 Open Access Article
Open Access ArticleSimultaneous control of regioselectivity and enantioselectivity in the hydroxycarbonylation and methoxycarbonylation of vinyl arenes†
Tina M.
Konrad
a,
Jamie T.
Durrani
a,
Christopher J.
Cobley
b and
Matthew L.
Clarke
*a
aSchool of Chemistry, University of St Andrews, EaStCHEM, St Andrews, Fife, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463850
bChirotech Technology Ltd. Dr Reddys Laboratories (EU) Limited, 410 Cambridge Science Park, Milton road, Cambridge, UK CB4 0PE
First published on 5th March 2013
Abstract
Using a family of novel mononuclear and dinuclear palladium complexes of phanephos ligands, the simultaneous control of regioselectivity and enantioselectivity in the hydroxycarbonylation and alkoxycarbonylation of styrene derivatives has been realised for the first time.
Chiral carboxylic acids are key building blocks for the synthesis of chiral drugs and natural products. To be viable at larger scale, a synthesis of enantiomerically enriched small molecules needs to use cheap reagents. Hydroxycarbonylation is a reaction that converts a generally cheap class of starting material (an alkene) into an acid, using two of the cheapest chemicals known: water and carbon monoxide; in an idealised case there are no by-products.1,2 There are several processes to produce achiral or racemic acids (or esters by alkoxycarbonylation) at large scale.
Since the birth of asymmetric catalysis, attempts have been made to realise alkene carbonylation enantioselectively. Although some excellent research has helped understand the nature of this reaction, until recently enantioselective hydroxycarbonylation of the model substrate styrene always gave near racemic products, often with no control of regioselectivity and unfavourable reaction conditions.3 Enantioselective methoxycarbonylation of styrene has recorded better results, but none are really synthetically useful and the use of high temperatures with >1 equivalents of acid cocatalyst is common.4 In general monophosphines do control regioselectivity, but give low ee, while diphosphines give mainly linear products. A few papers report on branched-selective alkoxy-carbonylation of styrene with diphosphine species with electron deficient or bulky phosphorus centres;1h,2c,h,4a these show that this type of modification can improve branched selectivity. However, the only chiral catalysts in these studies gave 30% ee or less with a b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :l of 2:1 (or less) in methoxycarbonylation of styrene.4a
:l of 2:1 (or less) in methoxycarbonylation of styrene.4a
We recently discovered that Pd complexes of the Phanephos ligands gave high enantioselectivity in these reactions (up to 95% ee for norbornene; 80% ee for styrene).5 None-the-less, even these catalysts did not control regioselectivity in the hydroxycarbonylation of styrene. Since this class of reaction and the Phanephos ligands6 have been proven to be adaptable to commercial production, we have initiated a project aiming to transform these promising initial findings into a synthetically useful catalyst. Here we show that new phanephos ligands generate catalysts that display exquisite control of regioselectivity while maintaining good levels of enantioselectivity in both hydroxycarbonylation and methoxycarbonylation of styrenes.
Before embarking on a programme of making new catalysts, a range of other reaction variables were investigated for the hydroxycarbonylation of styrene using the Xyl-Phanephos–Pd system. These results are archived in the ESI† and can be simply summarised; the dipalladium species outperforms the monomeric catalysts significantly in terms of productivity, an acid at least as strong as trifluoroacetic acid is needed, and chloride is required for the higher enantioselectivity. Under any conditions examined, the regioselectivity is quite close to 1:1. A range of new Phanephos–Pd catalysts were therefore prepared (Scheme 1 and ESI†).
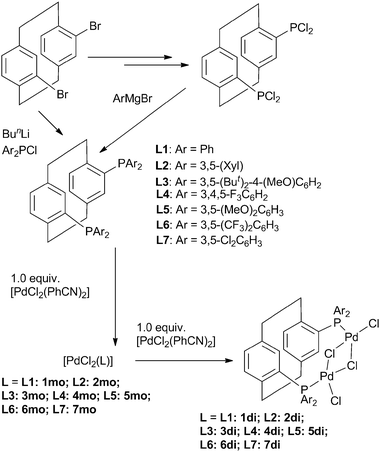 | ||
| Scheme 1 Synthesis of Phanephos ligands and their mononuclear and dinuclear palladium complexes. (S)-Phanephos derivatives are shown above, but in some cases both enantiomers were prepared. | ||
The ligands were either prepared using commercially available chlorophosphines or from the known precursor bis-dichlorophosphine paracyclophane6e The choice of meta-substitution patterns stemmed from repeatedly finding better results with meta-xylyl-Phanephos relative to Ph-Phanephos in our prior studies. The procedures reported reproducibly gave crude ligands of high purity, that were then fully purified in workable yields. The monomeric complexes are readily prepared in pure form. If these are treated with one further equivalent of [PdCl2(PhCN)2], then immediate formation of the dipalladium species was observed, or the dipalladium complexes are more conveniently prepared from two equivalents of [PdCl2(PhCN)2]. Some of the dipalladium complexes have sparing solubility, and complex 3di was completely insoluble in common solvents, possibly influencing its performance in catalysis. A family of 14 Phanephos-derived catalysts were available to test in the carbonylations. All the complexes are air stable for extended periods of time, and as testified by the catalytic application in water, are tolerant of moisture.
