
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt–benzimidazole swapped metal–organic macrocycle with reduced graphene oxide as a hybrid electrocatalyst for highly efficient oxygen evolution reaction†
C. P. Keshavananda
Prabhu‡
 *a,
Kenkera Rayappa
Naveen‡
b and
Jaehyun
Hur
*a
*a,
Kenkera Rayappa
Naveen‡
b and
Jaehyun
Hur
*a
aDepartment of Chemical and Biological Engineering, Gachon University, Seongnam-si 13120, Gyeonggi-do, South Korea. E-mail: keshavmgm@gmail.com; jhhur@gachon.ac.kr
bInstitut für Anorganische Chemie and, Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 27th November 2023
Abstract
The increasing global demand for sustainable and efficient energy-conversion technologies necessitates the development of high-performance electrocatalysts for the oxygen evolution reaction (OER). Herein, we present a novel electrocatalyst, namely a cobalt-benzimidazole substituted metal–organic phthalocyanine with reduced graphene oxide (CoBImPc/rGO) hybrid, for boosting the catalytic activity and stability for the OER, with potential applications in water splitting and energy-storage systems. The synthesis of CoBImPc/rGO involved integrating cobalt–benzimidazole substituted metal–organic phthalocyanine onto reduced graphene oxide, resulting in a well-defined hybrid structure. Based on comprehensive characterization, including scanning electron microscopy (SEM) and energy-dispersive X-ray (EDX) spectroscopy, we confirmed the homogeneous distribution of CoBImPc particles on the rGO sheets and the successful incorporation of the elements cobalt, carbon, nitrogen, bromine, and oxygen in the final hybrid material. Electrochemical evaluations based on cyclic voltammetry demonstrated that the CoTBImPc/rGO hybrid manifested a significantly improved current density and lower overpotential than pure CoTBImPc and rGO, indicating its enhanced electrocatalytic activity. Furthermore, the Tafel slope obtained from the Tafel plot was substantially reduced, suggesting a faster kinetics for the OER on the hybrid electrocatalyst. Notably, the CoTBImPc/rGO hybrid electrocatalyst revealed excellent stability and durability during long-term electrolysis tests, maintaining its catalytic activity even after prolonged operation. When CoTBImPc/rGO was coated with Ni foam and used as an OER catalyst, it showed a lower overpotential of 350 mV at a current density of 10 mA cm−2. This impressive electrocatalytic performance and stability of the CoTBImPc/rGO hybrid make it a promising candidate for clean energy applications, with the potential to contribute to sustainable energy-conversion technologies and a greener future.
1. Introduction
The energy-rich carriers that supply the current world energy needs are primarily fossil fuels (such as petroleum, coal, and natural gas).1,2 Nevertheless, the essential reserves of these fossil fuels are mostly limited and diminished by continuous consumption. Moreover, the combustion of fossil fuels creates environmental problems, such as CO2 gas emissions, which contribute significantly to global warming.2 Highly efficient and alternative energy sources are urgently needed to avoid such emissions and to store clean and sustainable energy as chemical energy. Among the various energy-conversion technologies, splitting water into hydrogen (H2) and oxygen (O2) by electrochemical techniques is considered a green and renewable innovation to meet the enormous energy demand in the forthcoming years with several applications.3–6 However, the electrolysis of water to O2 and H2 through oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) is a more complicated process as it requires a high overpotential.7–9 Subsequently, noble materials, such as Ru, Ir, and Pt, are considered efficient catalysts for water splitting due to their lower operating potential, high electrical conductivity, and good chemical stability. However, these materials are very costly, rare, and restricted from large-scale applications, making them less superior.10,11 Therefore, developing low-cost, environmentally friendly, earth-abundant, non-toxic, highly stable, and non-noble electrode materials for the water-splitting process is a hot topic in today's research. Currently, the OER is pivotal for sustainable hydrogen and oxygen production from water.12 Usually, electrochemical water splitting comprises two half-cells, where the HER occurs at the cathode, while the OER takes place at the anode.13–15 In the water-splitting electrochemical process, the water molecules are dissociated into oxygen and hydrogen gas using an electrolyte and suitable catalysts at the anode and cathode.16–20 Electrochemical water splitting holds immense potential as a clean and renewable method for hydrogen production, offering versatility as a sustainable energy carrier. In this landscape, the OER is a significant process in energy-storage devices, like metal–air batteries and electrochemical water splitting.21 Efforts to enhance OER electrocatalysts stem from the demand for clean energy sources. The OER, involving water transformation into oxygen and protons, underpins renewable energy technologies.22 Its sluggish kinetics and high overpotential, however, limit the efficiency of the OER due to the multi-step proton-coupled process; however, the use of electrocatalysts could expedite the reaction.An effective OER electrocatalyst is required to show a lower overpotential. Currently, ruthenium(IV) oxide (RuO2) and iridium(IV) oxide (IrO2) are the benchmark catalysts for the OER. Due to these materials' high cost, rare, and expensive nature, efficient, high-performance materials for the OER are still needed to solve the present situation. After these, different molecular compounds have been studied as effective and efficient catalysts for the OER. The first report of a metal phthalocyanine for use as an electrocatalyst for the OER was in 1964.23 Besides the low price and better commercial applications of such a catalyst, it is also the most attractive catalyst in many electrochemical reactions. It contains an N4 coordination site for metal ions and an induced redox reaction macrocyclic ring at the site of the coordinated metal ions.24 The catalytic activity of the metal phthalocyanine strongly depends on the nature of the central metal ion. These central metal ions form bonds with –OH and O2. Additionally, the functionalization of rGO and CoTBImPc is usually carried out under an organic solvent medium and with additional processes, like ultrasonication, centrifugation, washing, and drying. So far, a series of earth-abundant transition metal-based materials have been revealed to exhibit promising OER electrocatalysis performance in the water-splitting process.25–28 For example, Co-MOF nanorods (NRs) were synthesized by Kelong Ao et al., and then the MOF was converted into CoS NRs on carbon cloth (CoS/CC) by calcination and a hydrothermal sulfurization.29 The fabricated CoS@CoNi-LDH/CC demonstrated exceptional electrocatalytic activity for the HER by fusing the benefits of its structure and composition. Later, a hybrid nickel phosphonate (NiPPA) material was developed by Bhanja Piyali et al., which could serve as an electrocatalyst for the OER in alkaline conditions.30 Similarly, stable and effective transition bimetallic chalcogenides were introduced as precious metal electrocatalysts for the alkaline OER (with NiFe selenide as a catalyst).31 Hu Shengnam et al. developed bifunctional catalysts for hydrogen evolution and urea oxidation, whereby they effectively produced a unique MOF-derived nickel nitride on nickel foam catalysts at three distinct temperatures (300 °C, 350 °C, and 400 °C).32 Using these catalysts, the urea oxidation process (UOR) was shown to be highly active and could be durably catalyzed at 350 °C, with a necessary potential of 1.337 V at 10 mA cm−2. With a low overpotential of 47 mV at 10 mA cm−2, the 350 °C temperature sample exhibited better catalytic activity toward the HER than the other materials. Septiani Ni Luh Wulan et al. also used similar bimetallic nickel–cobalt (Ni–Co) hydrogen phosphate nanotubes as catalysts, revealing the superior activity of the Ni-rich Ni–Co hydrogen phosphate electrode for the OER compared to its Co-rich and equimolar counterparts, with a smaller overpotential.33 Even though several complexes have been developed,34 the Co phthalocyanine derivatives could serve most effectively as catalysts in practical applications in the water electrolysis industry.35–37 Notably, benzimidazole-functionalized CoTBImPc could enhance the catalytic properties, while the incorporation of rGO could improve the charge transport and structural stability among the developed macrocycles. The synergistic effects between CoTBImPc and rGO were expected to lower the OER overpotential. This research contributes to understanding the CoTBImPc/rGO hybrid's intrinsic improvement in OER activity.26 Despite advancements, developing efficient and low-cost OER catalysts remains a challenge. The unique features of metal phthalocyanines, such as their N4 coordination sites and redox-active macrocycles, make them attractive candidates for the OER. The choice of the central metal ion affects the catalytic activity, with CoPc often outperforming FePc.24 Generally, benzimidazole-substituted cobalt phthalocyanine derivatives have shown promise as active catalysts for water electrolysis; however, they are limited due to their synthetic flaws.38–41
In the present work, the cobalt tetrabenzimidazole phthalocyanine macrocycle was successfully synthesized through easy and scalable synthetic protocols. The synthesized molecule and its amalgamation with rGO were coated on a nickel (Ni) foam surface and used as an OER catalyst. When amalgamated with rGO, it demonstrated enhanced activity compared to conventional Ni foam and separate rGO catalysts. The results were compared with the hybrid composite of CoTBImPc with and without rGO. The application of rGO supported the improved conductivity, surface properties, and stability of the proposed phthalocyanine. The successful development of this CoTBImPc/rGO hybrid electrocatalyst could open up new possibilities for advancing the field of electrocatalysis and clean energy technologies. By contributing to the understanding of the structure–activity relationships in metal–organic phthalocyanine-based electrocatalysts and their synergistic interactions with 2D materials, this research seeks to realize efficient and sustainable energy-conversion and -storage systems. The potential applications of such hybrid electrocatalysts extend beyond the OER to other electrochemical processes, making them precious components in the quest for cleaner and greener energy for the future.
