
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzylic C–H isocyanation/amine coupling sequence enabling high-throughput synthesis of pharmaceutically relevant ureas†
Sung-Eun
Suh‡
 a,
Leah E.
Nkulu
a,
Shishi
Lin
b,
Shane W.
Krska
b and
Shannon S.
Stahl
*a
a,
Leah E.
Nkulu
a,
Shishi
Lin
b,
Shane W.
Krska
b and
Shannon S.
Stahl
*a
aDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA. E-mail: stahl@chem.wsic.edu
bChemistry Capabilities for Accelerating Therapeutics, Merck & Co., Inc., 2000 Galloping Hill Road, Kenilworth, New Jersey 07033, USA
First published on 2nd July 2021
Abstract
C(sp3)–H functionalization methods provide an ideal synthetic platform for medicinal chemistry; however, such methods are often constrained by practical limitations. The present study outlines a C(sp3)–H isocyanation protocol that enables the synthesis of diverse, pharmaceutically relevant benzylic ureas in high-throughput format. The operationally simple C–H isocyanation method shows high site selectivity and good functional group tolerance, and uses commercially available catalyst components and reagents [CuOAc, 2,2′-bis(oxazoline) ligand, (trimethylsilyl)isocyanate, and N-fluorobenzenesulfonimide]. The isocyanate products may be used without isolation or purification in a subsequent coupling step with primary and secondary amines to afford hundreds of diverse ureas. These results provide a template for implementation of C–H functionalization/cross-coupling in drug discovery.
Introduction
Synthetic methods that join two fragments derived from abundant molecular building blocks, such as amide coupling and Pd-catalyzed cross-coupling, are the most widely used in drug-discovery.1–5 Implementation of these methods using high-throughput experimentation (HTE) enables rapid creation of diverse complex molecules for biological screening.6,7 Reactions capable of using C(sp3)–H bonds as sites for cross-coupling would be very appealing in this context.8,9 In order to find broad utility, such methods should (1) use the C–H coupling partner as the limiting reagent, (2) exhibit high and predictable site selectivity, (3) show broad functional group tolerance, (4) use readily available catalysts and reagents, and (5) be compatible with HTE methods.Benzylic ureas are common motifs in FDA-approved drugs and bioactive targets in medicinal chemistry (Fig. 1A),10–16 and aromatic compounds with (hetero)benzylic C(sp3)–H bonds are ubiquitous potential precursors to these structures. Recognizing that ureas are readily accessed via coupling of amines and organic isocyanates,17 two C(sp3)–H functionalization/cross-coupling strategies were considered to access benzylic ureas (Fig. 1B). A C–H amination/isocyanate coupling sequence (Fig. 1B, bottom) would leverage C–H amination methods to prepare a benzylic amine intermediate;18–25 however, these methods use ammonia surrogates (e.g., azide, sulfonamides) that require an additional step to obtain the amine. Moreover, the small number of commercially available organic isocyanates (Fig. 1B, right) limits the number of structures that could be readily accessed. These factors complicate the use of HTE and limit the synthetic scope and utility.26 The alternative sequence, involving C–H isocyanation/amine coupling (Fig. 1B, top) offers important advantages. This route leverages the vast number of commercially available amines (Fig. 1B, right) and affords the desired structures in only two steps. On the other hand, C(sp3)–H isocyanation methods have much less precedent.
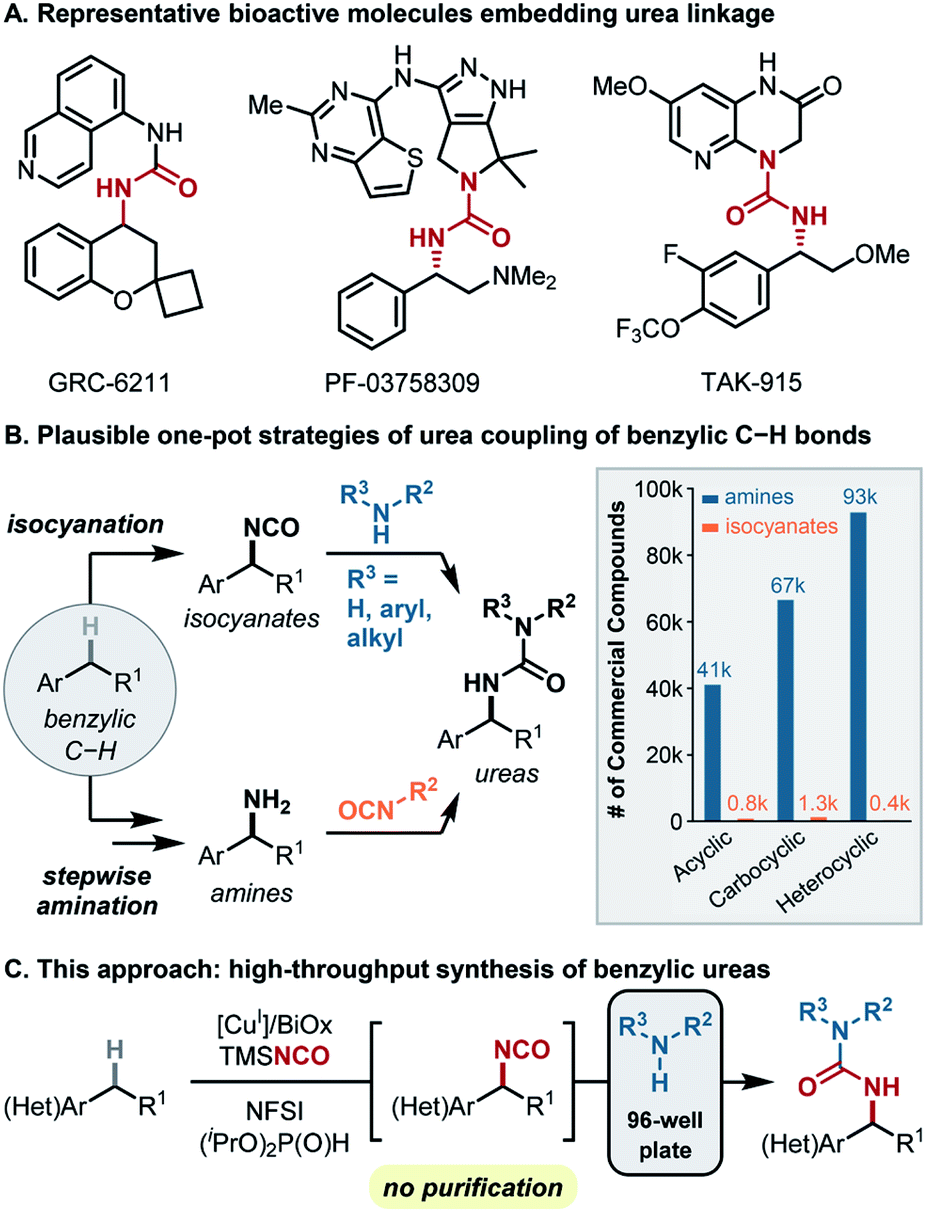 | ||