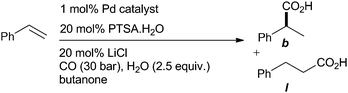
The new catalysts were then investigated in the enantioselective hydroxycarbonylation of styrene (Table 1 and Table S1, ESI†). It can be seen from Table 1 that all of the new functionalised catalysts give enhanced regioselectivity in hydroxycarbonylation relative to the parent Ph-Phanephos system. The results strongly suggest that steric bulk and electronegative substituents both have a separate positive effect on the branched regioselectivity.
| Entrya | Catalyst | Time (h) | % Acidb {yield} | B/Lb | eec,d % |
|---|---|---|---|---|---|
|
a Reactions were carried out according to table, equation and ESI unless stated otherwise. % Product determined against 1-methylnaphthalene internal standard using 1H-NMR spectroscopy.
b B/L ratio determined by 1H NMR. Conversion of alkene is identical or very similar to production of acid quoted. Yields in {} refer to yield of pure acid isolated after acid–base extraction.
c Enantiomeric excess determined by chiral HPLC.
d (R)-catalysts give (R) product and vice versa.
e Dipalladium species formed from monomer in situ.
|
|||||
| 1 | 1mo | 42 | 4 | 0.8 | 62 |
| 2 | 1di | 42 | 35 | 1.1 | 69 |
| 3 | 2mo | 42 | 8 | 0.4 | 50 |
| 4 | 2di | 42 | 71 | 1.1 | 80 |
| 5 | 3mo | 20 | 10 | 1.7 | 25 |
| 6 | 3di | 20 | 7 | 3.0 | 48e |
| 7 | 4mo | 20 | 98 | 6.0 | 50 |
| 8 | 4di | 20 | >99 | 16 | 55 |
| 9 | 5mo | 20 | 38 | 2.9 | 69 |
| 10 | 5di | 20 | >99 | 4.2 | 53 |
| 11 | 6mo | 24 | 44 {40} | 48 | 54 |
| 12 | 6di | 19 | 90 {82} | >100 | 77 |
| 13 | 7mo | 24 | 97 {91} | 20 | 60 |
| 14 | 7di | 24 | >99 {94} | 29 | 65 |
The most regioselective catalyst, which gave essentially a single regioisomer, was the dipalladium complex derived from the most electron withdrawing ligand, L6. This catalyst achieves similar ee values as the Xyl-Phanephos dipalladium catalyst, and is also more reactive than 2di. The somewhat insoluble catalysts derived from the bulkiest ligand, L3 displays relatively poor activity in these reactions and while some ligands display very significant improvements if introduced as a dipalladium species, this is not the case for the insoluble 3di. Extension of the reaction to some other vinyl arenes was carried out with (R)-6di. This was successful although slight variations in the reaction conditions were required. It may be worth noting that the more electron donating alkene gives measureable traces of the linear acid in this reaction, consistent with reduced levels of electron density on palladium being significant in the control of regioselectivity. All of the acid products could be isolated in pure form by simple acid–base extraction (Table 2).

| Entry | R group in substrate | T (°C) | Time (h) | % Acida [yield] | B/Lb | eec,d |
|---|---|---|---|---|---|---|
|
a Reactions were carried out according to equation and ESI unless stated otherwise. % Product determined against 1-methylnaphthalene internal standard using 1H-NMR spectroscopy. Yields in [] refer to yield of pure acid isolated after acid–base extraction.
b B/L ratio determined by 1H NMR.
c Enantiomeric excess determined by chiral HPLC.
d (R)-catalysts give (R) product and vice versa except where sign is given: (—) enantiomers are assigned as (R) by analogy.
|
||||||
| 1 | 4-Cl | 70 | 22 | >99 [93] | >100 | 66 |
| 2 | 4-Cl | 40 | 22 | 43 | >100 | 75 |
| 3 | 4-t-Bu | 40 | 22 | 40 [24] | 68 | 62 |
| 4 | 4-CO2H | 50 | 22 | 91 [74] | >100 | 66 (—) |
| 5 | 2-Cl | 60 | 22 | 89 [71] | 57 | 60 |
| 6 | 3-F | 60 | 22 | >99 [90] | 95 | 73 |
| 7 | 4-Ph | 60 | 22 | 95 [81] | 61 | 62 |
The alkoxycarbonylation of alkenes is also a potentially important reaction, since it produces esters in a single step without coupling agents. All the new catalysts were examined in methoxycarbonylation of styrene as a model reaction. A full table showing all the pre-catalysts under a variety of conditions can be found in the ESI.†Scheme 2 shows the most useful results. We were delighted to find that 6di delivers high yields of an essentially regiochemically pure branched ester with similarly good enantioselectivity as it does in hydroxycarbonylation, providing methanol is used as a reagent (2.5 equiv.), rather than solvent. The catalysts derived from ligand L4 also show very different behaviour in neat methanol to using a small excess (ESI†), with quite respectable 93% regioselectivity and 4:1 ratio of enantiomers (Scheme 2). The bulky ligand L3, which gave very poor catalysts in styrene hydroxycarbonylation, gives good reactivity near room temperature using 0.5% catalyst, reasonable control of regioselectivity (80%) and over 20:1 ratio of enantiomers. In this case, the more soluble monomeric catalyst is slightly more active than the (less soluble) dimer and the performance of the former is shown in Scheme 2.
 | ||
| Scheme 2 The leading catalysts for enantioselective and regioselective methoxycarbonylation of styrene. Results from a full catalyst screen can be found in the ESI.† | ||
A range of mono- and dinuclear palladium complexes of a family of Phanephos ligands have been prepared. Investigating this group of catalysts in the hydroxycarbonylation and methoxycarbonylation of styrene and a few of it's derivatives has uncovered a remarkable improvement in regioselectivity from around 1:1 in the parent Ph-substituted system to over 100:1. While the results referred to in the literature enabled us to predict some increase in branched selectivity using the fluorinated ligands, the ability to tune from 1:1 to essentially perfect regioselectivity, whilst retaining decent levels of enantioselectivity is completely unexpected. The parent system, 2di while giving good enantioselectivity delivers less than 50% yield of the desired enantiomer once conversion, regioselectivity and ee are taken into account, but 6di produces around 90% enantiomer yield in these reactions, not too dissimilar from the best examples of enantioselective hydroformylation.7 It is therefore hoped that asymmetric hydroxy- and methoxy-carbonylation of alkenes may be capable of becoming a practical reaction for scaleable asymmetric synthesis. Mechanistic studies, catalyst recycling, further optimisation of catalysts, and new synthetic applications seem to be the most pressing topics to be dealt with and investigations are underway.
JTD and TMK are joint major contributors to this research project. The authors thank Chirotech-Dr Reddys Laboratories and the European Union ITN, NANOHOST for funding. The contributions of the St Andrews technical staff and the EPSRC National Mass spectrometry Service are gratefully acknowledged.
Notes and references
- (a) I. del Rio, C. Claver and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 2001, 2719 CrossRef CAS; (b) H. Neumann, A. Brennführer and M. Beller, Adv. Synth. Catal., 2008, 350, 2437 CrossRef CAS; (c) A. Seayad, S. Jayasree and R. V. Chaudhari, Org. Lett., 1999, 1, 459 CrossRef CAS; (d) A. Ionescu, G. Laurenczy and O. F. Wendt, Dalton Trans., 2006, 3934 RSC; (e) M. S. Goedheijt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Chem. Commun., 1998, 2431 RSC; (f) J. J. R. Frew, M. L. Clarke, U. Mayer, H. van Rensburgh and R. P. Tooze, Dalton Trans., 2008, 1976 RSC; (g) A. Grabulosa, J. A. Fuentes, J. J. R. Frew, A. M. Z. Slawin and M. L. Clarke, J. Mol. Catal. A: Chem., 2010, 330, 18 CrossRef CAS; (h) J. J. R. Frew, M. L. Clarke, K. Damian, A. M. Z. Slawin, H. van Rensburgh and R. P. Tooze, Chem.–Eur. J., 2009, 15, 10504 CrossRef CAS and ref's therein.
- (a) W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. L. Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC; (b) C. Jimenez Rodriguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720 RSC. For related processes: (c) H. Ooka, T. Inoue, S. Itsuno and M. Tanaka, Chem. Commun., 2005, 1173 RSC; (d) R. I. Pugh, E. Drent and P. G. Pringle, Chem. Commun., 2001, 1476 RSC; (e) J. A. Fuentes, A. M. Z. Slawin and M. L. Clarke, Catal. Sci. Technol., 2012, 2, 715 RSC; (f) T. Fanjul, G. Eastham, M. F. Haddow, A. Hamilton, P. G. Pringle, A. G. Orpen, T. P. W. Turner and M. Waugh, Catal. Sci. Technol., 2012, 2, 937 RSC; (g) V. de la Fuente, M. Waugh, G. R. Eastham, J. a. Iggo, S. Castillon and C. Claver, Chem.–Eur. J., 2010, 16, 6919 CrossRef CAS; (h) L. Diab, M. Gouygou, E. Manoury, P. Kalck and M. Urrutigoïty, Tetrahedron Lett., 2008, 49, 5186 CrossRef CAS.
- (a) I. del Rio, N. Ruiz, C. Claver, L. A. van der Veen and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 2000, 161, 39 CrossRef CAS; (b) The selectivity of an enantioselective carbonylation using chiral phosphonic acid ligands seems to be difficult to reproduce and transfer to related reactions (H. Alper, personal communication). Other problems that preclude application are the use of >10% catalyst along with 3 promoters (CuCl2, HCl and oxygen); H. Alper and N. Hamel, J. Am. Chem. Soc., 1990, 112, 2803 CrossRef CAS; (c) 8–38% ee was realised in alkoxycarbonylations using the same system: Y. Kewu and J. Zuanzhen, Chem. J. Internet, 2005, 7, 14 Search PubMed; (d) M. Dolors Miqual-Serrano, A. Aghmiz, M. Diéguez, A. M. Madeu-Bultó, C. Claver and D. Sinou, Tetrahedron: Asymmetry, 1999, 10, 4463 CrossRef; (e) K. Damian and M. L. Clarke, unpublished results.
- (a) E. Guiu, M. Caporali, B. Muñoz, C. Muller, M. Lutz, A. L. Spek, C. Claver and P. W. N. M. Van Leeuwen, Organometallics, 2006, 25, 3102 CrossRef CAS; (b) C. Goddard, A. Ruiz and C. Claver, Helv. Chim. Acta, 2006, 89, 1610 CrossRef; (c) I. del Rio, N. Ruiz and C. Claver, Inorg. Chem. Commun., 2000, 3, 166 CrossRef CAS; (d) B. Muñoz, A. Marinetti, A. Ruiz, S. Castillon and C. Claver, Inorg. Chem. Commun., 2005, 8, 1113 CrossRef; (e) S. Oi, M. Nomura, T. Aiko and Y. Inoue, J. Mol. Catal. A: Chem., 1997, 115, 289 CrossRef CAS; (f) G. Cometti and G. P. Chiousoli, J. Organomet. Chem., 1982, 236, C31 CrossRef CAS; (g) B. C. Zhu and X. Z. Jiang, Appl. Organomet. Chem., 2006, 20, 277 CrossRef CAS; (h) C. Blanco, A. Ruiz, C. Godard, N. Fleury-Brégeot, A. Marinetti and C. Claver, Adv. Synth. Catal., 2009, 351, 1813 CrossRef CAS.
- (a) T. M. Konrad, J. A. Fuentes, A. M. Z. Slawin and M. L. Clarke, Angew. Chem., Int. Ed., 2010, 49, 9197 CrossRef CAS; (b) M. L. Clarke and T. M. Konrad, WO2011030110, University of St Andrews, 2009.
- (a) P. J. Pye, K. Rossen, R. A. Reamer, N. N. Tsou, R. P. Volante and P. J. Reider, J. Am. Chem. Soc., 1997, 119, 6207 CrossRef CAS; (b) P. J. Pye, K. Rossen, R. A. Reamer, R. P. Volante and P. J. Reider, Tetrahedron Lett., 1998, 39, 4441 CrossRef CAS; (c) M. J. Burk, W. Hems, D. Herzberg, Ch. Malan and A. Zanotti-Gerosa, Org. Lett., 2000, 2, 4173 CrossRef CAS; (d) B. Dominguez, A. Zanotti-Gerosa and W. Hems, Org. Lett., 2004, 6, 1927 CrossRef CAS; (e) A. Zanotti-Gerosa, Ch. Malan and D. Herzberg, Org. Lett., 2001, 3, 3687 CrossRef CAS.
- Some of the most regioselective enantioselective hydroformylation catalysts for styrene and related substrates: (a) T. P. Clark, C. R. Landis, S. L. Freed, J. Klosin and K. A. Abboud, J. Am. Chem. Soc., 2004, 127, 5040 CrossRef; (b) G. M. Noonan, J. A. Fuentes, C. J. Cobley and M. L. Clarke, Angew. Chem., Int. Ed., 2012, 51, 2477 CrossRef CAS (see also WO2012016147); (c) A. T. Axtell, C. J. Cobley, J. Klosin, G. T. Whiteker, A. Zanotti-Gerosa and K. A. Abboud, Angew. Chem., Int. Ed., 2005, 44, 5834 CrossRef CAS; (d) N. Sakai, S. Mano, K. Nozaki and H. Takaya, J. Am. Chem. Soc., 1993, 115, 7033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and analytical data. See DOI: 10.1039/c3cc41291a |
| This journal is © The Royal Society of Chemistry 2013 |