2. Experimental section
2.1. Materials
The chemicals used for the synthesis of CoTBImPc were either obtained from Sigma Aldrich or TCI and used as such without any purification. All the chemicals are tabulated below with their chemical formula and purity.| Chemicals | Chemical formula | Purity |
|---|---|---|
| Cobalt chloride | CoCl2 | 99.9% |
| p-Bromo benzoic acid | C7H5BrO2 | 97% |
| o-Phenylenediamine | OPDA: C6H4(NH2)2 | 99.2% |
| 4-Nitrophthalonitrile | 4NPN: C6H4NO2(CN)2 | 97% |
| 1,8-Diazabicyclo [5.4.0]undec-7-ene | DBU: C9H16N2 | 98% |
| n-pentanol | C5H11OH | 99.0% |
| Dimethylformamide | DMF: C3H7NO | 99.9% |
| Dimethylsulfoxide | DMSO: C2H6OS | 99.9% |
| Hydrochloric acid | HCl | 95–99% |
| Sulfuric acid | H2SO4 | 98% |
| Tetrabutylammonium perchlorate | TBAP: C16H36ClNO4 | 98–100% |
| Potassium carbonate | K2CO3 | 99.9% |
| Sodium bicarbonate or baking soda | NaHCO3 | 99% |
| Sodium hydroxide | NaOH | 80% |
| Potassium ferrocyanide | K4[Fe(CN)6]·3H2O | 98% |
| Methanol | CH3OH | 99.85% |
| Ethanol | C2H5OH | 99.9% |
| Acetone | CH3COCH3 | 99.60% |
| n-Hexane | C6H14 | 96% |
| Reduced graphene oxide | rGO | 99.5% |
2.2. Synthetic procedures and characterization
2.3. Characterization methods
The synthesized CoTBImPc/rGO hybrid electrocatalyst was characterized using various techniques, including scanning electron microscopy (SEM), X-ray diffraction (XRD), and energy-dispersive X-ray spectroscopy (EDX) for the morphological and elemental analyses. A Vario EL III CHNS elemental analyzer was utilized to analyze the carbon, hydrogen, and nitrogen content. The metal content in the CoTBImPc complex was determined using a gravimetric procedure. UV-vis spectra were obtained using an Ocean Optics spectrometer with the serial number FLMSO 4808, in the range from 300–800 nm, with a 0.1 mM CoTBImPc complex in DMSO. FT-IR spectra were obtained in the 4000–500 cm−1 range using the KBr pellet sampling technique and an FT-IR Perkin Elmer Spectrum-Two spectrophotometer. The mass analysis was performed using an Agilent Technologies 6100 series GC/MS instrument. The XRD pattern of the complex was recorded using a Bruker D8 Advance X-ray diffraction instrument. The thermal stability of the complex was monitored using a STA6000 machine, in the temperature range of 30–700 °C and a heating rate of 10 °C min−1. The CHI6005E electrochemical analyzer was employed for the electrochemical studies using a three-electrode system. Nickel foam was used as the working electrode, a saturated Ag/AgCl electrode was the reference, and a platinum wire was the counter electrode. Before each experiment, the system was purged in a N2 atmosphere for 20 min.42–462.4. Preparation of the electrocatalysts
A suitable support material is often used to enhance the stability and surface area of electrocatalysts. Common support materials include carbon-based materials, like reduced graphene oxide, carbon nanotubes or graphene, metal oxides, or conductive polymers. The support material can be prepared through different methods, such as hydrothermal synthesis, chemical vapor deposition, or physical mixing. The metal phthalocyanine is typically deposited onto the rGO support material to produce the electrocatalyst. There are several techniques for catalyst deposition, including drop-casting, spin-coating, electrodeposition, or inkjet printing. The choice of deposition method depends on factors such as the desired catalyst loading, film thickness, and uniformity. Here, we used the drop-coating method. After deposition, the electrocatalyst was characterized by assessing its performance using electrochemical techniques.2.5. Catalytic ink preparation
For the preparation of the catalyst ink dedicated to each specific electrocatalyst, a precisely measured 0.5 g of the powdered catalytic material was carefully blended with Nafion binder in a well-calibrated mass ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The resultant amalgamation was subsequently dispersed into a medium of 2-propanol and subjected to a rigorous 30 min sonication process. In addition to this meticulous preparation, the resulting ink was delicately applied onto a substrate composed of a high-porosity nickel (Ni) foam, configured at dimensions of 1 × 1 cm2 (Fig. 1). The choice of the Ni foam substrate was deliberate, as it endows the system not only with outstanding electrical conductivity properties but also with the advantageous ability to promote efficient gas evolution and enhance the diffusion of the electrolyte within the electrocatalytic system.
:5. The resultant amalgamation was subsequently dispersed into a medium of 2-propanol and subjected to a rigorous 30 min sonication process. In addition to this meticulous preparation, the resulting ink was delicately applied onto a substrate composed of a high-porosity nickel (Ni) foam, configured at dimensions of 1 × 1 cm2 (Fig. 1). The choice of the Ni foam substrate was deliberate, as it endows the system not only with outstanding electrical conductivity properties but also with the advantageous ability to promote efficient gas evolution and enhance the diffusion of the electrolyte within the electrocatalytic system.
