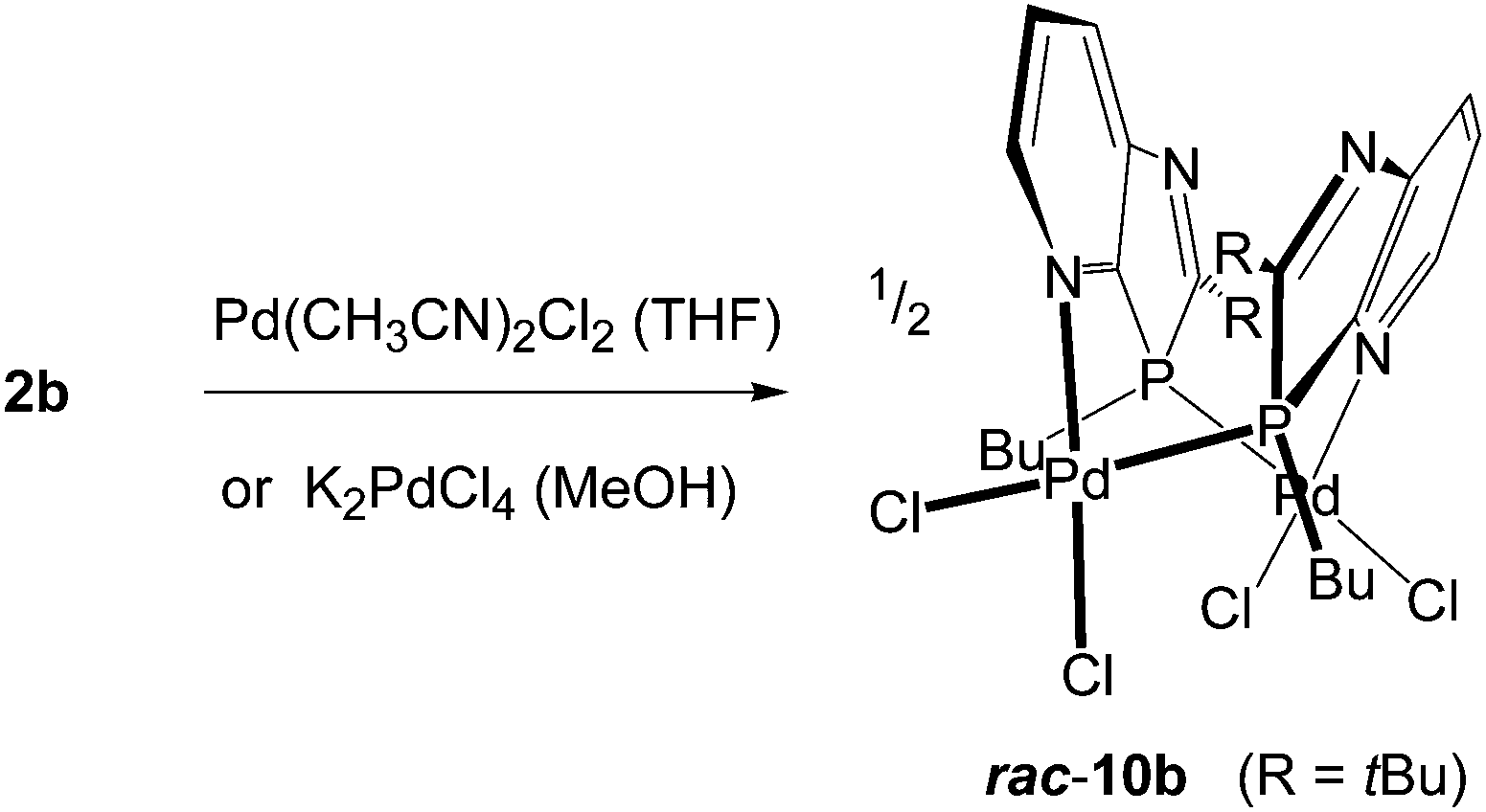
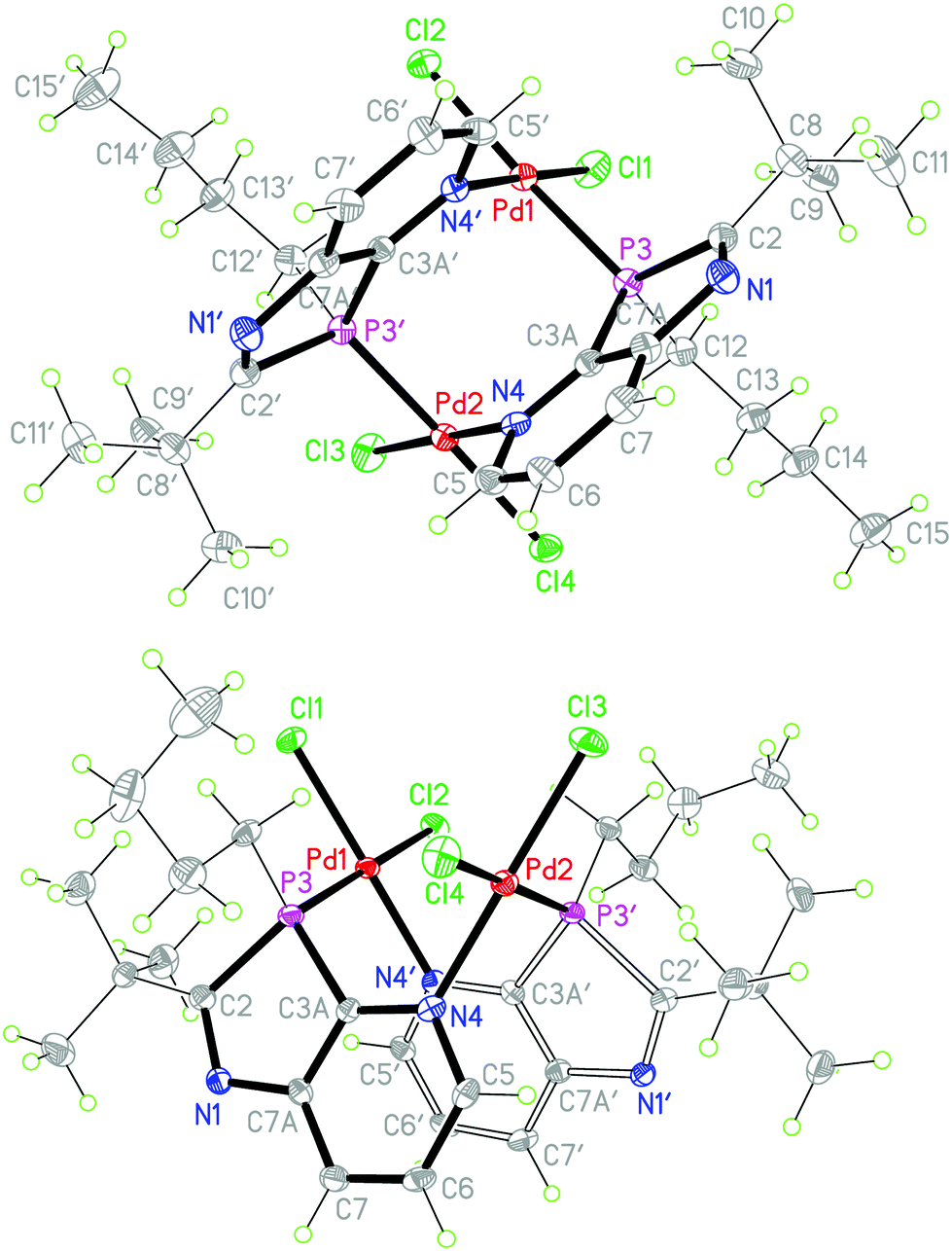
To obtain information on the nature of palladium complexes containing only PdCl2 units and azaphospholo[4,5-b]pyridines, ligand 2b was treated with bis(acetonitrile)palladium dichloride in THF and in a second experiment with potassium tetrachloropalladate in methanol, each in a 1:1 molar ratio (Scheme 4). The rigidity of the aromatic pyridine ring, coordinating the metal at nitrogen within the ring plane, together with the rigid position of the tetrahedrally coordinated phosphorus in the five-membered ring with the metal outside the pyridine ring plane are inappropriate for the formation of stable mononuclear four-membered N,P-chelate complexes. At best, labile intermediates with some stabilization by π-interactions with the pyridine N-atom outside the ring plane are conceivable. These should either dimerize or polymerize to more stable products with coordination at nitrogen within the ring plane. The orange-yellow and yellow complexes, formed from the two different precursors, differed in their solubility, the first being well soluble in THF or CDCl3, the second rather sparingly soluble in these solvents and better soluble only in more polar solvents such as methanol or acetone. The NMR spectra indicated complex formation by downfield shifts of the phosphorus resonance, Δδ = 33.5 and 29.2 ppm, respectively, slightly (Cq of 2-tBu) and clearly (C7a) increased 2JPC coupling constants, typically for complexes of 2b, and a somewhat downfield shifted 2-tBu proton singlet, while the majority of the 13C and proton signals is scarcely changed by the coordination. However, XRD analyses of crystals of both products, grown by slow diffusion of hexane into the complex solution in THF or by slow concentration of the D6-acetone solution, respectively, revealed in both cases a dimeric structure consisting of [cis-Cl2Pd(L-P,N′)]2 moieties with Pd(II) bound at P and pyridine-N′ or P′ and pyridine-N, one from each azaphospholopyridine bridging ligand. Both structures exhibit the two ligands on the same side (syn-form) of the connecting Cl–Pd–P axes with opposite orientation of the five- and six-membered rings, so that they are different solvates of the same dinuclear complex 10b. Neither complex displays imposed crystallographic symmetry, but the THF solvate has approximately twofold symmetry (r.m.s. deviation 0.04 Å), while the acetone solvate does not, because of different orientations of the substituents. The ligand ring systems remain planar (mean deviations 0.01–0.02 Å) and the interplanar angles are 32° and 27° respectively for the THF and acetone solvates. The square planar configuration around Pd(II), with cis-orientation of the two chloride ligands, causes an almost perpendicular orientation of the Cl–Pd–P axis towards the plane of the N-coordinated pyridine ring (angles between Cl⋯P vector and normal to the ligand plane 87–89°), while the coordination of the distorted tetrahedral phosphorus outside the pyridine-ring plane of the respective ligand enforces a considerable mutual twisting of the N–Pd–Cl axes of the two Cl2PdNP fragments in the dimer (angle between vectors N4′⋯Cl1 and N4⋯Cl3: 62, 61°) and a “twisted boat structure” for the central eight-membered ring. The mutual orientation of the two azaphospholopyridine ligands, together with the local twofold symmetry, implies the same configuration for the two P atoms in the dimers and leads for both 10b·4.5THF and 10b·2D6-acetone (Fig. 4), to pairs of (3S,3′S)- and (3R,3′R)-diastereoisomers, co-crystallising as racemic products. Different packing in the crystals of 10b·4.5THF and 10b·2D6-acetone must thus be caused by the different solvent molecules; the large number of H⋯Cl and H⋯O contacts however makes an exact analysis difficult. The Pd–P, Pd–N and Pd–Cl and intraligand bond lengths and angles exhibit no special features and are similar in both complexes and also in (R,S)-9b·2CDCl3. The somewhat longer Pd–Cl bond trans to phosphorus compared to that trans to nitrogen reflects the known stronger trans influence of P versus N donors.
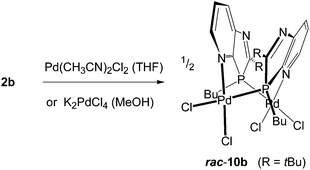 |
| | Scheme 4 Formation of dimeric (1,3-azaphospholopyridine-κ1P, κ1N′)PdCl2 complexes 10b with schematic presentation of its “twisted boat structure”. | |
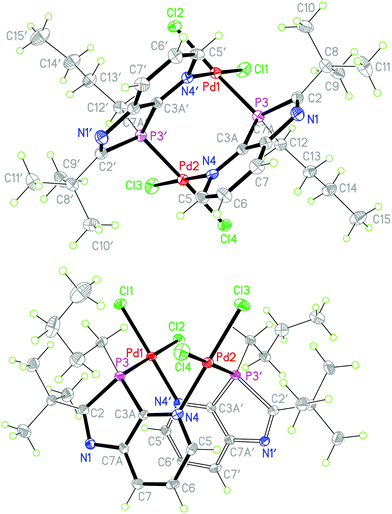 |