| Fig. 1 Copper-catalyzed benzylic C(sp3)–H isocyanation/amine coupling. (A) Drug candidates embedding benzylic urea. (B) Two possible one-pot synthetic approaches toward ureas from benzylic C–H substrates. (C) Strategy for high-throughput synthesis of ureas. NFSI, N-fluorobenzenesulfonimide; BiOx, 2,2′-bis(oxazoline); TMSNCO, (trimethylsilyl)isocyanate. | ||
Existing C(sp3)–H isocyanation methods27,28 lack the features necessary for medicinal chemistry applications. The first precedent, reported by Hill and coworkers in 1983, used MnIII(TPP)NCO (TPP = tetraphenylporphyrin) with iodosylbenzene (PhIO) as the oxidant and sodium cyanate as the coupling partner. This reaction provided important proof-of-concept, but the reaction was only demonstrated with cyclohexane, used as a cosolvent in CH2Cl2, and afforded the Cy-NCO product in only 7% yield with respect to PhIO. Groves and coworkers significantly improved upon this method using Mn(TMP)F (TMP = tetramesitylporphyrin) as the catalyst. While this method employs C–H substrates as the limiting reagent and affords isocyanate products in useful yields (∼40–60%), the complex protocols involved in catalyst preparation and the isocyanation reaction limit the utility of the method. For example, each reaction must be monitored by GC-MS to discern appropriate timing for 9–18 sequential additions of the PhIO oxidant and 3–6 additions of trimethylsilyl isocyanate (TMSNCO) over the course of the reaction.
In light of the above considerations, we sought an isocyanation/amine coupling protocol that combines operational simplicity with broad substrate scope to access diverse synthetic libraries (Fig. 1C). The isocyanation/coupling sequence outlined herein meets key criteria for use in medicinal chemistry, including use of the C–H substrate as the limiting reagent, predictable benzylic site selectivity, no requirement for isolation/purification of the reactivity isocyanate intermediate, compatibility with HTE methods, and exclusive use of commercially available catalysts and reagents. These methods demonstrate the use of (hetero)benzylic C–H bonds as latent functional groups that may be engaged in cross-coupling reactions and, more generally, provide a framework for further development of C–H functionalization/coupling methods that could find utility in medicinal chemistry.
Results and discussion
Mechanistic considerations and reaction optimization
Cu-catalyzed radical-relay reactions that use N-fluorobenzenesulfonimide (NFSI) as the oxidant have expanded rapidly in recent years19,29–39 and offered a possible strategy to achieve C–H isocyanation as these methods often support oxidative coupling with the C–H substrate as the limiting reagent. Several mechanistic features of these reactions merit consideration. CuI-mediated activation of NSFI is proposed to generate an N-centered radical (˙NSI) that promotes hydrogen-atom transfer (HAT), and the resulting benzylic radical undergoes oxidative functionalization (Fig. 2A, top cycle). The imidyl radical generated in this process can alternatively react with a second equivalent of CuI, “wasting” the imidyl radical and consuming CuI, which is needed for activation of NFSI (dashed gray pathway in Fig. 2A). Recent studies show that this deleterious side-reaction can be offset by including a mild sacrificial reductant, such as a dialkylphosphite, in the reaction. This additive serves as a “redox buffer” to rescue catalytic activity via in situ regeneration of CuI (Fig. 2A, green portion of bottom cycle).35–37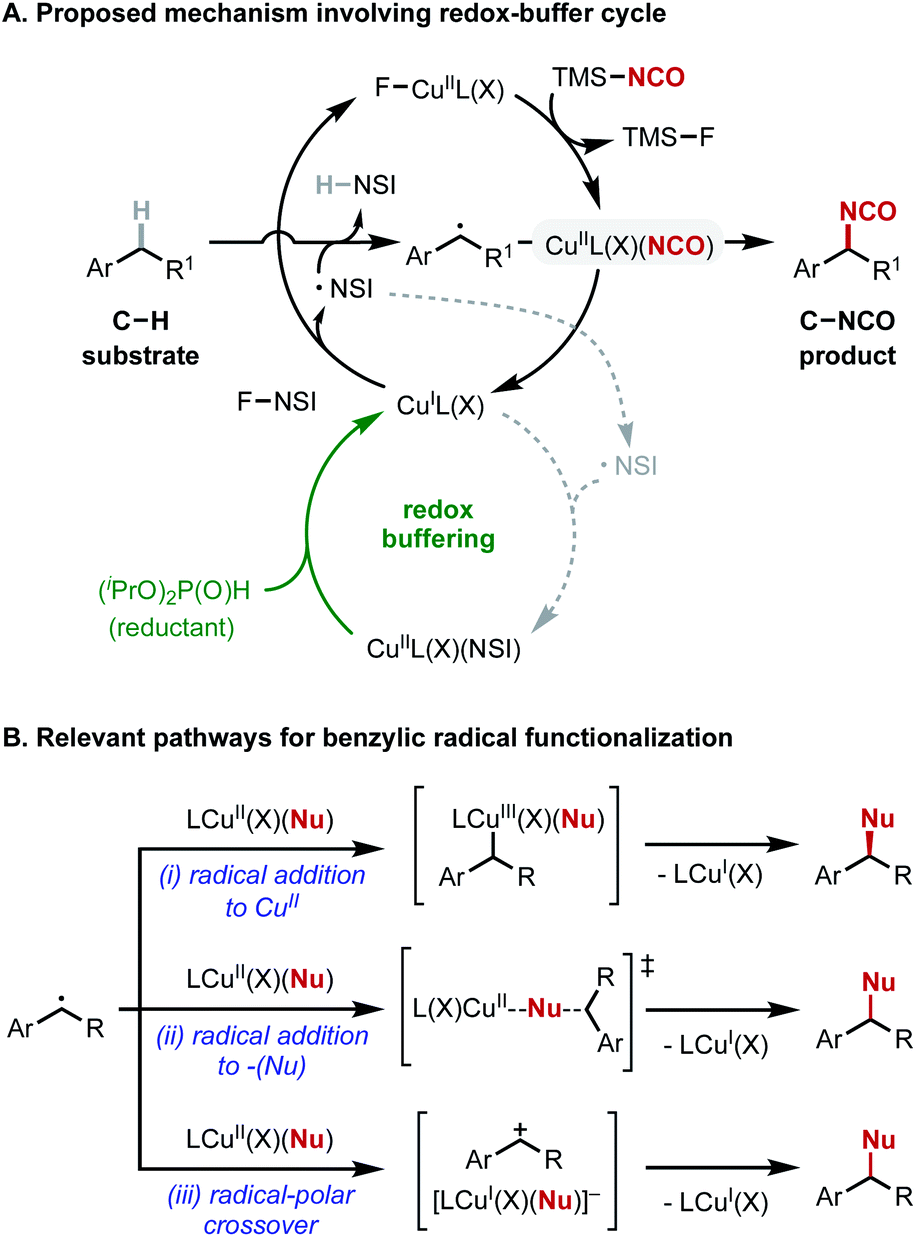 | ||
| Fig. 2 Mechanistic features of copper-catalyzed benzylic C(sp3)–H isocyanation with N-fluorobenzenesulfonimide (NFSI). (A) Catalytic cycles for formation of C–NCO bond and regeneration of copper(I). (B) Possible mechanisms for benzylic radical functionalization in Cu/NFSI systems. | ||
A second mechanistic feature involves radical functionalization, which can proceed by several different pathways. CuI can initiate radical-chain fluorination of C–H bonds36,37,40–43 with NFSI, but Cu/NFSI methods more commonly promote oxidative coupling of C–H bonds with nucleophilic reaction partners (Nu = CN, N3, OR, Ar, among others). The mechanism of coupling can vary and depends on the coupling partner (Fig. 2B(i)–(iii)). For example, enantioselective benzylic cyanation has been demonstrated, and mechanistic data support radical addition to CuII followed by reductive elimination from a benzyl–CuIII intermediate (Fig. 2B(i)).30 Azidation affords racemic products, even with the same chiral ligands used for cyanation. Experimental and computational data implicate a radical-polar crossover mechanism, involving a benzyl cation intermediate (Fig. 2B(iii)), but radical addition to the distal N-atom of a CuII-bound azide is also possible (Fig. 2B(ii)).19 C–H etherification with alcohol coupling partners affords racemic products, and mechanistic studies support a radical-polar crossover mechanism (Fig. 2B(iii)).35
With these mechanistic considerations in mind, screening efforts were initiated to identify effective conditions for benzylic C–H isocyanation. TMSNCO was selected as the source of isocyanate because the affinity of the TMS group for fluoride provides a driving force for isocyanate transfer to the copper(II) center, where it could undergo coupling with the benzylic radical. 1-Bromo-4-ethylbenzene (1a), which incorporates an aryl bromide fragment suitable for subsequent elaboration, was selected as the benzylic substrate. Relevant optimization data is summarized in Table 1, with additional details provided in the ESI (see Tables S1–S10†). A solvent screen with 10 mol% copper(I) acetate and 2,2′-bis(oxazoline) L1 (entries 1–4) revealed a moderate yield of the benzyl isocyanate 2a from the reaction in acetonitrile (36%). Varying the ligand identity (L1–L4, entries 4–7) did not improve the yields (26–30%, see Table S4† for additional ligands). Notable improvement in C–H isocyanation was observed upon adding di(isopropyl)phosphite [(iPrO)2P(O)H] as a redox buffer (entries 8–11). Optimal yield of 2a was achieved with 0.5 equiv. (iPrO)2P(O)H (56% yield). The substrate is almost fully consumed in the reaction, with the sole identified byproduct arising from C–N coupling with the sulfonimide fragment of NFSI (9%). Other potential side products, such as those with C–OiPr, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–F fragments, were not identified. The remaining mass balance is attributed to the high reactivity of organic isocyanates, which are susceptible to various side reactions.17 The lack of cyanate (i.e., the C–OCN) product formation is consistent with the reported instability of this isomer and ability of cyanates to isomerize spontaneously into isocyanates.28,44,45 Racemic products were formed, even when using chiral bis(oxazoline) ligands L2 and L3 (see Fig. S1 of the ESI†). Thorough mechanistic studies were not pursued; however, this observation and the similarity between isocyanate and azide is consistent with radical-polar crossover pathway (Fig. 2B(iii)) involving oxidation of the benzylic radical to afford a benzylic cation intermediate.
O, and C–F fragments, were not identified. The remaining mass balance is attributed to the high reactivity of organic isocyanates, which are susceptible to various side reactions.17 The lack of cyanate (i.e., the C–OCN) product formation is consistent with the reported instability of this isomer and ability of cyanates to isomerize spontaneously into isocyanates.28,44,45 Racemic products were formed, even when using chiral bis(oxazoline) ligands L2 and L3 (see Fig. S1 of the ESI†). Thorough mechanistic studies were not pursued; however, this observation and the similarity between isocyanate and azide is consistent with radical-polar crossover pathway (Fig. 2B(iii)) involving oxidation of the benzylic radical to afford a benzylic cation intermediate.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Additive (mol%) | Yieldb (%) |
| a 0.4 mmol 1a. b NMR yield, ext. std. = mesitylene. c no e.e. d 2.0 mol% CuOAc/L1, 3.0 eq. TMSNCO, 2.5 eq. NFSI, 0.5 eq. (iPrO)2P(O)H, 0.12 M CH3CN, 30 °C, 2 h. | ||||
| 1 | L1 | Benzene | — | 0 |
| 2 | L1 | CH2Cl2 | — | 7 |
| 3 | L1 | CH3NO2 | — | 15 |
| 4 | L1 | CH3CN | — | 36 |
| 5 | L2 | CH3CN | — | 30c |
| 6 | L3 | CH3CN | — | 26c |
| 7 | L4 | CH3CN | — | 28 |
| 8 | L1 | CH3CN | (iPrO)2P(O)H (40) | 39 |
| 9 | L1 | CH3CN | (iPrO)2P(O)H (50) | 42 |
| 10 | L1 | CH3CN | (iPrO)2P(O)H (60) | 39 |
| 11 | L1 | CH3CN | (iPrO)2P(O)H (50) | 56 |
|
||||

Synthetic scope of benzylic isocyanation
The conditions identified for the preparation of 2a were then evaluated with a number of other benzylic substrates (Fig. 3). Given the aforementioned instability of isocyanates, only two of the isocyanate intermediates were isolated and fully characterized (2t and 2u, see ESI†), in order to validate the in situ1H NMR yields obtained with the other substrates. In the other cases, the isocyanation reactions were conducted in parallel, and the crude reaction mixtures were then added to neat m-anisidine (5.0 equiv.) under N2, without any intermediate work-up. The urea products (3a–3w) obtained from this protocol were isolated and fully characterized. Analysis of the data in Fig. 3 shows that the majority of benzylic substrates afford the desired isocyanate in yields of 40–60%, with the subsequent urea formation proceeding in 60–99% yields.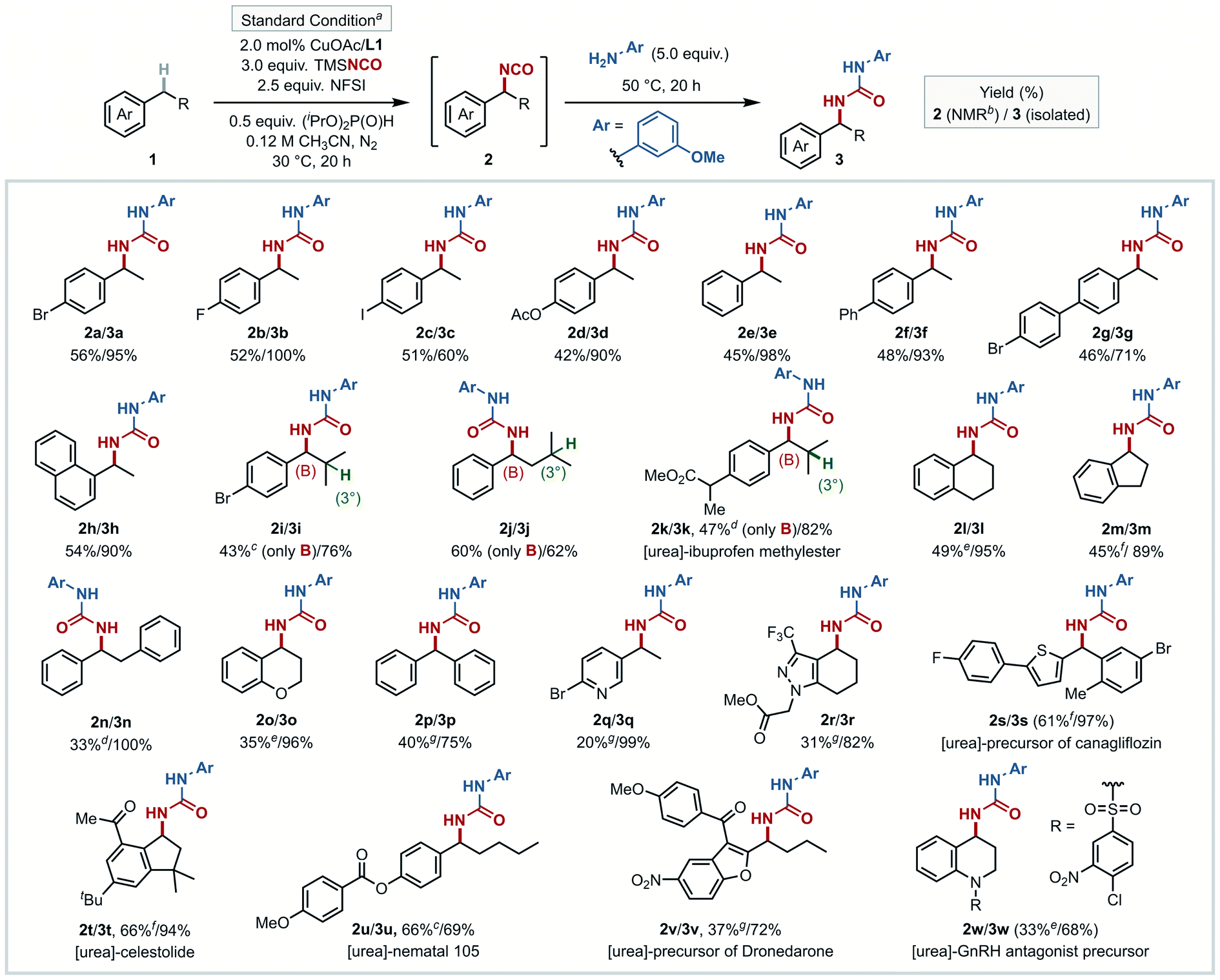 | ||
| Fig. 3 Scope of alkylarene for in situ benzylic C–H isocyanation and urea formation. a Substrate (0.4 mmol). b NMR yield, ext. std. = mesitylene. c CuOAc (10 mol%), L1 (10 mol%) at 30 °C for 20 h. d CuOAc (6.0 mol%), L1 (6.0 mol%) at 40 °C for 20 h. e Standard condition for 2 h. f Standard condition at 25 °C for 2 h. g CuOAc (6.0 mol%), L1 (6.0 mol%) at 30 °C for 20 h. | ||
Effective C–H substrates include relatively simple unsubstituted or para-substituted ethylbenzene derivatives (1a–1g), 1-ethylnaphthalene (1h), and diphenylmethane (1p), although isocyanation of the electron-deficient and electron-rich substrates 4-CN and 2-OMe ethylbenzene led to lower yields (24% and 34%; results provided in Table S11,† together with other less successful substrates). Reactions of substrates having both tertiary aliphatic and benzylic C–H bonds exclusively gave benzylic isocyanate products (2i–2k), with none of the corresponding tertiary product. Fused 6- and 5-membered ring cyclic substrates tetralin and indane showed good reactivity (2l and 2m), and only mono-isocyanation was observed with bibenzyl (2n) and chroman (2o). Isocyanation was achieved at heterobenzylic C–H sites of nitrogen heterocycles (2q and 2r), in addition to benzylic sites in several complex molecules (2s–2w). The precursor of canagliflozin46 having a thiophene heterobenzylic C–H site afforded 2s (61% yield), and both celestolide47 and nematal 105 (ref. 48) gave their respective C–H isocyanation products, 2t and 2u, in 66% yield. 1t was selected for a testing on larger preparative scale (5.0 mmol). Conversion to isocyanate intermediate 2t and trapping with m-anisidine proceeded in 75% and 79% yields, respectively, affording 1.2 g of urea 3t (55% yield over two steps). The precursor of dronedarone,49 with a benzofuran scaffold, afforded the corresponding product 2v in 37% yield. The tetrahydroquinoline-embedded GnRH antagonist50 precursor was functionalized at the benzylic site furthest from the nitrogen (2w, 33% yield). Collectively, this set of substrates shows that the isocyanation method is applicable to a broad cross-section of pharmaceutically relevant core fragments containing (hetero)benzylic C–H bonds.
High-throughput synthesis of ureas via sequential C–H isocyanation/amine coupling
Subsequent efforts explored the scope of amine coupling partners, emphasizing demonstration of the sequential C–H isocyanation/amine coupling in a high-throughput platform (Fig. 4A). A collection of 96 different amine coupling partners was selected for testing in 96-well plates (Fig. 4B). The amines were chosen to feature examples with both aliphatic and aromatic substituents, including those with different substitution patterns and diverse functional groups, in order to assess their compatibility with the isocyanation method. Overall, the amine building blocks consisted of 43 aromatic (a1–d7, Fig. 4B) and 53 aliphatic (d8–h12, Fig. 4B) amines, including primary/secondary and cyclic/acyclic derivatives. The substrates incorporated a wide array of functional groups such as halogens, nitro, (pinacolato)boronate, trifluoromethyl, carboxylic esters and amides, carbamate, sulfone, and sulfonamides, in addition to fused/non-fused and saturated/unsaturated heteroaromatic rings. Three C–H building blocks, including the canagliflozin precursor (1s), celestolide (1t), and nematal 105 (1u), were then subjected to the sequential protocol (Fig. 4C, left). Each C–H substrate was converted to the isocyanate on 1.3 mmol scale, after which the reactions were analyzed by 1H NMR spectroscopy to determine the yield of isocyanate. Then, aliquots of the benzylic isocyanate (0.01 mmol each) were dispensed into the wells of the 96-well plates, pre-loaded with the stock solutions of the amine coupling partners. The isocyanate/amine coupling reactions were maintained at 35 °C for 20 h, and then analyzed by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) to determine product identity by MS and an approximate yield from the product peak area vs. total peak areas obtained from UV absorption at 273 nm.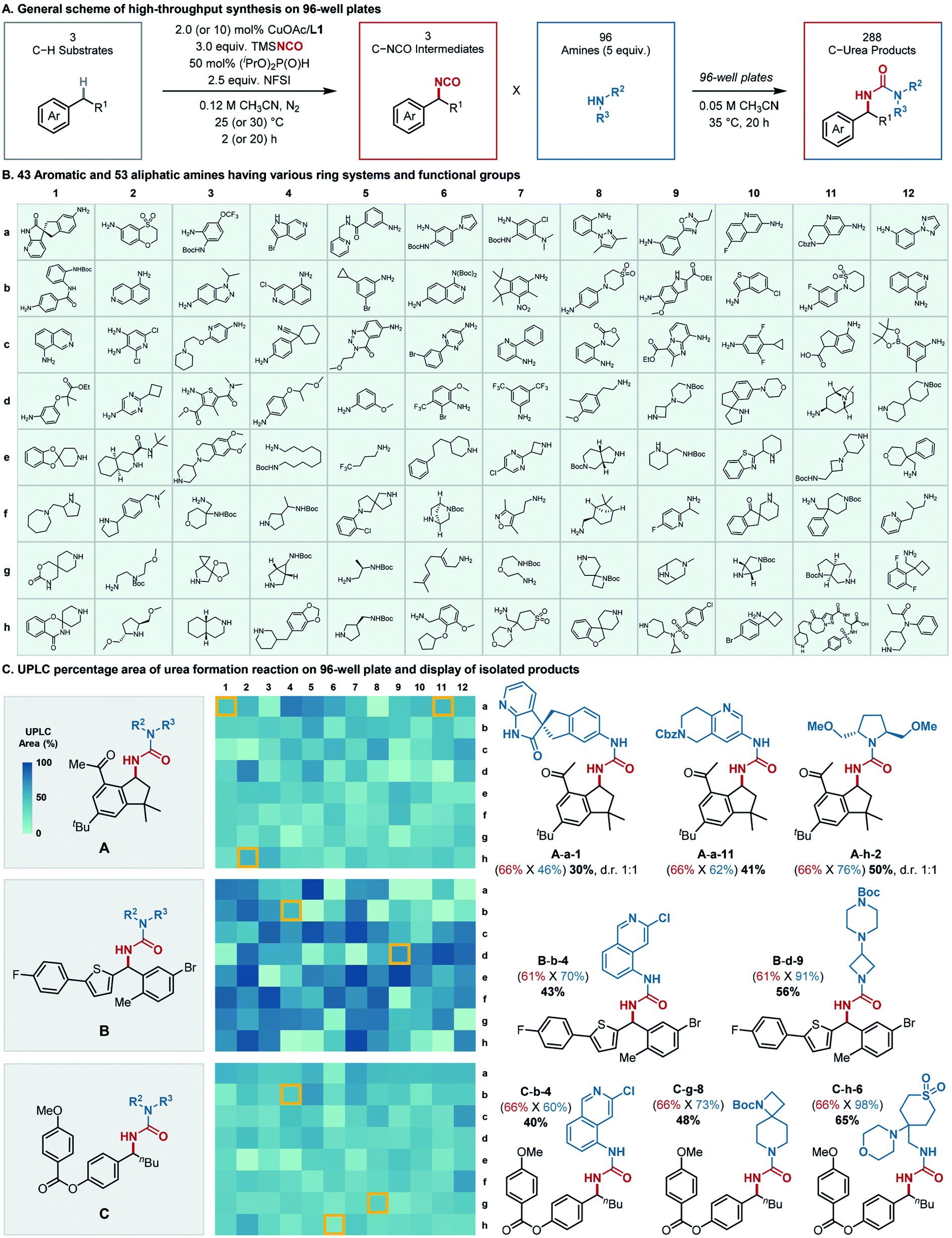 | ||
| Fig. 4 High-throughput synthesis of benzylic ureas. (A) Optimal reaction conditions for isocyanation/urea formation for synthesis of 288 ureas from 3 C–H substrates and 96 amines. 1.3 and 0.01 mmol scale for isocyanation and urea formation respectively. (B) Amines on 96 well plates. (C) Three representative C–H substrates (left), display of UPLC area percentages of urea products on three plates (center) and isolation result of selected ureas (right). Reactions were reproduced in 0.2 mmol scale for isolation. Yields are presented as follows: (NMR yield of C–H isocyanation X isolated yield of urea formation) overall isolated yield from scale-up reaction. The detection wavelength was 273 nm for UPLC analysis. | ||
The desired benzylic ureas were obtained from 274 of the 288 reactions. Little congruence was evident between the high- and low-yielding amines in the reactions with the three different C–H substrates (Fig. 4C, center). To validate the structures and yields of the products determined by UPLC-MS, eight representative reactions were reproduced on 0.2 mmol scale. The isolated yields of urea formation (Fig. 4C, right panel, blue-colored font in parenthesis) measured from the scale-up synthesis are almost always higher than those estimated from high-throughput UPLC assay (Fig. S4–S6†). A minor exception is the celestolide derivative A-a-1, which shows that the UPLC-based product yield (48%, Fig. S4†) of urea formation is nearly identical to that obtained from the scale-up experiment (46%, Fig. 4C). Collectively, these results validate the high-throughput results, and suggest they provide a lower bound for yields that may be expected from larger scale reactions.