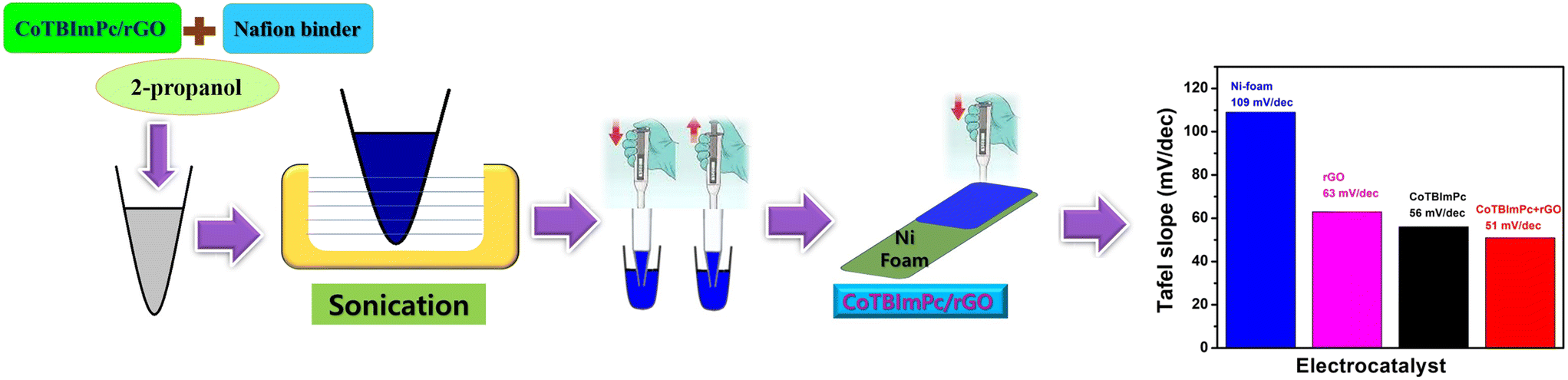 | ||
| Fig. 1 Graphical representation of the catalytic ink preparation and coating on Ni foam for assessing the influence on the overpotential values. | ||
3. Results and discussion
3.1. Synthesis
The synthesis of the target macrocycle, cobalt tetra benzimidazole phthalocyanine (CoTBImPc), was successfully accomplished through a multi-step process, as illustrated in Scheme 1. This synthetic pathway involved three distinct stages. In the initial step, the precursor (I) was generated by subjecting an acid and o-phenylene diamine (OPDA) to cyclization under acidic conditions. The resulting product was subsequently neutralized using bicarbonate. The progression of benzimidazole formation was tracked by thin-layer chromatography (TLC). Moving on to the second step, a nucleophilic substitution reaction took place between compound (I) and 4-nitrophthalonitrile (4-NPN) under mildly primary conditions facilitated by potassium carbonate (K2CO3). This interaction led to the creation of a benzimidazole-substituted phthalonitrile ligand (II). The substitution reaction was instigated by utilizing hydrogen from a secondary amine group. In the final step, the desired macrocyclic complex was synthesized by reacting four equivalents of compound (II) with cobalt salt and a bicyclic DBU catalyst in the presence of 1-pentanol at a temperature of 140 °C. The resulting crude product (III) exhibited a green-blue hue. To refine the product, it underwent a purification process involving successive washes with ethanol (EtOH), water, and hexane. The purified product was subsequently dried at 60 °C for 1 h in an oven and then stored in a vacuum desiccator containing silica gel. CoTBImPc complexes were purified and obtained in good yields. The elemental analysis data well agreed with the theoretical values. The synthesized cobalt phthalocyanine complex was amorphous and dark bluish-green in color. The CoTBImPc complexes were insoluble in protic solvents and soluble in aprotic solvents like DMSO, DMF, and DMA in addition to conc. H2SO4.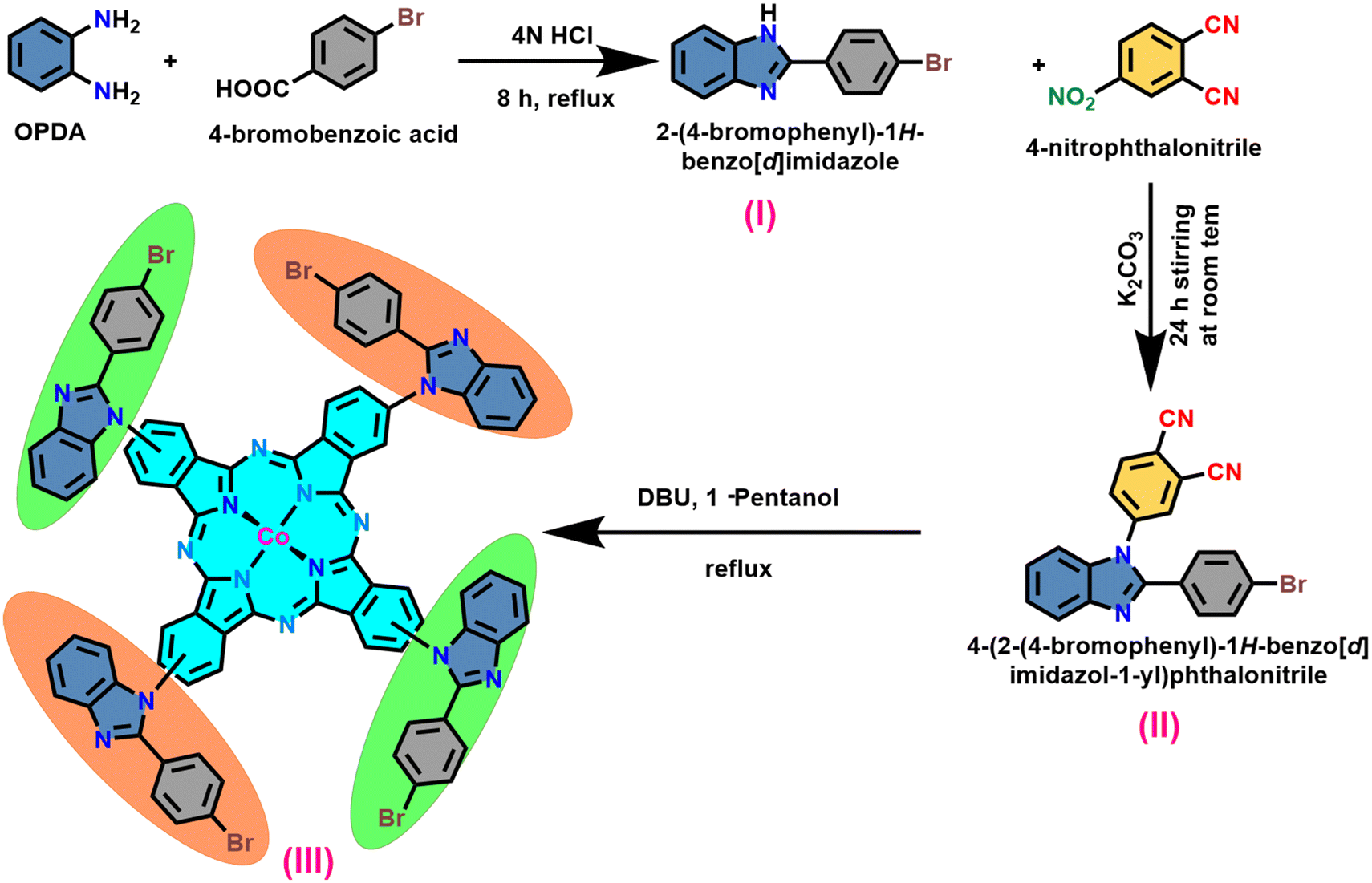 | ||
| Scheme 1 Synthetic route of the precursor (I) ligand (II) and CoTBImPc (III) complex. | ||
The prepared electrocatalyst was evaluated for its OER performance to assess its structure, composition, and electrochemical properties. Techniques such as scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) were employed to analyze the morphology, crystal structure, and elemental composition of the catalyst. Based on the obtained results, the electrocatalyst synthesis and preparation process could be optimized by adjusting various parameters, such as the metal phthalocyanine concentration, deposition method, catalyst loading, or support material properties. Additional studies can focus on understanding the reaction mechanisms, stability under prolonged conditions, and improving the overall efficiency.
3.2. UV-visible spectroscopy analysis
The obtained pure sample of CoTBImPc (III) (0.1 mM) was dissolved in DMSO and prepared as a concentrated, transparent solution for analysis in the UV-vis range using a UV-vis spectrophotometer equipped with a suitable light source. First, it was important to ensure that the spectrophotometer was appropriately calibrated, and the sample holder or cuvette was clean. The UV-vis spectrum of the CoTBImPc complex was recorded by measuring the absorbance or transmittance of light across the 250–850 nm wavelength range. The UV-vis spectrophotometer generated a spectrum showing the absorbance or transmittance as a function of wavelength. Both the complexes showed a B-band at 268 and 329 nm. Similarly, the Q-band was around 612 and 671 nm, respectively. The B-band was due to n to π* transitions, whereas the Q-band was due to π to π* transitions. A shoulder peak was observed at 612 nm in the Q-band region due to the vibronic fine structure and presence of dimer and oligomeric complex species in solution.47–49 Additionally, changes in the peak positions, peak intensities, or shape of the UV-vis spectrum can provide insights into the electronic structure, coordination, or other structural changes in a complex. The UV-vis spectrum of the CoTBImPc offered information about the compound's electronic transitions and absorption properties, allowing for its characterization and identification (Fig. 2(A)).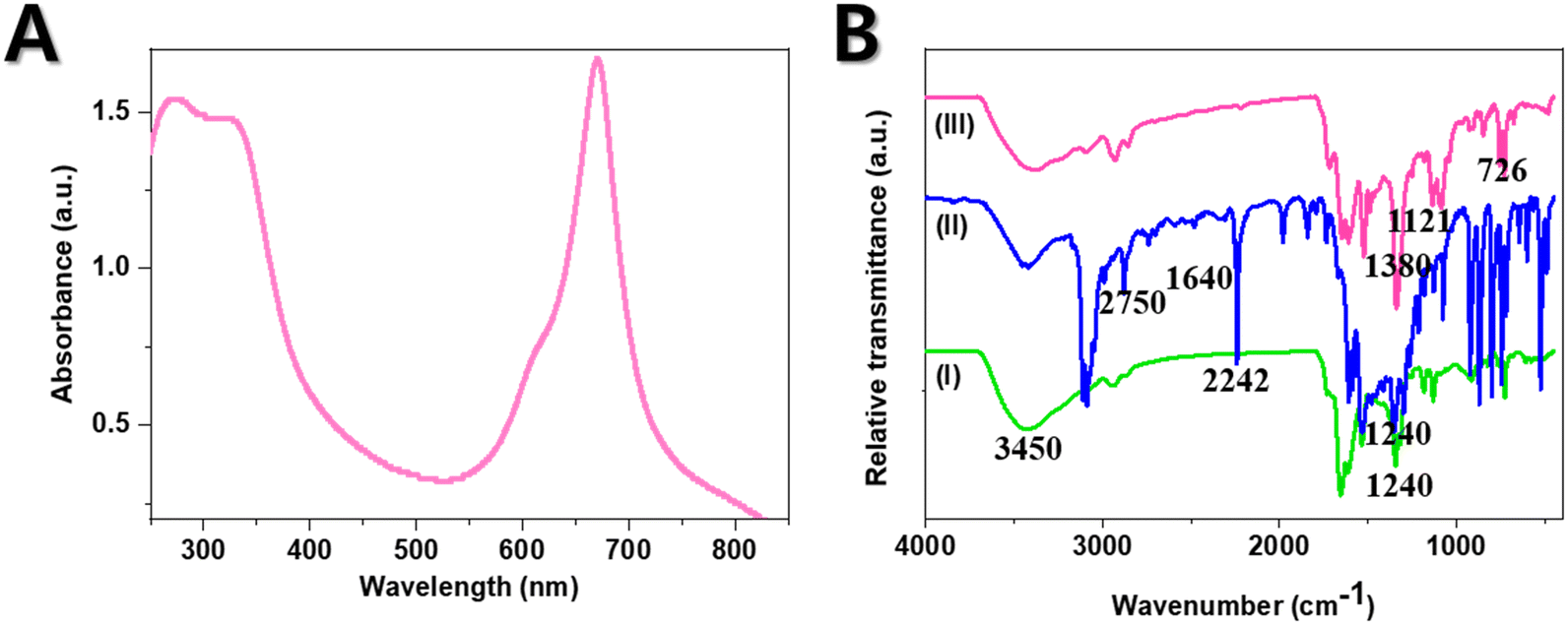 | ||
| Fig. 2 (A) UV-visible absorption spectrum for CoTBImPc; (B) FT-IR spectra of the precursor (I), ligand (II), and CoTBImPc (III). | ||
3.3. FT-IR spectra
For the FT-IR analysis, the synthesized complexes were ground with an IR-transparent matrix, such as potassium bromide (KBr), to create either pellets or thin films for measurement. The spectral range of 500–4000 cm−1 was utilized to record the IR spectra of the precursor (I), ligand (II), and Co complex (III), as depicted in Fig. 2(B). The resulting IR spectra illustrated the absorption intensity plotted against the wavenumber (cm−1). The peaks observed in the spectrum were assigned based on established vibrational modes and functional groups inherent to the complex. Interpretation of the IR data involved considering both the ligands' characteristics and the complex's structural attributes. The complex's identity and specific functional groups were validated by comparing the obtained spectrum with reference spectra and databases.The FT-IR spectrum of the precursor (I) exhibited a peak at 1660 cm−1 attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching, with additional peaks at 2750 cm−1 corresponding to aromatic –CH stretching, 1240 cm−1 for C–OH, and a broad peak spanning 3200–3450 cm−1 indicating –OH groups. The ligand (II) demonstrated