| | Fig. 4 (a) View of the (3S,3′S)-enantiomers in the crystal of 10b·4.5THF (above) and (b) in the crystal of 10b·2(D6-acetone) (below, alternative view direction); solvents omitted for clarity, ellipsoids with 50% probability. Selected bond lengths (Å) and angles (°) for 10b·4.5THF: Pd1–P3 2.2397(9), Pd1–N4′ 2.048(3), Pd1–Cl1 2.2725(9), Pd1–Cl2 2.3396(8), C2–P3 1.865(3), P3–C3A 1.808(3), P3–C12 1.821(3); N4′–Pd1–Cl1 178.70(8), N4′–Pd1–P3 90.62(8), N4′–Pd1–Cl2 89.47(8), P3–Pd1–Cl1 88.39(3), P3–Pd1–Cl2 176.55(3), Cl1–Pd1–Cl2 91.57(3), C2–P3–C3A 87.61(15), C12–P3–Pd1 122.68(11), N1–C2–P3 113.7(2); bond lengths and angles of 10b·2(D6-acetone) are similar (see deposited material for details). | |
Catalytic tests
Pyridylphosphines1 have found wide applications in transition-metal-catalysed organic syntheses. The hemilabile properties, known also from other P,N ligands,21 might be advantageous by enabling temporary substrate binding on replacement of the more labile donor and catalyst stabilization after reductive elimination by re-occupation of an empty coordination site. For the first test of the suitability of azaphospholopyridine complexes as catalysts the known N-arylation of 2,4,6-trimethylaniline (mesitylamine) with 2-bromopyridine22,23 was chosen, providing 2-mesitylaminopyridine (11) as a potential precursor for the synthesis of N-mesityl-substituted 1,3-azaphospholo[5,4-b]pyridine.4b C–N cross-couplings of this type, also known as Buchwald–Hartwig reactions,24 are important tools for the production of arylamine fine chemicals.25 The coupling according to Scheme 5 was carried out using a similar protocol to that used with the formerly applied catalysts,22,23 to allow a comparison of the catalytic performance. The reactants were heated in toluene in the presence of two equivalents of KOtBu with 5 mol% 10b or precatalysts, prepared in situ from 2a or 2b and Pd(OAc)2 or Pd2DBA3 (DBA – dibenzoylacetone) in a 1:1 or 2:1 molar ratio. With 10b the yield of 11 was rather low (Table 1, entry 1, 26%), comparable to that obtained with catalysts for comparison generated in situ from Pd2DBA3 and triphenyl- or tri-tolylphosphines (1:2 molar ratio), whereas the catalysts formed from 2a or 2b and Pd(OAc)2 or Pd2DBA3 gave higher yields, particularly for a 1:1 molar ratio of metal to ligand (Table 1, entries 2, 4 and 6). The maximum yield of 57% (after isolation) is not high, but is higher than the reported results with DPPE/Pd2DBA3 (51% yield for 2 mol% catalyst and heating with 2 equiv. NaOtBu overnight)22 and 2,3-bis(diphenyl)quinoxaline/Pd(OAc)2 or Pd2DBA3 (36–43% yield with 2–5 mol% catalyst, conditions as here).23
 |
| | Scheme 5 The Pd-catalysed C–N coupling reaction of 2-bromopyridine with mesitylamine. | |
Table 1 Screening of 10b and in situ generated azaphospholopyridine–Pd catalysts in the arylamination of 2-bromopyridinea
| Entry |
Pd compound |
Ligand |
Metal/ligand ratio |
Mol% ligand |
Yield (%) |
|
Reaction conditions: Heating of 2-bromopyridine, mesitylamine and KOtBu with the given amount of 10b or the given ligand and Pd-compound in toluene for 24 h at 100 °C; yields refer to isolated product after separation by column chromatography.
|
| 1 |
10b
|
— |
1:1 |
5 |
26 |
| 2 |
Pd2DBA3 |
2a
|
1:1 |
5 |
57 |
| 3 |
Pd2DBA3 |
2a
|
1:2 |
10 |
43 |
| 4 |
Pd2DBA3 |
2b
|
1:1 |
5 |
54 |
| 5 |
Pd2DBA3 |
2b
|
1:2 |
10 |
42 |
| 6 |
Pd(OAc)2 |
2b
|
1:1 |
5 |
51 |
| 7 |
Pd(OAc)2 |
2b
|
1:2 |
10 |
33 |
| 8 |
Pd2DBA3 |
Ph3P |
1:2 |
10 |
21 |
| 9 |
Pd2DBA3 |
(p-Tol)3P |
1:2 |
10 |
23 |
| 10 |
Pd2DBA3 |
(o-Tol)3P |
1:2 |
10 |
18 |
Conclusions and outlook
The first synthesis of 3H-1,3-azaphospholo-pyridines, which except for a single 4-aza-dibenzophosphole seem also to be the first representative of pyrido[b]-anellated five- and six-membered heterocycles containing one phosphorus atom, paves the way for investigations of transition metal complexes of these novel P,N ligands, closely related to 2-pyridylphosphines, but with restricted flexibility and lacking free rotation around the C–P bond at the pyridine ring. The first examples of such complexes, hints at dynamic properties and applicability to transition-metal-catalysed reactions were presented. The asymmetry at phosphorus and the potential for a wide range of different P- and/or N-coordinated complexes, including bridging or cluster compounds as known for pyridyl phosphine ligands, open a wide field for coordination chemical studies with the novel ligands. If the additional imino donor of the five-membered ring is also involved in transition metal coordination, even a three-dimensional metal–ligand network might become accessible. Last but not least, electronic delocalization within the effectively planar imino-conjugated pyridine π-system might lead to transition metal complexes with interesting redox and/or photophysical properties.
