
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonated polythiophene-interfaced graphene for water-redispersible graphene powder with high conductivity and electrocatalytic activity†
Tuan Sang
Tran
 a,
Rajkamal
Balu
a,
Liliana
de Campo
b,
Naba Kumar
Dutta
*a and
Namita Roy
Choudhury
*a
a,
Rajkamal
Balu
a,
Liliana
de Campo
b,
Naba Kumar
Dutta
*a and
Namita Roy
Choudhury
*a
aSchool of Engineering, RMIT University, Melbourne, Victoria 3000, Australia. E-mail: naba.dutta@rmit.edu.au; namita.choudhury@rmit.edu.au
bAustralian Centre for Neutron Scattering, ANSTO, Lucas Heights, NSW 2232, Australia
First published on 9th January 2023
Abstract
The production of redispersible graphene, especially in an aqueous medium, is highly desirable for its practical applications. In this study, we develop a simple route for the production of water-redispersible graphene powder by interfacing graphene with an amphiphilic polymer, poly[2-(3-thienyl)ethyloxy-4-butylsulfonate] sodium salt (PTEBS), via exfoliation-assisted noncovalent functionalisation. The PTEBS-interfaced graphene flakes can be self-dispersed in the aqueous phase without the presence of excessive stabilisers. The interfacial interactions between pristine graphene and amphiphilic PTEBS molecules and the intrinsic colloidal structure of their dispersions at the nanoscale were studied using ultra-small/small angle neutron scattering (U/SANS) with contrast-variation for the first time. It is found that the strong noncovalent π-stacking interaction between graphene and the hydrophobic thiophene rings of PTEBS disrupts the π–π interactions holding the graphite layers and promotes exfoliation; whereas, the sodium sulfonated moieties of PTEBS render hydrophilicity to the exfoliated graphene. A water-redispersible graphene powder based on π-stacking of PTEBS molecules is produced, which shows excellent capabilities for the formulation of graphene inks for the printing of flexible conductive circuits (∼30 Ω sq−1) and metal-free electrocatalyst layers for the oxygen reduction reaction (ORR), which reduce O2 molecules to OH− ions through the highly efficient four-electron pathway and exhibit superior durability under a methanol crossover effect.
1. Introduction
Graphene, a two-dimensional allotrope of carbon, has attracted enormous attention in the scientific and technological fields due to its unprecedented properties. Over the past decade, graphene has found significant utilities in the energy,1,2 biomedical,3,4 environmental5,6 and electronics fields.7,8 Nevertheless, the scalable production of pristine graphene in a processable form for industrial applications is still challenging, yet highly desirable.Among the available production strategies, liquid-phase exfoliation has the most promising track record for bulk production of high quality graphene due to its cost-effectiveness, simplicity and scalability.9,10 The fundamental principle of liquid-phase exfoliation is to overcome the π–π interactions (van der Waals forces) between stacked graphite layers for the extraction of individual graphene flakes in a liquid medium by applying energy, such as ultrasonication or high-shear.10,11 With respect to the London forces (dispersive interactions), the intermolecular forces between the graphite layers can be significantly reduced by liquid immersion in a medium with matching surface energy.12,13 Therefore, organic solvents with similar surface energies, such as N-methyl-2-pyrrolidone (NMP) and N,N-dimethylformamide (DMF) are extensively used for graphite exfoliation.13 However, these solvents are expensive and highly toxic. Their industrial use has raised significant environmental concerns and is subjected to strict regulations.14 For this reason, recent research has driven toward the use of water as a low-cost and eco-friendly dispersion medium for graphene dispersions.15 Due to the intrinsic hydrophobicity of graphitic carbon, an excess of stabilisers (or surfactants) is added to promote exfoliation and prevent aggregation.16 However, the presence of these compounds in the graphene dispersions is undesirable, especially for high-performance electronics, where dispersants become contaminants.17 Therefore, it is essential to develop new approaches to disperse graphene in an aqueous medium without the presence of excessive dispersants.
In attempts to incite the disruption of π–π interactions holding the adjacent graphite layers, various studies have been conducted on the adsorption of aromatic molecules on graphitic carbon.18 It has been reported that surface modification of graphite by sidewall noncovalent functionalisation via π-stacking with aromatic compounds can interrupt the π–π interactions between the adjacent graphite layers stacked together, thereby destabilising the adjacent layer structures and promoting exfoliation.19 For many years, the adsorption of aromatic molecules on graphite has been studied extensively,20 although the notion of exploiting this fundamental process as the driving force for graphite exfoliation has only recently emerged. A recent study by Denis et al.21 reported that thiophene groups can be strongly adsorbed on the surface of graphene. More recently, Gharib and co-workers22 demonstrated that the efficient exfoliation of graphite utilises organic acceptors in the solvent, driven by the charge transfer process from electron-rich graphene flakes to electron-withdrawing aromatic molecules via π-stacking. It is noted that the cleavage of the π–π interactions was likened to a solubilisation process and inextricably linked to the solubility behaviour of the organic molecules.22,23 To this end, we hypothesise that exfoliation relies on π-stacking of aromatic amphiphilic molecules, which can render hydrophilicity onto the graphene surface, and would allow for graphene to be dispersed in water. As a result, the noncovalently functionalised graphene sheets can remain individually stable whilst removing the un-adsorbed amphiphilic molecules from the aqueous dispersions.
Poly[2-(3-thienyl)ethyloxy-4-butylsulfonate] sodium salt (PTEBS), a water-soluble polythiophene derivative, is widely used as an efficient photo-induced charge transfer for polymer photovoltaic applications. PTEBS is an electrically conductive amphiphilic molecule composed of heterocyclic aromatic rings (thiophene groups) and appended sodium sulfonated functional groups (Fig. 1(a)). These sodium sulfonated moieties make PTEBS soluble in water, while the thiophene groups enable it to interact with graphene, allowing for PTEBS to act as an interfacial stabiliser in the aqueous solution. Therefore, it is expected that under the assistance of sonication, PTEBS can disrupt the π–π interactions, strongly adsorbs onto the graphene basal plane and extends the exfoliated flakes to the aqueous phase to form a stable homogenous graphene dispersion. As the noncovalently functionalised graphene flakes can be solvated in water by itself, the un-adsorbed PTEBS molecules can be removed without affecting the stability of the graphene dispersions. Successful exfoliation and stabilisation of graphene via this approach would allow for the emergence of a new class of self-dispersible pristine graphene and provide opportunities for further processing into dry water-redispersible graphene powder.