peaks like for precursor (I) but an extra peak at 2242 cm−1 indicative of –CN stretching. The emergence of this –CN peak indicated the formation of the phthalonitrile ligand (II). Upon analyzing the spectrum of CoTBImPc (III), distinctive peaks were identified at 720–750, 800, 910–957, 1010, 1060–1090, and 1118 cm−1, which were attributed to phthalocyanine skeletal vibrations. These findings aligned well with the literature values.50,51 Further, specific peaks in the infrared spectra were associated with distinct molecular vibrations and functional groups. For instance, the peak at 726 cm−1 corresponded to aromatic isoindole rings, while the 816 cm−1 peak was linked to macrocyclic planar vibration. The 925 cm−1 peak was assigned to phthalocyanine in-plane vibrations, and the peaks at 1050 and 1105 cm−1 indicated C–H in-plane deformation. An 1121 cm−1 band signified C–H in-plane bending vibration, and the 1330 cm−1 peak represented C–N stretching. Notably, the 1380 cm−1 peak denoted C–C stretching in isoindole, while the 1640 cm−1 peak was associated with CC macrocyclic ring deformation. Remarkably, the nitrile peak at 2242 cm−1 disappeared in the spectrum of the cobalt phthalocyanine complex (III), suggesting the complete conversion of the phthalonitrile molecule to the phthalocyanine complex.52,53
N stretching, with additional peaks at 2750 cm−1 corresponding to aromatic –CH stretching, 1240 cm−1 for C–OH, and a broad peak spanning 3200–3450 cm−1 indicating –OH groups. The ligand (II) demonstrated peaks like for precursor (I) but an extra peak at 2242 cm−1 indicative of –CN stretching. The emergence of this –CN peak indicated the formation of the phthalonitrile ligand (II). Upon analyzing the spectrum of CoTBImPc (III), distinctive peaks were identified at 720–750, 800, 910–957, 1010, 1060–1090, and 1118 cm−1, which were attributed to phthalocyanine skeletal vibrations. These findings aligned well with the literature values.50,51 Further, specific peaks in the infrared spectra were associated with distinct molecular vibrations and functional groups. For instance, the peak at 726 cm−1 corresponded to aromatic isoindole rings, while the 816 cm−1 peak was linked to macrocyclic planar vibration. The 925 cm−1 peak was assigned to phthalocyanine in-plane vibrations, and the peaks at 1050 and 1105 cm−1 indicated C–H in-plane deformation. An 1121 cm−1 band signified C–H in-plane bending vibration, and the 1330 cm−1 peak represented C–N stretching. Notably, the 1380 cm−1 peak denoted C–C stretching in isoindole, while the 1640 cm−1 peak was associated with CC macrocyclic ring deformation. Remarkably, the nitrile peak at 2242 cm−1 disappeared in the spectrum of the cobalt phthalocyanine complex (III), suggesting the complete conversion of the phthalonitrile molecule to the phthalocyanine complex.52,53
3.4. NMR spectra
Nuclear magnetic resonance (NMR) spectroscopy is a powerful technique for studying the structure, composition, and properties of materials, like the precursor (I) and ligand (II). NMR spectroscopy provides information about the chemical environment and interactions of the nuclei present in a molecule, allowing for the characterization of a complex. The compounds were dissolved in deuterated dimethyl sulfoxide (DMSO-d65) for the NMR analysis. The choice of the solvents depends on the solubility of the complex and the desired NMR techniques. One-dimensional (1D) NMR spectroscopy, such as proton (1H NMR) or carbon-13 (13C NMR) NMR, can provide information about the chemical shifts and coupling patterns of the nuclei present in the materials. The 1H NMR of the precursor (I) and ligand (II) materials were examined and are given in Fig. S1 and S2.† These results were compared to reference spectra to identify the chemical moieties and confirm the structure. The precursor (I) showed a peak around δ 8.9 ppm for the –OH proton, whereas the phthalonitrile ligand (II) did not show the –OH proton peak, indicating the formation of the phthalonitrile ligand (II).3.5. Mass spectra
Mass spectrometry is a valuable technique and was used for studying the phthalocyanine complex, providing information about its molecular mass, fragmentation patterns, and structural elucidation. All three compounds' spectra are shown in Fig. S3–S5.† This was also used to confirm the formation of the precursor (I), ligand (II), and CoTBImPc (III). The compounds are typically dissolved in a suitable solvent, such as methanol or acetonitrile, to create an analysis solution. The sample concentration should be optimized to ensure a sufficient signal-to-noise ratio in the mass spectrum. The mass spectrometer records the ions relative abundance as a function of their m/z values, generating a mass spectrum. Computational methods, such as density functional theory (DFT) calculations, can also simulate the mass spectra and aid structure elucidation. The synthesized precursors (I) showed a peak at m/z = 269 corresponding to the M−2 molecular ion peak, while ligand (II) showed a peak at m/z = 397 corresponding to the M−1 molecular ion peak. The CoTBImPc (III) showed a peak at m/z = 1665 corresponding to the M+1 molecular ion peak. The data obtained from the mass spectra are shown in their respective synthetic data.3.6. X-Ray diffraction studies
