
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,2-Dithiolane/yne photopolymerizations to generate high refractive index polymers†
Marianela
Trujillo-Lemon
a,
Benjamin D.
Fairbanks
a,
Andrew N.
Sias
b,
Robert R.
McLeod
bc and
Christopher N.
Bowman
 *bc
*bc
aDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80303, USA. E-mail: Christopher.Bowman@colorado.edu
bDepartment of Electrical, Computer, and Energy Engineering, University of Colorado, Boulder, CO 80309, USA
cMaterials Science and Engineering Program, University of Colorado, Boulder, CO 80303, USA
First published on 3rd February 2025
Abstract
Copolymerization and conjugate addition of disulfides generally and dithiolanes particularly have been reported for various applications. Here, a new framework for preparing high refractive index polymeric materials through the photoinitiated addition of methyl ester of lipoic acid (LipOMe) or methyl 4-methyl-1,2-dithiolane-4-carboxylate (Me-AspOMe) with various alkynes is explored, and an infrared spectroscopy methodology was developed for understanding the dithiolane homopolymerization kinetics. The effects of the 1,2-dithiolane and alkyne chemical structures on reaction rates, polymer structures, and optical properties of the synthesized polymers were examined. Characterization of the photopolymerization products showed significant dependence on the specific structure of the 1,2-dithiolane and alkyne reactants. The ability of the 1,2-dithiolane/alkyne reaction to introduce a large amount of sulfide linkages resulted in differences in the polymer refractive index relative to that of the unreacted materials, reaching values up to 0.07. Furthermore, the application of these 1,2-dithiolane-alkyne systems into two-stage photopolymeric holography materials in a two-dimensional, high-refractive index structure was demonstrated.
1. Introduction
High-refractive index polymers (HRIPs) play a crucial role in many optical and optoelectronic technologies, including lenses, waveguides, nanoimprinting lithography, and holography.1–4 With the increasing advances in optical devices, the demand for high refractive index, high thermal stability, high transparency, and low birefringence polymers has increased accordingly. Practically, two common ways to achieve high refractive indices polymers include: (1) introducing high refractive index inorganic nanoparticles in the polymer matrix5–8 (2) introducing atoms or groups with high intrinsic molar refractions.3,9–13 Both nanocomposites and intrinsically high refractive index polymers offer advantages and disadvantages. While high refractive index nanocomposite polymers have a refractive index above 1.8, the nanoparticle type, size, and content can negatively affect the storage stability and processability.9 Alternative, intrinsically high refractive index polymers usually reach refractive indices below 1.8 but offer advantages of lower cost, high stability, and ease of processability. Many factors affect high refractive index polymer performance including molecular packing, molar volume, chain flexibility, and substituent content. The main strategy to develop intrinsically high refractive index polymers is the incorporation of high molar refraction groups such as sulfur,14 phosphorous,15 bromine, iodine,16 and/or aromatic structures.17 Due to its high polarizability, high stability, and straightforward insertion in the molecular structure, sulfur has attracted particular interest in increasing the refractive index. In recent years, various high refractive index polymers containing sulfur have been synthesized including poly (phenylene thioether)s, poly(thioether sulfone)s, thioether-bridge poly(phenyl quinoxaline)s,18–21 and polythiophenes.22–24 Unfortunately, these high refractive index sulfur-containing materials frequently require complex and elaborate synthesis, exhibit poor processability, and show low transparency, making them unsuitable for optical applications where large-scale production and/or thick films are required.Thiol–ene chemistry, which intrinsically forms high refractive index thioether linkages, has been explored for holography applications.12,25,26 However, despite the benefits of low shrinkage, reduced oxygen inhibition, and delayed vitrification,27 the nature of the reaction limits its high refractive index capabilities since each ene can bond to only one sulfur in the thiol–ene coupling. This limits the introduced sulfur content and, thereby, the ability to enhance the refractive index of such polymers via polymerization.
Another convenient route toward HRIPs consists of the radical-mediated addition of thiol to alkynes. Similar in mechanism to radical thiol–ene coupling, thiol–yne adds two thiols per alkyne to form 1,2-dithioether while preserving the advantages of the alkene-based reaction.28,29 Bader and coworkers’ early work on the reaction between thioacetic acid and various alkynes in the presence of peroxides demonstrated that the ability to obtain the mono- and disubstituted adducts depended on the alkyne structure.30 Blomquist and Wolinsky described a study on the addition of ethyl mercaptan to a range of acetylenic species under UV conditions.31 Bowman and Hoyle's groups demonstrated the bireactivity of terminal alkynes with thiols during the radical-mediated thiol–yne step-growth photopolymerization to form highly crosslinked polymer networks where each alkyne functional group was capable of reacting consecutively with two thiols.32,33 In addition, Chan et al. reported that the reaction of a series of dialkynes and dithiols rapidly produces highly crosslinked networks with the resulting refractive index values directly dependent on the sulfur content. Recently, Li et al. provided new insight into disulfide–yne photoreaction, demonstrating that unlike the stable product generated from thiol–yne chemistry, the vinyl dithioether structure obtained from the disulfide–yne reaction exhibits unique reactivity toward thiols and disulfides.34
Another important property that substantially influences the performance of optical materials is the volumetric shrinkage and the associated stress. These are known to affect the optical properties of the materials and cause both stress and birefringence.35,36 Volumetric shrinkage occurs during curing reactions since the van der Waals distance of two molecules is shortened to the length of a (significantly shorter) covalent bond.37
A particular class of sulfur-containing molecules 1,2-dithiolanes, which contain a saturated five-membered disulfide ring, has shown remarkable potential for the development of intrinsically high refractive index polymers with low volumetric shrinkage due to its ability to participate in a variety of radical and ionic ring-opening reactions.38 Each reaction yields thiolate or sulfur-centered radicals all of which initiate ring-opening oligomerization or polymerization with another cyclic disulfide producing disulfide-rich backbone of poly(dithiolane)s Scheme 1.39,40
 | ||
| Scheme 1 Schematic representation of the change in refractive index from monomer to polymer in 1,2-dithiolane monomers and polymers. | ||
Among the 1,2-dithiolane molecules, lipoic and asparagusic acids (naturally occurring molecules) are the best-known members of this group. The ring-opening reaction of 1,2-dithiolanes occurs thermally and photochemically, even without an initiator Fig. 1.41 Early work by Calvin and coworkers mentions the polymerization of lipoic acid using UV irradiation without the need for any photoinitiator,39 an interesting feature recently used to prepare photocurable hydrogels without a photoinitiator.42
 | ||
| Fig. 1 Thermal or light-activated homopolymerization of the dithiolanes (a) LipOMe and (b) Me-AspOMe. | ||
The ability of cyclic disulfides to participate in copolymerization reactions with vinyl monomers upon heating or irradiation was established around the 1950s. Lipoic acid (LA) copolymerizes with alkenes, including styrene and vinyl acetate.43,44 Endo and co-workers studied the thermally triggered radical copolymerization of lipoamide with various vinyl monomers and established a reactivity order. Lipoamide copolymerization with acrylate was readily demonstrated; however, no copolymerization was observed with methacrylates.45 Leveraging this feature allowed for the synthesis of a methacrylate functionalized 1,2-dithiolane monomer to produce methacrylate-based polymers with pendant 1,2-dithiolane moieties.46 Recently, lipoic acid and diallyl isosorbide were polymerized in the presence of a photoinitiator to produce UV-cured thiol–ene coatings47 and to permanently crosslink hydrogels with the formation of irreversible thioethers bonds.48
While dithiolane–ene reactions have been explored, dithiolane–yne polymerization reactions have yet to be reported, so the feasibility of 5-membered cyclic disulfides (LipOMe and Me-AspOMe) to copolymerize with various alkynes (phenylacetylene, 1-octyne, propargyl acetate, and diphenylacetylene) affording polymers with disulfide-rich backbones is demonstrated here. Fig. 2 shows chemical structures, acronyms, names for the dithiolanes, and refractive indices of the materials used during this research. Photopolymerization kinetics and comparison of the curing rates of various compositions and stoichiometric ratios were evaluated together with the properties of the obtained polymers.
 | ||
| Fig. 2 Chemical structures, acronyms, names, and refractive indices for 1,2 dithiolane and alkyne monomers used in this study. Refractive index measurements were taken at the wavelength (λ) of 589 nm using an Abbe refractometer. | ||
Although 1,2-dithiolanes have been studied since the 1950s, to our knowledge, their copolymerization with alkynes, research concerning the refractive indices of the 1,2-dithiolane monomers and their polymers, and evaluation as optical materials have not been addressed until now.