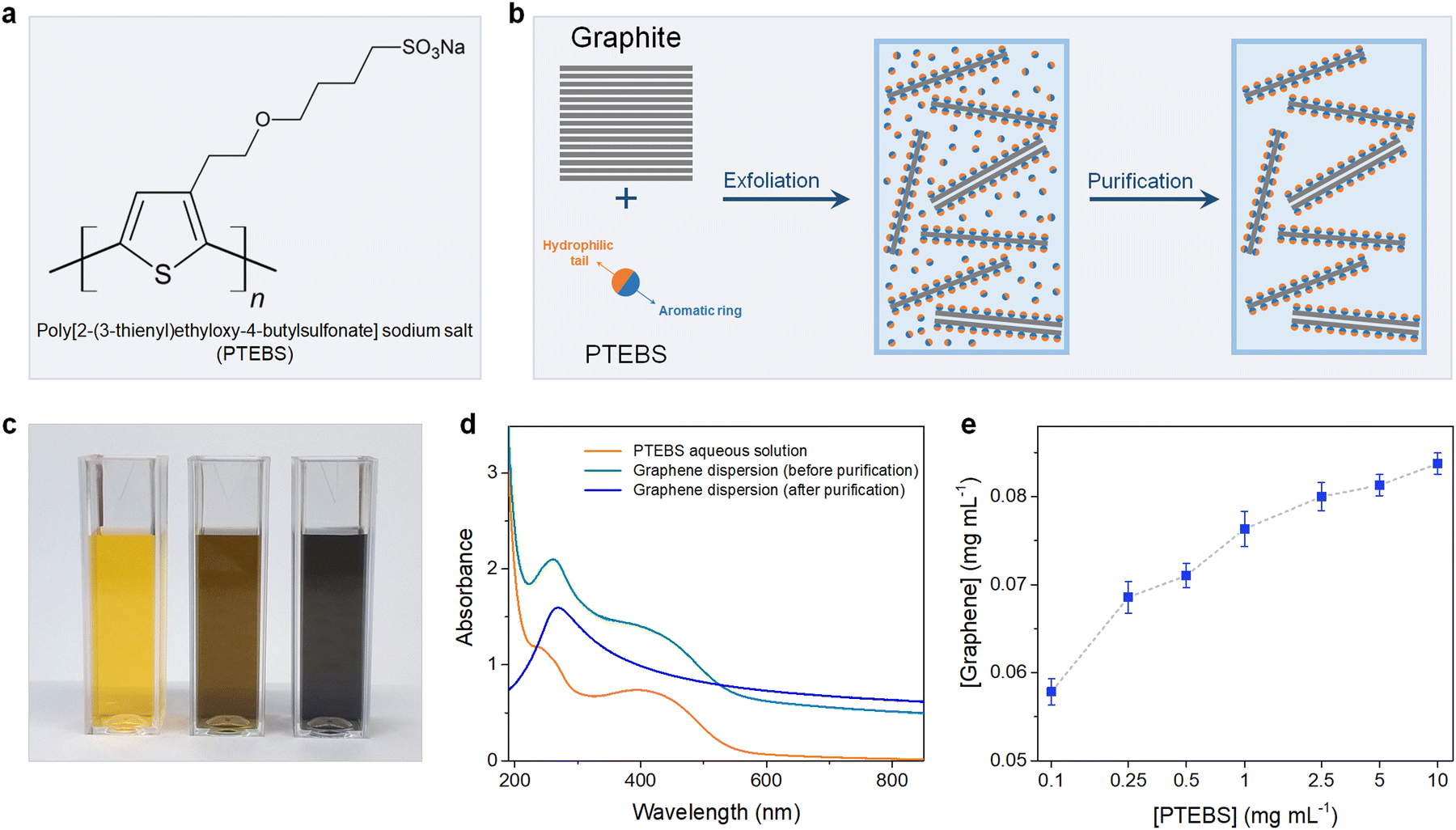 | ||
| Fig. 1 (a) Chemical structure of PTEBS. (b) Schematic of the preparation of PTEBS-interfaced graphene dispersions. (c) Photograph of the diluted solution of the aqueous PTEBS solution (left), and the graphene dispersion stabilised by PTEBS before purification (middle), and after purification (right). (d) The UV-vis spectra of aqueous PTEBS solution (orange trace), and the PTEBS-interfaced graphene dispersion before purification (cyan trace), and after purification (blue trace). (e) Graphene concentration plotted as a function of PTEBS concentration. | ||
In this study, we demonstrate such a process. We show that the adsorption of PTEBS molecules can disrupt the π–π interactions between graphite layers to incite exfoliation without generating defects or changing the chemical structure of graphene. The exfoliated flakes are self-dispersed in water without the presence of excessive dispersants. A water-redispersible graphene powder is produced, which shows excellent dispersibility in the aqueous phase. Highly concentrated water-based graphene inks (up to 10 mg mL−1) were formulated and utilised for printing of flexible conductive circuits, which exhibit an excellent electrical conductivity of ∼30 Ω sq−1 without the requirement for thermal treatment. The PTEBS-interfaced graphene was also employed for the fabrication of metal-free electrocatalyst layers for the oxygen reduction reaction (ORR), which reduce O2 molecules to OH− ions through the highly efficient four-electron pathway and exhibit superior durability under the methanol crossover effect. The described water-dispersible graphene can be mass-produced in an industrial manner and holds great potential for widespread applications.
2. Results and discussion
2.1. The interfacial interactions between graphene and PTEBS
The chemical structure of PTEBS is shown in Fig. 1(a). PTEBS is a water-soluble amphiphilic polymer consisting of a heterocyclic aromatic ring (thiophene group) appended with a hydrophilic sodium sulfonated moiety. The thiophene groups of PTEBS, owing to its planar and aromatic structure, can be adsorbed onto the surface of graphene in a parallel configuration by π-stacking;21,24 whereas, the hydrophilic sodium sulfonated moieties can be extended to the aqueous phase, providing water-dispersibility to grafted graphene flakes. Such a structure allows for PTEBS to act as an interfacing stabiliser for graphite exfoliation. In fact, ultrasonication of graphite in PTEBS aqueous solutions resulted in stable black-coloured graphitic supernatants, indicating the successful exfoliation and functionalisation. The resulted suspensions were purified through two cycles of sedimentation by ultra-centrifugation and redispersion in pure water to remove the un-adsorbed PTEBS molecules, yielding stable graphene dispersions without the presence of excessive dispersant in the solutions. The preparation of aqueous PTEBS-interfaced graphene dispersions is illustrated in Fig. 1(b).To gain more insight into the interfacial interactions between graphene and PTEBS molecules, we sought to compare the difference between the graphene suspensions before, and after purification. Fig. 1(c) shows the photograph of the aqueous PTEBS solution (left cuvette), graphene dispersion before purification (middle cuvette), and after purification (right cuvette). A unique orange colour was observed in the aqueous PTEBS solution, which was still apparent in the graphene dispersion before purification. In contrast, the graphene dispersion after purification showed a pristine black colour without any sight of the orange shade, suggesting the absence of free PTEBS molecules. The corresponding UV-vis absorption spectra of these three cuvettes are shown in Fig. 1(d). The absorption spectrum of the aqueous PTEBS solution (orange trace) is dominated by strong adsorption bands around ∼250 nm and ∼430 nm, corresponding to the π–π* interband and the n–π* transitions, respectively.25 The as-sonicated sample showed redshift of the ∼250 nm peak to ∼270 nm, indicating the presence of graphitic carbon and intermolecular interactions (π-stacking) between graphene and PTEBS;15,26 whereas, the characteristic bands at ∼430 nm of PTEBS remained apparent. In contrast, the absorption spectrum of the graphene dispersion after purification (blue trace) showed only one distinct maxima at ∼270 nm, corresponding to the π–π* conjugation of graphene.15,27,28 The signature bands of PTEBS had completely vanished, indicating the complete removal of the free PTEBS molecules after purification. These results suggest that the presence of excessive, free PTEBS molecules is not required to stabilise graphene in aqueous dispersions, unlike most surfactants reported in the literature.7,29–33 As a comparison, graphene dispersions stabilized by PVA and PTAA are also produced, in which the PVA-exfoliated graphene becomes unstable with significant aggregation upon the removal of excessive PVA molecules, demonstrating the interfacial effect associated with the solubility behaviour of the adsorbed amphiphilic molecules on the surface of graphene (Scheme S1 in the ESI†).