X-Ray diffraction (XRD) spectroscopy is a commanding technique for studying the crystal structure and molecular arrangement of phthalocyanine complexes. XRD provides information about the atomic positions, unit cell parameters, crystal symmetry, and crystalline phases present in the complex. The phthalocyanine complex was introduced as a powder sample for the XRD analysis. This powder sample could be obtained by grinding or milling the metal phthalocyanine complex. The XRD patterns for the CoTBImPc complex were recorded in the 2θ angle range of 5–70° and are shown in Fig. S6.† The diffraction pattern was processed to determine the positions and intensities of the diffraction peaks. Bragg's law was applied from the peak positions to calculate the interatomic distances and crystal lattice parameters. The diffraction pattern was found to be noisy without any major intense peaks. Less intense peaks were observed at 25.12°, 29.61°, 30.10°, 32.20°, 35.25°, 48.12°, and 52.10°. This indicated that both synthesized compounds were amorphous in nature.40,54,55 XRD can also be used to identify the presence of different crystalline phases in a mixture or to verify the purity of a phthalocyanine complex.3.7. Thermogravimetric studies
Thermogravimetric analysis (TGA) is a technique used to study a materials' thermal stability and decomposition behavior. CoTBImPc was typically prepared as a solid sample. The instrument was programmed to heat the sample at the desired heating rate while continuously measuring its mass. The change in mass was recorded as a function of temperature or time. During the TGA experiment, the complex undergoes thermal decomposition, and the TGA curve shows the weight loss as a function of temperature. This thermogravimetric analysis study was carried out in the temperature range between 0–700 °C and the thermograms obtained are shown in Fig. S7.† By analyzing the TGA curve, information about the thermal stability, decomposition temperatures, and the number of decomposition steps could be obtained. The TGA data were analyzed by examining the temperature at which weight loss occurred, the rate of the weight loss, and the extent of weight loss. The thermal stability of the CoTBImPc compounds showed excellent thermal stability and decomposition temperature. The nature of the curve indicated that the CoTBImPc had higher stability up to 380 °C due to the substituted benzimidazole groups at the terminal ends. The increased molecular weight and structural arraignment increased the thermal stability and decomposition temperature of the CoTBImPc complex. This information helps in understanding the thermal behavior of the complex.3.8. Morphological and structural analysis
Scanning electron microscopy (SEM) can be used to examine various materials' morphology, structure, and composition, including complexes. To ensure effective SEM analysis, samples must exhibit stability and conductivity or be coated with a thin conductive layer. Through SEM, high-resolution images of the sample surface are generated, enabling a meticulous investigation of complex morphology, surface topography, and particle size distribution. These images serve both qualitative and quantitative analytical purposes. The SEM images (Fig. 3) of the CoTBImPc and CoTBImPc/rGO complexes help elucidate the structural analysis and surface morphology of both the CoTBImPc and CoTBImPc/rGO hybrid electrocatalysts and provide essential insights into their composition and distribution, which directly impact their electrocatalytic performance. SEM imaging revealed the surface and internal morphology of both the CoTBImPc and CoTBImPc/rGO hybrid electrocatalysts, showing a uniform and well-dispersed distribution of both CoTBImPc particles and CoTBImPc/rGO on the surface of the electrode from high to low magnification. This homogeneous distribution is crucial for maximizing the exposure of catalytically active sites and ensuring efficient charge transfer during electrochemical reactions. | ||
| Fig. 3 High to low magnification SEM images of CoTBImPc (i) and CoTBImPc/rGO hybrid (ii). | ||
Further, we performed energy-dispersive X-ray spectroscopy (EDS), which allows for elemental analysis, revealing the composition of the complex. The EDX spectrum of the surface complex (depicted in Fig. S8†) illustrated the presence of C, N, O, Br, and Co. While consistent with the elemental composition of CoTBImPc, the hybrid composite demonstrated an increased carbon content, confirming the successful integration of rGO with CoTBImPc, as evident in Fig. S9.† This observation further validated the successful modification of the electrode surface with the Pc complex and its hybrid composite. The presence of increased carbon indicated the existence of rGO, while the oxygen could be attributed to the surface functional groups and oxygen-containing species on the rGO and CoTBImPc surfaces. The homogeneous distribution of CoTBImPc materials on rGO, as evidenced by SEM, and the successful incorporation of cobalt, carbon, and oxygen elements, as revealed by EDX, together verified the successful synthesis of the CoTBImPc/rGO hybrid electrocatalyst. These findings indicate the formation of a well-defined hybrid structure, wherein the CoTBImPc particles were integrated into the rGO matrix, potentially resulting in enhanced electrocatalytic properties. The observed homogeneity and elemental composition provide confidence for the performance of the CoTBImPc/rGO hybrid electrocatalyst, and these findings pave the way for further investigations into its electrocatalytic activity, stability, and practical application in the OER and other electrochemical processes.
4. Electrochemical performances
4.1. Oxygen evolution reaction along with the mechanism