The relevance of this C–H isocyanation/amine-coupling protocol for medicinal chemistry is reinforced by analysis of the molecular properties of the urea products. Six physicochemical properties were computed for each product obtained from the reactions in Fig. 4: number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), molecular weight (MW), calculated octanol–water partition coefficient (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), number of rotatable bonds (NRotB), and topological polar surface area (TPSA) (Fig. 5). The computed parameters show that the synthesized urea molecules51 closely align with desirable properties defined by the “oral druggable space beyond the rule of 5 (bRo5)” designations.52
P), number of rotatable bonds (NRotB), and topological polar surface area (TPSA) (Fig. 5). The computed parameters show that the synthesized urea molecules51 closely align with desirable properties defined by the “oral druggable space beyond the rule of 5 (bRo5)” designations.52
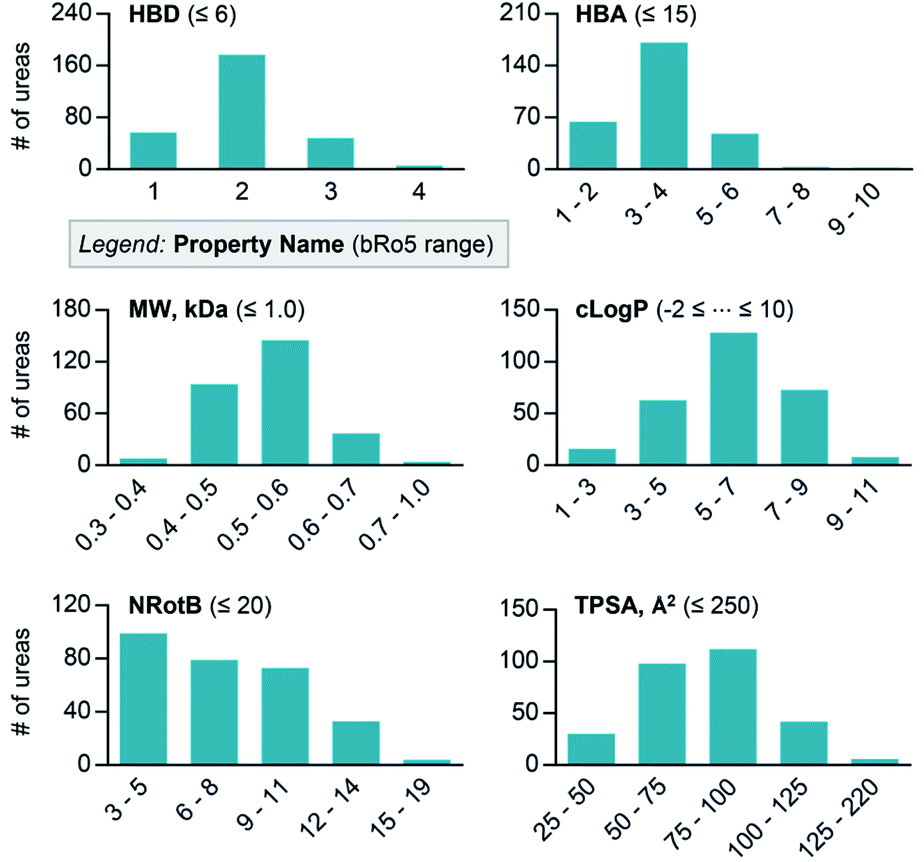 | ||
| Fig. 5 Distribution of synthesized ureas on the plots versus molecular properties. HBD, number of hydrogen bond donors; HBA, number of hydrogen bond acceptors; MW, molecular weight; clogP, calculated octanol–water partition coefficient; NRotB, number of rotatable bonds; TPSA, topological polar surface area. The ranges associated with the “beyond rule of five (bRo5)” designations for oral drugs are defined in parentheses. | ||
Conclusions
In summary, we have developed a practical new method for site-selective isocyanation of benzylic C–H bonds. This method may be combined with amine coupling to prepare benzylic ureas, without purification of the benzylic isocyanate intermediate. The sequential process is amenable to high-throughput experimentation, enabling rapid access to large numbers of diverse ureas with drug-like physicochemical properties using the benzylic C–H bond as the point of diversification. The selectivity for benzylic over many other C(sp3)–H bonds enables the vast array of (hetero)aromatic molecules to be viewed and used as cross-coupling partners for synthesis of bioactive compounds in drug discovery.Data availability
Experimental details with screening results, characterization data, NMR spectra, high throughput experimentation protocols, and theoretical parameters of compounds (PDF) are provided in the ESI.Author contributions
S.-E. S., S. W. K., and S. S. S. conceived and planned the study. S.-E. S. and L. E. N. carried out the synthesis and characterization. L. E. N. performed the calculations of the physicochemical properties. All authors contributed to the analysis of the results and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Cyndi He (Merck & Co., Inc.) for valuable discussions about cheminformatics during the course of this work. This work was supported by funding from the NIH (R35 GM134929) and Merck & Co., Inc. (Kenilworth, NJ USA). Spectroscopic instrumentation was supported by a gift from Paul. J. Bender, the NSF (CHE-1048642), and the NIH (1S10 OD020022-1).References
- M. Orita, K. Ohno and T. Niimi, Drug Discovery Today, 2009, 14, 321–328 CrossRef CAS PubMed.
- P. J. Hajduk, W. R. J. D. Galloway and D. R. Spring, Nature, 2011, 470, 42–43 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS.
- W. Jahnke, D. A. Erlanson, I. J. P. de Esch, C. N. Johnson, P. N. Mortenson, Y. Ochi and T. Urushima, J. Med. Chem., 2020, 63, 15494–15507 CrossRef CAS PubMed.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. E. de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS.
- W. K. Lau, D. Mercer, K. M. Itani, D. P. Nicolau, J. L. Kuti, D. Mansfield and A. Dana, Antimicrob. Agents Chemother., 2006, 50, 3556–3561 CrossRef CAS PubMed.
- A. Gomtsyan, E. K. Bayburt, R. C. Schidt, C. S. Surowy, P. Honore, K. C. Marsh, S. N. Hannick, H. A. McDonald, J. M. Wetter, J. P. Sullivan, M. F. Jarvis, C. R. Faltynek and C.-H. Lee, J. Med. Chem., 2008, 51, 392–395 CrossRef CAS PubMed.
- M. E. Bellizzi, A. V. Bhatia, S. C. Cullen, J. Gandarilla, A. W. Kruger and D. S. Welch, Org. Process Res. Dev., 2014, 18, 303–309 CrossRef CAS.
- E. A. Voight, J. F. Daanen, S. M. Hannick, B. H. Shelat, F. A. Kerdesky, D. J. Plata and M. E. Kort, Tetrahedron Lett., 2010, 51, 5904–5907 CrossRef CAS.
- A. Charrua, C. D. Cruz, S. Narayanan, L. Gharat, S. Gullapalli, F. Cruz and A. Avelino, Urology, 2009, 181, 379–386 CrossRef CAS PubMed.
- B. W. Murray, C. Guo, J. Piraino, J. K. Westwick, C. Zhang, J. Lamerdin, E. Dagostino, D. Knighton, C. M. Loi, M. Zager, E. Kraynov, I. Popoff, J. G. Christensen, R. Martinez, S. E. Kephart, J. Marakovits, S. Karlicek, S. Bergqvist and T. Smeal, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9446–9451 CrossRef CAS PubMed.