Experimental
All manipulations were carried out under an atmosphere of dry argon or nitrogen using Schlenk techniques. Solvents were dried over sodium ketyl and distilled before use. 3-Amino-2-chloropyridine, primary phosphines and transition metal compounds were used as purchased. NMR spectra were recorded on a multinuclear FT-NMR spectrometer ARX300 or Avance300 (Bruker) at 300.1 (1H), 75.5 (13C), and 121.5 (31P) MHz and 298 K. The 1H, 13C and 31P chemical shifts are δ values and given in ppm relative to Me4Si and H3PO4 (85%), respectively, as external standards or to solvents calibrated with the aforementioned standards. Coupling constants refer to H–H (1H NMR) or P–C couplings (13C NMR) unless stated otherwise. NMR assignment numbers follow the nomenclature, and are illustrated in Scheme 1 for the compounds 1 and 2a–c and used also for the coordinated ligands. For atom numbers in crystal structures see Fig. 1–4. Mass spectra were recorded on a single-focusing sector field mass spectrometer AMD40 (Intectra, EI 70 eV); HRMS spectra were recorded on a double-focusing sector field mass spectrometer MAT 95 (Fa. Finnigan, EI 70 eV) or an ion cyclotron resonance-mass spectrometer APEX IV (Bruker Daltonik, ESI in MeOH, NH4OAc). Elemental analyses (C, H, N) were carried out using a LECO elemental analyzer, Model CHNS-932, under standard conditions.
N-(2-Chloropyrid-3-yl)-trimethylacetimidoyl chloride (1)
(a) Trimethylacetylchloride (5.14 mL, 42.0 mmol) was added dropwise to a solution of 3-amino-2-chloropyridine (4.5 g, 35.0 mmol) and triethylamine (5.85 mL, 42.0 mmol) in THF (20 mL) and Et2O (20 mL) at 0 °C. The reaction mixture was stirred for 1 h at 0 °C, overnight at room temperature, and then treated with water to remove trimethylamine hydrochloride, trimethylacetic acid and pyridinium salts. The aqueous phase was separated, extracted with ether, the combined organic phase washed with a concentrated aqueous solution of NaHCO3, and then dried over MgSO4. The solvent was removed in a vacuum to give 6.2 g (83%) of NMR-spectroscopically pure N-2-chloropyrid-3-yl-trimethylacetamide, which was used without recrystallization in step (b). 1H NMR (CDCl3): δ = 1.36 (s, 9 H, CMe3), 7.27 (t, 3J = 8.2, 4.7 Hz, 1 H, H-5), 8.00 (v br, NH), 8.11 (dd, 3J = 4.7, 4J = 1.7 Hz, 1 H, H-6), 8.76 (dd, 3J = 8.2, 4J = 1.8 Hz, 1 H, H-4).
(b) Phosphorus pentachloride (5.1 g, 24.5 mmol) was added to a solution of N-2-chloropyrid-3-yl-trimethylacetamide (5.1 g, 24.0 mmol) in dry toluene (50 mL). The reaction mixture was refluxed for 4 h, after which the solvent and volatile by-products were removed under vacuum. The residue was distilled at 10−5 mbar/96 °C to give 5.0 g (90%) of 1 as colourless oil. 1H NMR (CDCl3): δ = 1.30 (s, 9 H, CMe3), 7.08 (dd, 3J = 7.8, 4J = 1.6 Hz, 1 H, H-4), 7.14 (dd, 3J = 7.8, 4.8 Hz, 1 H, H-5), 8.08 (dd, 3J = 4.8, 4J = 1.6 Hz, 1 H, H-6). 13C{1H} NMR (CDCl3): δ = 27.90 (CMe3), 43.97 (CMe3), 122.57 (C-5), 129.06 (C-4), 141.38, 141.49 (Cq-2, Cq-3), 144.70 (C-6), 160.48 (CN). MS (EI 70 eV, 65 °C): m/z (%) = 232 (5), 230 (8) [M+], 197 (21), 195 (66), 141 (28), 139 (84), 57 (100). Anal. calcd for C10H12Cl2N2 (231.12): C 51.97, H 5.23, N 12.12; found: C 51.87, H 5.38, N 12.00.
DL-2-tert-Butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine (2a)
n-Butyl lithium solution (19.4 mL, 1.6 M in hexane, 31.4 mmol) was slowly added while stirring at −70 °C to phenylphosphine (1.70 mL, 15.44 mmol) dissolved in THF (20 mL). After 1 h a solution of 1 (2.90 g, 12.6 mmol) in THF (100 mL) was added dropwise at −70 °C to the reaction mixture. Stirring was continued for 1 h at low temperature and then overnight at room temperature. The solvent was removed under vacuum, the residue was extracted with Et2O (50 mL), and ether was evaporated. The residue was distilled at 10−5 mbar/95–100 °C (bath temp.) to give 0.71 g (21%) pale yellow oily 2a, which solidified on storage at room temperature. 1H NMR (CDCl3): δ = 1.27 (s, 9 H, CMe3), 7.20–7.38 (m, 6 H, phenyl, H-6), 8.05 (ddd, 3J = 8.1, 4J = 1.3, 4JPH = 1.7 Hz, 1 H, H-7), 8.48 (dt, 3J = 4.8, 4J = 1.3, 4JPH = 1.2 Hz, 1 H, H-5). 13C{1H} NMR (CDCl3): δ = 30.09 (d, 3J = 5.3 Hz, CMe3), 40.65 (d, 2J = 16.7 Hz, CMe3), 123.17 (s, C-6), 128.86 (d, 1J = 13.3 Hz, Cq-i), 129.10 (d, 3J = 9.2 Hz, 2 C-m), 129.45 (s, C-7) 130.46 (d, 4J = 1.9 Hz, C-p), 135.02 (d, 2J = 20.6 Hz, 2 C-o), 147.61 (d, 3J = 9.9 Hz, C-5), 151.93 (d, 2J = 18.9 Hz, Cq-7a), 164.68 (d, 1J = 20.3 Hz, Cq-3a), 200.64 (d, 1J = 35.1 Hz, Cq-2). 31P{1H} NMR: δ = −1.9 (CDCl3); −4.4 (D6-DMSO). MS (EI 70 eV, 25 °C): m/z (%) = 269 (17), 268 (100) [M+], 253 (71), 212 (45), 211 (30), 139 (34), 57 (88). HRMS (EI, 70 eV): calcd for [M]+ 268.1124; found: 268.1128. Anal. calcd for C16H17N2P (268.29): H 6.39, N 10.44; found: H 6.16, N 10.27.
DL-3-n-Butyl-2-tert-butyl-1,3-azaphospholo[4,5-b]pyridine (2b)
Compound 2b was prepared in analogy to 2a by lithiation of n-butylphosphine (1.47 mL, 13.1 mmol) in THF (10 mL) at −70 °C with n-butyl lithium (16.4 mL, 1.6 M in hexane, 26.16 mmol), and subsequent reaction with a solution of 1 (2.52 g, 10.9 mmol) in THF (80 mL) at −70 °C to room temperature and work-up as described above to give 1.6 g (59%) of an air sensitive pale yellow viscous oil, distilled at 10−5 mbar/90–96 °C (bath). 1H NMR (CDCl3): δ = 0.72 (t, 3J = 7.0 Hz, 3 H, CH3), 1.05–1.32 (m, 6 H, CH2), 1.34 (s, 9 H, CMe3), 7.21 (dd, 3J = 8.1, 4.8 Hz, 1 H, H-6), 7.91 (dt, 3J = 8.1, 4J = 1.3, 4JPH = 1.6 Hz, 1 H, H-7), 8.43 (dt, 3J = 4.8, 4J ≈ 4JPH = 1.2, 1.3 Hz, 1 H, H-5). 13C{1H} NMR (CDCl3): δ = 13.10 (s, Me), 23.47 (d, J = 8.0 Hz, CH2), 24.92 (d, J = 20.2 Hz, CH2), 28.40 (d, J = 1.5 Hz, CH2), 29.32 (d, 3J = 4.8 Hz, CMe3), 39.74 (d, 2J = 17.2 Hz, CMe3), 122.19 (s, C-6), 128.68 (s, C-7), 146.57 (d, 3J = 9.7 Hz, C-5), 151.13 (d, 2J = 17.5 Hz, Cq-7a), 163.38 (d, 1J = 17.2 Hz, Cq-3a), 200.13 (d, 1J = 38.3 Hz, Cq-2). 31P{1H} NMR (CDCl3): δ = −2.5. MS (EI 70 eV, 20 °C): m/z (%) = 249 (2), 248 (26) [M+], 192 (35), 191 (100) [M − Bu+], 165 (48), 149 (28), 148 (29), 137 (21), 136 (29), 57 (35). HRMS (ESI in MeOH, NH4OAc): C14H21N2P (248.30) calcd for [M + H]+ 249.15151; found 249.15161.
DL-2-tert-Butyl-3-isobutyl-1,3-azaphospholo[4,5-b]pyridine (2c)
Compound 2c was prepared in analogy to 2a by lithiation of isobutylphosphine (0.70 mL, 6.22 mmol) in THF (10 mL) at −70 °C with n-butyl lithium (7.81 mL, 1.6 M, 12.5 mmol), reaction with a solution of 1 (1.20 g, 5.19 mmol) in THF (80 mL) at −70 °C to room temperature and work-up as described above to give at 10−5 mbar/80–85 °C (bath) 0.81 g (33%) air sensitive slightly contaminated pale yellow oily 2c. 1H NMR (C6D6): δ = 0.75–1.0 (m, 6 H, CHMeAB), 1.37 (s, 9 H, CMe3), 1.6–2.05 (br m, 2 H, PCH2), 2.10–2.30 (m, 1 H, CH), 6.72 (dd, 3J = 8.0, 4.8 Hz, 1 H, H-6), 7.83 (dt, 3J = 8.0, 4J = 4JPH = 1.5 Hz, 1 H, H-7), 8.40 (ddd, 3J = 4.8, 4J = 1.5, 4JPH = 1.0 Hz, 1 H, H-5). 13C{1H} (CH-COSY, DEPT135) NMR (C6D6): δ = 24.38 (d, 3J = 7.3 Hz, CMeA), 24.67 (d, 3J = 8.4 Hz, CMeB), 28.17 (d, 2J = 9.0 Hz, CH), 30.79 (d, 3J = 5.1 Hz, CMe3), 35.72 (d, 1J = 19.3 Hz, PCH2), 40.98 (d, 2J = 17.3 Hz, CMe3), 123.33 (s, CH-6), 129.90 (s, CH-7), 147.79 (d, 3J = 10.0 Hz, CH-5), 152.26 (d, 1J = 18.6 Hz, Cq-3a), 166.17 (d, 2J = 17.4 Hz, Cq-7a), 201.48 (d, 1J = 37.8 Hz, Cq-2). 31P{1H} NMR (C6D6): δ = −10.2. MS (EI 70 eV, 250 °C): m/z (%) = 249 (8), 248 (31) [M+], 192 (62), 191 (89), 177 (100). HRMS (ESI in MeOH/NH4OAc): C14H21N2P (248.30), calcd for [M + H]+ 249.15151, found: 249.15160.
DL-(2-tert-Butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)pentacarbonyl chromium(0) (3a)
A solution of Cr(CO)5(THF), prepared by irradiation of Cr(CO)6 (351 mg, 1.60 mmol) in THF (30 mL; 36 mL of CO evolved), was added to a solution of 2a (214 mg, 0.797 mmol) in THF (10 ml) at −10 °C. The solution was warmed to room temperature and stirred for 2 d. The solvent was evaporated under vacuum, excess Cr(CO)6 was removed under high vacuum, and the residue was extracted with ether/hexane yielding 330 mg of an air-sensitive pale brown powder with rather low solubility, still containing some Cr(CO)6. 1H NMR (CDCl3): δ = 1.31 (s, 9 H, CMe3), 7.37–7.51 (m, 6 H, phenyl and H-6), 8.07 (ddd, 3J = 8.1, 4JPH = 2.4, 4J = 1.2 Hz, 1 H, H-7), 8.63 (dd, 3J = 4.3, 4J ≈ 1 Hz, 1 H, H-5). 13C{1H} NMR (CDCl3): δ = 30.10 (d, 3J = 2.3 Hz, CMe3), 41.03 (d, 2J = 18.0 Hz, CMe3), 125.05 (C-6), 129.43 (d, 3J = 10.6 Hz, 2 C-m), 131.7, 131.8 (C-7, C-p), 133.15 (d, 2J = 12.0 Hz, 2 C-o), 149.09 (d, 3J = 14.2 Hz, C-5), 215.53 (d, 2J = 11.9 Hz, 4 cis-CO); Cq signals except for cis-CO at the noise level. 31P{1H} NMR (CDCl3): δ = 58.1. MS (EI 70 eV, 90 °C): m/z (%) = 460 (4), 349 (5), 348 (20) [M+ − 4CO], 321 (22), 320 (100) [M+ − 5CO], 52 (98) [Cr+]. HRMS (ESI in MeOH + NH4OAc): C21H17CrN2O5P (460.34) calcd for: [M + H]+ 461.03530, found: 461.03530. IR (KBr): ν(CO) = 2068 (w) cm−1; the very strong band at 1950 cm−1 is superimposed by the absorption of the Cr(CO)6 contamination.
Detection of DL-(2-tert-butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)pentacarbonyl molybdenum(0) (4a)
A solution of Mo(CO)5(THF), prepared by irradiation of Mo(CO)6 (208 mg, 0.79 mmol) in THF (20 mL; 18 mL of CO evolved), was added to a solution of 2a (141 mg, 0.53 mmol) in THF (5 mL) at −10 °C. Filtration, removal of the solvent after 2 d at room temperature and repeated extraction of unconverted 2a from the crude product with ether/hexane afforded 105 mg (40%) of an air-sensitive pale brown powder with rather low solubility. 1H NMR (D6-acetone): δ = 1.33 (s, 9 H, CMe3), 7.46–7.64 (m, 6 H, phenyl and H-6), 8.19 (ddd, 3J = 8.0, 4JPH = 2.6, 4J = 1.5 Hz, 1 H, H-7), 8.65 (ddd, 3J = 4.9, 4J = 1.5, 4JPH = 0.6 Hz, 1 H, H-5). 13C{1H} NMR (D6-acetone): δ = CMe3 superimposed by solvent signals, 41.66 (d, 2J = 18.9 Hz, CMe3), 126.50 (C-6), 130.57 (d, 3J = 10.6 Hz, 2 C-m), 131.18 (C-7), 133.00 (d, 4J = 2.4 Hz, C-p), 134.38 (d, 2J = 13.5 Hz, 2 C-o), 150.13 (d, 3J = 13.9 Hz, C-5), 205.52 (d, 2J = 8.2 Hz, 4 cis-CO); Cq signals except for cis-CO at the noise level. 31P{1H} NMR (D6-acetone): δ = 34.9. IR (KBr): ν(CO) = 2076 (w), 1944 (vs) cm−1.