2. Results and discussion
2.1. Development and implementation of an FTIR-based technique for the homopolymerization kinetics of LipOMe and Me-AspOMe
The methyl ester of lipoic acid (LipOMe) was synthesized following an analogous procedure to the one previously published (S1).49 The synthesis was carried out using a commercially available α-lipoic acid. At the same time, the methyl 4-methyl-1,2-dithiolane-4-carboxylate (Me-AspOMe) was synthesized in a two-step procedure that involved the preparation of methyl-asparagusic acid (S2), and then the methyl ester product (S3).50 Homopolymers of the methyl ester of lipoic acid (LipOMe) and methyl 4-methyl-1,2-dithiolane-4-carboxylate (Me-AspOMe) were obtained via dithiolane ring opening photopolymerization by irradiating in bulk the low viscosity monomers containing 1 wt% TPO as the photoinitiator with 400–500 nm filtered mercury arc lamp at 40 mW cm−2 for 600 s. For the first time, FTIR was demonstrated to be suitable for monitoring the homopolymerization kinetics of the dithiolane unit of these two monomers. ESI, Fig. S4a and S4b† show the FTIR spectra region between 800 and 500 cm−1 for LipOMe and Me-AspOMe before and after polymerization obtained by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) spectroscopy using the smart ITX accessory shown in Fig. S5,† here a drop of LipOMe/or Me-AspOMe were placed over the diamond ATR crystal and covered with a glass slide, and the corresponding before and after polymerization spectra of the in situ polymerization were collected using 64 scans and 4 cm−1 resolution. It's important to mention that the ATR-FTIR with the IXT accessory allowed for the collection of spectra from 4000 to 500 cm−1, and the background obtained with the smart accessory is quite stable due to the controlled environment. High degrees of conversion, 92% (LipOMe) and 75% (Me-AspOMe) were determined by measuring the intensity of the peak area corresponding to C–S stretching at 676 cm−1 for LipOMe and 671 cm−1 for Me-AspOMe before and after polymerization. The FTIR technique and the corresponding conversions for the two 1,2-dithiolanes under study were validated by 1H NMR (32 scans, d1 = 10 s) by comparing the integrals of the polymer peak and monomer peak using the signals for the CH2 hydrogen in the dithiolane ring. For LipOMe, the peak used for conversion calculations for the unreacted monomer was found at 2.50 ppm, and for the polymer at 2.80 ppm (Fig. S6a†), while for the Me-AspOMe monomer, the peak chosen was at 3.70 ppm (or 2.97 ppm) for the monomer and at 3.17 ppm for the polymer (Fig. S6b†). Table S1, in ESI,† summarizes, among other properties, conversion results obtained for ATR-FTIR and 1H NMR, showing agreement between the two techniques and verifying that the FTIR technique is suitable to monitor the ring-opening of the 1,2-dithiolane during homopolymerizations.The reversible nature of the ring-opening polymerization reaction for LipOMe and Me-AspOMe was studied by ATR-FTIR, monitoring the recovery of the monomer peaks at 676 cm−1 and 671 cm−1 for LipOMe and Me-AspOMe after heating the polymer samples at 140 °C for 3 hours (Fig. S7†). The percentages of LipOMe and Me-AspOMe recovered after heating the respective polymers under the same conditions were 84% for LipOMe and 10% for Me-AspOMe evidencing the more stable nature of poly(Me-AspOMe) toward depolymerization, compared to poly(LipOMe). It may seem counterintuitive that the monomer that exhibits lower reaction rates would also yield lower recovery upon depolymerization. It must be noted that it cannot be assumed that equilibrium, as governed by the thermodynamics of the system, will correlate with kinetic reaction rates. Alternative experimental conditions may prove more or less favorable to the asparagusate monomer recovery. Table S2† summarizes the conversion values obtained from peak area values for the initial monomers, the polymer following photopolymerization, and the sample after heating using eqn (1):
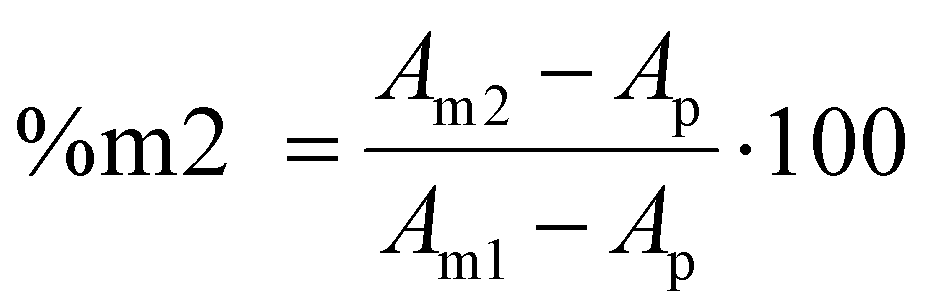 | (1) |
Time-dependent photopolymerization reaction kinetics were also measured by FTIR spectroscopy during the photopolymerization using a previously reported in-house fabricated device described in Fig. S8.†51 The horizontal accessory allowed for mounting the sample between KBr discs in a horizontal orientation and irradiating it from an almost perpendicular position without interfering with the IR beam through the use of optical mirrors. The polymerization rate was calculated from the rate at which the monomer is being consumed using the equation Rp = −d[M]/dt, where Rp is the rate of polymerization, [M] concentration of monomer at any given time, and d[M]/dt is the rate of decrease in monomer concentration with respect to time. Polymerization delay was observed for Me-AspOMe but not for LipOMe (Fig. 3), demonstrating a difference in reactivity for the 1,2-dithiolanes. As expected, LipOMe reacted 12 times faster (Rp = 0.05 s−1) than Me-AspOMe (Rp = 0.006 s−1). This data correlates with ring substitution patterns being determined to be an important factor in the reactivity of 1,2-dithiolanes due to ring strain, which was found to be more significant for LipOMe than Me-AspOMe.50 Earlier, Whiteside and coworkers showed that higher-substituted 1,2-dithiolanes are more resistant to reduction and ring-opening.52
 | ||
| Fig. 3 Real-time measurement of conversion as a function of time for LipOMe (black squares) and Me-AspOMe (red circles) showing that LipOMe reacts 12 times faster than Me-AspOMe upon exposure filtered 450 ± 50 nm light at an intensity of 40 mW cm−2 (1 wt% TPO photoinitiator). The dotted line indicates when the light was turned on at 30 seconds. Delayed polymerization was observed for Me-AspOMe. | ||
A modular gel permeation chromatography (GPC/SEC) system equipped with a refractive index detector was employed to determine the weight average molecular weight (Mw), number average molecular weight (Mn), and dispersity Đ = Mw/Mn of the photopolymer obtained based on polystyrene standards. LipOMe and Me-AspOMe homopolymers show molecular weights of 14 kDa and 12 kDa with a relatively narrow dispersity of Đ = 1.21 and 1.23, respectively (Table S1†). Both polymers were soluble in commonly used organic solvents. The index of refraction of the synthesized monomers and their polymers was measured at 20 °C using an Abbe refractometer (589 nm). Table S1† shows that the ring-opening polymerization of the 1,2-dithiolanes increases the refractive index by 0.02 for LipOMe and 0.04 for Me-AspOMe. The high refractive index values of the polymers obtained are a direct result of the high inherent atomic refraction of the sulfur-containing polymers.