- S. Mikami, S. Nakamura, T. Ashizawa, I. Nomura, M. Kawasaki, S. Sasaki, H. Oki, H. Kokubo, I. D. Hoffman, H. Zou, N. Uchiyama, K. Nakashima, N. Kamiguchi, H. Imada, N. Suzuki, H. Iwashita and T. Taniguchi, J. Med. Chem., 2017, 60, 7677–7702 CrossRef CAS PubMed.
- J. H. Saunders and R. J. Slocombe, Chem. Rev., 1948, 43, 203–218 CrossRef CAS PubMed.
- X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300–5303 CrossRef CAS PubMed.
- S.-E. Suh, S.-J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS PubMed.
- K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef CAS PubMed.
- D. A. Powell and H. Fan, J. Org. Chem., 2010, 75, 2726–2729 CrossRef CAS PubMed.
- J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583–591 CrossRef CAS PubMed.
- A. Nasrallah, V. Boquet, A. Hecker, P. Retailleau, B. Darses and P. Dauban, Angew. Chem., Int. Ed., 2019, 58, 8192–8196 CrossRef CAS PubMed.
- A. E. Bosnidou and K. Muñiz, Angew. Chem., Int. Ed., 2019, 58, 7485–7489 CrossRef CAS PubMed.
- Z.-W. Hou, D.-J. Liu, P. Xiong, X.-L. Lai, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef CAS PubMed.
- Structure search was performed on Reaxys on December 18, 2020. It was drawn that “Y–NCO” locking N, C, and O atoms for isocyanates or “Y–N(AH)H” locking H atom for amines (AH = any including H). Y was defined as ACY (any acyclic), CBC (carbocyclic), or CHC (heterocyclic). Each search excluded hits of tautomers, mixtures, and isotopes. Availability was limited to “product for purchase” only.
- C. L. Hill, J. A. Smegal and T. J. Henly, J. Org. Chem., 1983, 48, 3277–3281 CrossRef CAS.
- X. Huang, T. Zhuang, P. A. Kates, H. Gao, X. Chen and J. T. Groves, J. Am. Chem. Soc., 2017, 139, 15407–15413 CrossRef CAS PubMed.
- Z. Ni, Q. Zhang, T. Xiong, Y. Zheng, Y. Li, H. Zhang, J. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2012, 51, 1244–1247 CrossRef CAS PubMed.
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed.
- W. Zhang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 7709–7712 CrossRef CAS PubMed.
- H. Xiao, Z. Liu, H. Shen, B. Zhang, L. Zhu and C. Li, Chem, 2019, 5, 940–949 CAS.
- W. Zhang, L. Wu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2019, 58, 6425–6429 CrossRef CAS PubMed.
- L. Fu, Z. Zhang, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2020, 142, 12493–12500 CrossRef CAS PubMed.
- H. Hu, S.-J. Chen, M. Mandal, S. M. Pratik, J. A. Buss, S. W. Krska, C. J. Cramer and S. S. Stahl, Nat. Catal., 2020, 3, 358–367 CrossRef CAS PubMed.
- J. A. Buss, A. Vasilopoulos, D. L. Golden and S. S. Stahl, Org. Lett., 2020, 22, 5749–5752 CrossRef CAS PubMed.
- A. Vasilopoulos, D. L. Golden, J. A. Buss and S. S. Stahl, Org. Lett., 2020, 22, 5753–5757 CrossRef CAS PubMed.
- For leading references to related, intramolecular reactivity, see the following and ref. 39: Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207–1211 RSC.
- Z. Zhang, X. Zhan and D. A. Nagib, Chem, 2019, 5, 3127–3134 CAS.
- S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. A. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef CAS.
- M. B. Nodwell, A. Bagai, S. D. Halperin, R. E. Martin, H. Knust and R. Britton, Chem. Commun., 2015, 51, 11783–11786 RSC.
- C. R. Pitts, B. Ling, R. Woltornist, R. Liu and T. Lectka, J. Org. Chem., 2014, 79, 8895–8899 CrossRef CAS PubMed.
- C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed.
- K. A. Jensen, M. Due and A. Holm, Acta Chem. Scand., 1965, 19, 438–442 CrossRef CAS.
- K. A. Jensen, M. Due, A. Holm and C. Wentrup, Acta Chem. Scand., 1966, 20, 2091–2106 CrossRef CAS.
- V. Perkovic, M. J. Jardine, B. Neal, S. Bompoint, H. J. L. Heerspink, D. M. Charytan, R. Edwards, R. Agarwal, G. Bakris, S. Bull, C. P. Cannon, G. Capuano, P.-L. Chu, D. de Zeeuw, T. Greene, A. Levin, C. Pollock, D. C. Wheeler, Y. Yavin, H. Zhang, B. Zinman, G. Meininger, B. M. Brenner and K. W. Mahaffey, N. Engl. J. Med., 2019, 380, 2295–2306 CrossRef CAS PubMed.
- A. M. Peck, J. R. Kucklick and M. M. Schantz, Anal. Bioanal. Chem., 2007, 387, 2381–2388 CrossRef CAS.
- J. P. Van Meter and B. H. Klanderman, Mol. Cryst. Liq. Cryst., 1973, 22, 271–284 CrossRef CAS.
- S. H. Hohnloser, H. J. G. M. Crijns, M. van Eickels, C. Gaudin, R. L. Page, C. Torp-Pedersen and S. J. Connolly, N. Engl. J. Med., 2009, 360, 668–678 CrossRef CAS PubMed.
- S. Yonekubo, T. Miyagi, K. Ohno, M. Kambara and N. Fushimi, Int. Pat., 2008129994A1, 2008 Search PubMed.
- The original urea structures had Boc or Cbz on amino groups. These N-protecting groups were deprotected to conduct meaningful assessment of their drug-likeness before calculation of molecular properties.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02049h |
| ‡ Present address: Department of Chemistry, Ajou University, Suwon 16499, Korea. |
| This journal is © The Royal Society of Chemistry 2021 |