The exfoliation process was optimised under different processing parameters, in which the concentration of graphene produced was estimated according to the Lambert–Beer law.9,11 The initial precursor graphite concentration was set at 10 mg mL−1, which was found to be optimal for exfoliation as increasing graphite concentrations did not yield equally more graphene (Fig. S1 in the ESI†). We then studied the effect of PTEBS concentration on graphene exfoliation by varying the initial PTEBS concentration from 0.1 to 10 mg mL−1, as shown in Fig. 1(e). It is noted that with 100 times increase in the initial PTEBS concentrations, the concentration of obtained graphene was only slightly increased by ∼1.4 times (i.e. from 0.58 to 0.84 mg mL−1). This suggests that the excessive amount of free PTEBS molecules did not play a significant role in graphene exfoliation, since only a limited amount of these amphiphilic molecules could be adsorbed on the graphene surface. Furthermore, the graphene concentration in the dispersions increased gradually until 4 h of sonication time and longer ultrasound treatment (up to 12 h) did not result in a reasonable higher concentration (Fig. S2 in the ESI†). Overall, the yield of graphene was close to ∼1% in a typical experiment setting, which could be further increased up to ∼3.5% by decreasing the initial graphite concentration, superior compared to previous studies.9,11,13,29
2.2. The interfacial graphene–PTEBS interactions at the nanoscale
Neutron scattering techniques, such as SANS and USANS, are powerful experimental tools to study the hierarchical structure of colloidal dispersions of the order of 1 nm to 50 μm.34,35 Due to the particular sensitivity of neutrons toward hydrogen, SANS and USANS offer a unique capability of contrast-matching one component over others (with different neutron SLD) and studying their individual structure in mixed/complex dispersions.36 Although SANS and USANS have been used to study the bulk structure of GO-based dispersions,37 the colloidal structure of pristine graphene dispersions and their interfacial interactions with stabilising molecules have never been explored.In this work, the interfacial interactions and intrinsic colloidal structure of the PTEBS-interfaced graphene dispersions at the nanoscale were studied by combined SANS and USANS under different contrast-matching conditions for the first time. Fig. 2(a) shows the schematic of the visible structures of the produced graphene (comprising graphene and PTEBS) that can be studied under different contrast-matching conditions. Fig. 2(b) shows the combined SANS and USANS intensity profiles of the aqueous graphene dispersion under different contrasts. With 100% H2O as the dispersion medium, scattering contributions from both PTEBS and graphene can be seen (i.e. without any contrast matching); whereas, with 30% D2O and 100% D2O the individual scattering contributions of only graphene and PTEBS were highlighted, respectively. The scattering intensity profile (within the experimental range) of the graphene dispersion prepared with 100% H2O showed three major Porod regions: a high-q (0.001 < q < 0.3 Å−1), a mid-q (0.000175 < q < 0.001 Å−1), and a low-q (0.00007 < q < 0.000175 Å−1). The Guinier and mid-q Porod regions provide the radius-of-gyration (Rg), which estimates the overall shape and size of the colloidal structures36,38 and the high-q Porod region provides information about the “fractal dimensions” of the scattering object or quality of the interface.39 When compared to the 100% H2O sample, a decrease in overall scattering intensity with no significant change in the shape of the scattering curve was observed for the contrast-matched samples. Moreover, the square root of the scattering intensity, I(q), as a function of the dispersion medium SLD (Fig. 2(c)) provides the overall contrast match point of the PTEBS-interfaced graphene in the dispersion, which is between those of pristine PTEBS (∼1.52 × 10−6 Å−2) and graphene (∼6.40 × 10−6 Å−2). This demonstrates that PTEBS is absorbed on the graphene surface and acts as an interfacing bridge between graphene and the aqueous interfaces. Thus, the noncovalently functionalised graphene derived from π-stacking of amphiphilic PTEBS molecules on the graphene surface in the present work is established as graphene interfacing PTEBS molecules in an aqueous dispersion.
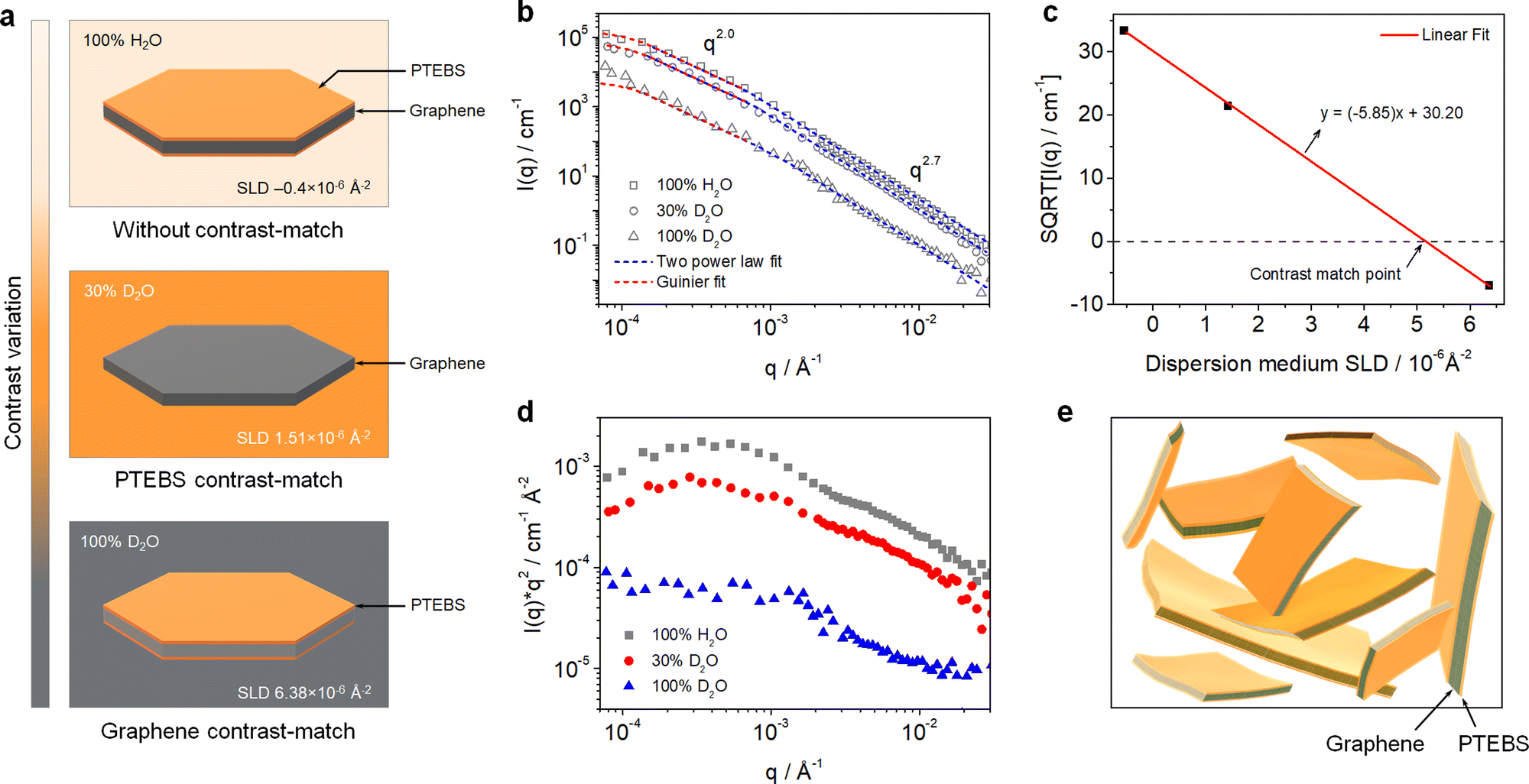 | ||
| Fig. 2 (a) Schematic of visible structures in the PTEBS-interfaced graphene dispersion under different contrast-matching conditions. (b) Combined SANS and USANS intensity profiles of the PTEBS-interfaced graphene dispersions with shape-independent model fits under different contrast-matching conditions. (c) Square root of the measured intensity (at q = 0.001) as a function of the dispersion medium SLD. (d) Kratky plots of the PTEBS-interfaced graphene dispersion under different contrast-matching conditions. (e) Schematic of the colloidal structure of the PTEBS-interfaced graphene dispersion. | ||
To determine the overall size and shape and fractal dimensions of the colloids in the PTEBS-interfaced graphene dispersion, a combination of two shape-independent models was fitted to the respective scattering data, as shown in Fig. 2(b): a Guinier–Porod fit at low-q to mid-q and a two power law fit at mid-q and high-q, where the power law for the two models in the mid-q region was identical.40 The Guinier model fit (with mid-q Porod slope ∼2.0) returned an Rg value of 1.20 ± 0.19 μm, consistent with the size values estimated from the SEM results. The mid-q Porod slope of ∼2.0 corresponded to the overall sheet-like structure of the PTEBS-interfaced graphene;41 where, both the graphene and PTEBS contrast-matched samples exhibited a sheet-like structure, indicating a monolayer-like coverage of PTEBS on graphene. The two power law model fit returned mid-q and high-q Porod slopes of ∼2.0 and ∼2.7, respectively, for all the samples. The high-q Porod slope of ∼2.7 for all the samples suggests that the local structure of graphene in the aqueous dispersion is not a perfectly flat sheet and must have some variations in the thickness, as would be observed for locally varying numbers of graphene layers or wrinkles.41 The overall and local structure of the graphene in the dispersion is also supported by the Kratky plots of the 100% H2O and 30% D2O samples (Fig. 2(d)); where, two distinct regions can be observed in the mid-q and high-q regions, respectively. Therefore, the hierarchical structure of the produced dispersion is established as a colloidal dispersion of graphene sheets stabilised by monolayers of PTEBS adsorbed on their surface, where graphene is interfacing PTEBS molecules in an aqueous dispersion. A schematic of the colloidal structure of the PTEBS-interfaced graphene dispersion is shown in Fig. 2(e).