To understand the OER kinetics, we measured the current density and potential of the individual and hybrid complexes. The CoTBImPc/rGO hybrid electrocatalyst exhibited notably enhanced current density compared to the pure CoTBImPc and rGO, which indicated the improved electrocatalytic activity of the hybrid material for the OER. Fig. 4A presents the overpotential graphs for rGO/Ni, CoTBImPc/Ni, and CoTBImPc/rGO/Ni and Ni-foam electrodes under a current density of 10 mAcm−2. Fig. 4B illustrates the overpotential graphs for rGO/Ni, CoTBImPc/Ni, and CoTBImPc/rGO/Ni and Ni-foam electrodes under a current density of 10 mAcm−2. The enhanced current density could be attributed to the synergistic effects between the CoTBImPc and rGO components. Embedding the benzimidazole substituents on the phthalocyanine ligands may facilitate substantial active sites for oxygen evolution. Meanwhile, rGO's high surface area and excellent electrical conductivity improve the charge transfer and aid efficiently utilizing the catalytically active centers. The overpotential is the additional voltage required to drive the OER. A lower overpotential indicates more favorable kinetics for the OER. The CoTBImPc/rGO hybrid electrocatalyst displayed a lower overpotential than pure CoTBImPc and rGO, suggesting that the hybrid material enabled faster electrocatalytic kinetics, leading to a more efficient OER. The improved overpotential was due to the unique combination of active sites and charge transfer facilitation provided by the hybrid architecture.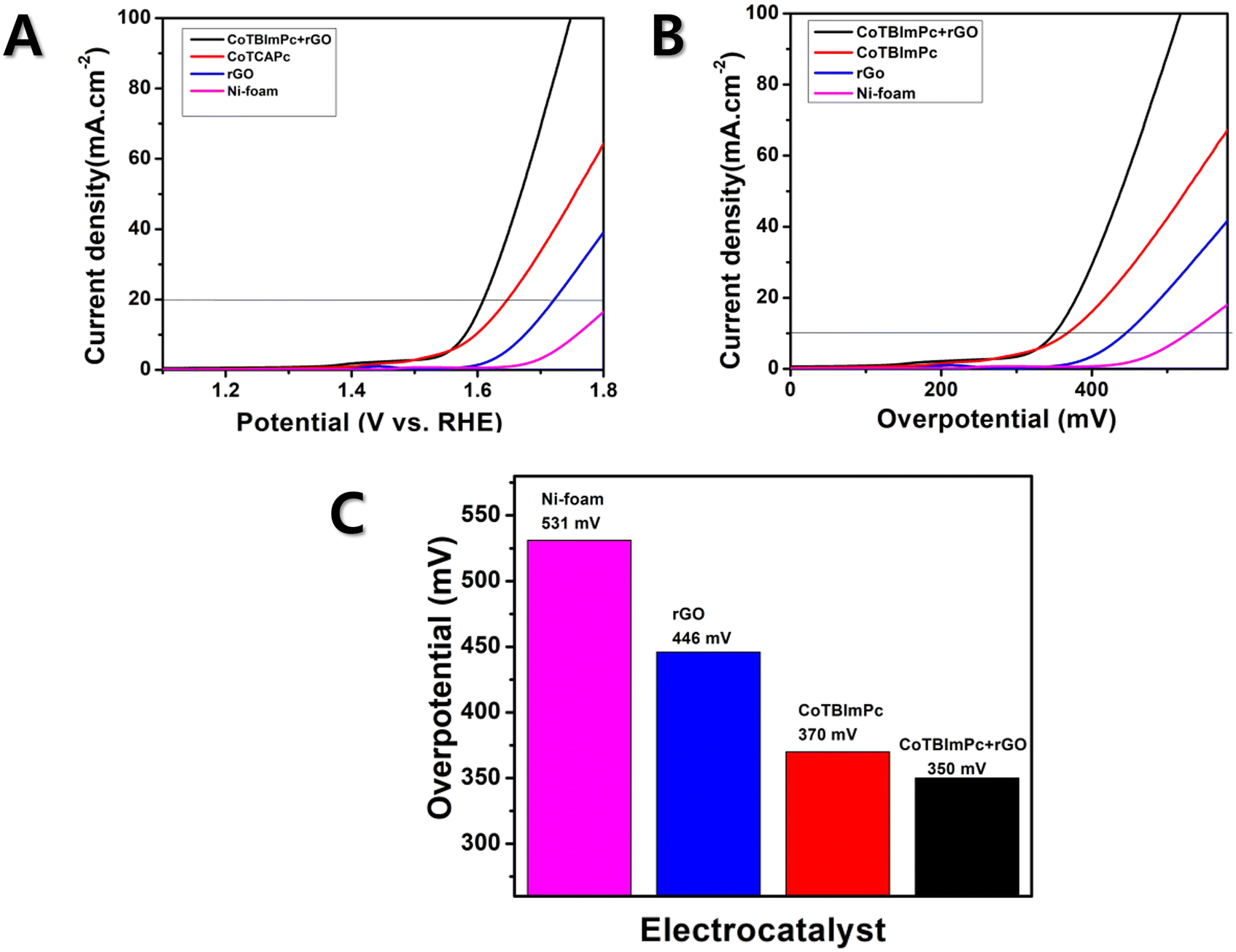 | ||
| Fig. 4 OER performances of the CoTBImPc/rGO hybrid catalyst. | ||
Moreover, the electrochemical performance of the CoTBImPc/rGO hybrid electrocatalyst was analyzed using a three-electrode configuration. The working electrode consisted of CoTBImPc/rGO coated on Ni foam. In contrast, the reference electrode was Ag/AgCl (immersed in 3 M saturated KCl solution), and the counter electrode was Pt wire measuring 1 cm × 1 cm. To ensure perfect functioning, the Ni-foam was reactivated before the experiment. This involved sonicating it separately with 1.0 M HCl and 1.0 M KOH for 30 min each. The performances of the different modified electrodes, namely Ni-foam, CoTBImPc/Ni, rGO/Ni, and CoTBImPc/rGO/Ni, were evaluated for the OER in 1.0 M KOH solution. Linear sweep voltammetry was conducted within the potential range of 1.2–1.8 V, using a lower scan rate of 5 mV s−1. The obtained potentials from the Ag/AgCI electrodes were then converted to reversible hydrogen electrode (RHE) potentials using the following equation,
| E(RHE) = E(Ag/AgCl) + 0.059 (pH) + 0.197 V |
The Ni-foam electrode manifested a lower catalytic activity, with a higher onset potential of 531 mV vs. RHE in 1.0 M KOH solution at a current density of 10 mA cm2 (Fig. 4C). However, when rGO was coated on Ni foam (rGO/Ni-foam electrode), it exhibited an overpotential of 446 mV. Furthermore, when CoTBImPc was coated on the Ni-foam electrode, it showed an improvement in the overpotential of 370 mV. However, during the OER, the hybrid rGO was attached on cobalt phthalocyanine (CoTBImPc/rGO) assured superior catalytic activity, attributed to the Co center and peripheral ligands, which further furnished active sites for OER catalysis. The Co ion undergoes redox reactions, cycling between different oxidation states (Co(II) to Co(III)/Co(IV)/Co(V)) during the electrochemical redox reaction. CoPc constituted a central cobalt ion surrounded by a macrocyclic phthalocyanine ligand. This unique structure could deliver active sites for catalysis and enabled the 4e− transfer processes. Hence, the Ni foam modified with CoTBImPc/GO/Ni-foam exhibited excellent catalytic performance at 350 mV, near the benchmark catalysts RuO2 and IrO2, while also being more cost-effective. This indicates that the synthesized macrocycle demonstrated excellent active participation in oxygen evolution. Notably, this hybrid catalyst showed the potential to replace the benchmark catalysts due to the collaborative interaction of cobalt phthalocyanine and the ligand, which likely contributes to its superior activity.
Initially, in 1 M KOH solution, KOH dissociates into K+ ions and OH− ions. The OH− ions play a crucial role in the OER. CoTBImPc/rGO could serve as an electrocatalyst, promoting the OER. OH− ions adsorb onto the catalyst's surface (CoTBImPc/rGO). This step involves the interaction of OH− with the catalyst's active sites. The adsorption of OH− ions leads to the formation of active sites on the catalyst surface, which are conducive to the OER. In contrast, the water molecules (H2O) in the KOH solution are oxidized at the active sites on the catalyst surface. Two H2O molecules are oxidized to form one molecule of O2 gas. The other product is protons (H+ ions), which can be released into the solution. During the OER, electrons are transferred from the H2O molecules to the catalyst's active sites. The catalyst facilitates this electron transfer, which acts as a mediator. After the OER, the catalyst may change its oxidation state. In some cases, the catalyst needs to be regenerated by gaining electrons, returning it to its active state for subsequent OER cycles. The overall reaction can be represented as:
| 4OH− → O2 + 2H2O + 4e− |
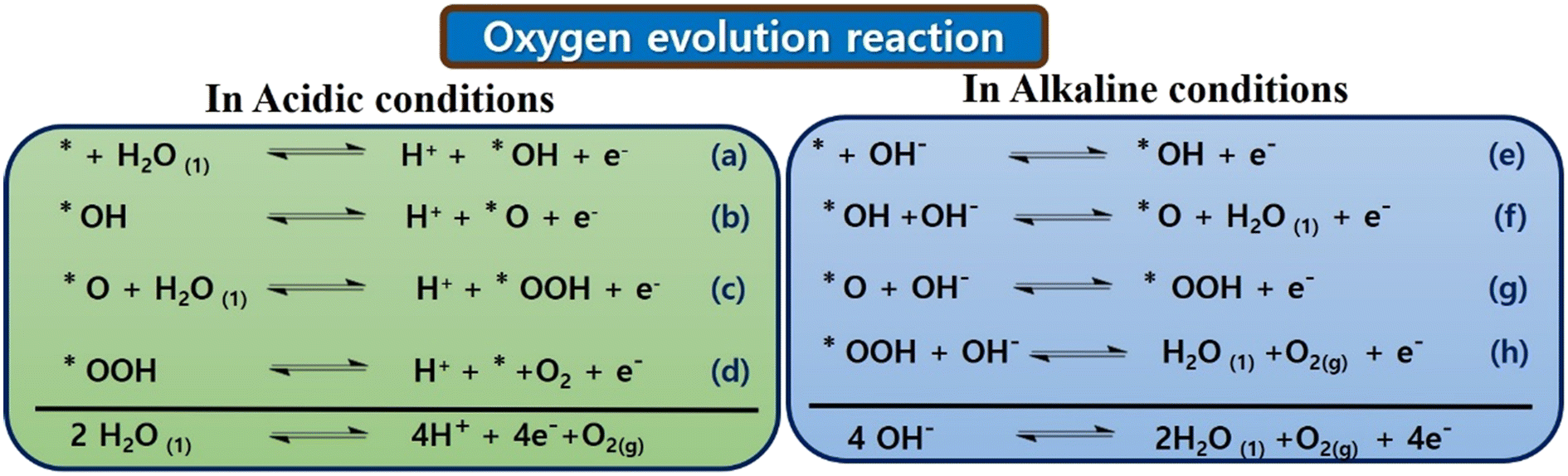 | ||
| Fig. 5 Probable mechanisms of OER in both acidic and alkaline conditions. | ||
4.2. Tafel slope, stability, and durability tests
The Tafel slope is a parameter obtained from a Tafel plot, representing the relationship between the logarithm of the current density and the overpotential. Usually, a reduced Tafel slope signifies a faster OER rate on the electrocatalyst surface. The Tafel slope value offers reliable information about the sensitivity of the current response to a specific voltage setting. The current density is plotted against E to obtain the Tafel plot, resulting in a linear plot with a slope of 1/b. If a catalyst exhibits a higher current density and a lower Tafel slope value, it is considered a suitable designed catalyst for electrochemical OER. Herein, the Tafel plots generated for Ni foam, rGO/Ni, CoTBImPc/Ni, and CoTBImPc/rGO/Ni electrode were extracted from the linear polarization curves shown in Fig. 6. The bare Ni foam had a Tafel slope of 109 mV dec−1. In comparison, rGO/Ni and CoTBImPc/Ni had Tafel slopes of 63 and 56 mV dec−1, respectively. The designed CoTBImPc/rGO/Ni electrode showed a lower value of 51 mV dec−1. The significantly lower Tafel slope value observed for the OER at the CoTBImPc/rGO/Ni electrode indicated the favorable kinetics of the OER at this fabricated electrode. These findings suggest that the hybrid composite CoTBImPc/rGO/Ni electrode is a promising material for OER activity and could potentially replace the expensive benchmark catalysts, such as IrO2 and RuO2. The collected Tafel data were contrasted with those from other previously documented metal phthalocyanine studies as shown in Table 1.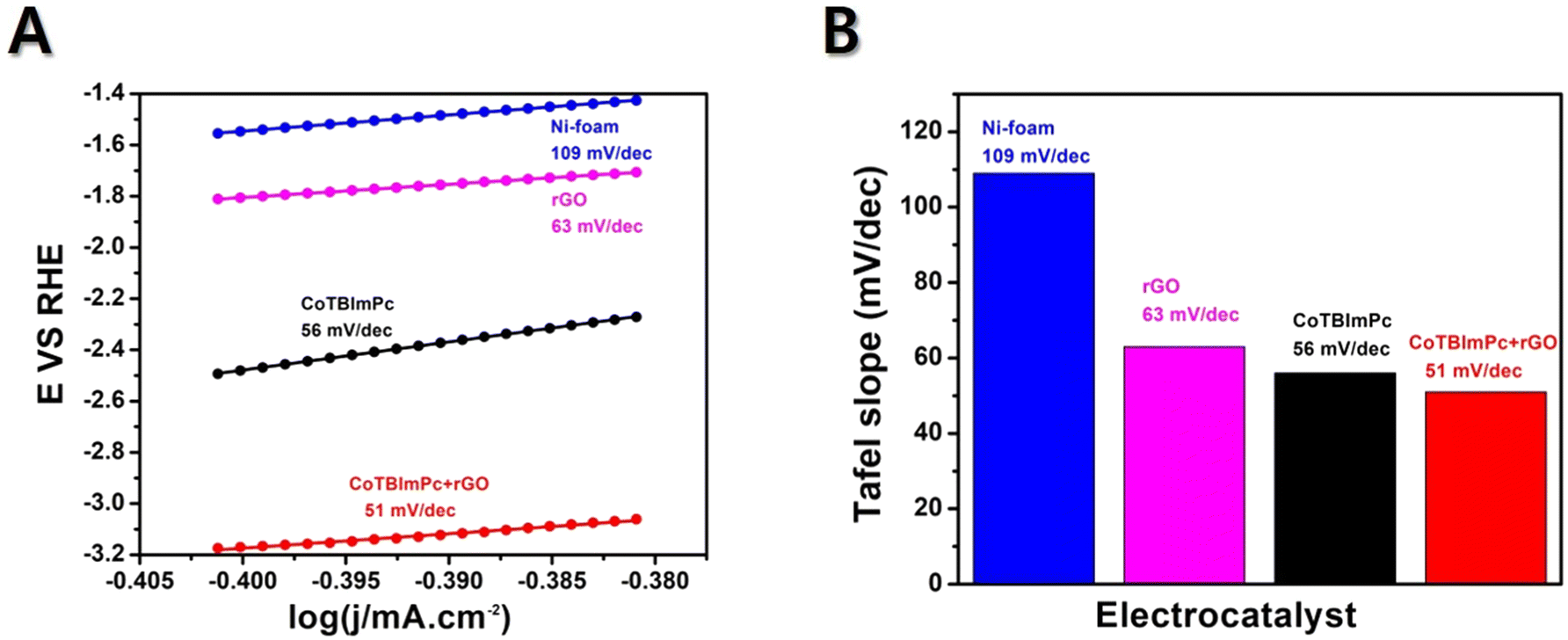 | ||
| Fig. 6 Tafel slope plots for CoTBImPc/rGO hybrid. | ||
| Catalyst | Substrate | Electrolyte | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|
| NiPc | SWCNTs | 1 M KOH | 56.05 | 56 |
| CoPc | SWCNTs | 1 M KOH | 75.36 | |
| CoPc | FTO | 0.1 M NaPO3 | 120.00 | 57 |
| FePc | MWCNT | 0.1 M KOH | 108.50 | 58 |
| NiPc | MWCNT | 0.1 M KOH | 132.90 | |
| NiPc | MOF | 1 M KOH | 74.00 | 59 |
| MnPc | ITO/PNI | 4-Azidoaniline | 300.00 | 60 |
| FePc | Hollow carbon spheres | 0.1 M KOH | 83.00 | 61 |
| NiPc/CoP | Porous carbon nanosheets | 1 M KOH | 71.10 | 62 |
| CoTBImPc | Without rGO | 1.0 M KOH | 56.00 | This work |
| CoTBImPc | With rGO | 1.0 M KOH | 51.00 |
An electrocatalyst's stability is crucial for its commercial applications in energy-conversion and -storage systems. We conducted continuous electrolysis tests at a constant current density over an extended period to evaluate the long-term stability of the CoTBImPc/rGO/Ni-foam hybrid electrocatalyst. The stability of the CoTBImPc/rGO/Ni-foam catalyst was assessed by chronoamperometry with a current density of 10 mA cm−2. The hybrid catalyst exhibited stability for 5.5 h, as shown in Fig. S10a.† The catalyst exhibited electrochemical performance in the current response even after 5.5 h. Usually, N4-macrocycles exhibit exceptional thermal stability up to 380 °C, and prolonged electrochemical cycling without further degradation. Notably, they are insoluble but maintain stability in aqueous environments. Incorporating the rGO moiety augmented the conjugation, increasing the molecule's overall stability. This observation implies that the structural integrity of the organic macrocycle remained unaltered even after enduring the OER durability test. The developed catalyst was thus found to be incredibly stable toward the OER catalytic activity. Fig. 3 and S10b† show that the morphologies of both the fresh and after-stability hybrid composite catalysts did not show any changes, and there was not much difference, indicating the hybrid catalysts' high stability. In summary, the suggested electrocatalyst is among the best catalysts for the OER with good onset potential and overpotential, which are like the benchmark catalysts but with significantly higher stability and current density. In terms of the simplicity of synthesis, electrode fabrication, and outstanding HER activity with less overpotential and remarkable stability, the CoTBImPc/rGO catalyst was shown to be among the top electrocatalysts.
5. Conclusion
The successful development of a CoTBImPc/rGO hybrid electrocatalyst marks a significant step toward advancing efficient and sustainable energy-conversion technologies. Through a facile and scalable synthesis method, the hybrid electrocatalyst was synthesized by integrating cobalt–benzimidazole substituted metal–organic phthalocyanine (CoTBImPc) with reduced graphene oxide (rGO). The integration of CoTBImPc with rGO resulted in a homogeneous distribution of CoTBImPc particles on the rGO, enabling efficient charge transfer and access to abundant catalytic active sites. The hybrid electrocatalyst demonstrated significantly enhanced current density and lower overpotential than the individual components, showcasing the synergistic effects between CoTBImPc and rGO. The reduced Tafel slope further indicated the faster kinetics for the OER on the hybrid electrocatalyst. These characteristics show the CoTBImPc/rGO hybrid's tremendous electrocatalytic performance for the OER. Notably, scaling up the synthesis process of the CoTBImPc/rGO hybrid electrocatalyst for commercial production could also be quickly achieved due to their accessible and prominent synthetic protocols. Developing cost-effective and large-scale manufacturing methods would be essential for its widespread adoption in energy technologies. The CoTBImPc/rGO hybrid electrocatalyst holds excellent promise as a high-performance electrocatalyst for the oxygen evolution reaction and other electrochemical processes. This hybrid material's successful synthesis and remarkable electrocatalytic properties open up exciting opportunities for advancing sustainable energy technologies and accelerating the transition toward a greener and more sustainable future.Author contributions
CPKP and KRN contributed to write and revise this manuscript. Both CPKP and JH equally supervised this work. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Capacity Enhancement Project through Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education (2019R1A6C1010016). K. R. N. gratefully acknowledges the generous financial and infrastructural support from Prof. Agnieszka Nowak-Król (University of Würzburg) and her Emmy-Noether Fellowship (NO1459/1-1) from the German Research Foundation (DFG).References
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed
.