The higher Δn value for Me-AspOMe is not a surprise as the refractive index is predicted by the Lorentz–Lorenz equation (eqn (2)):
 | (2) |
2.2. Copolymerization of LipOMe with alkynes
Having demonstrated the effect of the homopolymerization of 1,2-dithiolanes on polymer characteristics and optical properties of their polymers, the monomers were then copolymerized with alkynes. The consumption of the alkynes as a function of time during photopolymerization with LipOMe at various stoichiometric ratios was monitored using Fourier Transform Infrared (FTIR) spectroscopy with the horizontal device accessory previously described.51 The peaks corresponding to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H stretching absorption, at 3301, 3313, and 3294 cm−1 for phenylacetylene, 1-octyne, and propargyl acetate, were selected to track the consumption of alkynes during polymerization (Fig. 4a–d). In the absence of CC–H stretching for diphenylacetylene, the most prominent band of the alkynes in the IR spectrum centered at 2219 cm−1, which is associated with CC, was used for the internal alkyne. Even though the CC stretching (∼2200 cm−1) appears as a weak band, the resultant noise was not problematic as the standard deviations were consistent with other trials (Fig. 4d). Unfortunately, the ring-opening reaction of the 1,2-dithiolane ring could not be monitored simultaneously because the corresponding peak areas for C–S overlap with the absorption peaks of components in the reaction mixtures; however, they were confirmed by 1H NMR on the same samples analyzed by FTIR. The corresponding conversions reached for the LipOMe during copolymerization with alkynes relate to the reaction with the alkynes as well as homopolymerization (Table S3†). Fig. 4 and Table S3† display conversion versus time for the different compositions and ratios, final alkyne conversion (by FTIR), calculated reaction rates, and dithiolane conversion obtained from the 1H NMR. The rapid consumption of alkynes occurs in the first seconds of the reaction. Moreover, a higher degree of alkyne conversions is observed at higher LipOMe concentrations (dithiolane
C–H stretching absorption, at 3301, 3313, and 3294 cm−1 for phenylacetylene, 1-octyne, and propargyl acetate, were selected to track the consumption of alkynes during polymerization (Fig. 4a–d). In the absence of CC–H stretching for diphenylacetylene, the most prominent band of the alkynes in the IR spectrum centered at 2219 cm−1, which is associated with CC, was used for the internal alkyne. Even though the CC stretching (∼2200 cm−1) appears as a weak band, the resultant noise was not problematic as the standard deviations were consistent with other trials (Fig. 4d). Unfortunately, the ring-opening reaction of the 1,2-dithiolane ring could not be monitored simultaneously because the corresponding peak areas for C–S overlap with the absorption peaks of components in the reaction mixtures; however, they were confirmed by 1H NMR on the same samples analyzed by FTIR. The corresponding conversions reached for the LipOMe during copolymerization with alkynes relate to the reaction with the alkynes as well as homopolymerization (Table S3†). Fig. 4 and Table S3† display conversion versus time for the different compositions and ratios, final alkyne conversion (by FTIR), calculated reaction rates, and dithiolane conversion obtained from the 1H NMR. The rapid consumption of alkynes occurs in the first seconds of the reaction. Moreover, a higher degree of alkyne conversions is observed at higher LipOMe concentrations (dithiolane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :yne = 4:1 > 2:1 > 1:1 > 1:2) for all the terminal alkynes. It is essential to mention that, for the same stoichiometric ratios, the order of reaction rates observed for the different terminal alkynes studied here follows the trend of phenylacetylene > 1-octyne > propargyl acetate (Table S3†). The slow addition rate of LipOMe to propargyl acetate is rationalized in terms of electron density, like in the case of thiol–ene reactions, where photoaddition of thiols to electron-poor vinyl groups is unfavorable.54 Here, the thiyl radicals known for their electrophilic character react slowly with propargyl acetate as the electron-withdrawing carbonyl decreases the electron density of the π bonds. This effect was also observed for the photoinitiated addition of thiols to propargyl acetate and methyl propargyl ether.54,55 It is important to mention that while no homopolymerization of alkynes was observed in the absence of dithiolane (Fig. S9†), higher than anticipated alkyne consumption, presumably due to chain addition reactions between alkynes and unsaturated carbon–carbon bonds (i.e. alkynes, vinyl sulfides), was observed at low relative ratios of dithiolane, as monitored by FTIR and 1H NMR(Table S3†). Similar behavior was reported in thiol-limited thiol–yne addition reactions.32
:yne = 4:1 > 2:1 > 1:1 > 1:2) for all the terminal alkynes. It is essential to mention that, for the same stoichiometric ratios, the order of reaction rates observed for the different terminal alkynes studied here follows the trend of phenylacetylene > 1-octyne > propargyl acetate (Table S3†). The slow addition rate of LipOMe to propargyl acetate is rationalized in terms of electron density, like in the case of thiol–ene reactions, where photoaddition of thiols to electron-poor vinyl groups is unfavorable.54 Here, the thiyl radicals known for their electrophilic character react slowly with propargyl acetate as the electron-withdrawing carbonyl decreases the electron density of the π bonds. This effect was also observed for the photoinitiated addition of thiols to propargyl acetate and methyl propargyl ether.54,55 It is important to mention that while no homopolymerization of alkynes was observed in the absence of dithiolane (Fig. S9†), higher than anticipated alkyne consumption, presumably due to chain addition reactions between alkynes and unsaturated carbon–carbon bonds (i.e. alkynes, vinyl sulfides), was observed at low relative ratios of dithiolane, as monitored by FTIR and 1H NMR(Table S3†). Similar behavior was reported in thiol-limited thiol–yne addition reactions.32
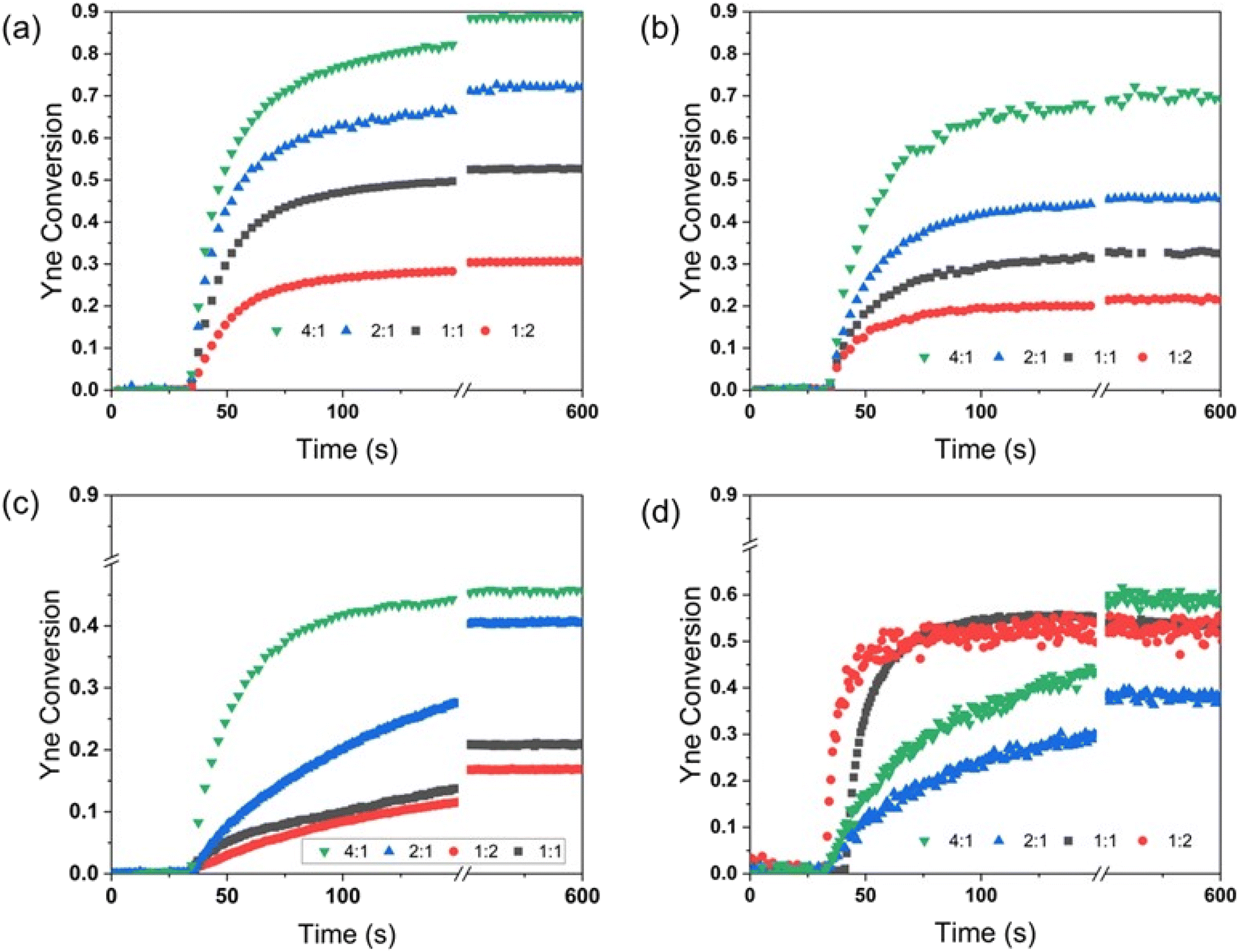 | ||
| Fig. 4 FTIR conversion versus time plots for alkyne reactive groups (λ = 405 nm, I = 40 mW cm−2, t = 600 s). (a) LipOMe:phenylacetylene, (b) LipOMe:1-octyne, (c) LipOMe:propargyl acetate, and (d) LipOMe:diphenylacetylene. Black squares (dithiolane:yne = 1:1), red circles (dithiolane:yne = 1:2), blue triangles (dithiolane:yne = 2:1), green triangles (dithiolane:yne = 4:1). | ||
The results obtained for the internal alkyne were unexpected in that at higher concentrations of diphenylacetylene 1:1 and 1:2 (dithiolane:yne) ratios (Fig. 4d), the final yne consumption reached values of 53% and 52%, respectively, which is similar to that found for the 4:1 ratio of dithiolane:yne (56%). Furthermore, the cure rates obtained exceed that of formulations with lower ratios of diphenylacetylene and even the ones of terminal alkynes. A more careful inspection of the KBr windows for the formulations before and after polymerization showed the presence of an opaque waxy solid for the higher concentrations of diphenylacetylene. The observed results may suggest a level of ordering or templating in the crystalline state, significantly enhancing the propagation reaction while minimizing termination, a phenomenon that has been reported previously.56,57 All of the conclusions drawn from this research correlate to liquid formulations, as the observed formation of the waxy/crystalline state for the 1:1 and 1:2 ratios (LipOMe:diphenylacetylene) will require further and deeper studies outside the scope of this publication.
The copolymerization of LipOMe with terminal alkynes studied resulted in the emergence of a new peak at 1545 cm−1 in the FT-IR spectrum and an approximate chemical shift of 6.0 ppm in the 1H NMR spectrum (Fig. S10†). This observation indicates the formation of a vinyl sulfide intermediate, similar to the one observed in the case of thiol–yne photopolymerization.32 However, the shift to higher frequencies for LipOMe-yne, compared to thiol–yne—which typically shows peaks in the FT-IR spectrum between 1620 and 1600 cm−1—suggests that the vinyl sulfide intermediate found during this research has a distinct structure. A more detailed discussion on the formation of the vinyl sulfide intermediate will follow in the next section.
GPC results of the polymerized comonomer mixtures are presented in Table S4.† Interestingly, the polymers obtained through this route are characterized by narrow dispersity Đ < 1.4. The index of refraction for 1,2-dithiolane and alkyne mixtures before and after polymerization are summarized in Table S4.† It is important to mention that a portion of 1,2-dithiolane monomer and its homopolymers are left in the mix after polymerization, so it is expected that the refractive index R.I. here showed are lower than expected due to incomplete polymerization and homopolymers formed.
2.3. Copolymerization of Me-AspOMe with alkynes
Polymerization reactions of Me-AspOMe with alkynes were also studied using FTIR using the horizontal attachment. Similar to LipOMe, the FTIR spectra for the photopolymerization reactions of Me-AspOMe with alkynes showed, as expected, a decrease of the peaks at ∼3281 and ∼2110 cm−1 corresponding toC–H and CC stretching upon polymerization (Fig. S11a†). Also, as expected, the appearance of the vinyl sulfide peak at 1530 cm−1 (Fig. S11b†) was observed at an even higher frequency than the one observed for the vinyl sulfide derivate of LipOMe (which was located around 1545 cm−1).
The photopolymerization of Me-AspOMe with alkynes highlighted differences in the reactivity between the two dithiolanes studied (Fig. S12 and Table S5†). The final conversion reached for the two dithiolanes resulted in values within the same order of magnitude. At the same time, the rates of polymerization for Me-AspOMe were reduced by a factor of two for the alkynes studied here. In general, the Me-AspOMe conversions obtained from 1H NMR analysis for the copolymerization products showed values that indicate the reaction of the dithiolane with alkynes was favored over dithiolane homopolymerization. This is in contrast to the behavior observed for LipOMe:yne systems, wherein dithiolane homopolymerization was significant during the mixed mode copolymerizations.
Table S5† shows GPC results for Me-AspOMe and alkynes studied. Surprisingly low molecular weights for the copolymerization of Me-AspOMe and alkynes were obtained despite the high degree of conversion (Table S6†) and changes in refractive indices from resin to polymer, which reached values of 0.06 for the Me-AspOMe:phenylacetylene 4:1, 2:1, and 1:1 ratio. The reaction products for the composition Me-AspOMe:phenylacetylene in a 2:1 ratio were separated using a Biotage automatic chromatography column instrument with hexane:ethyl acetate (80:20) as eluent, two fractions were obtained and analyzed by 1H and 13C NMR. As expected for polymeric structures, the 1H NMR of one minority product isolated (Fig. S13†) is quite complex and shows broad signals. Peaks found between 8.0–7.5 ppm corroborated the presence of phenyl rings, while signals at 3.7–3.5 ppm, 3.3–3.0 ppm, and 1.5–1.2 ppm ranges correspond to CH2 and CH3 groups associated with the opening of the dithiolane ring. It is noteworthy to mention the peak around 6.5 ppm, which is attributed to the presence of a vinyl sulfide unit within the polymer chain. The obtained integration of phenyl groups to methyl groups indicates a polymeric structure with one phenyl group per two disulfide groups and a molecular weight of ∼13 kDa (Table S5†). We suggest that the mechanism of incorporation of an alkyne into a dithiolane polymer involves the propagation of a single thiyl radical within a growing polydisulfide across the triple bond, resulting in a carbon-centered radical that adds to another dithiolane molecule (Scheme 2a). The 1H NMR in CDCl2 (Fig. S14†) of the second product isolated showed aside from the presence of unreacted Me-AspOMe, a singlet centered at 6.09 ppm, which is the region of 1H NMR spectra associated with vinylic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) protons, indicating the potential addition of one dithiolane to one yne. Furthermore, the presence of 5 aromatic protons in the range of 7.51–7.29 ppm, signals at 3.97–3.92 (m), 3.74 (s), 3.543.48 (m), and 1.44 (s) together with the corresponding 13C NMR are consistent with the mono-addition product (intramolecular cyclization), indicating the presence of the vinyl sulfide [6-methyl,6′-methylesther-2-phenyl-5,7dihydro-1,4-dithiepin] with a chemical structure similar to the one shown on Scheme 2b. Findings were verified by LC-MS (Fig. S15†) as the mass for charge ratio obtained m/z: 280.06 (41.6%), 281.07 (M+, 100%), 282.07 (20.8%), 283.06 (10.4%), 283.14 (3.2%) matches the expected values for the proposed chemical structure.
CH) protons, indicating the potential addition of one dithiolane to one yne. Furthermore, the presence of 5 aromatic protons in the range of 7.51–7.29 ppm, signals at 3.97–3.92 (m), 3.74 (s), 3.543.48 (m), and 1.44 (s) together with the corresponding 13C NMR are consistent with the mono-addition product (intramolecular cyclization), indicating the presence of the vinyl sulfide [6-methyl,6′-methylesther-2-phenyl-5,7dihydro-1,4-dithiepin] with a chemical structure similar to the one shown on Scheme 2b. Findings were verified by LC-MS (Fig. S15†) as the mass for charge ratio obtained m/z: 280.06 (41.6%), 281.07 (M+, 100%), 282.07 (20.8%), 283.06 (10.4%), 283.14 (3.2%) matches the expected values for the proposed chemical structure.
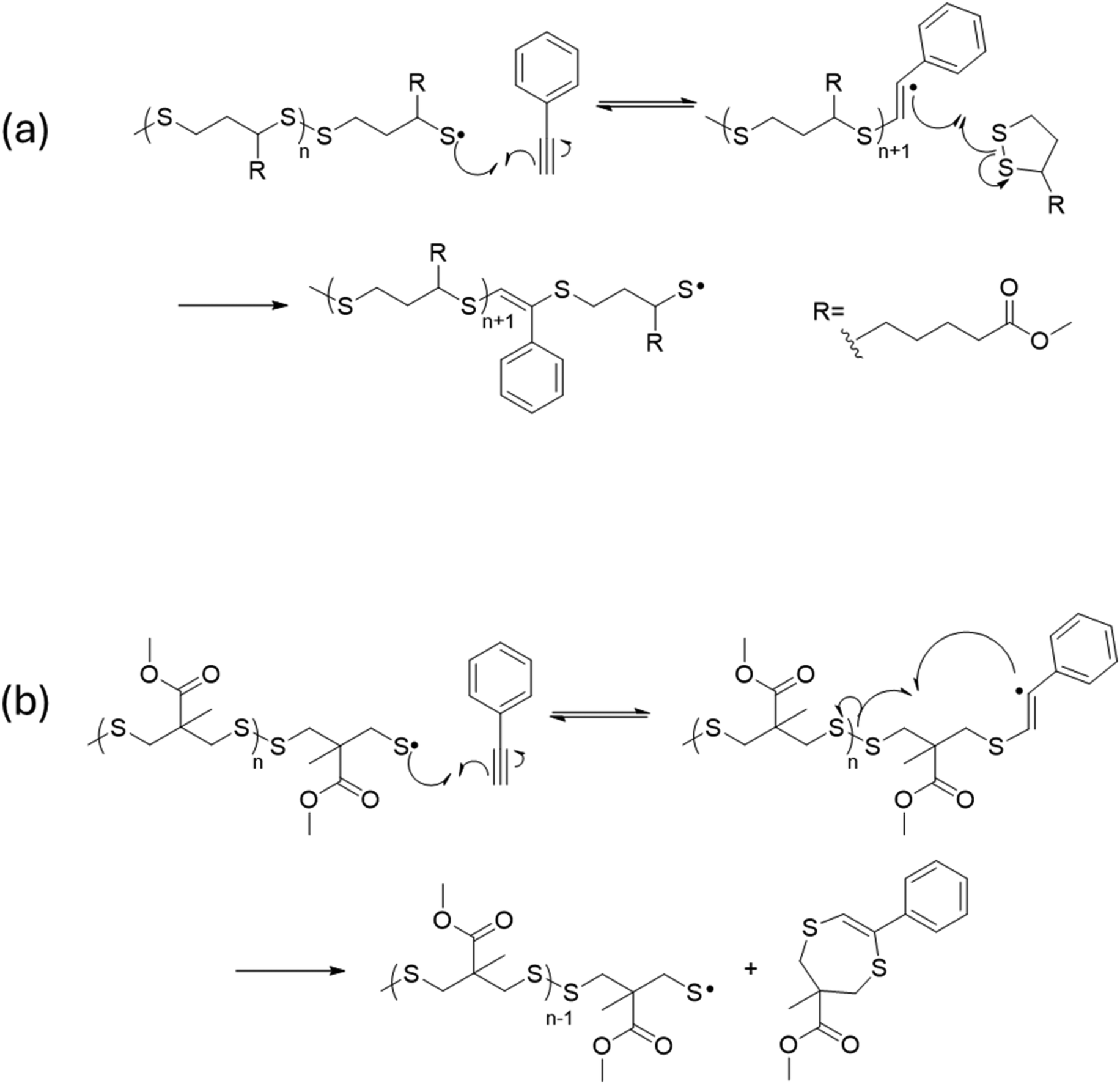 | ||
| Scheme 2 (a) Mechanism for copolymerization of alkynes and 1,2-dithiolanes via propagation of thiyl radical across triple bond and subsequent radical addition to new dithiolane. (b) Mechanism for the formation of isolated cyclic vinyl sulfide via intramolecular radical disulfide scission. | ||
The 0.4 ppm difference in the chemical shift of the vinyl proton (CCH) between the polymer (6.5 ppm) and the vinyl sulfide (dithiepin) (6.1 ppm) is not unexpected. This variation is likely due to the positioning of the vinyl proton in relation to the phenyl ring. In the isolated dithiepin, the vinyl proton is positioned cis to the phenyl ring. Thus, the vinyl proton appears at a slightly higher field, meaning its chemical shift is lower due to the shielding effect of the aromatic ring when compared to the vinyl proton trans to the phenyl ring in the proposed mechanism for polymer formation.
Vinyl sulfides (dithiepins) analogous to the one obtained here, have been reported for the reaction of 4,4-disubstituted 1,2-dithiolanes derivatives (polymerization-resistant) with acetylenide anions in tert-butyl alcohol in the presence of catalytic amounts of tert-BuOK58 or lithium butyl acetylenide which gave the corresponding ring-opening products that isomerize to vinylene-insertion products in protic solvents.59 We propose that the most likely mechanism for forming the isolated vinyl sulfide (dithiepin) is analogous to the one described in Scheme 2b. Just like before, a carbon-centered radical is formed by the propagation of a single thiyl radical within the growing polydisulfide across the triple bond, but in this case, the radical undergoes chain transfer, kicking out a thiyl radical and forming the vinyl sulfide.
Other alkynes studied here are presumed to add similarly to LipOMe and Me-AspOMe, as a closer observation of their FT-IR spectra during copolymerization showed the vinyl sulfide peak around 1530 and 1545 cm−1, respectively.
2.4. Holographic recording using 1,2-dithiolane-yne chemistry in a two-stage photopolymer
After investigating the photopolymerization reactions involving 1,2-dithiolanes (LipOMe or Me-AspOMe) and alkynes and having identified several intriguing properties, including rapid polymerization kinetics, medium to high final conversions, and refractive indices reaching values of up to 1.60 upon polymerization, the next step involved exploring the potential applications of this chemistry in holography.A photopolymer material containing a low refractive index polyurethane matrix of polyol 2000/Desmodur N3900 (TNCO) trifunctional isocyanate (nD = 1.478, measured at 20 °C and λ = 589 nm), alkyne pendant groups of 4-ethynyl benzyl alcohol (10 wt% of total matrix weight), 30 wt% of LipOMe (based on total weight) which correspond to a molar ratio of 2:1 dithiolane to alkyne and 1 wt% TPO as a radical photoinitiator (based on monomer weight) (Fig. S16†) was prepared by casting a ∼30 μm thick layer between glass microscope slides and tested by recording volume holographic phase transmission gratings into the material using a two-beam interference setup (Fig. S17†). It is worth mentioning that a higher ratio of dithiolane to yne (4:1) was also explored; however, it resulted in material compositions that do not fully form transparent films suitable for holographic recording.
Fig. 5a shows a photo of holograms illuminated near the Bragg incidence. Fig. 5b shows the angular playback diffraction efficiency (η) response about the Bragg condition of a representative transmission grating with fits to Kogelnik's coupled wave theory. This unslanted transmission hologram has a period of Λ = 650 nm and was recorded using s-polarized λ0 = 405 nm light at an irradiance of 3.7 mW cm−2 for 110 s. The quality of the fit (R2 = 0.996) indicates minimal fringe distortion and high fringe fidelity. The peak-to-mean refractive index contrast (Δn) of 0.006 obtained is comparable with the value described by Hata et al. of 0.008 utilizing silica nanoparticle–polymer composites based on thiol–yne photopolymers.26 More recently, Bowman and coworkers recorded transmission holograms in a thiol–yne formulation which exhibited Δn of 0.02 (λ0 = 405 nm, I = 16 mW cm−2); however, diffusional blurring induced by the incomplete conversion of the writing monomers reduced the overall index contrast and remaining a limitation in achieving high index modulation.13Fig. 5 shows the grating contrast development by non-destructively monitoring the diffraction efficiency at the Bragg condition during and after the optical exposure. No signal decay was observed after the exposure, indicating stable gratings. Holograms are stable at room temperature after a year of storage (Fig. 5d).
 | ||
| Fig. 5 (a) Recorded holograms for a two-stage photopolymer sample of LipOMe and yne attachment (between glass slides with 30 μm spacers from a Bragg-matched viewing angle). (b) Bragg-angle playback curve of a representative hologram showing excellent agreement with Kogelnik's coupled-wave theory (CWT) for volume-phase transmission holograms. (c) Growth development curve of diffraction efficiency during exposure, monitored nondestructively at the Bragg condition of the grating. (d) Recorded holograms after 1 year of storage at room temperature. | ||
A step further in evaluating the optical films was measuring the material birefringence. Birefringence is the polarization-dependent response of a material's interaction with light due to an anisotropic refractive index orientation. The difference in the material refractive index was evaluated by measuring s-polarized (TE) and p-polarized (TM) incident light via prism coupling.
Photopolymer samples with 30 wt% based on the total formulation weight of a 1,2-dithiolane–yne system composed of LipOMe and 4-ethyl benzyl alcohol (in a 2:1 ratio) within a urethane matrix exhibited birefringence values at the lower limits of detection for the prism coupler, δn = 7 ± 8 × 10–4 at 636 nm, indicating that the cured medium was essentially non-birefringent. Birefringence is known to be proportional to internal stress distributions within a material and is often related to stress-inducing processes like volume shrinkage. The low birefringence values potentially result from shrinkage compensation during polymerization due to the ring opening of the 1,2-dithiolane monomer or other contributing factors that can mitigate the development of internal stresses like dynamic bond exchange during polymerization of the disulfides noticed by Keyser et al. during their research.38
3. Conclusions
This work has demonstrated a methodology to explore the reaction kinetics of dithiolane homopolymerization in real time via FTIR spectroscopy. It also demonstrated the effective copolymerization of dithiolanes with terminal aryl and aliphatic alkynes and internal bisaryl alkynes. These reactions allow the selective bis addition of the dithiolane to the yne, resulting in disulfide vinyl intermediate species. Materials with different properties were prepared by adjusting the dithiolane to alkyne composition and ratio. Significantly, all the materials are readily soluble in common organic solvents. Still, most importantly, the high sulfur content of some of the materials prepared using this framework results in high refractive indices, which can be tuned by varying the architecture and monomers used. The low-cost and easily prepared materials combined with the optical properties obtained indicate that these polymers are suitable candidates for the manufacturing of modern optoelectronic devices, including displays and light sensors, as was shown during this investigation by writing stable, low birefringence holograms at very low light intensity. The development of high refractive index materials is a crucial factor for the advancements of optical devices with advanced features.4. Experimental section
4.1 Materials
The structures of materials used in this study are shown in Fig. 2. α-Lipoic acid, phenylacetylene, 1-octyne, propargyl acetate, diphenylacetylene, diphenyl(2,4,6-trimethyl(benzoyl)phosphine oxide) (TPO, purity: 98%), polycaprolactone-block-polytetrahydrofuran-block-polycaprolactone (polyol 2000), and pentaerithiritol ethoxylate 15:4 (PE 15/4) were purchased from Aldrich. N,N-Diisopropylcarbodiimide (DIC) was purchased from 1PlusChem, and dimethyl aminopyridine (DMPA) was purchased from Oakwood Chemical. Celite, magnesium sulfate (MgSO4), sodium sulfate (Na2SO4), and sodium carbonate (Na2CO3), were purchased from Fisher Scientific. Covestro LLC donated Desmodur N3900 (TNCO). Methyl ester of lipoic acid (LipOMe),49 4-methyl-1,2-dithiolane-4-carboxylic acid (Me-AspAc) and methyl 4-methyl-1,2-dithiolane-4-carboxylate (Me-AspOMe) were synthesized according to previous procedures described in the literature.60
4.2 Formulation preparation
Each formulation was prepared by placing the appropriate amount of monomer(s) in a 20 mL vial and mixing it with 1 wt% (based on the total monomer concentration) of TPO photoinitiator. The mixtures were stirred using a vortex until a fully homogeneous mixture was formed. Next, vacuum was applied to remove air bubbles from the formulations.4.3 Nuclear magnetic resonance (NMR)
NMR spectra were recorded on a Bruker AVANCE-III 400 NMR spectrometer at 25 °C in a deuterated solvent. All chemical shifts are reported in ppm relative to TMS. Eqn (3) was used to calculate conversion (C); Ip is the integral of the peak associated with the polymer and Im is the integral of the peak associated with the monomer. | (3) |
4.4 Refractive index measurements
The refractive index of monomers and polymers was measured using an Abbe refractometer (Abbemat MW) at the Fraunhofer lines-sodium-D (589 nm) at room temperature.4.5 Fourier transform infrared spectroscopy
ATR-FTIR (Thermo Scientific Nicolet IS FTIR with iTX smart accessory spectrometer) or FTIR (Thermo Scientific Nicolet 6700 FTIR spectrometer) were employed to monitor the dithiolanes or alkyne monomers concentrations before and after (static measurements, 64 scans, 2 resolution) or during polymerization (real-time measurements, 2 scans, 4 resolution). Samples were irradiated using an EXFO Acticure A 4000 high-pressure mercury vapor short arc lamp with a 400–500 nm bandpass filter insert. Light intensity was adjusted to the desired value (40 mW cm−2 for all the experiments conducted during this research) using a radiometer from Thorlabs, model PM100D at 405 nm wavelength.The ATR-FTIR spectrometer with ITX accessory (Fig. S5†) was used to obtain the FTIR spectra of the materials synthesized as well as carried out static and real-time measurements for homopolymerization of the 1,2-dithiolanes under study by placing the monomer samples directly over the diamond crystal-covered with glass slide, this arrangement allowed for monitoring of the C–S peak for the dithiolanes 689–660 cm−1 with a timed and defined illumination of 40 mW cm−2. To follow the copolymerization (1,2-dithiolane:alkynes) in real-time, optically thin samples were prepared between two salts (KBr) plates and irradiated from the top using a horizontal device inserted in the FTIR spectrometer (Fig. S8†). This setup has been described previously in the literature.51 Real-time spectra at a resolution of 4 cm−1 and 2 scans were collected.
Terminal alkyne concentrations were determined by integrating the peak at ∼3280 cm−1 corresponding to CC–H stretching or the CC peak at ∼2100 cm−1 for the internal alkyne. Eqn (4) was used to calculate the percentage of conversion (C); A0 is the area under the peak associated with the unconsumed functional group (689–660 cm−1 for dithiolanes, 3552–3162 cm−1 for internal alkynes and 2235–2192 for the internal alkyne) and At is the area under the functional group peak at time “t”.
 | (4) |
4.6 Gas permeation chromatography (GPC) – size exclusion chromatography (SEC)
Molecular weights and molecular weight distribution dispersity (Đ = Mw/Mn) were determined by size exclusion chromatography (SEC) on a Tosoh EcoSEC system equipped with two columns in series (SuperH4000 and SuperH2500) using refractive index (RI) detector. Chloroform was used as the eluent at a flow rate of 0.35 mL min−1 (40 °C). The SEC calibration was based on linear polystyrene standards.4.7 Liquid chromatography-mass spectrometry (LC-MS)
Samples were dissolved in dichloromethane at 10 mM. 25 ml samples were injected into Waters™ SYNAPT G2 mass spectrometer by a manually controlled syringe system. The mass spectrometry data were collected under MS mode with positive polarity. All samples were acquired in resolution mode. The capillary voltage was set to 3.0 kV for the sample sprayer. Data were acquired at a scan time of 1 s with a range of 100–1500 m/z. Mass correction was done using Leucine Enkephalin (556.2771 Da) as a reference.4.8 Film preparation for holography
Two-stage photopolymer film samples were prepared by premixing in a vial the writing component, LipOMe, measured at a set weight percentage (30 wt%) (relative to the entire formulation) and 10 wt% of 4-ethyl benzyl alcohol (yne component of the writing system) for a dithiolane:yne ratio of 2:1, in a matrix consisting of a mixture of tetrafunctional polyols (PE 15/4) in 13 wt% and polyol 2000 (in a 25 mL vial) in 22 wt% and with 1% TPO photoinitiator (based on writing monomers concentration), after mixing with vortex until homogeneous a stoichiometric amount (OH:NCO = 1:1) of Desmodur N3900 (TNCO) trifunctional isocyanate (20 wt%) was added into the vial and stirred until fully homogeneous, to continue the mixture was vacuumed to remove air bubbles created during the mixing procedure. The mixture was sandwiched between glass slides with a spacer of 30 μm and placed in the oven at 40 °C for 18 hours to form a polyurethane film with the writing system on it.
4.9 Holographic recording
The holographic recording was performed using the two-beam interference setup shown in Fig. S17† to pattern unslanted holographic volume phase transmission gratings. A spatially filtered laser diode (Ondax CP-405-PLR-40, λrecord = 405 nm) was used to record the holographic gratings. Each recording beam had a circular diameter of drecord = 9.5 mm and a slowly varying transverse intensity profile. The two recording beams were power matched for a total optical intensity of 3.7 mW cm−2 at the plane of the sample. The s-polarized beams were incident upon the media sample at an external recording half-angle of θ = 18.2° to produce an unslanted, sinusoidal interference pattern with a fringe spacing of Λ ≈ 650 nm. A probe laser (λread = 635 nm) was aligned to the center of the exposure area at the Bragg-matching angle to nondestructively monitor holographic fringe formation during the exposure (Thorlabs PL202, 1 mW). The probe beam has a circular diameter of dread = 0.5 mm. The transmitted and diffracted probe beams were monitored with power meters (Newport 2936-R and 918D-UV x2). The grating-development time dynamics were measured during the exposure by pre-aligning the read beam to the Bragg condition of the grating and monitoring diffraction efficiency throughout the grating development. Then, the hologram is rotated 12 degrees about the Bragg angle using a rotary stage (Newport URS100BPP) to acquire the Bragg-angle detuning response. Each hologram's diffraction efficiency (η) was calculated by taking the quotient of the measured diffracted power to the total power. This can be written as eqn (5):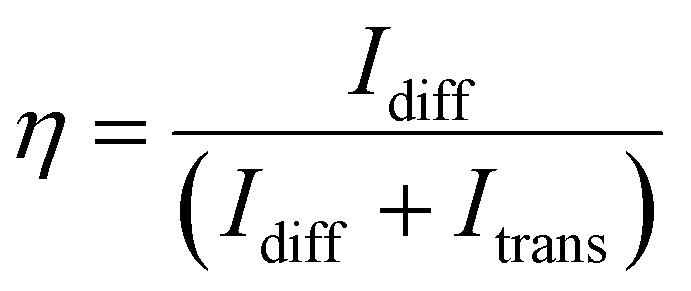 | (5) |
4.10 Birefringence measurements
The s-polarized (nTE) and p-polarized (nTM) refractive indices of the two-stage photopolymer films were measured using a prism coupler (Metricon, model PC-2000) equipped with a He–Ne laser (λ = 636 nm) with polarization modulated by a half-waveplate in the light path. The δn values were calculated as a difference between nTE and nTM according to eqn (6):| δn = |nTE − nTM| | (6) |
Author contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors do not have any conflicts of interest to declare.Acknowledgements
The authors would like to thank Covestro LLC for the generous donation of Desmodur N3900 and Meagan N. Arguien and Alexander J. Osterbank for their input on editing the manuscript.References
- Y. Tang, S. Cabrini, J. Nie and C. Pina-Hernandez, Chin. Chem. Lett., 2020, 31, 256–260 CrossRef CAS.
- R. Fernández, S. Bleda, S. Gallego, C. Neipp, A. Márquez, Y. Tomita, I. Pascual and A. Beléndez, Opt. Express, 2019, 27, 827–840 CrossRef PubMed.
- M. D. Alim, D. J. Glugla, S. Mavila, C. Wang, P. D. Nystrom, A. C. Sullivan, R. R. McLeod and C. N. Bowman, ACS Appl. Mater. Interfaces, 2018, 10, 1217–1224 CrossRef CAS.
- J. Liu and M. Ueda, J. Mater. Chem., 2009, 19, 8907–8919 RSC.
- C.-C. Chang and W.-C. Chen, J. Polym. Sci. Part A: Polym. Chem., 2001, 39, 3419–3427 CrossRef CAS.
- T. Itoh, T. Uchida, N. Izu and W. Shin, Materials, 2017, 10, 710 CrossRef PubMed.
- G.-S. Liou, P.-H. Lin, H.-J. Yen, Y.-Y. Yu and W.-C. Chen, J. Polym. Sci. Part A: Polym. Chem., 2010, 48, 1433–1440 CrossRef CAS.
- M. Takafuji, M. Kajiwara, N. Hano, Y. Kuwahara and H. Ihara, Nanomaterials, 2019, 9, 514 CrossRef.
- E. K. Macdonald and M. P. Shaver, Polym. Int., 2015, 64, 6–14 CrossRef CAS.
- T. Higashihara and M. Ueda, Macromolecules, 2015, 48, 1915–1929 CrossRef CAS.
- J. Zhang, T. Bai, W. Liu, M. Li, Q. Zang, C. Ye, J. Z. Sun, Y. Shi, J. Ling, A. Qin and B. Z. Tang, Nat. Commun., 2023, 14, 3524 CrossRef CAS PubMed.
- Y. Hu, B. A. Kowalski, S. Mavila, M. Podgórski, J. Sinha, A. C. Sullivan, R. R. McLeod and C. N. Bowman, ACS Appl. Mater. Interfaces, 2020, 12, 44103–44109 CrossRef CAS PubMed.
- S. Mavila, J. Sinha, Y. Hu, M. Podgórski, P. K. Shah and C. N. Bowman, ACS Appl. Mater. Interfaces, 2021, 13, 15647–15658 CrossRef CAS PubMed.
- D. H. Kim, W. Jang, K. Choi, J. S. Choi, J. Pyun, J. Lim, K. Char and S. G. Im, Sci. Adv., 2020, 6, eabb5320 CrossRef CAS PubMed.
- L. Fang, J. Sun, X. Chen, Y. Tao, J. Zhou, C. Wang and Q. Fang, Macromolecules, 2020, 53, 125–131 CrossRef CAS.
- H. Maekawa, H. Amano, I. Nishina and H. Kudo, ChemistrySelect, 2022, 7, e202201543 CrossRef CAS.
- R. Khaleghi Qusheh Bolagh, A. Salimi, K. Kabiri and A. Pourali, J. Appl. Polym. Sci., 2023, 140, e53375 CrossRef CAS.
- K. Nakabayashi, T. Imai, M.-C. Fu, S. Ando, T. Higashihara and M. Ueda, Macromolecules, 2016, 49, 5849–5856 CrossRef CAS.
- H. Yeo, J. Lee, M. Goh, B.-C. Ku, H. Sohn, M. Ueda and N.-H. You, J. Polym. Sci. Part A: Polym. Chem., 2015, 53, 944–950 CrossRef CAS.
- R. Okutsu, Y. Suzuki, S. Ando and M. Ueda, Macromolecules, 2008, 41, 6165–6168 CrossRef CAS.
- C. Li, Z. Li, J. Liu, X. Zhao, H. Yang and S. Yang, Polymer, 2010, 51, 3851–3858 CrossRef CAS.
- K. Mazumder, B. Voit and S. Banerjee, ACS Omega, 2024, 9, 6253–6279 CrossRef CAS PubMed.
- Y. S. Kochergin, Y. Noda, R. Kulkarni, K. Škodáková, J. Tarábek, J. Schmidt and M. J. Bojdys, Macromolecules, 2019, 52, 7696–7703 CrossRef CAS.
- Y. Zhou, Z. Zhu, K. Zhang and B. Yang, Macromol. Rapid Commun., 2023, 44, 2300411 CrossRef CAS PubMed.
- L. V. Natarajan, C. K. Shepherd, D. M. Brandelik, R. L. Sutherland, S. Chandra, V. P. Tondiglia, D. Tomlin and T. J. Bunning, Chem. Mater., 2003, 15, 2477–2484 CrossRef CAS.
- E. Hata, K. Mitsube, K. Momose and Y. Tomita, Opt. Mater. Express, 2011, 1, 207–222 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, AIChE J., 2008, 54, 2775–2795 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- H. Bader, L. C. Cross, I. Heilbron and E. R. H. Jones, J. Chem. Soc. Resumed, 1949, 619–623 RSC.
- A. T. Blomquist and J. Wolinsky, J. Org. Chem., 1958, 23, 551–554 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS PubMed.
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS.
- Y. Li, S. Li, X. Du and Z. Gu, RSC Adv., 2021, 11, 21023–21028 RSC.
- P. Marx and F. Wiesbrock, Polymers, 2021, 13, 806 CrossRef CAS PubMed.
- F. Ay, A. Kocabas, C. Kocabas, A. Aydinli and S. Agan, J. Appl. Phys., 2004, 96, 7147–7153 CrossRef CAS.
- C. Charton, P. Colon and F. Pla, Dent. Mater., 2007, 23, 911–920 CrossRef CAS PubMed.
- S. P. Keyser, M. Trujillo-Lemon, A. N. Sias, B. D. Fairbanks, R. R. McLeod and C. N. Bowman, ACS Appl. Mater. Interfaces, 2024, 16, 45577–45588 CrossRef CAS PubMed.
- J. A. Barltrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76, 4348–4367 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, M. Chen and D.-H. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 44860–44867 CrossRef CAS PubMed.
- G. M. Scheutz, J. L. Rowell, F.-S. Wang, K. A. Abboud, C.-H. Peng and B. S. Sumerlin, Org. Biomol. Chem., 2020, 18, 6509–6513 RSC.
- A. V. Tobolsky and B. Baysal, J. Am. Chem. Soc., 1953, 75, 1757–1757 CrossRef CAS.
- W. H. Stockmayer, R. O. Howard and J. T. Clarke, J. Am. Chem. Soc., 1953, 75, 1756–1757 CrossRef CAS.
- T. Suzuki, Y. Nambu and T. Endo, Macromolecules, 1990, 23, 1579–1582 CrossRef CAS.
- S. Maes, V. Scholiers and F. E. Du Prez, Macromol. Chem. Phys., 2023, 224, 2100445 CrossRef CAS.
- L. Pezzana and M. Sangermano, Prog. Org. Coat., 2021, 157, 106295 CrossRef CAS.
- B. R. Nelson, B. E. Kirkpatrick, C. E. Miksch, M. D. Davidson, N. P. Skillin, G. K. Hach, A. Khang, S. N. Hummel, B. D. Fairbanks, J. A. Burdick, C. N. Bowman and K. S. Anseth, Adv. Mater., 2024, 36, 2211209 CrossRef CAS PubMed.
- United States, US20070055070A1, 2007.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS.
- L. G. Lovell, K. A. Berchtold, J. E. Elliott, H. Lu and C. N. Bowman, Polym. Adv. Technol., 2001, 12, 335–345 CrossRef CAS.
- J. Houk and G. M. Whitesides, J. Am. Chem. Soc., 1987, 109, 6825–6836 CrossRef CAS.
- H. Dislich, Angew. Chem., Int. Ed. Engl., 1979, 18, 49–59 CrossRef.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS.
- D. Melandri, P. C. Montevecchi and M. L. Navacchia, Tetrahedron, 1999, 55, 12227–12236 CrossRef CAS.
- K. A. Berchtold, B. Hacioğlu, J. Nie, N. B. Cramer, J. W. Stansbury and C. N. Bowman, Macromolecules, 2009, 42, 2433–2437 CrossRef CAS PubMed.
- Y. Jian, Y. He, J. Wang, W. Yang and J. Nie, Polym. Int., 2013, 62, 1692–1697 CrossRef CAS.
- M. Tazaki, M. Kumakura, S. Nagahama and M. Takagi, J. Chem. Soc., Chem. Commun., 1995, 1763–1764 RSC.
- D. V. Demchuk and G. I. Nikishin, Russ. Chem. Bull., 1997, 46, 199–201 CrossRef CAS.
- United States, US8617524B2, 2013.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01337a |
| This journal is © The Royal Society of Chemistry 2025 |