2.3. Morphology and quality of the PTEBS-interfaced graphene
The morphology of the PTEBS-interfaced graphene flakes was assessed by TEM, SEM, and AFM. The TEM images show that thin flakes of graphene were successfully produced, with different lateral sizes ranging from 500 to 2500 nm (Fig. 3(a)–(c), and Fig. S3 in the ESI†), indicating the successful exfoliation of graphite. The selected-area electron diffraction (SAED) pattern obtained from the central part of the exfoliated flake displays a six-fold symmetric diffraction pattern (inset in Fig. 3(c)), indicating the presence of monolayer graphene.42 For statistical analysis of the flake size, graphene sheets were transferred on to an alumina membrane by vacuum filtration of a diluted graphene dispersion and observed under SEM (Fig. 3(d)). We measured the lateral size (the largest dimension) of more than 250 flakes and discovered that they predominantly (∼85%) ranged from 1 to 3 μm (Fig. S4a in the ESI†). Some larger flakes with sizes up to 5 μm were also observed. Fig. 3(e) shows the AFM image of the produced graphene. The height profile acquired across the flake revealed its corresponding thickness of close to ∼1 nm (Fig. 3(f)), similar to that of monolayer surfactant-exfoliated graphene.9,29 The statistical analysis of 100 graphene sheets revealed that most flakes were thinner than 5 nm, indicating that ∼95% of the exfoliated flakes were composed of less than 5 layers (Fig. S4b in the ESI†).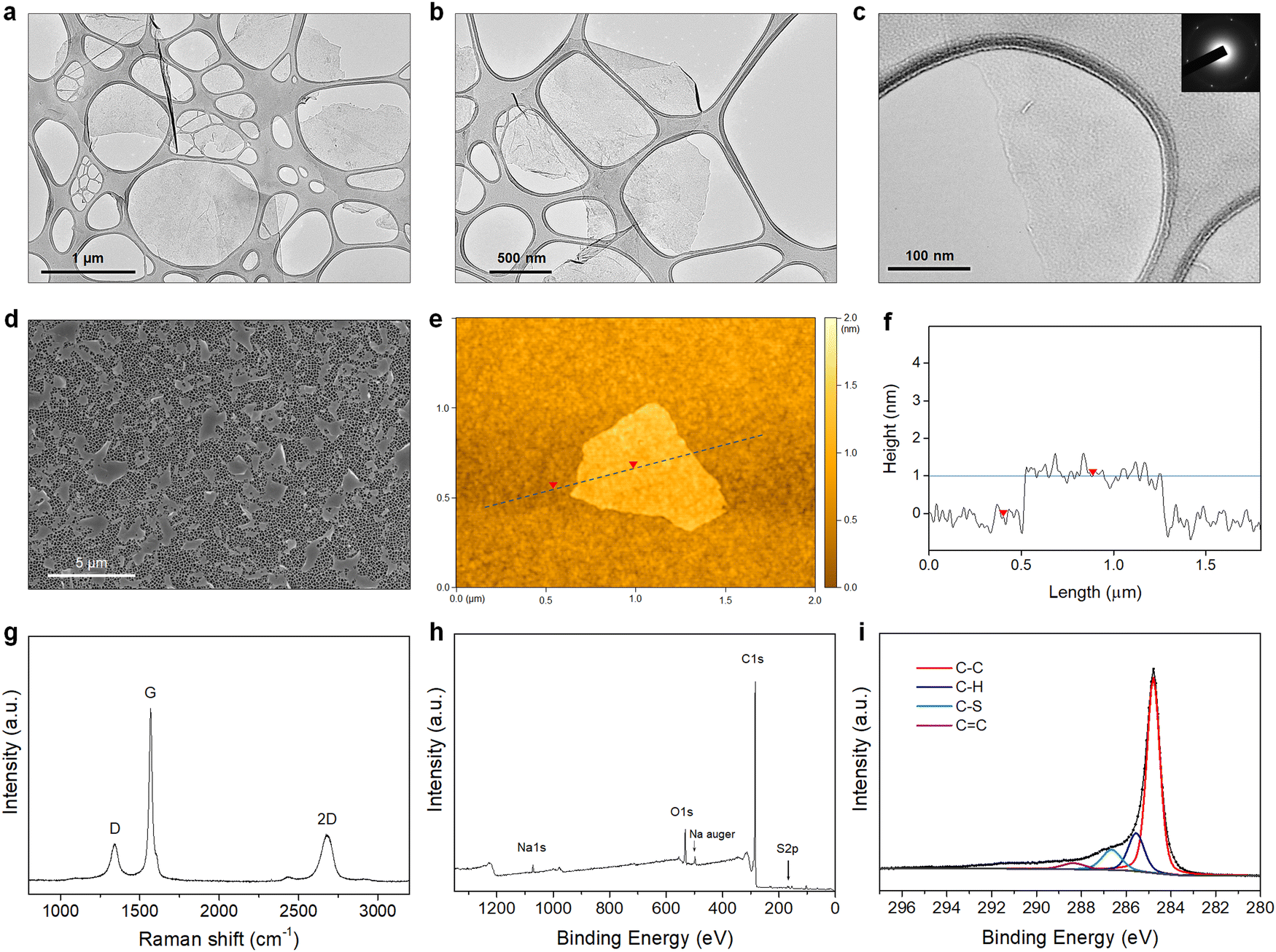 | ||
| Fig. 3 (a)–(c) The TEM images of the exfoliated graphene flakes (inset in [c]: the selected-area electron diffraction pattern). (d) The SEM image of the graphene flakes laid on the alumina membrane. (e) The AFM image of the graphene flake. (f) The height profile (marked by the dashed line in [e]). (g) The Raman spectrum, (h) XPS survey spectrum, and (i) C 1s core-level spectrum of the PTEBS-interfaced graphene. | ||
Fig. 3(g) shows the Raman spectrum of the exfoliated graphene, with three typical characteristic bands at ∼1350 cm−1 (D-band), ∼1580 cm−1 (G-band), and ∼2700 cm−1 (2D-band). As defects (such as sp3 defects, edges, or vacancies) of the graphene lattice incite the emergence of the D-band, the intensity ratio (ID/IG) is used to establish the degree of defects of the graphene lattice.43 The PTEBS-interfaced graphene flakes exhibited a relatively weak D-band with (ID/IG) of ∼0.2, comparable to solvent- and surfactant-exfoliated pristine graphene.9,11,44 The XPS survey spectrum of PTEBS-interfaced graphene is shown in Fig. 3(h), where only carbon, oxygen, sodium, and sulphur were detected. The presence of sulphur and sodium could only originate from the thiophene and sodium sulfonated groups of the PTEBS molecules, since neither the starting graphite material nor the liquid medium contains these atoms. A sodium Auger peak was observed at ∼497 eV, suggesting the strong adsorption of PTEBS molecules on the graphene surface.45 The high-resolution S 2p spectrum of the PTEBS-interfaced graphene (Fig. S5 in the ESI†) shows two peaks at ∼163 and ∼165 eV originating from the thiophene group adsorbed on the surface of graphene and the doublet asymmetric peaks due to the effect of spin–orbit splitting of sulfur elements.46Fig. 3(i) shows the C 1s core level spectrum of the produced graphene, which shows a dominant peak at ∼284.8 eV, representing the sp2 graphitic carbon (C–C).11,47 The additional small peaks located at 285.6, 286.7 and 288.5 eV can be assigned to the sp3 carbon (C–H), sulfonated carbon (C–S) and (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) double bond, respectively.47–51 As Raman spectroscopy confirmed that there were no significant basal plane defects, these minor peaks are attributed to the adsorbed PTEBS molecules on the graphene surface. Therefore, it is suggested that the graphene powder produced in this work is of high quality, non-oxidative and free of defects with comparable characteristics to the pristine graphene produced by other solvent/surfactant liquid-exfoliation processes.9,11,29,44,52
C) double bond, respectively.47–51 As Raman spectroscopy confirmed that there were no significant basal plane defects, these minor peaks are attributed to the adsorbed PTEBS molecules on the graphene surface. Therefore, it is suggested that the graphene powder produced in this work is of high quality, non-oxidative and free of defects with comparable characteristics to the pristine graphene produced by other solvent/surfactant liquid-exfoliation processes.9,11,29,44,52
2.4. Printing application of the PTEBS-interfaced graphene dispersions
For printing applications, formulation of graphene inks in aqueous suspensions is simple and straightforward as the prepared graphene powder is easily dispersed in water by ultrasonication (Fig. S6 in the ESI†). To explore the printing potential of the prepared G/PTEBS dispersions (at concentrations varying from 1 to 10 mg mL−1), the viscoelastic properties of the inks are investigated. The surface tensions of the formulated graphene inks were observed to be similar to that of water (∼72 mN m−1) and showed no significant difference while increasing the graphene concentration from 1 to 10 mg mL−1 (Fig. S7 in the ESI†). This is due to the fact that the total solid content of the tested inks was less than 1 wt%, thereby not affecting the density and intermolecular attractions of the dispersions. Fig. S8 in the ESI† displays the viscosity profiles of graphene inks at different concentrations; where, the inks with higher concentrations possess higher inherent viscosities.The printing indicator (a measure of likeliness of successful ejection of a fluid through a nozzle for printing) estimated from the surface tension and viscosity of the dispersion provides good guidance for choosing suitable nozzle sizes and conditions feasible for printing. The flow of the inks through the nozzle and the inertia of the fluid relative to its surface tension can be predicted using the Reynolds number (Re) and Weber number (We). Several research groups have determined that if the ratio of the surface tension energy dissipation to the internal viscosity (the square root of the Weber number divided by the Reynolds number) has a value ranging from 1 to 10, the ejection of the fluid through the nozzle is likely to be successful (eqn (S6) in the ESI†). The printing-induced shear rate (γmax) was estimated using the below equation:53
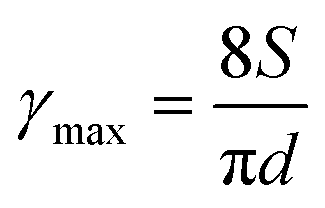 | (1) |
 | ||
| Fig. 4 (a) Printing indicator of the graphene inks (10 mg mL−1) at different shear rates. (b) Printing of multiple flexible conductive circuit patterns. (c) Bending of the printed flexible conductive circuit without failure. (d) Lighting up of an LED chip on the printed flexible conductive circuit. (e) The optical microscopy image of a printed line. (f) The corresponding SEM image of the printed pattern. | ||
Fig. 4(b) shows the fabrication of the multiple circuit patterns printed by a single continuous printing process. The printed circuits are highly flexible, which allows for repetitious and aggressive bending without failure (Fig. 4(c)) and exhibits exceptional electrical performance with a sheet resistance of ∼30 Ω sq−1 without thermal annealing. The excellent conductivity of the printed patterns is due to the fact that there are no excessive stabilisers sandwiched between the graphene sheets. Only a minimal amount of highly conductive PTEBS molecules (adsorbed on graphene surface) act as an interfacing constituent, providing a highly conductive transport network for accelerated charge mobility. The printed circuit was utilised to power an LED chip, as shown in Fig. 4(d). The printed patterns were studied under an optical microscope (Fig. 4(e)) to estimate the printing accuracy; where, it shows a rather straight and smooth printed line while printing in the linear direction and broader corners when the printing direction suddenly changed (90°) due to lateral spreading. The corresponding SEM image of the printed patterns (Fig. 4(f)) shows a dense layer of 2D graphene sheets uniformly arranged into a tile-like structure, indicating the formation of an efficient network for high electrical conductivity. The highly conductive PTEBS-interfaced graphene may find importance in various printed electronic applications.
2.5. Electrocatalytic activity of the functionalised graphene towards the ORR
In fuel cells, the hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR) take place at the anode and cathode, respectively, generating electricity and water (by-product). During the ORR, oxygen is reduced to water (via a four-electron pathway, which is fast and preferred) or hydrogen peroxide (via a two-electron pathway, which is slow and undesirable). The ORR is relatively sluggish, and it requires the use of electrocatalysts to enhance the half-reactions.55 As the π-interactions between graphene and PTEBS can induce intermolecular charge transfer at the interface toward PTEBS molecules, resulting in positively charged atoms at the central site of graphene lattice to facilitate the ORR,56 we use the produced PTEBS-functionalised graphene (denoted as G/PTEBS) to fabricate electrocatalyst layers for the ORR and compare its performance to that of rGO and commercial Pt/C.Fig. 5(a) displays the CV curves (at 50 mV s−1) of the G/PTEBS, rGO, and commercial Pt/C electrocatalyst. While the reduction peak of rGO seemed to be relatively unclear because of the thick electrochemical double layer, the presence of PTEBS as a dopant in the G/PTEBS clearly improved its electrocatalytic activity with more distinct characteristic peaks. The reduction peaks were observed around −0.53 and −0.21 V for the G/PTEBS and Pt/C electrodes, respectively. Fig. 5(b) compares the LSV curves at 1500 rpm of G/PTEBS with those of rGO and commercial Pt/C (scan rate of 50 mV s−1). The onset potentials were established as −0.18 for rGO and −0.11 V for commercial Pt/C, respectively. Similar to rGO, the G/PTEBS catalyst also exhibited similar two wave oxygen reduction characteristics with an onset of oxygen reduction potential observed at −0.21 V and the half-wave potential of the first wave of oxygen reduction observed at −0.27 V, which indicates that the active sites for the ORR are populated on the surface of graphene. Additionally, the LSV of the G/PTEBS catalyst exhibits a stronger limiting diffusion current at −0.8 V (−2.34 mA cm−2), which is more than 40% higher than that of rGO (−1.64 mA cm−2). This indicates that the intermolecular charge transfer between graphene and PTEBS enhances the reaction kinetics for efficient electrocatalytic oxygen reduction. To further investigate the mechanism of the ORR, we performed RDE voltammetry measurements on the G/PTEBS at various rotation rates, as shown in Fig. 5(c). The limiting current density of the G/PTEBS catalyst is found to steadily increase with the rotation rate, indicating mass diffusion behavior. Fig. 5(d) displays the Koutecky–Levich (K–L) plots of G/PTEBS, which show good linearity at increasing potentials from −0.4 to −0.7 V. The K–L equation was utilised to calculate the electron transfer numbers of the G/PTEBS, rGO and commercial Pt/C catalyst, as shown in Fig. 5(e). At potentials ranging from −0.4 to −0.7 V, the electron transfer number of the G/PTEBS catalyst was estimated to be 2.30–3.99, which is always above that of rGO (2.24–2.87). These results indicate that the ORR process through the rGO electrocatalyst follows the two-electron pathway (a sluggish two-step reaction with the generation of intermediate peroxide species), whereas the ORR process through the G/PTEBS electrocatalyst follows the efficient four-electron pathway (one-step reduction of O2 directly to OH−) at a potential range from −0.6 to −0.7 V vs. Ag/AgCl, reaching that of the commercial Pt/C catalyst. In contrast, the ORR reaction on rGO follows the two-electron pathway for all the tested potentials.
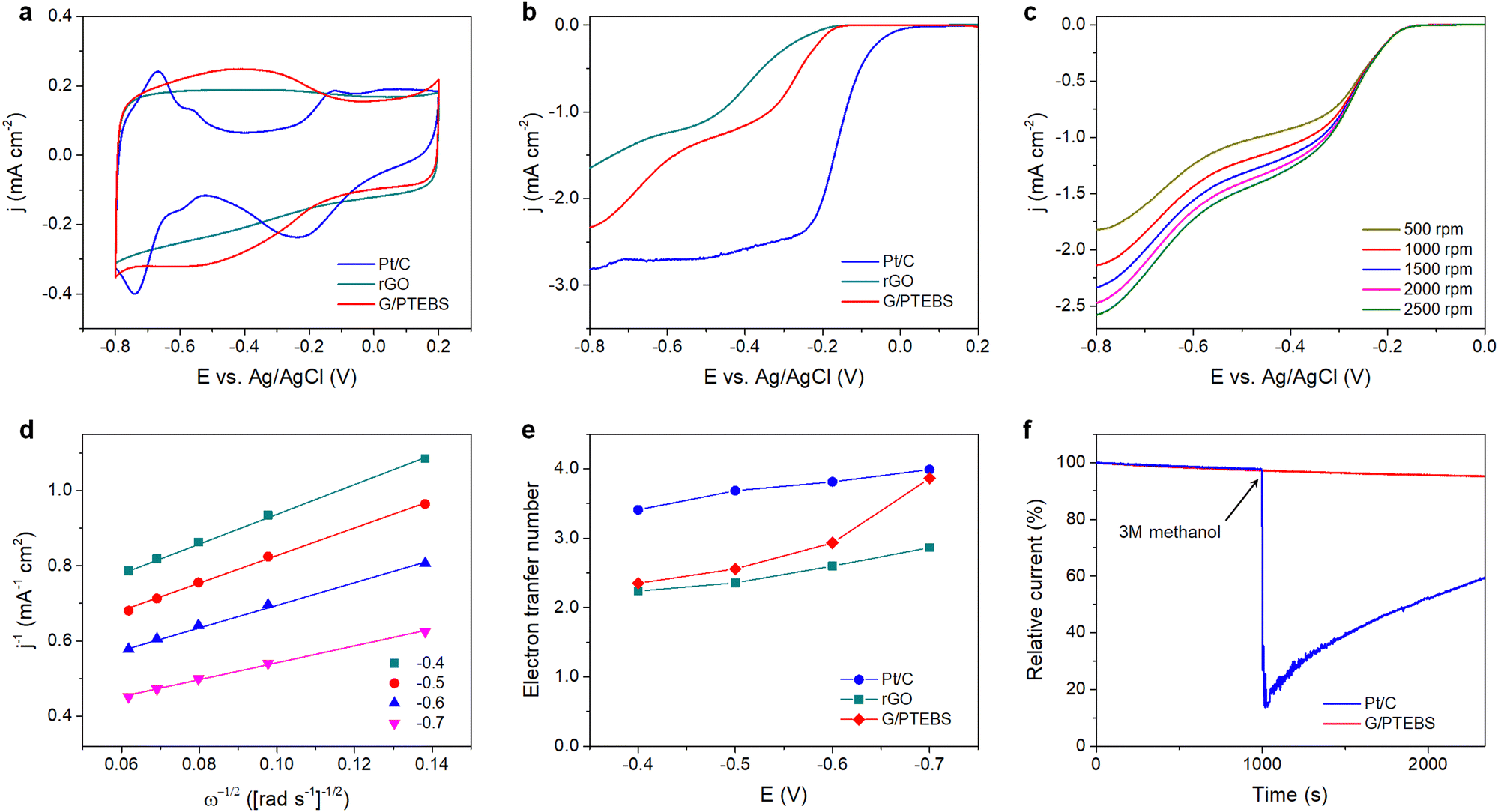 | ||
| Fig. 5 (a) CV curves and (b) LSV curves (at 1500 rpm) of G/PTEBS, rGO and the commercial Pt/C electrocatalysts measured in 0.1 M KOH (scan rate: 50 mV s−1). (c) RDE voltammograms (LSV curves) of G/PTEBS at increasing rotation speeds (scan rate: 10 mV s−1). (d) K–L plots of the G/PTEBS electrocatalysts at different potentials. (e) Electron transfer numbers of G/PTEBS compared to those of rGO and the commercial Pt/C. (f) Chronoamperometric responses of G/PTEBS and the Pt/C catalyst with the addition of methanol at 1000 s. | ||
As G/PTEBS appears to be a promising electrocatalyst for efficient ORR, we sought to study its durability under various conditions. The G/PTEBS electrocatalyst was cycled 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times at 200 mV s−1 (Fig. S9 in the ESI†), which retained more than ∼95% of its initial electrochemical surface area, demonstrating excellent long-term stability (Fig. S10 in the ESI†). To explore the potential of G/PTEBS as a metal-free electrocatalyst for organic fuels, we also study its durability under methanol crossover conditions. The chronoamperometric responses (at −0.5 V and 1500 rpm) of G/PTEBS and Pt/C for the ORR are compared in Fig. 5(f). Upon addition of methanol, the commercial Pt/C exhibits a significant drop (∼85%) in cathodic current; whereas, the G/PTEBS catalyst shows superior durability with no significant decrease in cathodic current, demonstrating its high tolerance to methanol crossover. The demonstrated G/PTEBS catalyst may find importance in large-scale and low-cost fuel cell power systems.
000 times at 200 mV s−1 (Fig. S9 in the ESI†), which retained more than ∼95% of its initial electrochemical surface area, demonstrating excellent long-term stability (Fig. S10 in the ESI†). To explore the potential of G/PTEBS as a metal-free electrocatalyst for organic fuels, we also study its durability under methanol crossover conditions. The chronoamperometric responses (at −0.5 V and 1500 rpm) of G/PTEBS and Pt/C for the ORR are compared in Fig. 5(f). Upon addition of methanol, the commercial Pt/C exhibits a significant drop (∼85%) in cathodic current; whereas, the G/PTEBS catalyst shows superior durability with no significant decrease in cathodic current, demonstrating its high tolerance to methanol crossover. The demonstrated G/PTEBS catalyst may find importance in large-scale and low-cost fuel cell power systems.
3. Conclusions
We have successfully developed a facile and scalable route to produce water-redispersible graphene powder by interfacing graphene with amphiphilic PTEBS molecules via exfoliation-assisted noncovalent functionalisation utilising the adsorption of PTEBS on the surface of graphene. The produced graphene powder is of high quality and can be easily dispersed in water. Aqueous graphene inks were formulated and demonstrated for printing of flexible conductive circuits, providing superior conductivity without the need for thermal treatment. The produced PTEBS-interfaced graphene also exhibited outstanding electrocatalytic activity for the ORR via the highly efficient four-electron reaction pathway, demonstrating its promising prospects for green energy applications.4. Experimental section
4.1. Preparation of water-redispersible graphene powder
PTEBS (MW = 40–70 kDa) was obtained from Solaris Chem (Canada) and used as provided. The precursor graphite, electrochemically exfoliated graphite, was prepared following our previously reported method.57 In a typical experiment, 1 g of the precursor graphite was added to 100 mL of the aqueous PTEBS solution and ultrasonicated at 10 °C for 2 h. The mixture was then centrifuged at 2000 rpm for 30 min to sediment down the unexfoliated graphite particles and the supernatant was collected for further purification. To remove the excessive free PTEBS molecules from the graphene dispersion, the suspension was subjected to two cycles of purification by centrifugation at 20000 rpm for 60 min to sediment down the graphene flakes and then redispersing them in deionised water by sonication for 2 min. The purified graphene suspension was finally lyophilised to yield a lightweight, water-redispersible dry graphene powder.
As control experiments, graphene dispersions stabilised by poly(3-thiophene acetic acid) (PTAA) and poly(vinyl alcohol) (PVA) were also prepared. The detailed experimental procedures and comparison are provided in Scheme S1 in the ESI.†
4.2. SANS and USANS experiments
The interfacial interactions and colloidal structure of graphene dispersions were studied using the SANS (BILBY) and USANS (KOOKABURRA) instruments at ANSTO. 100% D2O and 30% D2O were used as dispersion media to individually highlight the scattering of PTEBS and graphene, by contrast matching the neutron scattering length density (SLD) of graphene (∼6.38 × 10−6 Å−2)58 and PTEBS (∼1.51 × 10−6 Å−2; calculated using the NIST Center for Neutron Research neutron activation and scattering calculator), respectively; whereas, 100% H2O was used as the dispersion medium to collect scattering from both graphene and PTEBS. Samples were loaded into a custom-built quartz cell with 1 mm path length and equilibrated at 25 °C for scattering measurements. BILBY was operated in time-of-flight mode using a neutron wavelength range of 2–20 Å with a single detector setup and the neutron scattering data were collected against the scattering vector (q) in the range of 0.001–0.3 Å−1. KOOKABURRA was operated using a neutron wavelength of 4.74 Å and the neutron scattering data were collected against scattering vector (q) in the range of 0.00007–0.003 Å−1: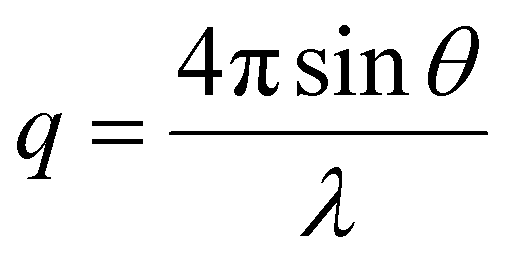 | (2) |
4.3. Printing of flexible conductive circuits
For the formulation of graphene inks, a controlled amount of the as-prepared dry graphene powder was humidified (95%) overnight at 25 °C using an ESPEC SH-222 environmental chamber and dispersed in water by ultrasonication for 10 min. Flexible polyethylene terephthalate (PET) substrates (0.1 mm, Goodfellow) were treated with O2 plasma (Solarus 950, Gatan) for 60 s to render surface hydrophilicity. Printing of flexible conductive circuits was carried out using a GeSiM BioScaffolder 3.1 precision dispensing system. The circuit patterns were designed on AutoCAD and converted to G-code using Gesim Robotics software. Printing was carried out using a Nordson precision dispensing needle (25 GA) with an applied pressure of 20 kPa and a printing rate of 20 mm s−1. After printing, the printed patterns were allowed to dry for 1 h under ambient conditions before being transferred into a vacuum oven to dry overnight at 25 °C.4.4. ORR experiments
For the preparation of the working electrode, 20 μL of the as-prepared graphene dispersion (2 mg mL−1) was spin-coated onto a glassy carbon rotating disk electrode (denoted as G/PTEBS) under ambient conditions at 700 rpm for 1 h using an electrode rotator (WaveVortex 10, Pine Research). Then, 5 μL of perfluorinated resin solution (Nafion®, Sigma-Aldrich) was drop cast on top of the above-fabricated electrode (for fixing the fabricated G/PTEBS thin-film, i.e. electrocatalyst layer) and dried at ambient temperature for 30 min. As control experiments, the electrocatalytic activities of reduced graphene oxide (rGO) and commercial Pt/C electrocatalyst were also investigated. The detailed experimental procedures are provided in the ESI.†ORR experiments were performed using a three-electrode setup comprising a reference electrode (Ag/AgCl saturated with KCl), a counter electrode (graphite rod), a rotating disc working electrode (G/PTEBS-coated glassy carbon) and an electrolyte solution (0.1 M KOH). Cyclic voltammetry (CV) experiments (at 0 rpm) and linear sweep voltammetry (LSV) experiments (at 500, 1000, 1500, 2000 and 2500 rpm) were performed using a Gamry Reference 3000 potentiostat and a Pine Research WaveVortex 10 electrode rotator. For CV and LSV experiments, the electrolyte was pre-purged with N2 and O2, respectively for 30 min, and kept purging throughout the experiments.
Conflicts of interest
Part of this study was used for a patent application. The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the School of Graduate Research, RMIT University for supporting Tuan Sang Tran with the PhD scholarship. The research has been supported by the Australian Research Council (ARC) Industry Transformation research hub funding (IH 150100003). The SANS and USANS experiments were supported by the ANSTO beam time award (P5871). This work was performed in part at the RMIT Micro Nano Research Facility (MNRF) in the Victorian Node of the Australian National Fabrication Facility (ANFF). The authors acknowledge the facilities and the technical assistance of the RMIT Microscopy & Microanalysis Facility (RMMF) at RMIT University. This work benefited from the use of the SasView application (https://www.sasview.org/), originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement no 654000.References
- M. F. El-Kady, Y. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS
.
- T. S. Tran, K. M. Tripathi, B. N. Kim, I.-K. You, B. J. Park, Y. H. Han and T. Kim, Mater. Res. Bull., 2017, 96, 395–404 CrossRef CAS
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833–3842 RSC
- D. Bitounis, H. Ali-Boucetta, B. H. Hong, D. H. Min and K. Kostarelos, Adv. Mater., 2013, 25, 2258–2268 CrossRef CAS PubMed
- F. Perreault, A. F. De Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC
- Y. Myung, S. Jung, T. T. Tung, K. M. Tripathi and T. Kim, ACS Sustainable Chem. Eng., 2019, 7, 3772–3782 CrossRef CAS
- T. S. Tran, N. K. Dutta and N. R. Choudhury, Adv. Colloid Interface Sci., 2018, 261, 41–61 CrossRef CAS PubMed
- H. Jang, Y. J. Park, X. Chen, T. Das, M. S. Kim and J. H. Ahn, Adv. Mater., 2016, 28, 4184–4202 CrossRef CAS PubMed
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O’Neill, C. Boland, M. Lotya, O. M. Istrate and P. King, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC
- T. S. Tran, S. J. Park, S. S. Yoo, T.-R. Lee and T. Kim, RSC Adv., 2016, 6, 12003–12008 RSC
- F. M. Fowkes, J. Adhes., 1972, 4, 155–159 CrossRef CAS
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. McGovern, B. Holland, M. Byrne and Y. K. Gun’Ko, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed
- E. C. A. (ECHA), 2018 https://echa.europa.eu/documents/10162/5662605/FINAL_MB_03_2019_%282%29_Annex_General_Report_2018_MB53.pdf/8cf36986-ad19-c96e-1644-d1a73886a598.
- L. Guardia, M. Fernández-Merino, J. Paredes, P. Solís-Fernández, S. Villar-Rodil, A. Martinez-Alonso and J. Tascón, Carbon, 2011, 49, 1653–1662 CrossRef CAS
- A. Mohamed, T. Ardyani, S. A. Bakar, P. Brown, M. Hollamby, M. Sagisaka and J. Eastoe, Adv. Colloid Interface Sci., 2016, 230, 54–69 CrossRef CAS PubMed
- T. S. Tran, N. K. Dutta and N. R. Choudhury, Materials, 2019, 12, 978 CrossRef CAS PubMed
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef
- A. Ghosh, K. V. Rao, R. Voggu and S. J. George, Chem. Phys. Lett., 2010, 488, 198–201 CrossRef CAS
- A. Groszek, Faraday Discuss. Chem. Soc., 1975, 59, 109–116 RSC
- P. A. Denis and F. Iribarne, J. Mol. Struct., 2010, 957, 114–119 CrossRef CAS
- D. H. Gharib, S. Gietman, F. Malherbe and S. E. Moulton, Carbon, 2017, 123, 695–707 CrossRef CAS
- S. Amemori, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2013, 52, 4174–4178 CrossRef CAS PubMed
- F. Rodríguez-Ropero, J. Casanovas and C. Alemán, J. Comput. Chem., 2008, 29, 69–78 CrossRef PubMed
- S. R. P. Gnanakan, M. Rajasekhar and A. Subramania, Int. J. Electrochem. Sci., 2009, 4, 1289–1301 CAS
- S. Santra, G. Hu, R. Howe, A. De Luca, S. Ali, F. Udrea, J. Gardner, S. Ray, P. Guha and T. Hasan, Sci. Rep., 2015, 5, 1–12 Search PubMed
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed
- Z. Sun, J. Masa, Z. Liu, W. Schuhmann and M. Muhler, Chem. – Eur. J., 2012, 18, 6972–6978 CrossRef CAS PubMed
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang and I. McGovern, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS PubMed
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288–3294 CrossRef CAS
- J. N. Coleman, Acc. Chem. Res., 2012, 46, 14–22 CrossRef PubMed
- A. Y. Sham and S. M. Notley, Soft Matter, 2013, 9, 6645–6653 RSC
- M. J. Hollamby, Phys. Chem. Chem. Phys., 2013, 15, 10566–10579 RSC
- D. W. Schaefer and M. M. Agamalian, Curr. Opin. Solid State Mater. Sci., 2004, 8, 39–47 CrossRef CAS
- R. Balu, N. R. Choudhury, J. P. Mata, L. de Campo, C. Rehm, A. J. Hill and N. K. Dutta, ACS Appl. Mater. Interfaces, 2019, 11, 9934–9946 CrossRef CAS PubMed
- T. M. McCoy, L. De Campo, A. V. Sokolova, I. Grillo, E. I. Izgorodina and R. F. Tabor, Phys. Chem. Chem. Phys., 2018, 20, 16801–16816 RSC
- R. Balu, S. Reeder, R. Knott, J. Mata, L. de Campo, N. K. Dutta and N. R. Choudhury, Langmuir, 2018, 34, 9238–9251 CrossRef CAS PubMed
-
L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Springer, US, 2013 Search PubMed
- B. Hammouda, J. Appl. Crystallogr., 2010, 43, 716–719 CrossRef CAS
- D. Saurel, J. Segalini, M. Jauregui, A. Pendashteh, B. Daffos, P. Simon and M. Casas-Cabanas, Energy Storage Mater., 2019, 21, 162–173 CrossRef
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS PubMed
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed
- U. Khan, A. O’Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed
- Z. Luo, J. Wang, L. Qu, J. Jia, S. Jiang, X. Zhou, X. Wu and Z. Wu, New J. Chem., 2017, 41, 12596–12603 RSC
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS
- P. Merel, M. Tabbal, M. Chaker, S. Moisa and J. Margot, Appl. Surf. Sci., 1998, 136, 105–110 CrossRef CAS
- T. Leung, W. Man, P. Lim, W. Chan, F. Gaspari and S. Zukotynski, J. Non-Cryst. Solids, 1999, 254, 156–160 CrossRef CAS
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS
- N. I. Kovtyukhova, Y. Wang, A. Berkdemir, R. Cruz-Silva, M. Terrones, V. H. Crespi and T. E. Mallouk, Nat. Chem., 2014, 6, 957 CrossRef CAS PubMed
- S. Hong and F. Boerio, Surf. Interface Anal., 1994, 21, 650–658 CrossRef CAS
- B. Hou, H. Liu, S. Qi, Y. Zhu, B. Zhou, X. Jiang and L. Zhu, J. Colloid Interface Sci., 2018, 510, 103–110 CrossRef CAS PubMed
- A. M’Barki, L. Bocquet and A. Stevenson, Sci. Rep., 2017, 7, 6017 CrossRef PubMed
-
I. Gibson, D. W. Rosen and B. Stucker, Additive manufacturing technologies, Springer, 2014 Search PubMed
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed
- M. Zhang, W. Yuan, B. Yao, C. Li and G. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 3587–3593 CrossRef CAS PubMed
- T. S. Tran, N. K. Dutta and N. R. Choudhury, ACS Appl. Nano Mater., 2020, 3, 11608–11619 CrossRef CAS
- E. M. Milner, N. T. Skipper, C. A. Howard, M. S. Shaffer, D. J. Buckley, K. A. Rahnejat, P. L. Cullen, R. K. Heenan, P. Lindner and R. Schweins, J. Am. Chem. Soc., 2012, 134, 8302–8305 CrossRef CAS PubMed
- O. Arnold, J.-C. Bilheux, J. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. F. Leal and M. Gigg, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS
- T. Lam, N. Hauser, A. Götz, P. Hathaway, F. Franceschini, H. Rayner and L. Zhang, Phys. B: Condens. Matter, 2006, 385, 1330–1332 CrossRef
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00298a |
| This journal is © The Royal Society of Chemistry 2023 |