- X. Long, H. Lin, D. Zhou, Y. An and S. Yang, ACS Energy Lett., 2018, 3, 290–296 CrossRef CAS
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 6217 CrossRef PubMed
- H. I. Yang, K. R. Naveen, S. M. Cho, J. Y. Kim, Y. H. Jung and J. H. Kwon, Org. Electron., 2023, 115, 106761 CrossRef CAS
- Y. Gawale, R. Ansari, K. R. Naveen and J. H. Kwon, Front. Chem., 2023, 11, 1211345 CrossRef CAS PubMed
- B. Mohanty, P. Bhanja and B. K. Jena, Mater. Today Energy, 2022, 23, 100902 CrossRef CAS
- Y. Liu, Y. Wang, S. Zhao and Z. Tang, Small Methods, 2022, 6, 2200773 CrossRef CAS PubMed
- Y. Liu, C. Li, C. Tan, Z. Pei, T. Yang, S. Zhang, Q. Huang, Y. Wang, Z. Zhou, X. Liao, J. Dong, H. Tan, W. Yan, H. Yin, Z.-Q. Liu, J. Huang and S. Zhao, Nat. Commun., 2023, 14, 2475 CrossRef CAS PubMed
- Z. Wu, Y. Zhao, H. Wu, Y. Gao, Z. Chen, W. Jin, J. Wang, T. Ma and L. Wang, Adv. Funct. Mater., 2021, 31, 2010437 CrossRef CAS
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2021, 31, 2009070 CrossRef CAS
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC
- J. M. van der Zalm, J. Quintal, S. A. Hira, S. Chen and A. Chen, Electrochim. Acta, 2023, 439, 141715 CrossRef CAS
- S. Zhang, C. Tan, R. Yan, X. Zou, F.-L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202302795 CrossRef CAS PubMed
- F. Qureshi, M. Yusuf, H. Kamyab, S. Zaidi, M. Junaid Khalil, M. Arham Khan, M. Azad Alam, F. Masood, L. Bazli, S. Chelliapan and B. Abdullah, Sustain. Energy Technol. Assess., 2022, 53, 102677 Search PubMed
- Y. Wu, G.-D. Li, Y. Liu, L. Yang, X. Lian, T. Asefa and X. Zou, Adv. Funct. Mater., 2016, 26, 4999 CrossRef
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS
- W. Zhou and D. B. Leznoff, Chem. Commun., 2018, 54, 1829–1832 RSC
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS
- X. Wang, L. Zhuang, T. He, Y. Jia, L. Zhang, X. Yan, M. Gao, A. Du, Z. Zhu, X. Yao and S. H. Yu, iScience, 2018, 7, 145–153 CrossRef CAS PubMed
- X. Zhu, T. Jin, C. Tian, C. Lu, X. Liu, M. Zeng, X. Zhuang, S. Yang, L. He, H. Liu and S. Dai, Adv. Mater., 2017, 29, 1704091 CrossRef PubMed
- K. Ao, Q. Wei and W. A. Daoud, ACS Appl. Mater. Interfaces, 2020, 12, 33595–33602 CrossRef CAS PubMed
- P. Bhanja, Y. Kim, B. Paul, Y. V. Kaneti, A. A. Alothman, A. Bhaumik and Y. Yamauchi, Chem. Eng. J., 2021, 405, 126803 CrossRef CAS
- Y. Guo, C. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Shi, ACS Sustainable Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS
- S. Hu, S. Wang, C. Feng, H. Wu, J. Zhang and H. Mei, ACS Sustainable Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, Y. Guo, Y. Ide, B. Yuliarto, X. Jiang, Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 3035–3047 RSC
- J. L. Gómez Urbano, M. Enterría, I. Monterrubio, I. Ruiz de Larramendi, D. Carriazo, N. Ortiz Vitoriano and T. Rojo, ChemSusChem, 2020, 13, 1203–1225 CrossRef PubMed
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS
- C. T. Bowen, H. J. Davis, B. F. Henshaw, R. Lachance, R. L. LeRoy and R. Renaud, Int. J. Hydrogen Energy, 1984, 9, 59–66 CrossRef CAS
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS
- K. P. Cp, S. Aralekallu, V. A. Sajjan, M. Palanna, S. Kumar and L. K. Sannegowda, Sustainable Energy Fuels, 2021, 5, 1448–1457 RSC
- S. Aralekallu, V. A. Sajjan, M. Palanna, C. P. Keshavananda Prabhu, M. Hojamberdiev and L. K. Sannegowda, J. Power Sources, 2020, 449, 227516 CrossRef CAS
- C. P. Keshavananda Prabhu, K. R. Naveen, S. Aralekallu, Shivalingayya and L. K. Sannegowda, RSC Sustainability, 2023, 1, 128–138 RSC
- S. Hadimane, S. Aralekallu, K. P. Cp, M. Hojamberdiev and L. K. Sannegowda, ACS Appl. Energy Mater., 2021, 4, 10826–10834 CrossRef CAS
- C. P. Keshavananda Prabhu, M. Nemakal, S. Aralekallu, I. Mohammed, M. Palanna, V. A. Sajjan, D. Akshitha and L. K. Sannegowda, J. Electroanal. Chem., 2019, 847, 113262 CrossRef CAS
- N. Manjunatha, M. Imadadulla, K. S. Lokesh and K. R. Venugopala Reddy, Dyes Pigm., 2018, 153, 213–224 CrossRef CAS
- V. A. Sajjan, I. Mohammed, M. Nemakal, S. Aralekallu, K. R. Hemantha Kumar, S. Swamy and L. K. Sannegowda, J. Electroanal. Chem., 2019, 838, 33–40 CrossRef CAS
- M. Nemakal, S. Aralekallu, I. Mohammed, C. P. Keshavananda Prabhu and L. Koodlur Sannegowda, Microchem. J., 2018, 143, 82–91 CrossRef CAS
- C. P. Keshavananda Prabhu, S. Aralekallu, M. Palanna, V. Sajjan, B. Renuka and L. K. Sannegowda, J. Appl. Electrochem., 2022, 52, 325–338 CrossRef CAS
- P. Gaudillat, F. Jurin, B. Lakard, C. Buron, J.-M. Suisse and M. Bouvet, Sensors, 2014, 14, 13476–13495 CrossRef PubMed
- K. S. Lokesh and A. Adriaens, Dyes Pigm., 2013, 96, 269–277 CrossRef CAS
- C. P. Keshavananda Prabhu, N. Manjunatha, A. Shambhulinga, M. Imadadulla, K. H. Shivaprasad, M. K. Amshumali and K. S. Lokesh, J. Electroanal. Chem., 2019, 834, 130–137 CrossRef CAS
- M. Palanna, S. Aralekallu, C. P. Keshavananda Prabhu, V. A. Sajjan, Mounesh and L. K. Sannegowda, Electrochim. Acta, 2021, 367, 137519 CrossRef CAS
- S. Aralekallu, M. Palanna, S. Hadimani, C. P. Keshavananda Prabhu, V. A. Sajjan, M. O. Thotiyl and L. K. Sannegowda, Dalton Trans., 2020, 49, 15061–15071 RSC
- M. Nemakal, S. Aralekallu, I. Mohammed, S. Swamy and L. K. Sannegowda, J. Electroanal. Chem., 2019, 839, 238–246 CrossRef CAS
- C. P. Keshavananda Prabhu, M. Nemakal, M. Managa, T. Nyokong and L. Koodlur Sannegowda, Front. Chem., 2021, 9, 647331 CrossRef PubMed
- O. O. Fashedemi and K. I. Ozoemena, Sens. Actuators, B, 2011, 160, 7–14 CrossRef CAS
- I. Mohammed, M. Nemakal, V. A. Sajjan, D. B. Puttappashetty and L. K. Sannegowda, J. Electroanal. Chem., 2018, 826, 96–103 CrossRef CAS
- N. Alzate-Carvajal, L. M. Bolivar-Pineda, V. Meza-Laguna, V. A. Basiuk, E. V. Basiuk and E. A. Baranova, ChemElectroChem, 2020, 7, 428–436 CrossRef CAS
- N. Morlanés, K. S. Joya, K. Takanabe and V. Rodionov, Eur. J. Inorg. Chem., 2015, 2015, 49–52 CrossRef
- A. Abbaspour and E. Mirahmadi, Electrochim. Acta, 2013, 105, 92–98 CrossRef CAS
- H. Jia, Y. Yao, J. Zhao, Y. Gao, Z. Luo and P. Du, J. Mater. Chem. A, 2018, 6, 1188–1195 RSC
- B. Öztaş, D. Akyüz and A. Koca, Phys. Chem. Chem. Phys., 2017, 19, 26121–26131 RSC
- M. Y. Song, D.-S. Yang, K. P. Singh, J. Yuan and J.-S. Yu, Appl. Catal., B, 2016, 191, 202–208 CrossRef CAS
- H.-J. Niu, A.-J. Wang, L. Zhang, J.-J. Guo and J.-J. Feng, Mater. Chem. Front., 2019, 3, 1849–1858 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00193h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |