
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual redox catalysts for oxygen reduction and evolution reactions: towards a redox flow Li–O2 battery†
Yun Guang
Zhu
,
Chuankun
Jia
,
Jing
Yang
,
Feng
Pan
,
Qizhao
Huang
and
Qing
Wang
*
Department of Materials Science and Engineering, Faculty of Engineering, NUSNNI-NanoCore, National University of Singapore, 117576, Singapore. E-mail: msewq@nus.edu.sg; Fax: +65-6776-3604; Tel: +65-6516-7118
First published on 27th April 2015
Abstract
A redox flow lithium–oxygen battery (RFLOB) by using soluble redox catalysts with good performance was demonstrated for large-scale energy storage. The new device enables the reversible formation and decomposition of Li2O2via redox targeting reactions in a gas diffusion tank, spatially separated from the electrode, which obviates the passivation and pore clogging of the cathode.
With the increasing demand for high-density energy storage, various electrochemical energy storage technologies have been proposed, of which the lithium–oxygen (Li–O2) battery is believed to be one of the most promising solutions.1–5 The Li–O2 battery “breathes” oxygen from air as a reactant, which greatly enhances the gravimetric energy density and reduces the cost of the cell.6 Therefore, Li–O2 batteries have recently attracted considerable attention from all over the world. Despite the great promise, Li–O2 batteries confront several critical issues before they become a credible solution for next generation energy storage, for instance, stability of the aprotic electrolyte and electrode in the presence of O2˙− radical, effectiveness of catalyst with the passivation of Li2O2, microstructures of the cathode to accommodate Li2O2 while allowing the access of O2 and Li+, and cyclability of the lithium anode upon prolonged cycling, etc. While profound studies have been done in searching stable electrolytes7–12 and efficient oxygen electrocatalysts,13–20 challenges relating to surface passivation and pore clogging by the insoluble Li2O2 in the gas diffusion cathode remain.21 This severely impairs the round-trip energy efficiency and limits the achievable capacity of the cell. Recently, redox catalysis was introduced to mitigate the overpotentials for oxygen reduction or evolution reactions by utilizing redox mediators dissolved in the electrolyte.22–29 These soluble redox active species could either reduce O2 forming Li2O2 in the presence of Li+ in the electrolyte, or oxidize Li2O2 releasing oxygen. Since the “catalysts” exist in the electrolyte, the adverse effect of surface passivation is alleviated. By contrast, these redox-catalyzed reactions towards O2 in essence resemble the “redox targeting” reactions recently proposed for lithium-ion battery materials.30–32 In the presence of suitable redox mediators in the electrolyte, battery materials could be charged and discharged through reversible chemical lithiation and delithiation without attaching to the current collector, which intuitively results in a novel battery system – redox flow lithium-ion batteries (RFLBs). We notice that a Li-air fuel cell system with a circulating catholyte has been demonstrated by H. Zhou and co-workers.33 The same group also proposed a flow system using a soluble catalyst in an aqueous catholyte for the oxygen reduction reaction,34 which in principle results in a redox flow Li–O2 primary cell.
Here we demonstrate a new implementable solution – rechargeable redox flow Li–O2 battery (RFLOB) – to tackle the critical issues confronted by non-aqueous Li–O2 batteries. As illustrated in Fig. 1, the RFLOB has a gas diffusion tank (GDT) connected to the electrochemical cell stack. The electrolyte fluid is circulated between the tank and the cell using a peristaltic pump, in which for the first time we concurrently introduced two different redox mediators to catalyze the O2 reduction and evolution reactions during discharging and charging processes, respectively. As a result, the use of conventional electrocatalysts on the cathode has been completely avoided. The GDT tank is filled with porous material allowing the easy access of redox fluid and O2, in which the O2 pressure is kept constant through a gas inlet and outlet. During the discharging process, redox mediator RM1 is reduced at the cathode and flows into the GDT tank where it is oxidized by O2 in the presence of Li+:
| RMox1 + e− → RMred1 (electrochemical reaction on the cathode) | (1) |
| Li+ + RMred1 + O2 → Li2O2 + RMox1 (chemical reaction in GDT) | (2) |
| RMred2 → RMox2 + e− (electrochemical reaction on the cathode) | (3) |
| Li2O2 + RMox2 → Li+ + RMred2 + O2 (chemical reaction in GDT) | (4) |
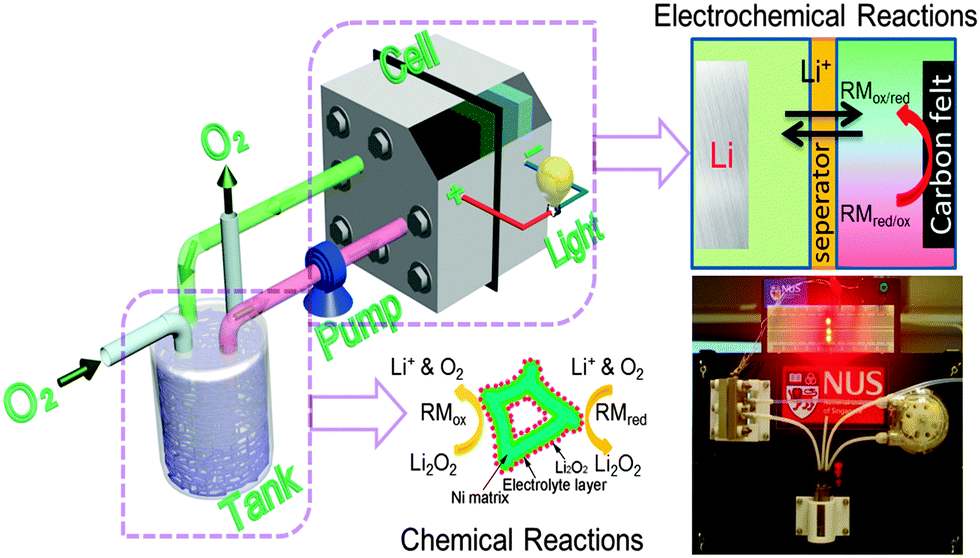 | ||
| Fig. 1 Schematic illustration of the configuration and working process of a redox flow lithium–oxygen battery (RFLOB). The cell stack constitutes a lithium metal anode and a carbon felt cathode (2 cm × 2 cm), separated by a membrane. A gas diffusion tank (GDT) is connected to the cathodic compartment through a pump. During the discharging process, oxygen flows into the tank and is reduced to form Li2O2 while the electrolyte fluid containing redox mediators and Li+ circulates between GDT and the cell. The photo at the lower right corner shows a RFLOB single cell powering three light-emitting diodes. | ||
The redox potential of Li2O2 in aprotic solvent is ∼2.96 V vs. Li/Li+. Considering the redox potentials of ethyl viologen (EV) and iodide, which are ∼2.65 V for EV+/EV2+ and ∼3.10/3.70 V for I−/I3−/I2 (Fig. 2a), just straddle that of Li2O2, these two redox species were identified as the mediators for oxygen reduction and evolution reactions in RFLOB, respectively. The potential difference between the mediators and Li2O2 provides the necessary thermodynamic driving force for the formation and decomposition of Li2O2via redox targeting reactions,35 of which the lower potential of EV+ enables the reduction of O2 forming Li2O2 during the discharge process (reaction 2), while the relatively positive potential of triiodide or iodine facilitates the oxidation Li2O2 during the charging process (reaction 4). Both redox mediators have been tested in static cells and showed good reversibility (Fig. S1, ESI†).
 | ||
| Fig. 2 (a) Cyclic voltammograms of EV and LiI in TEGDME. The scan rate is 0.10 V s−1. The redox potential of Li2O2 is indicated for reference. (b) Discharge/charge curves of a RFLOB in the first 3 cycles. The current density is 0.05 mA cm−2. (c) The discharge curve of a RFLOB primary cell showing the high utilization ratio of the lithium anode in the presence of 10 mM EV2+. The current density is 0.125 mA cm−2. (d) Discharge/charge curves and Coulombic efficiency of a RFLOB employing a PVDF–Nafion membrane at different cycle numbers. The current density is 0.125 mA cm−2. The electrolyte was 1.0 M LiTFSI in TEGDME containing 10 mM EV2+/10 mM I−, with a volume of 8 ml in (b), (c), and 4 ml in (d). | ||
As seen from the photograph shown in Fig. 1, RFLOB was fabricated with a GDT tank filled with 8 ml redox electrolyte consisting of 10 mM EV2+/10 mM I− and 1.0 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) in tetraethyleneglycol dimethylether (TEGDME). The O2 pressure in the tank was kept at 1 atm. Vinylene carbonate pretreated lithium foil was used as an anode in the electrochemical cell to preclude the reaction with redox mediators, since the Celgard® separator is unable to block the crossover of the redox mediators.36,37 The cell was discharged and charged in the galvanostatic mode and the voltage profiles are shown in Fig. 2b. In the first discharging process, only a single voltage plateau at ∼2.70 V was observed, which matches the reduction of EV2+. However, the theoretical discharge capacity of EV2+ to EV+ is noted to be only ∼2 mA h, the much-extended capacity (here the cell capacity was controlled at 6 mA h) implies the reduction of O2 by EV+ in the GDT tank, forming EV2+ and Li2O2 as revealed later. The regenerated EV2+ then flows back to the cell and starts a second round of reduction meanwhile electricity is generated. In theory, the above discharging process could carry on until Li metal in the anode is used up and reaches the theoretical specific energy of the cell. For instance, in a non-constrained discharging process close to 80% lithium was converted into Li2O2 with relatively low overpotential loss in the presence of 10 mM EV2+ (Fig. 2c), which paves a way for making low-cost and extremely high-energy density Li–O2 primary cells.
During the charging process, two voltage steps appeared at ∼3.55 and 3.75 V (Fig. 2b). The oxidation of EV+ was not observed since most of EV+ have been oxidized to EV2+ by the surplus O2 in the tank. Hence the cell voltage shoots directly up to that for the oxidation of I− to I3− upon charging, at which there seemed to be very limited reaction between I3− and Li2O2 since the capacity extension at this voltage is rather small. Thereafter the voltage rises steadily until it reaches the second voltage plateau. Brown color appeared gradually in the electrolyte, indicating that more and more polyiodide was produced. The high voltage plateau corresponds to the further oxidation of I3− to higher order polyiodides and eventually to iodine. The extended capacity at the high voltage indicates that the reaction between iodine and Li2O2 was efficient which resulted in the oxidation of the latter and release of O2.
The charge transfer process between Li2O2 and I2 is corroborated by theoretical calculations. Fig. S2 (ESI†) shows the electron density difference maps of I3− and I2 on the Li2O2 (0001) surface in parallel adsorption geometry, where the blue and yellow zones correspond to electron density deduction and enhancement regions, respectively. Apparently there is a tendency of electron transfer from Li2O2 to both I3− and I2. The charge transfer is quantified by the Bader charge calculation to be 0.49 e− for Li2O2/I2, relative to that for Li2O2/I3−, indicating much more effective electron transfer in the former as compared to the latter.
In the subsequent discharging process, a short voltage plateau appeared at ∼3.30 V, attributed to the reduction of iodine in the electrolyte. After that, the cell voltage remained stable at ∼2.70 V, exhibiting good reversibility of EV. In order to rule out the capacity from the direct reduction of dissolved O2 on the cathode, the cell was also tested in the absence of both redox mediators (Fig. S3, ESI†). It is obvious that given the extremely low capacity, the reaction of dissolved O2 on the cathode has negligible contribution to the overall cell capacity.
The above results are very encouraging that, since the deposition of Li2O2 mainly occurs in the GDT tank, which intrinsically obviates the passivation and pore clogging of cathode in the cell, very stable voltage profiles with relatively low overpotentials were achieved in the first three cycles, even in the absence of electrocatalysts. To prove the formation and decomposition of Li2O2 in the GDT tank, X-ray photoelectron spectroscopy (XPS) was employed to investigate the chemical states of Li and O for species formed in the tank at the end of discharge and charge. The signal associated with the Li–F bond (56.6 eV) is presented for all the samples (Fig. 3), presumably from the remaining LiTFSI.38 The peak of Li–O–O–Li (55.0 eV) is clearly seen in the Li 1s spectra after discharging while nearly disappeared after charging. The existence of Li2O2 is further corroborated by O 1s spectra, where two large peaks assigned to the O1s (532.3 eV) from LiTFSI and Li–O–O–Li (531.2 eV) in Li2O2 are evidently observed.38,39 The latter then vanished after charging, in agreement with the Li 1s spectra. In order to confirm the formation of Li2O2 on the Ni foam in the GDT tank, XRD measurement was carried out with the sample after full discharging. As shown in the diffraction pattern in Fig. S4 (ESI†), the characteristic peaks of Li2O2 such as (101) and (100) are clearly seen. These peaks are relatively broad in width in contrast to those from the Ni substrate, indicating the nanocrystalline nature of the formed Li2O2. In addition, some minor peaks, which may be assigned to LiOH and Li2CO3 phases, are also visible. These by-products may plausibly be introduced during sample transfer and/or XRD measurement, which were conducted in air. The XPS and XRD results were further substantiated by scanning and transmission electron microscopic measurements.
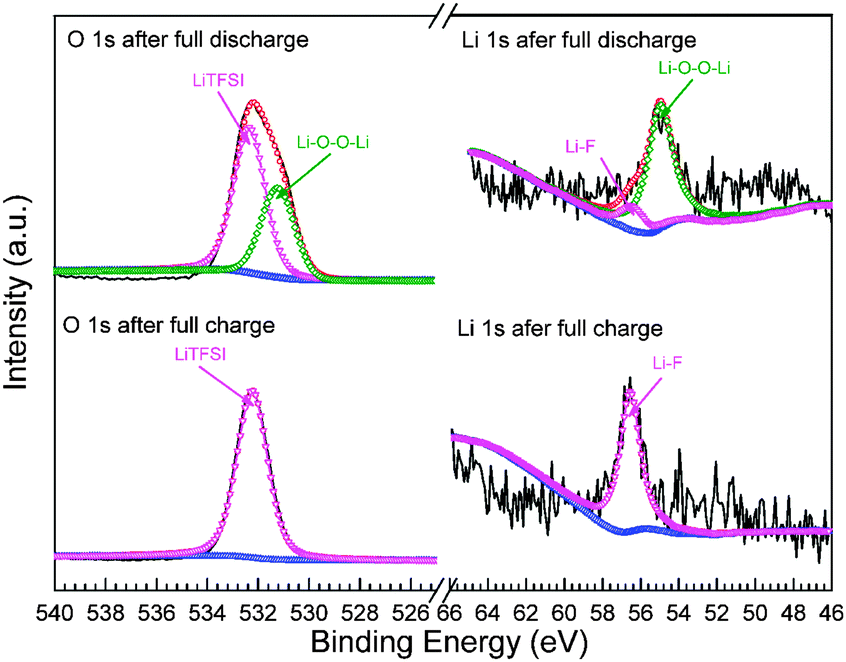 | ||
| Fig. 3 X-ray photoemission spectroscopy (XPS) measurement of Li 1s and O 1s spectra and the corresponding peak deconvolutions. The upper and lower panels display those of the discharged and charged species in the GDT tank, respectively. | ||
As seen from the SEM images shown in Fig. 4, after discharging the smooth surface of the pristine Ni foam (Fig. S5a, ESI†) was covered by a layer of agglomerated particles (Fig. 4a), which nearly disappeared with only little residual left after charging (Fig. 4b). This is consistent with the XPS measurement should the particles be Li2O2. The cell in Fig. 2c was also examined after full discharging, in which we expect that much more product would be formed in the GDT tank. As revealed in Fig. S5b (ESI†), not surprisingly, a much thicker layer of a particulate precipitate was observed on the Ni foam. The particles are in round shape and 10–20 nm in diameter (Fig. 4c), which are crystalline in nature as revealed by high resolution TEM. The lattice fringes of (101) and (100) crystal planes of Li2O2 are clearly identified (Fig. 4d), with d-spacings of 0.25 nm and 0.27 nm, respectively. This unambiguously confirms the formation of Li2O2 upon discharging.
 | ||
| Fig. 4 (a, b) Field emission scanning electron microscopy (FESEM) images showing the morphology evolution of the Ni foam after discharging (a) and re-charging (b) in the GDT tank. The insets illustrate the formation and decomposition of Li2O2 on the surface of the Ni foam. (c, d) Transmission electron microscopy (TEM) and high-resolution TEM images showing the agglomerated nanoparticles (c) of Li2O2 and the lattice fringes (d). | ||
The above results have convincingly validated the working principle of RFLOB. In order to assess the viability of the cell for long-term cycling, despite that it has yet been optimized and there are a few other critical issues to be addressed (such as the poor cyclability of the lithium anode), we tested the cycling performance of the above RFLOB cell at a controlled discharging capacity (Li2O2 to redox molecule ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A PVDF–Nafion composite membrane was employed to protect the lithium anode from being attacked by the redox mediators and dissolved O2 upon repeated striping and plating in a long cycling process. From the voltage profiles shown in Fig. 2d, relatively large overpotential was observed due to the large IR drop across the membrane. Interestingly, with increasing cycle number, the overpotential of the cell decreased gradually, largely a result of the reduced resistance of the membrane over cycling. There is no deterioration of charging capacity in the first 30 cycles. Instead, due to improved conductivity of the membrane, the charging capacity was even enhanced with the Coulombic efficiency reaching nearly 100% after the 10th cycle (Fig. 2d).
:1). A PVDF–Nafion composite membrane was employed to protect the lithium anode from being attacked by the redox mediators and dissolved O2 upon repeated striping and plating in a long cycling process. From the voltage profiles shown in Fig. 2d, relatively large overpotential was observed due to the large IR drop across the membrane. Interestingly, with increasing cycle number, the overpotential of the cell decreased gradually, largely a result of the reduced resistance of the membrane over cycling. There is no deterioration of charging capacity in the first 30 cycles. Instead, due to improved conductivity of the membrane, the charging capacity was even enhanced with the Coulombic efficiency reaching nearly 100% after the 10th cycle (Fig. 2d).
The above results provide compelling evidence and concertedly validate the functionality of RFLOB. That is, with the assistance of redox mediators, the discharging product Li2O2 could be remotely formed in the GDT tank and reversibly oxidized in the charging process without depositing onto the cathode inside the cell. Such decoupled reactions of Li2O2 provide great flexibility to circumvent the issues confronted by the conventional Li–O2 batteries. The surface passivation and pore clogging of the cathode resulting from Li2O2 precipitation, which are inevitable in conventional cells, are essentially avoided in the new operation mode. While the overpotential persists during the charging process, the intolerably large voltage hysteresis could in theory be mitigated by using suitable redox mediators even in the absence of electrocatalysts, which on the other hand is expected to also improve the cycling stability of the cell. In addition, the capacity of the cathode could be expanded by simply enlarging the size of the GDT tank, which is however constrained by the pore volume of the cathode and catalysts deposited on it in the conventional Li–O2 batteries. Moreover, as the reaction of O2 in GDT is far apart from the electrodes, the tolerance of the cell towards air would be enhanced as well.
While promising, to develop RFLOB into a viable device for advanced large-scale energy storage, the large voltage hysteresis would have to be further reduced. In the present study, the stagnant reaction between triiodide and Li2O2 and the resistive Li+-conducting membrane represent the main causes of the large overpotential during the charging process. Faster redox mediators with matching potential to the oxidation of Li2O2 are desired to expedite the reactions. In addition, optimization of the three-phase interface in the GDT tank to facilitate the reactions of O2 and Li+ and more effectively utilize the volume to accommodate Li2O2 is also required. Highly porous low-weight materials with good affinity to the deposition of Li2O2 and superior chemical resistance would be the ideal option. We are currently pursuing the above aspects to develop RFLOB into a low-cost and durable alternative to the Li–O2 batteries for large-scale energy storage applications.
This research was supported by the National Research Foundation, Prime Minister's Office, Singapore, under its Competitive Research Programs (CRP Award No. NRF-CRP8-2011-04 and NRF-CRP10-2012-06).
Notes and references
- E. L. Littauer and K. C. Tsai, J. Electrochem. Soc., 1976, 123, 771–776 CrossRef CAS PubMed.
- E. L. Littauer and K. C. Tsai, J. Electrochem. Soc., 1977, 124, 850–855 CrossRef CAS PubMed.
- K. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS.
- N. Imanishi, A. C. Luntz and P. Bruce, The Lithium Air Battery: Fundamentals, Springer, 2014 Search PubMed.
- M. Bhatt, H. Geaney, M. Nolan and C. Odwyer, Phys. Chem. Chem. Phys., 2014, 16, 12093–12130 RSC.
- Y. Chen, S. A. Freunberger, Z. Peng, F. Bardé and P. G. Bruce, J. Am. Chem. Soc., 2012, 134, 7952–7957 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J. M. Tarascon and P. G. Bruce, Angew. Chem., 2011, 123, 6475–6479 CrossRef PubMed.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- D. Kwabi, N. Ortiz-Vitoriano, S. Freunberger, Y. Chen, N. Imanishi, P. Bruce and Y. Shao-Horn, MRS Bull., 2014, 39, 443–452 CrossRef CAS.
- Y.-C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- M. M. O. Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef PubMed.
- B. Sun, X. Huang, S. Chen, P. R. Munroe and G. Wang, Nano Lett., 2014, 14, 3145–3152 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2012, 135, 494–500 CrossRef PubMed.
- J. Ming, W. J. Kwak, J. B. Park, C. D. Shin, J. Lu, L. Curtiss, K. Amine and Y. K. Sun, ChemPhysChem, 2014, 15, 2070–2076 CrossRef CAS PubMed.
- S. Liu, Y. Zhu, J. Xie, Y. Huo, H. Y. Yang, T. Zhu, G. Cao, X. Zhao and S. Zhang, Adv. Energy Mater., 2014, 4, 1301960 Search PubMed.
- A. K. Thapa and T. Ishihara, J. Power Sources, 2011, 196, 7016–7020 CrossRef CAS PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- H. D. Lim, H. Song, J. Kim, H. Gwon, Y. Bae, K. Y. Park, J. Hong, H. Kim, T. Kim and Y. H. Kim, Angew. Chem., Int. Ed., 2014, 53, 3926–3931 CrossRef CAS PubMed.
- D. Sun, Y. Shen, W. Zhang, L. Yu, Z. Yi, W. Yin, D. Wang, Y. Huang, J. Wang and D. Wang, J. Am. Chem. Soc., 2014, 136, 8941–8946 CrossRef CAS PubMed.
- W. Walker, V. Giordani, V. S. Bryantsev, J. Uddin, S. Zecevic, D. Addison and G. V. Chase, Meeting Abstracts, 2012.
- T. H. Yoon and Y. J. Park, RSC Adv., 2014, 4, 17434–17442 RSC.
- L. Yang, J. T. Frith, N. Garcia-Araez and J. R. Owen, Chem. Commun., 2014 Search PubMed.
- B. J. Bergner, A. Schürmann, K. Peppler, A. Garsuch and J. r. Janek, J. Am. Chem. Soc., 2014, 136, 15054–15064 CrossRef CAS PubMed.
- G. V. Chase, S. Zecevic, W. T. Wesley, J. Uddin, K. A. Sasaki, G. P. Vincent, V. Bryantsev, M. Blanco and D. D. Addison, US Pat., 20,120,028,137, 2012 Search PubMed.
- F. Pan, J. Yang, Q. Huang, X. Wang, H. Huang and Q. Wang, Adv. Energy Mater., 2014, 4, 1400567 Search PubMed.
- Q. Huang, H. Li, M. Gratzel and Q. Wang, Phys. Chem. Chem. Phys., 2013, 15, 1793–1797 RSC.
- Q. Wang, S. M. Zakeeruddin, D. Wang, I. Exnar and M. Grätzel, Angew. Chem., Int. Ed., 2006, 45, 8197–8200 CrossRef CAS PubMed.
- P. He, Y. Wang and H. Zhou, Electrochem. Commun., 2010, 12, 1686–1689 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Adv. Energy Mater., 2012, 2, 770–779 CrossRef CAS PubMed.
- M. J. Lacey, J. T. Frith and J. R. Owen, Electrochem. Commun., 2013, 26, 74–76 CrossRef CAS PubMed.
- H. Ota, K. Shima, M. Ue and J.-i. Yamaki, Electrochim. Acta, 2004, 49, 565–572 CrossRef CAS PubMed.
- L. El Ouatani, R. Dedryvere, C. Siret, P. Biensan, S. Reynaud, P. Iratcabal and D. Gonbeau, J. Electrochem. Soc., 2009, 156, A103–A113 CrossRef CAS PubMed.
- S. Oswald, D. Mikhailova, F. Scheiba, P. Reichel, A. Fiedler and H. Ehrenberg, Anal. Bioanal. Chem., 2011, 400, 691–696 CrossRef CAS PubMed.
- D. Ensling, M. Stjerndahl, A. Nytén, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2009, 19, 82–88 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; Fig. S1–S4, CV curves of two redox molecules at various scan rates, the charge and discharge curves of RFLOB in the absence of redox mediators in the electrolyte, XRD and SEM micrographs of the formed Li2O2 in a GDT tank. See DOI: 10.1039/c5cc01616a |
| This journal is © The Royal Society of Chemistry 2015 |