DL-(2-tert-Butyl-3-isobutyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)pentacarbonyl molybdenum(0) (4c)
Mo(CO)4(NBD) (117.1 mg, 0.39 mmol) and 2c (96.8 mg, 0.39 mmol) were placed into a Schlenk flask, THF (10 mL) was added and the mixture heated at 40 °C for 4 h. Insoluble impurities were filtered off and washed with ether. Removal of the solvent under vacuum provided a yellow-brown oil. 31P NMR monitoring displayed the product (δ = 30.6) along with two contaminants (δ = 29.1, −7.3). Purification by column chromatography on silica gel (hexane/2% ethyl acetate) furnished 61 mg (32%) of an air-sensitive pale yellow viscous oil. δ = 1H NMR (CDCl3): δ = 0.49 (d, 3J = 6.4 Hz, 3 H, CHMeA), 0.86 (d, 3J = 6.8 Hz, 3 H, CHMeB), 1.54 (s, 9 H, CMe3), 1.45–1.61 (superimposed m, PCH2), 2.35 (m, 1 H, CH), 7.38 (ddd, 3J = 7.9, 4.9, 5JPH = 1.5 Hz, 1 H, H-6), 7.99 (ddd, 3J = 7.9, 4JPH = 2.3, 4J = 1.5 Hz, 1 H, H-7), 8.64 (dd, 3J = 4.9, 4J = 1.5 Hz, 1 H, H-5). 13C{1H} and DEPT-135 NMR (CDCl3): δ = 23.52 (d, 3J = 5.3 Hz, CMeA), 24.97 (d, 3J = 9.3 Hz, CMeB), 28.25 (d, 2J = 8.0 Hz, CH), 29.86 (d, 3J = 2.7 Hz, CMe3), 39.52 (d, 1J = 11.9 Hz, PCH2), 40.43 (d, 2J = 18.6 Hz, CMe3), 124.68 (s, CH-6), 129.97 (s, CH-7), 147.92 (d, 2J = 29.2 Hz, Cq-7a), 148.52 (d, 3J = 13.3 Hz, CH-5), 160.05 (d, 1J = 65.0 Hz, Cq-3a), 195.25 (d, 1J = 9.3 Hz, Cq-2), 204.64 (d, 2J = 8.9 Hz, 4 COcis), 208.94 (d, 2J = 22.6 Hz, COtrans). 31P{1H} NMR (CDCl3): δ = 30.6. HRMS (DEI): C19H21MoN2PO5 (484.29); calcd for [M(98Mo)]+ 486.0237; found 486.0230; calcd for [M(98Mo) − CO]+ 458.0287; found: 458.0277.
DL-(2-tert-Butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)pentacarbonyl tungsten(0) (5a)
A solution of W(CO)5(THF), prepared by irradiation of W(CO)6 (160 mg, 0.455 mmol) in THF (30 mL; 10.2 mL of CO evolved), was added to a solution of 2a (0.121 g, 0.451 mmol) in THF (5 mL) at −10 °C. Work-up after 2 d as described for 5a afforded 128 mg (88%) of a pale green powder 5a, contaminated by a small amount of oligoethylene grease. 1H NMR (CDCl3): δ = 1.32 (s, 9 H, CMe3), 7.38–7.47 (m, 6 H, phenyl, H-6), 8.07 (ddd, 3J = 8.1, 4JPH = 2.6, 4J = 1.5 Hz, 1 H, H-7), 8.63 (dd, 3J = 4.8, 4JPH = 1.5 Hz, 1 H, H-5). 13C{1H} NMR (CDCl3): δ = 30.25 (d, 3J = 2.5 Hz, CMe3), 40.98 (d, 2J = 18.6 Hz, CMe3), 125.23 (C-6), 126.03 (d, 1J = 35.8 Hz, Cq-i), 129.49 (d, 3J = 10.6 Hz, 2 C-m), 130.17, 131.91 (2 d, J = 2.7 Hz, C-7, C-p), 133.48 (d, 2J = 11.9 Hz, 2 C-o), 148.26 (d, 2J = 31.8 Hz, Cq-7a), 149.04 (d, 3J = 14.6 Hz, C-5), 162.3 (low int. d, 1J ≈ 75 Hz, Cq-3a), 195.41 (d, 2J = 2.7 Hz, Cq-2), 195.58 (d, 2J = 6.6 Hz, 4 cis-CO), 197.23 (d, 1J = 22.6 Hz, 1 trans-CO). 31P{1H} NMR (CDCl3): δ = 18.2 (s, satl, 1JPW = 225 Hz). IR (KBr): ν(CO) = 2075 (w), 1936 (vs, br) cm−1. MS (EI 70 eV, 80 °C): m/z (%) = 593 (30) [M(184W) + H+], 453 (100%) [M(184W) + H+ − 5 CO]. HRMS (ESI in MeCN): calcd for [M(184W) + H+] 593.04571; found: 593.04569 (and correct isotopic pattern). Anal. calcd for C21H17N2O5PW (592.18): H 2.89, N 4.73; found: H 3.15, N 4.67.
DL-(2-tert-Butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)(η4-cycloocta-1,5-diene)rhodium(I)chloride (6a)
[RhCl(1,5-COD)]2 (32 mg, 0.065 mmol) in THF (5 mL) was added slowly at −20 °C to a solution of 2a (35 mg, 0.130 mmol) in THF (5 mL). The mixture was stirred overnight at room temperature, resulting in a colour change from yellow to deep orange. Insoluble material was removed by filtration, the solvent was evaporated under vacuum and the residue was washed several times with n-hexane and dried under vacuum to give ca. 60 mg (90%) of an orange-yellow solid. 1H NMR (CDCl3): δ = 1.61 (s, 9 H, CMe3), 1.9, 2.4 (vbr s, 8 H, CH2, COD), 4.2, 4.6, 5.6 (3 vbr s, 2 H, 1 H, 1 H, CH, COD), 7.27–7.40 (m, 4 H, H-6, H-p, H-o), 7.65 (vbr t, 3J ≈ 8.1 Hz, 2 H, H-m), 8.0 (vbr d, 3J = 7.8 Hz, 1 H, H-7), 8.45 (vbr, 1 H, H-5). 13C{1H} and DEPT135 NMR (CDCl3): δ = 30.21 (superimposed s, CMe3), 30.8 (superimposed vbr, 4 CH2, COD), 41.42 (d, 2J = 18.4 Hz, CMe3), 78.7 (vbr, CH, COD), 85.8 (vbr, CH, COD), 105.9 (vbr, 2 CH, COD), 124.5 (superimposed d, Cq-i), 124.79 (CH-6), 128.80 (d, 3J = 10.6 Hz, 2 CH-m), 130.4, 131.2 (2 br s, CH-7, CH-p), 134.16 (d, 2J = 12.1 Hz, 2 CH-o), 148.0 (br d, 3J = 14 Hz, CH-5), 150.08 (d, 2J = 27.6 Hz, Cq-7a), 159.56 (d, 1J = 68 Hz, Cq-3a), 192.6 (br, Cq-2). 31P{1H} NMR (CDCl3): δ = 27.2 (br d, half width each ≈25 Hz, 1JPRh = 139–143 Hz), −0.8 (minor solution species, 13–17%). Anal. calcd for C24H29ClN2PRh (514.83): C 55.99, H 5.68, N 5.44; found: C 55.58, H 5.43, N 5.35. Orange crystals suitable for XRD analysis, formed by slow diffusion of hexane into a solution of the solid in a small amount of THF, were selected from the mixture with mother liquor. Crystal data are compiled in Table 2, and the selected bond lengths and angles are shown Fig. 1.
Table 2 Crystal data and structure refinement
| Identification code |
6a
|
9b·2CDCl3 |
10b·4.5THF |
10b·2D6-acetone |
| Empirical formula |
C24H29ClN2PRh |
C36H52D2Cl10N4P2Pd3 |
C46H78Cl4N4O4.5P2Pd2 |
C34H42D12Cl4N4O2P2Pd2 |
| Formula weight |
514.82 |
1280.48 |
1175.66 |
979.43 |
| Temperature |
103(2) K |
100(2) K |
100(2) K |
100(2) K |
| Wavelength |
0.71073 Å |
0.71073 Å |
0.71073 Å |
0.71073 Å |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P |
P |
C2/c |
P21/c |
| Unit cell dimensions |
a = 9.1441(6) Å, α = 83.875(2)° |
a = 8.5594(7) Å, α = 101.959(4)° |
a = 27.727(3) Å, α = 90° |
a = 11.9584(4) Å, α = 90° |
|
b = 9.8885(6) Å, β = 81.588(2)° |
b = 9.6780(8) Å, β = 92.199(4)°, |
b = 16.625(2) Å, β = 90.891(6)° |
b = 29.4872(8) Å, β = 115.816(5)° |
|
c = 13.5572(8) Å, γ = 67.831(3)° |
c = 16.4206(13) Å, γ = 109.528(4)° |
c = 23.302(3) Å, γ = 90° |
c = 13.0738(4) Å, γ = 90° |
| Volume |
1121.25(12) Å3 |
1245.60(18) Å3 |
10739(2) Å3 |
4150.0(2) Å3 |
|
Z
|
2 |
1 |
8 |
4 |
| Density (calculated) |
1.525 Mg m−3 |
1.707 Mg m−3 |
1.454 Mg m−3 |
1.567 Mg m−3 |
| Absorption coefficient |
0.965 mm−1 |
1.702 mm−1 |
0.972 mm−1 |
1.236 mm−1 |
|
F(000) |
528 |
636 |
4864 |
1968 |
| Crystal size |
0.30 × 0.30 × 0.20 mm3 |
0.20 × 0.20 × 0.03 mm3 |
0.30 × 0.15 × 0.15 mm3 |
0.14 × 0.11 × 0.06 mm3 |
| Theta range for data collection |
1.52 to 31.06° |
2.30 to 30.58° |
2.27 to 30.03° |
2.70 to 28.28° |
| Index ranges |
−13 ≤ h ≤ 13, −14 ≤ k ≤ 14, −19 ≤ l ≤ 19 |
−12 ≤ h ≤ 12, −13 ≤ k ≤ 13, −23 ≤ l ≤ 23 |
−39 ≤ h ≤ 38, −23 ≤ k ≤ 23, −32 ≤ l ≤ 32 |
−15 ≤ h ≤ 15, −39 ≤ k ≤ 39, −17 ≤ l ≤ 17 |
| Reflections collected |
61697 |
38910 |
153859 |
76588 |
| Independent reflections |
7166 [R(int) = 0.0319] |
7630 [R(int) = 0.0307] |
15352 [R(int) = 0.0490] |
10287 [R(int) = 0.0800] |
| Completeness to theta |
= 30.00°, 99.9% |
= 30.50°, 100.0% |
= 30.00°, 97.7% |
= 28.28°, 99.8% |
| Absorption correction |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.830 and 0.694 |
0.951 and 0.808 |
0.868 and 0.732 |
1.000 and 0.952 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
7166/6/281 |
7630/0/274 |
15352/511/585 |
10287/266/445 |
| Goodness-of-fit on F2 |
1.04 |
1.03 |
1.18 |
0.82 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0166, wR2 = 0.0422 |
R
1 = 0.0248, wR2 = 0.0572 |
R
1 = 0.0409, wR2 = 0.0803 |
R
1 = 0.0304, wR2 = 0.0481 |
|
R indices (all data) |
R
1 = 0.0176, wR2 = 0.0429 |
R
1 = 0.0329, wR2 = 0.0612 |
R
1 = 0.0782, wR2 = 0.0982 |
R
1 = 0.0680, wR2 = 0.0516 |
| Largest diff. peak and hole |
0.52 and −0.32 e Å−3 |
2.53 and −1.59 e Å−3 |
1.33 and −0.68 e Å−3 |
0.76 and −0.47 e Å−3 |
DL-(3-n-Butyl-2-tert-butyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)(η4-cycloocta-1,5-diene)rhodium(I)chloride (6b) and detection of 7b
[RhCl(1,5-COD)]2 (177 mg, 0.36 mmol) in THF (5 mL) was added slowly at −20 °C to a solution of 2b (178 mg, 0.72 mmol) in THF (10 mL). The mixture was stirred for 1 d at room temperature (colour changed from yellow to deep orange and brownish-yellow) and worked up as described for 6a yielding 312 mg 6b (95 mol%, corr. yield 84%) as a yellow powder, containing 5 mol% 7b. 1H NMR (CDCl3): δ = 0.77 (t, 3J = 7.2 Hz, 3 H, CH3), 1.15–1.30 (m, 4 H, CH2), 1.68 (s, 9 H, CMe3), 1.79–1.93 (m, 4 H, CH2, COD), 2.15–2.40 (m, 2 H, PCH2), 2.33–2.52 (m, 4 H, CH2, COD), 3.67–3.77 (m, 2 H, CH, COD), 5.50–5.52 (m, 3J = 3.5 Hz, 1 H, CH, COD), 5.6 (vbr s, 1 H, CH, COD), 7.37 (ddd, 3J = 8.1, 4.9, 5JPH = 1.6 Hz, 1 H, H-6), 7.93 (ddd, 3J = 8.1, 4JPH = 2.2, 4J = 1.2 Hz, 1 H, H-7), 8.58 (dd, 3J = 4.8, 4J = 1.2 Hz, 1 H, H-5). 13C{1H} and DEPT135 NMR (CDCl3): δ = 13.45 (CH3), 23.25 (d, J = 17.5 Hz, CH2), 23.91 (d, J = 13.2 Hz, CH2), 26.44 (d, J = 6.1 Hz, CH2), 28.60, 28.97 (2 CH2, COD), 29.77 (CMe3), 32.36 (CH2, COD), 33.39 (d, J = 2.3 Hz, CH2, COD), 40.96 (d, 2J = 18.6 Hz, CMe3), 68.57 (d, J = 13.1 Hz, CH, COD), 72.64 (d, J = 13.2 Hz, CH, COD), 104.73 (dd, J = 10.5, 8.1 Hz, CH, COD), 105.28 (dd, J = 12.5, 7.3 Hz, CH, COD), 124.73 (s, CH-6), 129.85 (d, 3J = 2.0 Hz, CH-7), 147.99 (d, 3J = 13.2 Hz, CH-5), 150.78 (d, 2J = 26.6 Hz, Cq-7a), 156.53 (d, 1J = 67.4 Hz, Cq-3a), 192.29 (d, 1J = 6.1 Hz, Cq-2). 31P{1H} NMR (CDCl3): δ = 28.2 (d, 1JPRh = 141.7 Hz, 95 mol% 6b), 17.2 (d, 1JPRh = 151.1 Hz, 5 mol% 7b). HRMS (ESI in MeCN): calcd for [6b-Cl]+ 459.1431, calcd. for [7b-Cl]+ 705.1113; found 459.1431, 705.1111. Anal. calcd for 6b, C22H33ClN2PRh (494.84): C 53.40, H 6.72, N 5.66; calcd for 6b/7b (95/5 mol%): C 52.68, H 6.63, N 5.38; found: C 52.55, H 6.56, N 5.00.
Detection of 8a and [meso-bis{(η3-allyl)(2-tert-butyl-3-phenyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)palladium(II)chloride}-κ1N]palladium(II)dichloride (9a)
Allylpalladium chloride dimer (146 mg, 0.40 mmol) in THF (5 mL) was added slowly at −20 °C to a solution of 2a (145 mg, 0.54 mmol) in THF (5 mL) and stirred for 2 d at room temperature (colour changed from pale yellow to deep orange). The insoluble material was removed by filtration, the solvent was evaporated under vacuum and the residue was washed several times with n-hexane/Et2O and dried under vacuum to give 202 mg (53% referred to as 2a, 72% ref. to Pd) of an orange-yellow powder of crude 8a·THF with CHN analysis values roughly corresponding to a composition [(2a){(allyl)PdCl}1.2(PdCl2)0.8]·C4H8O. 1H NMR (CDCl3): δ = 1.27 (s, 9 H, CMe3), 2.8 (vbr, 2 H, allyl), 3.5, 5.5, 5.7 (vbr, 3 H, allyl), 7.45 (td, 3J ≈ 7.6, 4J ≈ 1.6 Hz, 2 H, H-m), 7.55 (t, 3J = 7.8, 7.7 Hz, 1 H, H-p), 7.60 (dd, 3J = 8.0, 5.8 Hz, 1 H, H-6), 7.9 (vbr s, 2 H, H-o), 8.18 (dt, 3J = 8.2, 4J ≈ 4JPH ≈ 1.5 Hz, 1 H, H-7), 8.8 (vbr, 1 H, H-5); THF: 1.85 (m, 4 H, CH2), 3.73 (m, 4 H, OCH2). 13C{1H} and DEPT-135 NMR (CDCl3): δ = 30.18 (d, 3J = 3.5 Hz, CMe3), 41.00 (d, 2J = 19.3 Hz, CMe3), ca. 60 (sh, minor, C′allyl), 62.8 (vbr, C-αallyl), 84.7 (minor, C′allyl), 114.8 (vbr, Callyl), 118.7 (vbr, Callyl), 123.56 (d, 1J = 31.1 Hz, Cq-i), 126.6 (br, CH-6), 129.76 (d, 3J = 10.9 Hz, 2 CH-m), 131.46 (s, CH-7), 132.54 (d, 4J = 1.9 Hz, CH-p), 134.61 (d, 2J = 12.1 Hz, 2 CH-o), 150.26 (d, 2J = 27.2 Hz, Cq-7a), 151.5 (vbr, CH-5), 160 (br d, 1J ≈ 60 Hz, Cq-3a), 195.4 (br, Cq-2); THF 25.60 (CH2), 67.96 (OCH2). 31P{1H} NMR (CDCl3): δ = 15.4 (vbr). Anal. calcd for 8a·THF (C23H30Cl3N2OPPd2, 700.67): C 39.43, H 4.32, N 4.00; calcd for [(2a)(allylPdCl)1.2(PdCl2)0.8]·C4H8O (C23.6H31Cl2.8N2OPPd2, 701.80): C 40.39, H 4.45, N 3.99; found: C 40.66, H 4.18, N 3.54. Slow diffusion of hexane into a concentrated solution of crude 8a in THF gave crystals of 9a·THF. Severe disorder of the allyl group and unexpected peaks in the residual electron density (possibly by twinning) did not allow satisfactory refinement of the XRD data, but allowed the identification of 9a·THF (Fig. S26, ESI†).
[meso-Bis{(η3-allyl)(3-n-butyl-2-tert-butyl-1,3-azaphospholo[4,5-b]pyridine-κ1P)palladium(II)chloride}-κ1N]palladium(II)dichloride (9b)
Reaction of allylpalladium chloride dimer (180 mg, 0.49 mmol) in THF (10 mL) with 2b (0.162 g, 0.652 mmol) in THF (15 mL) and workup as described for 8a gave 250 mg (74%) yellow powder. CHN analysis values are in accordance with THF-free 9b. Crude powder with residual THF – 1H NMR (CDCl3): δ = 0.80 (t, 3J = 7.2 Hz, 3 H, CH3), 1.28 (m, 3J = 7.8, 6.8 Hz, 4 H, CH2), 1.47 (br s, 9 H, CMe3), 2.6 (vbr s, 2 H, PCH2 and/or allyl), 2.7–3.4 (vbr m, 2 H, allyl and/or PCH2), 3.55–3.9 (superimposed by THF, vbr, Hallyl), 4.0–4.2 (vbr, Hallyl), 4.8, 5.3–5.8 (vbr, Hallyl), 7.5 (vbr, 1 H, H-6), 8.1 (vbr d, 3J ≈ 7 Hz, 1 H, H-7), 8.7 (vbr, 1 H, H-5); ca. 0.3 THF/9b: 1.85 (m, CH2), 3.75 (m, OCH2). 13C{1H} NMR (CDCl3): δ = 13.43 (s, CH3), 23.6–23.8 (br superimposed d, 2 CH2), 26.53 (d, J = 8.2 Hz, CH2), 29.68 (d, 3J = 2.9 Hz, CMe3), 40.54 (d, 2J = 19.6 Hz, CMe3), 58 (vbr, C′αγ-allyl), 62.2 (vbr, Cα-allyl), 80.5 (vbr, Cγ-allyl), 114.6 (br, C′β-allyl), 117.2, 118.8 (vbr, Cβ-allyl), 125.3 (vbr, C-6), 130.5 (vbr, C-7), 148.5–150.5 (vbr, C-5, Cq-7a), 158.6 (br d, 1J ≈ 72 Hz, Cq-3a), 192.4 (br, low int., Cq-2); int. Call: C′allca. 3:1. 31P{1H} NMR (CDCl3): δ = 17 (vbr), 25 (vbr), integral ratio 3:1. Anal. calcd for C34H52Cl4N4P2Pd3 (1039.82): C 39.27, H 5.04, N 5.39; found: C 38.87, H 5.12, N 5.14. Single crystals of 9b·2CDCl3, grown by slow concentration of the CDCl3 solution, were selected from the mixture with mother liquor for crystal structure determination. For selected bond lengths and angles see Fig. 3, and for crystal data see Table 2.
DL-anti-Bis[{(3-n-butyl-2-tert-butyl-1,3-azaphospholo[4,5-b]pyridine)-κ1N,κ1P}cis-palladium(II)dichloride] (10b) solvates
(A)
10b·4.5THF.
[Pd(CH3CN)2Cl2] (96.5 mg, 0.372 mmol) in THF (10 mL) was added slowly at −10 °C to a solution of 2b (92.4 mg, 0.372 mmol) in THF (10 mL). The reaction mixture was stirred for 2 d at room temperature (colour changed from pale yellow to orange) and filtered. Removal of the solvent under vacuum, washing the orange solid residue with n-hexane and drying under vacuum gave 120 mg (76%) yellow powder. 1H NMR (CDCl3): δ = 0.77 (t, 3J = 7.2 Hz, 3 H, CH3), 0.70–0.95 (superimposed m, 2 H, CH2), 1.13–1.28 (m, 2 H, CH2), 1.57 (s, 9 H, CMe3), 2.32–2.47 (m, 1 H, PCH), 3.67–3.84 (m, 1 H, PCH), 7.45 (ddt, 3J ≈ 8, 5, |5JPH + 5JP′H| = 3.5 Hz, 1 H, H-6), 7.94 (dt, 3J = 8.1, 4JPH = 2, 4J = 1.2 Hz, 1 H, H-7), 8.54 (tt, |3J + 4JP′H| = 4.2, 4J ≈ 4JPH = 1.2, 1.4 Hz, 1 H, H-5). 13C{1H} NMR (CDCl3): δ = 13.48 (s, CH3), 23.50 (d, J = 14.1 Hz, CH2), 24.79 (d, J = 17.4 Hz, CH2), 25.39 (d, J = 10.1 Hz, CH2), 29.74 (d, 3J = 3.8 Hz, CMe3), 40.70 (d, 2J = 21.1 Hz, CMe3), 127.59 (CH-6), 132.36 (CH-7), 150.6 (superimposed d, 3J = 10 Hz, CH-5), 150.7 (superimposed d, 2J = 28 Hz, Cq-7a), 157.3 (partly at noise level, Cq-3a), 190.4 (br, Cq-2). 31P{1H} NMR (CDCl3): δ = 26.7. Anal. calcd for THF free complex C28H42Cl4N4P2Pd2 (851.26): C 39.51, H 4.97, N 6.58; found: C (incomplete combustion), H 5.25, N 6.73. Single crystals of 10b·4.5THF were obtained by slow diffusion of n-hexane into the saturated solution in THF; the solvent was lost rapidly in air, and the crystal had to be handled under inert oil. Crystal data are given in Table 2, and the selected bond lengths and angles in Fig. 4a.
(B)
10b·2D6-acetone.
K2PdCl4 (120 mg, 0.368 mmol) in MeOH (10 mL) was added slowly at −20 °C to a solution of 2b (89 mg, 0.358 mmol) in MeOH (10 mL). The mixture was stirred for 3 d at room temperature, filtered, and the deep yellow precipitate was washed several times with water and MeOH, and then dried under vacuum yielding 108 mg (71%) of a yellow air-stable powder. This was insoluble in n-hexane and slightly soluble in acetone. 1H NMR (D6-acetone): δ = 0.75 (t, 3J = 7.3 Hz, 3 H, CH3), 1.0–1.43 (m, 4 H, CH2), 1.82 (s, 9 H, CMe3), 3.2–3.35, 3.7–3.85 (vbr m, 2 H, PCH), 7.69 (ddd, 3J = 8.2, 5.7, 5JPH = 1.4 Hz, 1 H, H-6), 8.08 (dt, 3J = 8.2, 4J ≈ 4JPH = 1.2 Hz, 1 H, H-7), 8.40 (dt, 3J = 5.6, 4J ≈ 4JPH = 1 Hz, 1 H, 5-H). 13C{1H} NMR (D6-acetone): δ = 14.26 (CH3), 24.64 (d, J = 16.6 Hz, CH2), 25.59 (d, J = 22.6 Hz, CH2), 26.96 (d, J = 20.4 Hz, CH2), 29 (br s superimposed with solvent, CMe3), 43.23 (d, 2J = 20.4 Hz, CMe3), 131.3 (vbr, CH-6), 135.2 (vbr, CH-7), 152.5 (vbr, CH-5); Cq-signals in noise. 31P{1H} NMR (D6-acetone): δ = 31.0. LRMS (ESI in MeCN): calcd for most abundant fragment [M − Cl]+ 815.00; found: 815.00 (and correct isotopic pattern). Anal. calcd for C28H42Cl4N4P2Pd2 (851.26): C 39.51, H 4.97, N 6.58; found: C 40.06, H 5.06, N 6.34. Slow diffusion of n-hexane into a saturated solution in D6-acetone provided single crystals of 10b·2D6-acetone. Crystal data are given in Table 2, and the structure is shown in Fig. 4b.
Catalytic tests – 2-mesitylaminopyridine 11
A Schlenk bottle was charged with 2-bromopyridine (212 mg, 1.34 mmol), mesitylamine (269 mg, 1.99 mmol), KOtBu (310 mg, 2.76 mmol), 10b (prepared from Pd(CH3CN)2Cl2, 29 mg) or the given amount of ligand and Pd-compound (see Table 1) and toluene (10 mL) and heated under nitrogen for 24 h at 100 °C. The mixture was filtered, washed with toluene, the solution was transferred to a silica gel column and compound 11 was separated using ethyl acetate/hexane 2:8. The results are compiled in Table 1. The 1H and 13C NMR data of 11 were in accordance with known values.22
Crystal structure analysis
Crystals of 6a, 9b·2CDCl3, 10b·2D6-acetone and 10b·4.5THF were mounted on glass fibres in an inert oil. Data were recorded at low temperature on an Oxford Diffraction Xcalibur E (10b·2(D6-acetone)) or a Bruker APEX2 diffractometer using MoKα-radiation (λ = 0.71073 Å). Crystal data are summarized in Table 2. The structures were refined anisotropically on F2 using the program SHELXL-97.26 Hydrogen atoms were included using a riding model or rigid methyl groups, except for hydrogens of coordinated allyls or coordinated double bonds, which were refined freely. For 10b·4.5THF, one THF is disordered over two positions and one lies on a twofold axis.
Crystallographic data for 6a, 9b·2CDCl3, 10b·4.5THF and 10b·2D6-acetone have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1423102, 1423105, 1423103 and 1423104 respectively.
Acknowledgements
A fellowship (M.S.S.A.) from the Deutscher Akademischer Austauschdienst (DAAD) and financial support by the Deutsche Forschungsgemeinschaft (HE 1997/12-2) are gratefully acknowledged. We thank B. Witt, G. Thede and M. Steinich for NMR and LRMS and Dr H. Frauendorf and G. Sommer-Udvarnoki (Georg-August-Universität Göttingen, Institut für Organische und Biomolekulare Chemie) for HRMS measurements.
References
-
(a) G. R. Newkome, Chem. Rev., 1993, 93, 2067–2089 CrossRef CAS;
(b) Z. Z. Zhang and H. Cheng, Coord. Chem. Rev., 1996, 147, 1–39 CrossRef CAS;
(c) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499–556 CrossRef CAS.
-
(a) M. M. Olmstead, A. Maisonnat, J. P. Farr and A. L. Balch, Inorg. Chem., 1981, 20, 4060–4064 CrossRef CAS;
(b) J. P. Farr, M. M. Olmstead, F. Wood and A. L. Balch, J. Am. Chem. Soc., 1983, 105, 792–798 CrossRef CAS;
(c) A. Ecke, W. Keim, M. C. Bonnet, I. Tkatchenko and F. Dahan, Organometallics, 1995, 14, 5302–5307 CrossRef CAS.
- S. Durben and T. Baumgartner, Inorg. Chem., 2011, 50, 6823–6836 CrossRef CAS PubMed.
-
(a) M. S. S. Adam, O. Kühl, M. K. Kindermann, J. W. Heinicke and P. G. Jones, Tetrahedron, 2008, 64, 7960–7967 CrossRef CAS;
(b) M. S. S. Adam, P. G. Jones and J. W. Heinicke, Eur. J. Inorg. Chem., 2010, 3307–3316 CrossRef CAS.
-
(a) R. K. Bansal and J. Heinicke, Chem. Rev., 2001, 101, 3549–3578 CrossRef CAS PubMed;
(b) J. Heinicke, N. Gupta, S. Singh, A. Surana, O. Kühl, R. K. Bansal, K. Karaghiosoff and M. Vogt, Z. Anorg. Allg. Chem., 2002, 628, 2869–2876 CrossRef CAS;
(c) J. Heinicke, K. Steinhauser, N. Peulecke, A. Spannenberg, P. Mayer and K. Karaghiosoff, Organometallics, 2002, 21, 912–919 CrossRef CAS;
(d) M. Ghalib, B. Niaz, P. G. Jones and J. W. Heinicke, Heteroat. Chem., 2013, 24, 452–456 CrossRef CAS.
-
(a) M. Ghalib, L. Könczöl, L. Nyulászi, P. G. Jones, G. J. Palm and J. W. Heinicke, Dalton Trans., 2014, 43, 51–54 RSC;
(b) M. Ghalib, L. Könczöl, L. Nyulászi, G. J. Palm, C. Schulzke and J. W. Heinicke, Dalton Trans., 2015, 44, 1769–1774 RSC;
(c) M. Ghalib, P. G. Jones, C. Schulzke, D. Sziebert, L. Nyulászi and J. W. Heinicke, Inorg. Chem., 2015, 2117–2127 CrossRef CAS PubMed;
(d) J. Heinicke, Eur. J. Inorg. Chem., 2016 DOI:10.1002/ejic.201500941.
-
(a) S. O. Grim, D. A. Wheatland and W. McFarlane, J. Am. Chem. Soc., 1967, 89, 5573–5577 CrossRef CAS;
(b) F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 4432–4438 CrossRef CAS.
-
(a) E. O. Fischer, L. Knauss, R. L. Keiter and J. G. Verkade, J. Organomet. Chem., 1972, 37, C7–C10 CrossRef CAS;
(b) F. Mercier, F. Mathey, C. Afiong-Akpan and J. F. Nixon, J. Organomet. Chem., 1988, 348, 361–367 CrossRef CAS.
-
(a) M. A. M. Meester, R. C. J. Vriends, D. J. Stufkens and K. Vrieze, Inorg. Chim. Acta, 1976, 19, 95–103 CrossRef CAS;
(b) H. Daamen and A. Oskam, Inorg. Chim. Acta, 1978, 26, 81–89 CrossRef CAS.
- W. Partenheimer and E. F. Hoy, Inorg. Chem., 1973, 12, 2805–2809 CrossRef CAS.
- E. Rotondo, G. Battaglia, C. G. Arena and F. Faraone, J. Organomet. Chem., 1991, 419, 399–402 CrossRef CAS.
-
(a) J. Tiburcio, S. Bernes and H. Torrens, Polyhedron, 2006, 25, 1549–1554 CrossRef CAS;
(b) K. Vrieze, H. C. Volger and A. P. Praat, J. Organomet. Chem., 1968, 14, 185–200 CrossRef CAS.
-
(a) H. L. M. van Gaal and J. P. J. Verlaan, J. Organomet. Chem., 1977, 133, 93–106 CrossRef CAS;
(b) I. J. Colquhoun and W. McFarlane, J. Magn. Reson., 1982, 46, 525–528 CAS.
-
(a) B. R. Aluri, M. K. Kindermann, P. G. Jones and J. W. Heinicke, Chem. – Eur. J., 2008, 14, 4328–4335 CrossRef CAS PubMed;
(b) M. Ghalib, P. G. Jones, S. Lysenko and J. W. Heinicke, Organometallics, 2014, 33, 804–816 CrossRef CAS.
- A. Brück and K. Ruhland, Organometallics, 2009, 28, 6383–6401 CrossRef.
- S. Chen, E. Manoury and R. Poli, Eur. J. Inorg. Chem., 2014, 5820–5826 CrossRef CAS.
- J. Rajput, A. T. Hutton, J. R. Moss, H. Su and C. Imrie, J. Organomet. Chem., 2006, 691, 4573–4588 CrossRef CAS.
- L. Bettucci, C. Bianchini, J. Filippi, A. Lavacchi and W. Oberhauser, Eur. J. Inorg. Chem., 2011, 1797–1805 CrossRef CAS.
- B. Henc, P. W. Jolly, R. Salz, G. Wilke, R. Benn, E. G. Hoffmann, R. Mynott, G. Schroth, K. Seevogel, J. C. Sekutowski and C. Krüger, J. Organomet. Chem., 1980, 191, 425–448 CrossRef CAS.
- B. E. Mann, R. Pietropaolo and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1973, 2390–2393 RSC.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS.
- E. J. Crust, I. J. Munslow, C. Morton and P. Scott, Dalton Trans., 2004, 2257–2266 RSC.
- M. S. S. Adam, A. D. Mohamad, P. G. Jones, M. K. Kindermann and J. W. Heinicke, Polyhedron, 2013, 50, 101–111 CrossRef CAS.
-
(a) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS;
(b) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 853–860 CrossRef.
- T. H. Riermeier, A. Zapf and M. Beller, Top. Catal., 1997, 4, 301–309 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1423102–1423105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04073f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence