
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted 1,3-dipolar cycloaddition with acrolein for cancer prodrug activation†
Ambara R.
Pradipta
 *ab,
Peni
Ahmadi
b,
Kazuki
Terashima
a,
Kyohei
Muguruma
a,
Motoko
Fujii
a,
Tomoya
Ichino
cd,
Satoshi
Maeda
cd and
Katsunori
Tanaka
*abe
*ab,
Peni
Ahmadi
b,
Kazuki
Terashima
a,
Kyohei
Muguruma
a,
Motoko
Fujii
a,
Tomoya
Ichino
cd,
Satoshi
Maeda
cd and
Katsunori
Tanaka
*abe
aDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro, 152-8552, Tokyo, Japan. E-mail: pradipta.a.aa@m.titech.ac.jp; tanaka.k.dg@m.titech.ac.jp
bBiofunctional Synthetic Chemistry Laboratory, Cluster for Pioneering Research, RIKEN, 2-1 Hirosawa, Wako, 351-1098, Saitama, Japan
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Kita 10 Nishi 8, Kita, 060-0815, Sapporo, Japan
dInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21 Nishi 10, Kita, 001-0021, Sapporo, Japan
eBiofunctional Chemistry Laboratory, Alexander Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Street, 420008, Kazan, Russian Federation
First published on 1st April 2021
Abstract
Cytotoxic anticancer drugs used in chemotherapy are often antiproliferative agents that preferentially kill rapidly growing cancer cells. Their mechanism relies mainly on the enhanced proliferation rate of cancer cells and is not genuinely selective for cancer cells. Therefore, these drugs can also significantly affect healthy cells. Prodrug therapy provides an alternative approach using a less cytotoxic form of anticancer drug. It involves the synthesis of inactive drug derivatives which are converted to an active form inside the body and, preferably, only at the site of cancerous tissues, thereby reducing adverse drug reaction (ADR) events. Herein, we demonstrate a prodrug activation strategy by utilizing the reaction between aryl azide and endogenous acrolein. Since acrolein is generally overproduced by most cancer cells, we anticipate our strategy as a starting point for further applications in mouse models with various cancers. Furthermore, cancer drugs that have had therapeutic index challenges might be reconsidered for application by utilizing our strategy.
Introduction
Adverse drug reactions (ADRs) are the decisive risk factor associated with cancer chemotherapy, leading to toxic side effects and poor efficacy.1 In general, to reduce ADRs, monoclonal antibody-directed enzyme therapy has been used in the targeted delivery of prodrugs to cancer cells.2 Alternatively, one could activate a systemic prodrug via a change in the pH environment of the cancerous tumor.3–5 In this case, the changes are often so modest that the selective activation of prodrug molecules is low, with the linkers susceptible to non-specific off-target hydrolysis. Another appealing alternative is the targeting of tumors through endocytosis of polymer–drug conjugates, followed by drug release.6–8 However, this approach could be inherently slow and lacks direct temporal and quantitative control over drug release.In the past decade, bioorthogonal chemistry has found broad applications, including prodrug activation strategies.9 For example, although no biological data was presented, Florent and co-workers described the initial proof of concept on the utilization of Staudinger ligation followed by a self-immolation reaction to release doxorubicin from its prodrug (ESI, Fig. S1†).10 Alternatively, Robillard and co-workers showed that the transformation of an azide to an amine group by a Staudinger reaction could be used to trigger drug release.11 Gamble and co-workers utilized trans-cyclooctene-azide cycloaddition to mitigate the limitations associated with phosphine reagents used for prodrug activation.12 Meanwhile, Robillard and co-workers demonstrated a drug release strategy by utilizing an inverse electron-demand Diels–Alder reaction between trans-cyclooctene and tetrazine cycloaddition.13 More recently, activation of a systemic prodrug via cleavage of a deactivating linker on the prodrug triggered by an overexpressed enzyme or localized activation agent on cancer cells has been reported.14–16 Nonetheless, in most cases, the enzyme is not naturally overexpressed at the cancer site, and enrichment or genetic modification is required to accumulate the activation agent. Despite this progress, co-administration of a prodrug with other exogenous activating agents could significantly induce immunogenicity and might hamper repeated administration.
Inspired by the initial bioorthogonal reaction strategies and the remaining in vivo application challenges, we set out to utilize a bioorthogonal prodrug activation reaction based on the 1,3-dipolar cycloaddition between endogenous acrolein and aryl azide. Acrolein, the most reactive α,β-unsaturated aldehyde,17 has long been known to be a critical biomarker associated with various types of disorders related to oxidative stress, including cancer and Alzheimer's disease.18 Acrolein is produced through the enzymatic oxidation of polyamines and is also generated during reactive oxygen species (ROS)-mediated oxidation of highly unsaturated lipids.19–21 It is sometimes generated on μM scales in oxidatively stressed cells.22
We previously described the uncatalyzed 1,3-dipolar cycloaddition between phenyl azide 1a and acrolein (Fig. 1a).23 The reaction proceeds to give 4-formyl-1,2,3-triazoline 2a, which immediately isomerized into the 4-formyl-2H-1,2,3-triazoline 3a or oxidized into 4-formyl-1,2,3-triazole 4a. The triazolines with electron-withdrawing substituents in the C4-position also undergo rearrangement to give the aliphatic α-diazocarbonyl compound 5a.24–28 The reaction of phenyl azide 1a, under physiological conditions, is highly chemoselective to acrolein. We utilized the TAMRA-labeled phenyl azide, namely click-to-sense (CTS) probe 1b (Fig. 1b(i)), for acrolein detection in cells undergoing oxidative stress and found that the CTS probe 1b could shuttle in and out of cells through transporter-mediated mechanisms (ESI, Fig. S2a†).29 If the CTS probe 1b encounters the endogenous intracellular acrolein, the 1,3-dipolar cycloaddition occurs to produce a triazoline compound, rearranges into a diazo derivative, and reacts with the nearest organelle to anchor the fluorophore via covalent attachment.
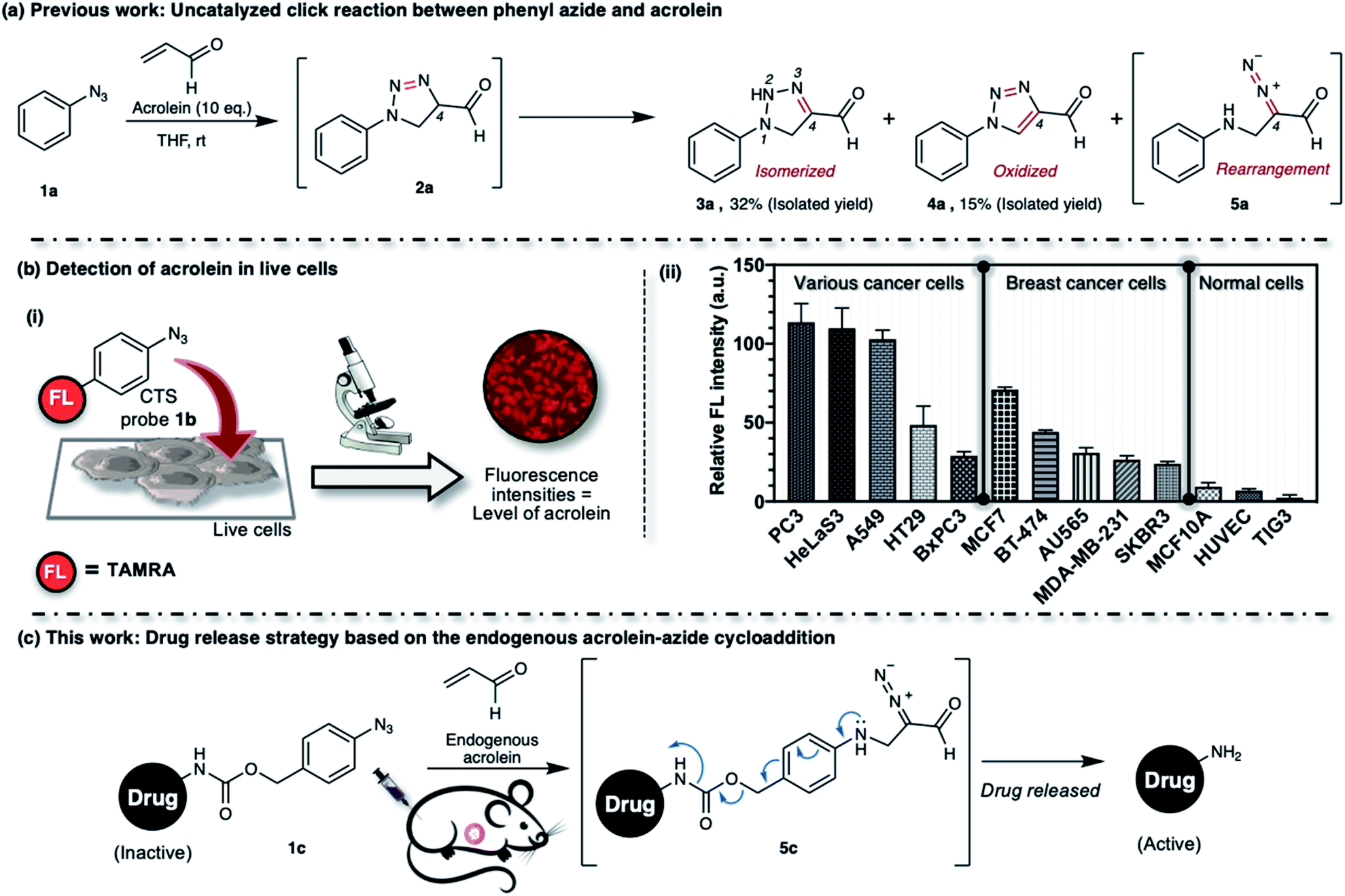 | ||
| Fig. 1 (a) Phenyl azide 1a reacts with acrolein through 1,3-dipolar cycloaddition to give intermediate 2a, which could isomerize, oxidize, or rearrange to 3a, 4a, or 5a, respectively. (b(i)) CTS probe 1b selectively reacts with intracellular acrolein. (b(ii)) Fluorescence intensity, which was proportional to the intracellular acrolein concentration, was observed for each cell line over the CTS probe 1b (22.5 μM). Various human cancer cell lines: PC3 (prostate cancer cell), HeLaS3 (cervical cancer cell), A549 (lung cancer cell), HT29 (colon cancer cell), BxPC3 (pancreatic cancer cell). Human breast cancer cell lines: MCF7 (ER+/PR+/HER2−), BT-474 (ER+/PR+/HER2+), AU565 (ER−/PR−/HER2+), MDA-MB-231 (ER−/PR−/HER2−), SKBR3 (ER−/PR−/HER2+). Human normal cell lines: MCF-10A (mammary cell), HUVEC (umbilical vein endothelial cell), TIG3 (diploid cell). Immunoprofiles of the breast cancer subtypes are shown in the brackets; ER = estrogen receptor; PR = progesterone receptor; HER2 = human epidermal growth factor receptor-2. TAMRA = 5(6)-carboxytetramethylrhodamine. (c) Our in vivo drug release strategy utilizes the 1,3-dipolar cycloaddition between aryl azide and endogenous acrolein. | ||
We found that the CTS probe 1b could be used selectively and sensitively to detect acrolein in live cells by observing the fluorescence intensity (Fig. 1b(i)). We demonstrated the feasibility of the method to determine acrolein levels in various human cell lines. In general, we found that cancer cells have a high level of acrolein, approximately between 50–250 nM, whereas healthy cells contain a negligible amount of acrolein (Fig. 1b(ii)).30 In other words, endogenous cellular acrolein could be used as a new cancer marker. Furthermore, we utilized CTS probe 1b to detect acrolein in breast cancer tissues resected from patients with breast cancer (ESI, Fig. S2b†).30 This method selectively labels the cellular contents of cancer cells within live tissues and visualizes their morphology. The CTS probe 1b has clinical significance in evaluating cancer morphology, and this method has been used in a clinical study as a highly selective, low-cost, and easy-to-perform method for cancer screening during breast cancer surgery.31 Considering all of the above, we aim to develop an aryl azide–acrolein reaction-based prodrug, which could be applied to most cancer types. We recognized that if a benzyl carbamate linker is attached to the phenyl azide at a para-position, such as in compound 1c (Fig. 1c), the reaction with endogenous acrolein will give the diazo compound 5c, which could have potential in cancer-targeted prodrug activation strategies. If it succeeds, this method eliminates the need for pretreatments to accumulate exogenous enzymes or other activating agents on the cancerous tissues before prodrug administration, making it more realistic for practical application.
Results and discussion
Thus far, azide is known to be one of the most effective and widely used bioorthogonal reagents.32 It was believed that azide is inert and will not react with any endogenous molecules in biosystems. Despite these assumptions, we found that aryl azide (CTS probe 1b, Fig. 1b(i)) could react with endogenous acrolein generated by cancer cells in the absence of a catalyst.23,29,30 Although the simple phenyl azide, such as in CTS probe 1b, is convenient for the azide–acrolein 1,3-dipolar cycloaddition, as demonstrated herein for the drug release application, we thought that a high reactive aryl azide derivative would be more suitable to ensure efficient payload release at a low concentration within a reasonable timescale.Previously, Hosoya and co-workers reported that the 2,6-diisopropylphenyl azide 1d (Fig. 2), despite its steric hindrance, reacts significantly faster than phenyl azide 1a in the 1,3-dipolar cycloaddition with an alkyne.33 While the azide–alkyne cycloaddition proceeds in a concerted manner,34,35 it is more likely that the azide–acrolein cycloaddition proceeds in a stepwise manner. Therefore, we thought it was necessary to analyze the reactivity of phenyl azide 1a and 2,6-diisopropylphenyl azide 1d toward acrolein. First, the acrolein–phenyl azide 1a (Fig. 1a and ESI, Fig. S3†) and the acrolein–2,6-diisopropylphenyl azide 1d (Fig. 2 and ESI, Fig. S4†) reactions were analyzed by reversed phased high-performance liquid chromatography (RP-HPLC). The results showed that, while phenyl azide 1a reacts with acrolein to give 4-formyl-2H-1,2,3-triazoline 3a and 4-formyl-1,2,3-triazole 4a, in the case of 2,6-diisopropylphenyl azide 1d, the possible product 4-formyl-2H-1,2,3-triazoline 3d could not be isolated from the reaction mixture (Fig. 2 and ESI, Fig. S4†). On the other hand, the reaction proceeds to give 4-formyl-1,2,3-triazole 4d and α-diazocarbonyl compound 5d, isolated as heterocycle 6 in good yields. It is reasonable to assume that diazo 5d reacts further through dimerization, cycloaddition with one more acrolein molecule, and intramolecular acetal formation to give the heterocycle 6.
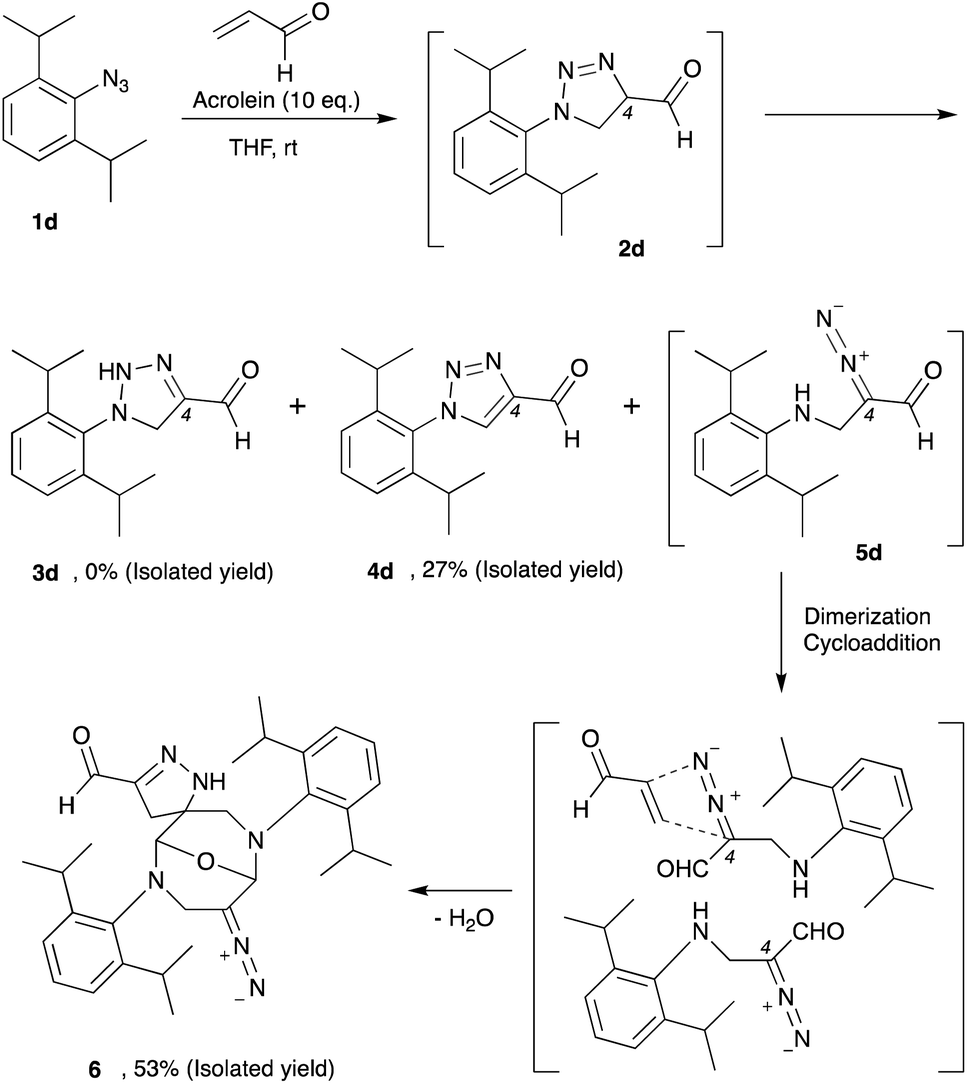 | ||
| Fig. 2 The 2,6-diisopropylphenyl azide 1d reacts with acrolein through 1,3-dipolar cycloaddition to give intermediate 2d, which could isomerize, oxidize, or rearrange to 3d, 4d, or 5d, respectively. The α-diazocarbonyl 5d was isolated as heterocycle 6. | ||
We then design an experiment to determine the reaction kinetics for these acrolein–azide 1,3-dipolar cycloadditions (ESI, Fig. S5†). Since the triazoline 2a and 2d are difficult to observe due to the rapid isomerization, oxidation, or rearrangement of the intermediates to the corresponding products, we then performed the quantitative analysis by observing the formation of compound 3a, 4d, and 6. We found that the second-order rate constant (k) for the reaction of 2,6-diisopropylphenyl azide 1d with acrolein to give heterocycle 6 (k = 3.8 × 10−1 M−1 min−1) was 9.7 times higher than the reaction of phenyl azide 1a with acrolein to give triazoline 3a (k = 3.9 × 10−2 M−1 min−1). Also, we observed that in the reaction of 2,6-diisopropylphenyl azide 1d and acrolein, the formation of heterocycle 6 was 6.6 times faster than the formation of 4-formyl-1,2,3-triazole 4d (k = 5.7 × 10−2 M−1 min−1).
Furthermore, we monitored the reaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1a and 1d with an excess amount of acrolein in CDCl3 by 1H-NMR spectroscopy and observed that the cycloaddition products from 1d predominate (ESI, Fig. S6†). In contrast, the cycloaddition products from 1a are hardly observed in the reaction mixture. Therefore, these results also demonstrate that the 2,6-diisopropylphenyl azide 1d reacts more quickly than phenyl azide 1a toward acrolein.
:1 mixture of 1a and 1d with an excess amount of acrolein in CDCl3 by 1H-NMR spectroscopy and observed that the cycloaddition products from 1d predominate (ESI, Fig. S6†). In contrast, the cycloaddition products from 1a are hardly observed in the reaction mixture. Therefore, these results also demonstrate that the 2,6-diisopropylphenyl azide 1d reacts more quickly than phenyl azide 1a toward acrolein.
To rationalize the enhanced reactivity of 2,6-diisopropyl derivatives, we performed a density functional theory (DFT) calculation to determine the cyclization's reaction barrier (Fig. 3). The activation energy of the 2,6-diisopropylphenyl azide 1d–acrolein cycloaddition (reaction 2) was lower than that of the phenyl azide 1a–acrolein cycloaddition (reaction 1) by 6.2 kJ mol−1 (Fig. 3a). We carried out a distortion/interaction analysis36 and a noncovalent interaction (NCI) analysis37 to uncover the activation energy difference. Fig. 3b shows that the smaller distortion energy and the larger interaction energy magnitude causes the lower activation energy in reaction 2. The smaller distortion energy in reaction 2 can be explained by the smaller structural change from INT1′ to TS2, as depicted in Fig. 3c. This substituent effect would be caused by the steric hindrance of the isopropyl groups, as discussed for the 1,3-dipolar cycloaddition of 1d with diyne.33 The NCI visualized in Fig. 3c demonstrates a weak attraction around the isopropyl groups in TS2, explaining the larger interaction energy magnitude in reaction 2. Therefore, compared to 1a, the reactivity of 1d with acrolein is enhanced by the isopropyl groups. The calculation result also explains that the regioselectivity of 2d over 2d′ (ESI, Fig. S7†) is induced by the kinetic advantage of the reaction between s-trans-acrolein and the 2,6-diisopropylphenyl azide major conformer. The DFT calculation result is in good agreement with the experimental observation.
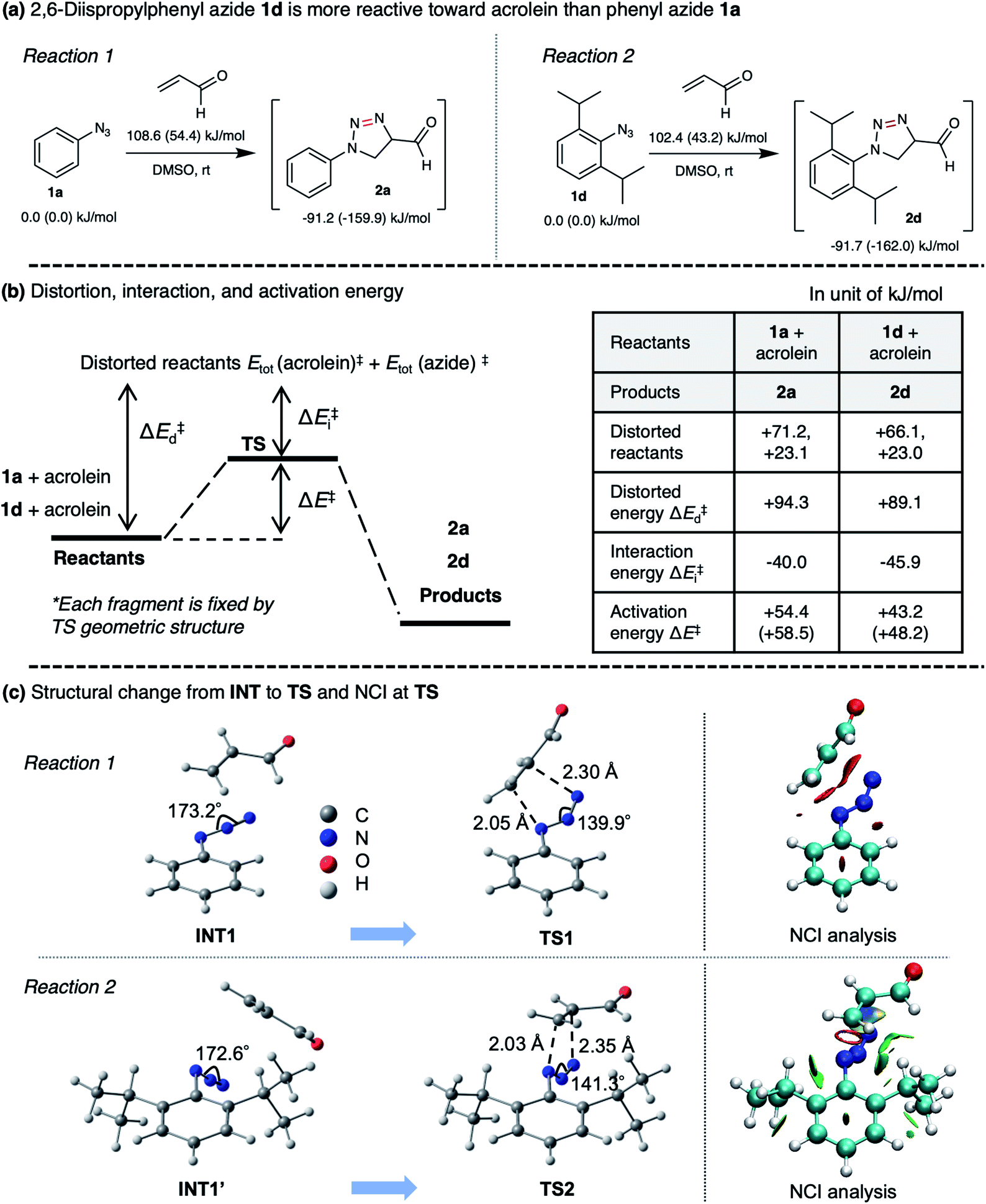 | ||
| Fig. 3 (a) Calculated activation energies and reaction energies in reactions 1 and 2 at the ωB97XD/D95V(d) level combined with the SMD solvation model (DMSO). Values outside and inside parentheses correspond to Gibbs energy at room temperature and electronic energy, respectively. (b) Results of the distortion/interaction analysis which decomposes the activation energy (ΔE) into distortion (ΔEd) and interaction (ΔEi) energies. (c) Structural changes from pre-reaction intermediate (INT) to transition state (TS) in the cyclization step and noncovalent interaction (NCI) at TS visualized by the NCI analysis. NCI depicted in red and green corresponds to steric repulsion and week van der Waals (vdW) attraction. | ||
The production of heterocycle 6 and hence the presence of diazo 5d in the reaction mixture, and the higher reactivity of 2,6-diisopropylphenyl azide 1d toward acrolein encourages us to test the azide–acrolein cycloaddition for our drug release strategy. As proof of concept, we designed coumarin-azidobenzylcarbamate (ABC) 7 (Fig. 4a). If successful, upon reaction with acrolein, the coumarin moiety will be released, producing an increase in fluorescence intensity that could be easily detected (ESI, Fig. S8a†). The synthesis started from 2,6-diisopropyl aniline, which was iodinated at the para-position to give 4-iodo-2,6-diisopropyl aniline S1 in 98% yield (Fig. 4b). Palladium-catalyzed carbonylation allows the introduction of the ester group to afford compound S2 in an 86% yield. An attempt to reduce the ester with LiAlH4 gave the desired alcohol S3 only in low yield while the undesired 4-methyl-2,6-diisopropyl aniline was obtained as the main product. We overcame this problem by utilizing DIBAL-H to smoothly reduce the ester to alcohol S3 in 99% yield. The amino moiety of compound S3 was then converted to an azide moiety using NaNO2 and NaN3 to give compound S4 in an 85% yield. The reaction of compound S4 with 7-amino-4-methylcoumarin in the presence of triphosgene and Hünig's base affords the desired coumarin-ABC 7 in 84% yield.
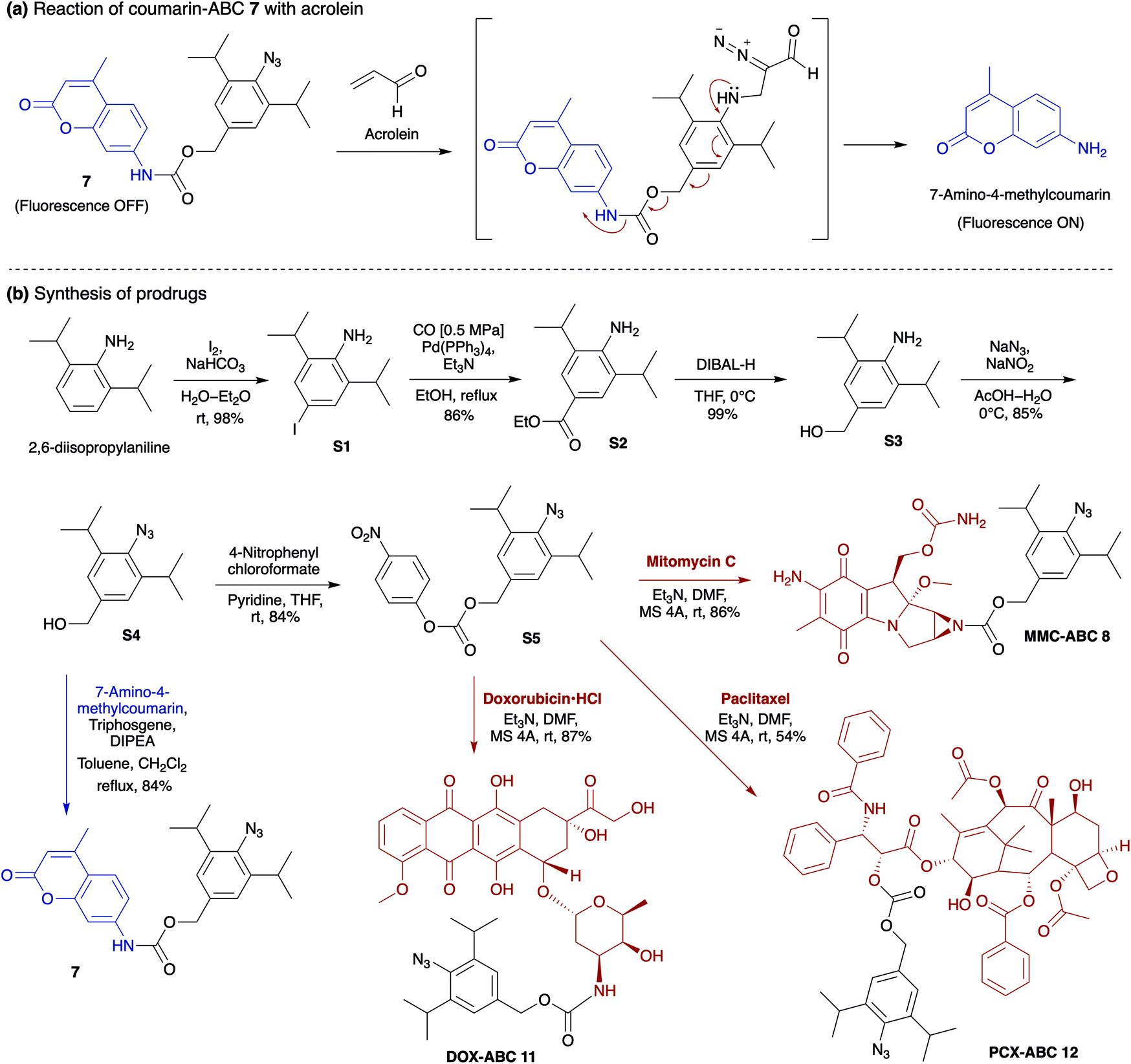 | ||
| Fig. 4 (a) The reaction of coumarin-ABC 7 with acrolein to give α-diazocarbonyl compound induces the coumarin release. (b) The coumarin-ABC 7 could be synthesized from 2,6-diisopropylaniline. The reaction of compound S4 with 4-nitrophenyl chloroformate proceeds to give intermediate S5, which could easily transform to prodrug MMC-ABC 8, DOX-ABC 11, and PCX-ABC 12. | ||
The coumarin-ABC 7 has good solubility in PBS with 1% DMSO (ESI, Fig. S8c†); also, the azide moiety is stable and not easily reduced under physiological conditions or by endogenous reductants, such as glutathione (GSH), in live cells. We examined the stability of coumarin-ABC 7 (20 μM) by incubating with GSH (20 μM in DMSO or 2 mM in PBS) and monitored the released coumarin by RP-HPLC analysis (Fig. 5a(i) and ESI, Fig. S8e(i)†). The result shows that even after 12 hours of incubation, the azide moiety was not susceptible to reduction, and the coumarin was not released as confirmed by RP-HPLC analysis. On the other hand, when coumarin-ABC 7 (20 μM) was incubated with acrolein (20 μM) in DMSO, the amount of released coumarin increases as the reaction time increases (Fig. 5a(ii)). We also examine the coumarin release kinetics and found that the second-order rate constant (k) was 5.1 × 10−2 mM−1 min−1 (ESI, Fig. S9†). A quantitative analysis estimated that approximately 16 μM of coumarin was released from 150 μM of coumarin-ABC 7 after 4 hours of incubation with acrolein (ESI, Fig. S12†). Furthermore, we incubated coumarin-ABC 7 (20 μM) in Dulbecco's Modified Eagle's Medium (DMEM) solution with or without acrolein at room temperature (Fig. 5b). In the presence of acrolein, the coumarin-ABC 7 reacts with acrolein to give a diazo derivative that could induce coumarin release and translate into fluorescence activation. In the absence of acrolein, we observed a slight increase of fluorescence signal, probably due to carbamate hydrolysis in DMEM solution. Nevertheless, the carbamate hydrolysis in DMEM solution proceeds slower than the coumarin release triggered by the azide–acrolein reaction.
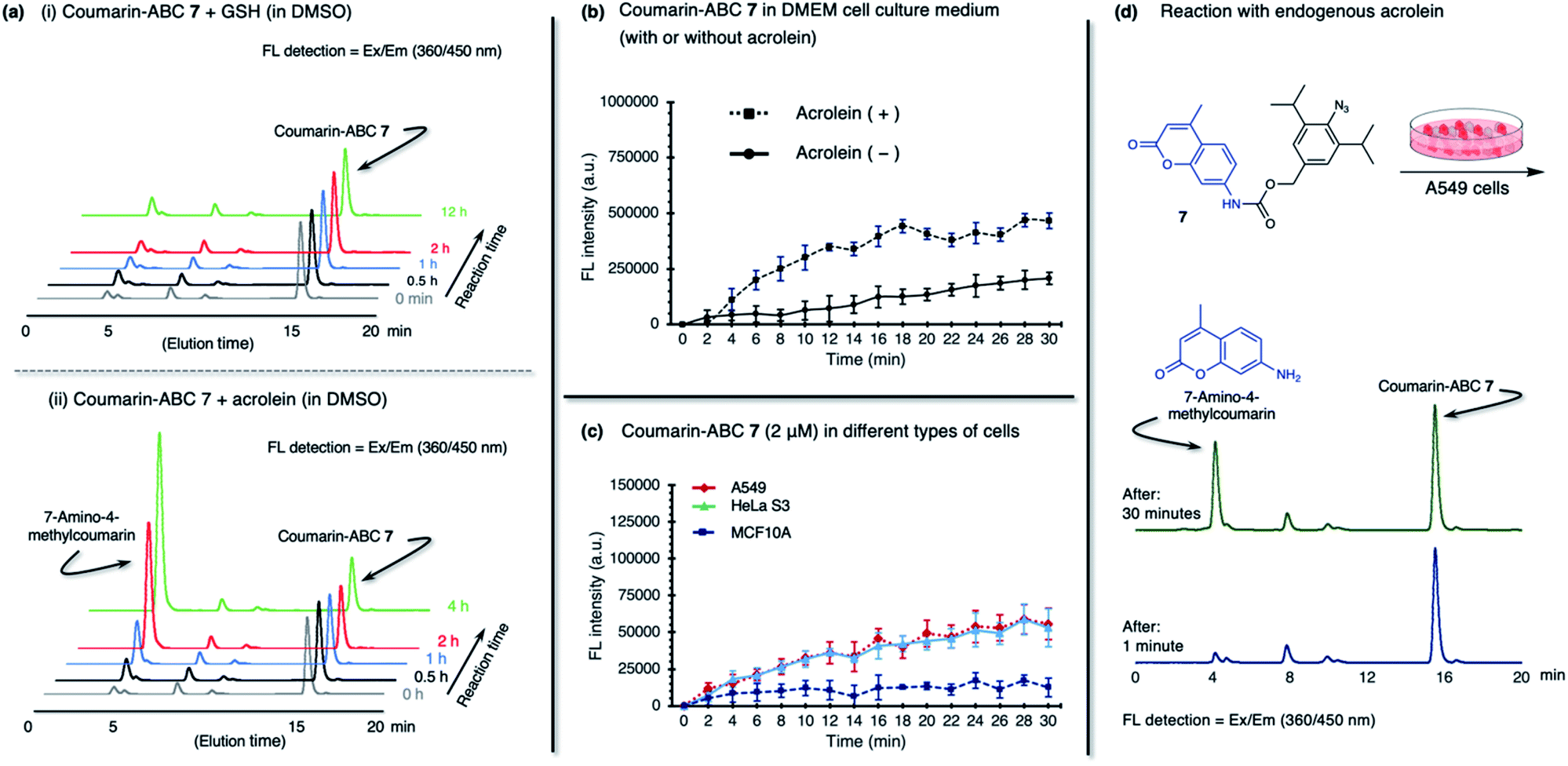 | ||
| Fig. 5 The reactions between coumarin-ABC 7 (1.0 eq.) and (a(i)) glutathione (GSH, 1.0 eq.), or (a(ii)) acrolein (1.0 eq.) in DMSO were monitored at a specific time, by directly injecting the reaction mixtures to HPLC. (b) The coumarin-ABC 7 (20 μM) is incubated in Dulbecco's Modified Eagle's Medium (DMEM) solution with the presence (dashed line) or absence (solid line) of acrolein at room temperature. (c) Two cancer cells (A549, HeLa S3); and one normal cell (MCF10A) were treated with 2 μM of coumarin-ABC 7. Fluorescence intensity was normalized for each cell line per 10000 cells. (d) The coumarin-ABC 7 (20 μM) was incubated in A549 cells. We used HPLC to analyze the cell culture medium at 1 min and 30 min of incubation. | ||
Based on the fact that acrolein is overproduced in cancer,30 we then incubated coumarin-ABC 7 in two types of cancer cells. Herein, we decided to utilize A549 (human lung cancer) and HeLa S3 (human cervical cancer) cells because they have a relatively higher acrolein level than other cancer cells, as shown in Fig. 1c(ii). We also used a normal cell, MCF10A (normal human mammary cells), as control cells that produce a negligible amount of acrolein. The acrolein generated by cancer cells might remain inside the cell membrane or excrete outside the cells,38 in this case to the culture medium. Therefore, in this in vitro cell experiment, we targeted both the intracellular and extracellular acrolein to react with coumarin-ABC 7. We treated the cells with coumarin-ABC 7 and directly observed the fluorescence change of cells in a culture medium every 2 minutes for 30 minutes. Incubation of 2 μM of coumarin-ABC 7 in cancer cells could initiate the reaction with the acrolein and trigger the coumarin release, as indicated by the increase in fluorescence intensity (Fig. 5c). On the contrary, the increase in fluorescence intensity in coumarin-ABC 7 treated normal cells was not significant. Following this procedure, A549 cells, HeLa S3 cells, and MCF10A cells were treated with coumarin-ABC 7 in five different concentrations in the presence or absence of 1 mM of N-acetyl cysteine (NAc-Cys), an acrolein scavenger, at room temperature for 60 minutes (ESI, Fig. S10†). The total fluorescence intensity of the cells was recorded using a spectrofluorometer. We observed decreased fluorescence intensity from the cells pretreated with NAc-Cys, indicating a lower amount of endogenous acrolein that could trigger the released coumarin.
We also confirmed the in vitro azide–endogenous acrolein 1,3-dipolar cycloaddition by observing the coumarin molecule's release with RP-HPLC analysis (Fig. 5d). We treated A549 cells with 20 μM of coumarin-ABC 7 and analyzed its HPLC profile at 1 minute and 30 minutes after incubation. As a result, the released 7-amino-4-methyl coumarin peak appeared in the analysis of the cell culture 30 minutes after incubation. Hence, these results prove that coumarin-ABC 7 selectively reacts with the endogenous acrolein produced by cancer cells. Also, we examined the stability of coumarin-ABC 7 in mouse blood serum and mouse liver microsome and found the half-life was 23.5 min and 22.8 min, respectively (ESI, Fig. S8e(ii) and (iii)†).
Taking into account the above results, for the in vivo cancer treatment study, we decided to replace the coumarin moiety with anticancer drugs, i.e., mitomycin C (MMC),39 doxorubicin (DOX),40 and paclitaxel (PCX)41 (Fig. 4b). MMC is an antibiotic that was initially approved as a cancer chemotherapeutic agent in 1974 by the Food and Drug Administration (FDA). MMC with an alkyl group on the aziridine ring's nitrogen atom is known to have lower cytotoxicity than free MMC.42–44 The mechanism of action of MMC is based on reductive activation and alkylation of DNA forming DNA adducts.45 Unlike many other chemotherapy agents, MMC is usually not associated with multidrug resistance. However, its problematic safety profile, mainly due to its sub-acute and cumulative toxicity to bone marrow and kidney, is a significant obstacle to exploiting its substantial tumors activity.42
On the other hand, although DOX has been reported to be effective against breast, lung, and ovarian cancers,46 it could also induce cardiotoxicity.47,48 DOX acts on cancer cells through two possible mechanisms: (i) intercalation into DNA and disruption of topoisomerase-II-mediated DNA repair, or (ii) generation of free radicals and their damage to DNA, proteins, and cellular membranes.49 Meanwhile, PCX, a microtubule-stabilizing drug used to treat ovarian, breast, and lung cancer, is recognized to induce mitotic arrest that leads to cell death in a subset of the affected population.50,51 Our prodrugs can be prepared by reacting intermediate compound S4 with 4-nitrophenyl chloroformate to give the activated carbonate S5 in 84% yield (Fig. 4b). Then, the ester–amide exchange reaction of S5 with the MMC, DOX, and PCX gives the desired prodrug MMC-ABC 8, DOX-ABC 11, and PCX-ABC 12 in 86%, 87%, and 54% yields, respectively.
In the in vitro cytotoxic assay, the free MMC and DOX nonspecifically showed cytotoxicity toward normal and cancer cells (Table 1 and ESI, Fig. S11a and b†). However, in MMC-ABC 8 or DOX-ABC 11, it showed good selectivity against cancer cell lines. Against MCF10A cells, both MMC-ABC 8 and DOX-ABC 11 displayed low cytotoxicity. On the other hand, when cancer cells (i.e., A549, HeLa S3) were treated with MMC-ABC 8 or DOX-ABC 11, the endogenous acrolein overexpressed in the cancer cells reacted with the azide moiety of MMC-ABC 8 or DOX-ABC 11 to induce the release of payload drugs. The cytotoxicity of prodrug MMC-ABC 8 and DOX-ABC 11 in these cancer cells increased substantially. Notably, in the case of MMC-ABC 8, it approached the cytotoxic level of free MMC. Nonetheless, the in vitro cytotoxic assay of PCX-ABC 12 showed cytotoxicity against the normal MCF10A cells (ESI, Fig. S11c†). The toxic nature of PCX and the drug-carbonate linkage with different properties with drug-carbamate linkage is not suitable for our strategy. Therefore, we excluded PCX-ABC 12 for further study.
| Compound | MCF10A (IC50, μM) | A549 (IC50, μM) | HeLaS3 (IC50, μM) |
|---|---|---|---|
| a 95% confidence interval (n > 5) is shown in the brackets. b No reduction in proliferation observed compared to untreated control. | |||
| MMC-ABC 8 | —b | 2.707 (1.929–3.798) | 0.671 (0.591–0.761) |
| MMC | 0.724 (0.437–1.200) | 0.901 (0.739–1.098) | 0.332 (0.273–0.405) |
| NAc-Cys + 8 | —b | 9.966 (7.934–12.52) | 6.069 (5.499–6.697) |
| Compound 9 | —b | 13.07 (7.780–21.95) | 13.11 (9.755–17.61) |
| DOX-ABC 11 | —b | 1.226 (0.979–1.535) | 0.858 (0.811–0.908) |
| DOX | 0.014 (0.002–0.135) | 0.194 (0.161–0.233) | 0.273 (0.167–0.446) |
Because MMC-ABC 8 has better drug release efficacy compared to DOX-ABC 11 to give selective cytotoxicity at a similar level as that of the free MMC, we decided to focus on prodrug MMC-ABC 8 for further investigation. When cancer cells were pretreated with NAc-Cys, the endogenous acrolein was scavenged; thus, treatment with MMC-ABC 8 showed no cytotoxicity against both A549 and HeLa S3 cells (Table 1 and ESI, Fig. S11a†). This result proved that MMC-ABC 8 reacts with endogenous acrolein produced by cancer cells to release the payload drug and produce its cytotoxic effect. As another control experiment, we also treated both normal and cancerous cells with 4-methyl-2,6-diisopropyl-phenyl azide 9 (chemical structure shown in Fig. 6a) and found that its cytotoxicity was negligible. In other words, without linkage to drugs, the aryl azide or the reaction of aryl azide with acrolein itself does not induce any cytotoxicity. Furthermore, through quantitative analysis, we found that incubation of MMC-ABC 8 (150 μM) with acrolein for 4 hours released approximately 18 μM of MMC, whereas in the case of DOX-ABC 11 (150 μM), approximately 13 μM of DOX was released (ESI, Fig. S12†).
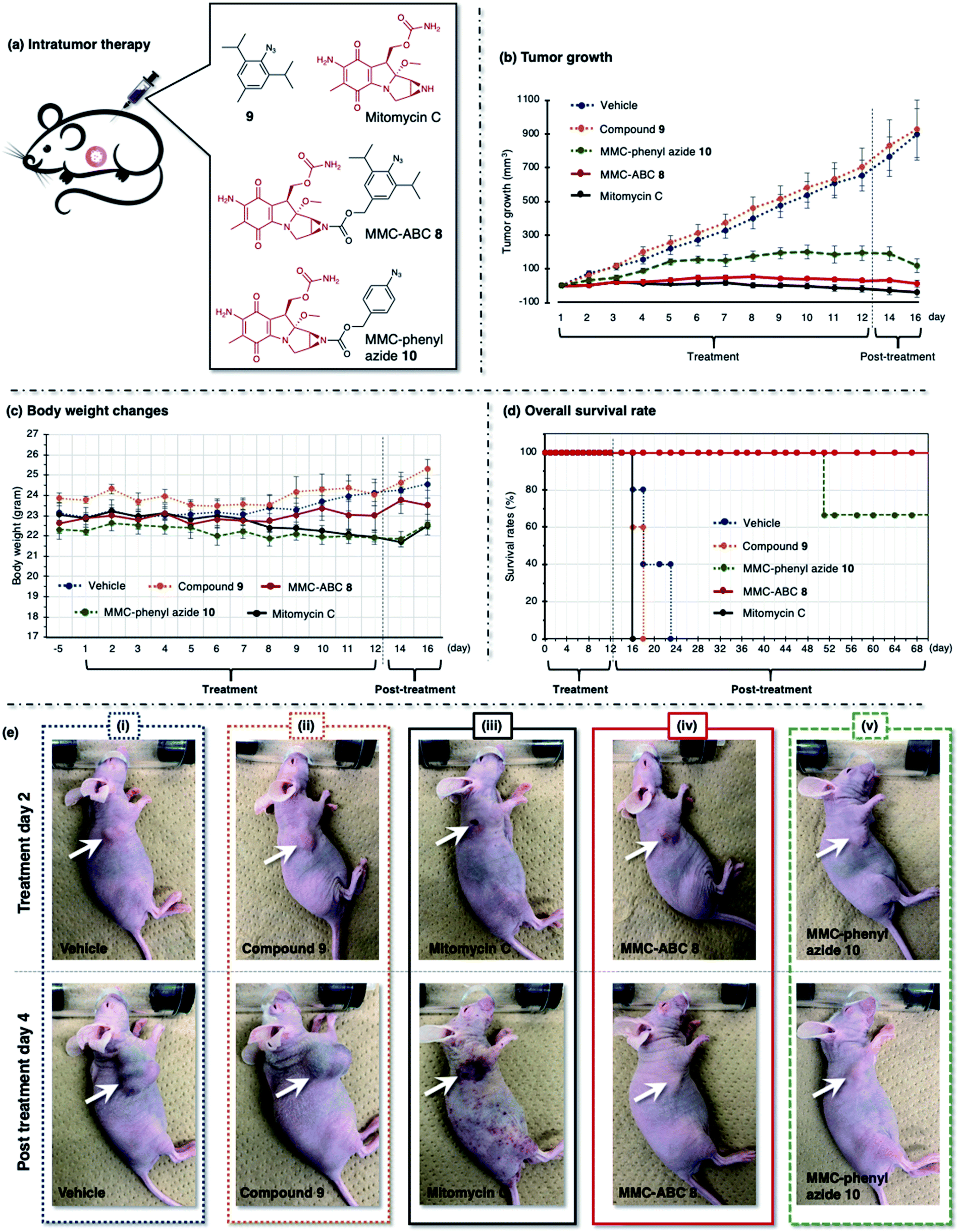 | ||
| Fig. 6 (a) The A549 cell xenograft-bearing nude mice were treated with vehicle, prodrug MMC-ABC 8, compound 9, MMC-phenyl azide 10, and free MMC. The compounds were administered intratumorally every day for 12 days, and the (b) tumor growth levels, (c) body weight changes, and (d) survival rates were observed. (e) The photos were taken during the second day of treatment and the fourth day after treatment. The arrow shows the location of cancer. See ESI Fig. S14 and S15† for other photos. | ||
To assess whether the strategy would work in vivo and result in the intended anticancer effect, we tested our prodrug system in living mice. For our first trial of in vivo study, we prepared A549 cell xenograft-bearing nude mice model. Our previous study estimated that approximately 200 nM of acrolein could be generated by A549 cells (2.5 × 105 cells per mL). However, before performing in vivo experiments, it was crucial to examine the acrolein levels in cancer tissues implanted into nude mice. The CTS probe 1b (Fig. 1b and ESI, Fig. S2†) has been utilized to differentiate cancer and normal tissues from tumor stumps resected from patients with breast cancer with high selectivity and sensitivity. We considered that CTS probe 1b might help determine the level of acrolein in the cancerous tissue surface of the mice. We therefore excised the mouse skin around the cancerous area to expose the tumor, including the border of cancerous and healthy tissue (area 1) and the healthy tissue (area 2) (ESI, Fig. S13†). The CTS probe 1b (20 μM) was sprayed carefully on the surface area, and the whole mouse body was directly analyzed by fluorescence imaging. We found that the tumor area's fluorescence intensity is higher than area 1 and area 2, which proved that acrolein is overproduced by the mouse xenograft model's cancer tissue. Based on the standard fluorescence intensity, approximately 10 μM of acrolein is generated on the mouse xenograft model's cancer tissue (1 × 1 cm). The amount is much higher than the acrolein generated by the A549 cell culture. Accordingly, we predicted that the in vivo endogenous acrolein–azide cycloadditions that trigger drug release would proceed more efficiently than in vitro cell experiments.
Therefore, we decided to intratumorally (Fig. 6) and intravenously (Fig. 7) treat a group of A549 cell xenograft-bearing nude mice with prodrug MMC-ABC 8 and compared them with other groups treated with vehicle, compound 9, MMC-phenyl azide 10, and free MMC. The MMC-phenyl azide 10 is derived from phenyl azide 1a and has lower reactivity toward acrolein (vide supra), and hence is expected to have a decreased ability to release the drug (see ESI† for MMC-phenyl azide 10 preparation). The appropriate dose of mitomycin C for mouse experiments has been reported previously, with the maximum non-lethal dose being 6.7 mg kg−1 and the rational dose being 1.7 mg kg−1 (120 nmol).52
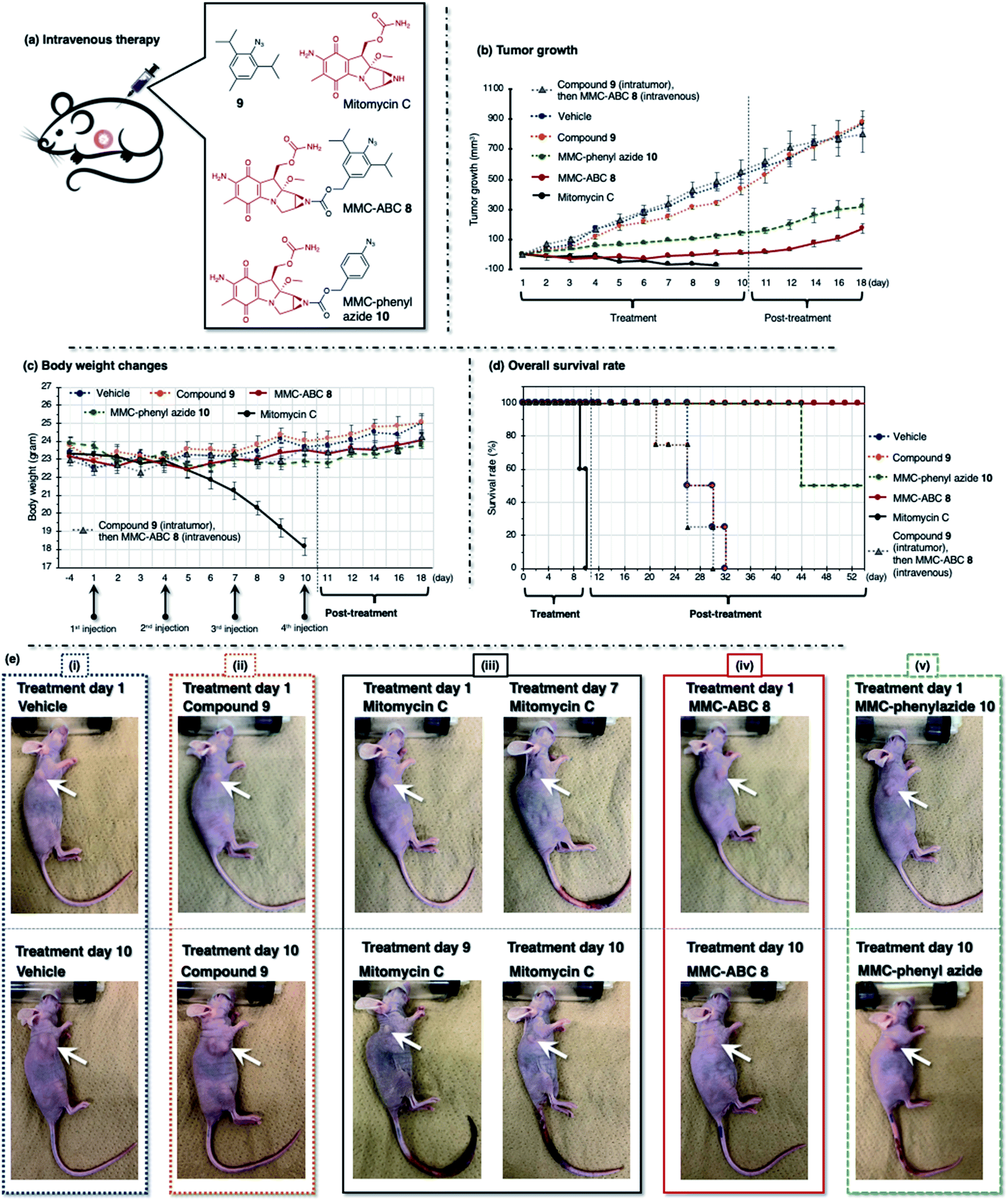 | ||
| Fig. 7 (a) The A549 cell xenograft-bearing nude mice were treated with vehicle, prodrug MMC-ABC 8, compound 9, MMC-phenyl azide 10, and free MMC. The compounds were administered intravenously every three days for ten days, and the (b) tumor growth levels, (c) body weight changes, and (d) survival rates were observed. (e) The photos were taken during the first and tenth day of treatment. The arrow shows the location of cancer. See ESI Fig. S16–S21† for other photos. | ||
First, we treated the cancer-bearing xenograft mouse with a repeated intratumoral injection every day for 12 days and observed tumor growth (Fig. 6 and ESI, Fig. S14 and S15†). We found that treatment with vehicle (DMSO:EtOH:saline = 1:4:5, 5 μL, n = 6) or compound 9 (5 μL, 120 nmol, 1.2 mg kg−1, n = 6) does not retard the tumor growth, and we observed a robust increase in tumor size (900 mm3) in two weeks (Fig. 6b, dashed blue and orange line). As a result, these groups survived less than twelve days after treatment (Fig. 6d, dashed blue and orange line). Meanwhile, treatment with free MMC (5 μL, 120 nmol, 1.8 mg kg−1, n = 6) significantly inhibits tumor growth (Fig. 6b, solid black line). However, despite its ability to inhibit tumor growth, this group survived less than five days after treatment (Fig. 6d, solid black line). The survival rate was even lower than that of the groups treated with vehicle and compound 9. This result might be caused by the ADR effect of free MMC on the mouse, as we observed that the mouse had more body weight loss (Fig. 6c, solid black line) and inflammation on the whole body (Fig. 6e(iii), photo of post-treatment day 4).
We found that cancer treatment was effective on the mouse subjected to prodrug MMC-ABC 8 (5 μL, 120 nmol, 3.2 mg kg−1, n = 6) (Fig. 6b, solid red line). Furthermore, we did not observe tumor regrowth even after the end of the treatment (ESI, Fig. S15b†). The average body weight could be maintained throughout the experiment (Fig. 6c, solid red line), and this group could survive up to two months after the treatment finished (Fig. 6d, solid red line). Inflammation and ADRs did not occur (Fig. 6e(iv), photo of post-treatment day 4), which proved that the prodrug specifically activated only at the desired cancer area and that cancer treatment with MMC-ABC 8 is promising. On the other hand, treatment with MMC-phenyl azide 10 (5 μL, 120 nmol, 2.8 mg kg−1, n = 6) could inhibit the tumor growth to some extent (Fig. 6b, dashed green line). Even though the survival rate (Fig. 6d, dashed green line) is higher than the control experiment, it is still lower than the group treated with MMC-ABC 8. We consider that this result is caused by the low reactivity of the MMC-phenyl azide 10 toward acrolein.
Alternatively, we treated the cancer-bearing xenograft mouse with a repeated intravenous injection every three days for ten days (four times injection) and observed tumor growth (Fig. 7 and ESI, Fig. S16–S21†). The drug concentration of the intravenous injection was two times higher than that of the intratumor injection. Herein, we could observe a similar tendency, as we observed from the intratumoral injection. We found that treatment with vehicle (DMSO:EtOH:saline = 1:4:5, 100 μL, n = 8) or compound 9 (100 μL, 240 nmol, 2.2 mg kg−1, n = 8) does not have any effect, as the tumor growth was not retarded, and we observed an increase of tumor size (800 mm3) by two weeks (Fig. 7b, dashed blue and orange line). As a result, these groups survived less than three weeks after treatment (Fig. 7d, dashed blue and orange line). Meanwhile, both prodrug MMC-ABC 8 (100 μL, 240 nmol, 6.2 mg kg−1, n = 8) (Fig. 7b, solid red line) and MMC-phenyl azide 10 (100 μL, 240 nmol, 5.2 mg kg−1, n = 8) (Fig. 7b, dashed green line) could inhibit the tumor growth, but the tumor inhibitory efficacy was more significant for prodrug MMC-ABC 8. We found that the group treated with prodrug MMC-ABC 8 (Fig. 7d, solid red line) has a higher survival rate than the group treated with MMC-phenyl azide 10 (Fig. 7d, dashed green line). The treatment of the mice with free MMC (100 μL, 240 nmol, 3.6 mg kg−1, n = 8) also significantly inhibited tumor growth (Fig. 7b, solid black line). Nevertheless, the body weight loss was very drastic (Fig. 7c, solid black line). If we compare the photos between the first day and the tenth day of treatment with MMC (Fig. 7e(iii)), we could notice the body size difference and inflammation in the tail area (ESI, Fig. S18†). Hence, the group treated with free MMC could not survive before the fourth injection (Fig. 7d, solid black line). Furthermore, to demonstrate that the observed therapeutic effect of MMC-ABC 8 is indeed mediated by acrolein-induced release, we treated another cancer-bearing xenograft mouse with compound 9 (5 μL, 120 nmol, 1.2 mg kg−1, n = 8) intratumorally to eliminate the endogenous acrolein before injecting MMC-ABC 8 (100 μL, 240 nmol, 6.2 mg kg−1, n = 8) intravenously. As a result, in this compound 9 and MMC-ABC 8 co-treated mouse group, we observed that the tumor growth increased significantly as the control group (Fig. 7b, dashed grey line, and photos in ESI, Fig. S21†).
To make a rough prediction on the stability of the prodrug in vivo, we performed RP-HPLC analysis of tumor and urine extracts from the mice models, two hours after prodrug MMC-ABC 8 and free MMC were intratumorally administered (ESI, Fig. S22†). In mice treated with MMC-ABC 8, we found that some prodrug was remaining on the tumor (Fig. S22b†), and almost no prodrug or released MMC was detected in urine (Fig. S22c†). Simultaneously, in mice treated with free MMC, the free drug could not be detected from the tumor (Fig. S22b†), but the secreted MMC drug was present in the urine (Fig. S22c†). This result suggests that MMC-ABC 8, which stays for longer time in the cancerous tissue, might gradually release MMC and gives its cytotoxic effect only at the desired target area. Based on the equilibrium dialysis method, we found that serum protein binding notably increased by derivatizing the parent compounds to the ABC compounds (e.g., 44% to >95% in the case of coumarin). Nevertheless, we also observed that the free ligands could be released upon treatment with acrolein (ESI, Fig. S8d†). Therefore, similar to the clinically used drugs (e.g., vinblastine, warfarin)53,54 with high serum binding affinity, we considered that the stabilized ABC prodrugs–protein complex is efficiently carried to the cancer region and activated by acrolein.
Herein, we have described the stability and selectivity of prodrug MMC-ABC 8 and shown that intratumor and intravenous administration does not affect its efficacy. Moreover, since acrolein is generally overproduced by most cancer cells, we anticipate that our strategy might be useful for further applications in other different cancer types. In the literature (ESI, Fig. S1b and c†),11,12 drug-conjugated phenyl azide, such as 1c (Fig. 1c), did not show cytotoxicity in vitro experiments with cancer cells. Moreover, to trigger the drug release, it is typically co-treated with other activating agents, such as phosphine reagent.11 Based on our results, the lower acrolein level in cancer cell cultures compared to living mice (vide supra) might affect the azide reaction toward endogenous acrolein. Also, we found that replacing the phenyl azide moiety with the more reactive 2,6-diisopropyl azide could increase its reaction kinetics toward endogenous acrolein in the mouse model's cancerous tissues, triggering the drug release.
Conclusions
In conclusion, we have developed a strategy for an in vivo anticancer drug delivery by utilizing endogenous acrolein–azide cycloaddition to trigger the drug release. This method enabled controlled prodrug activation by targeting cancer cells that produced a high acrolein level, including HeLa S3 and A549. The 2,6-diisopropylphenyl azide moiety of MMC-ABC 8 was efficiently reacted with endogenous acrolein to trigger drug release. The overall in vitro results showed a significant increase in the potency of prodrug 8 against cancerous tissues and satisfactory selectivity for cancerous over non-cancerous cells. Animal studies further validated our prodrug's excellent efficiency for in vivo tumor inhibition, as well as its biosafety. This method was applied to the A549 cancer-bearing xenograft mouse model, and it shows promising results, where cancer growth was inhibited, and the ADR effect was reduced. As shown in this study, the MMC-ABC 8 remains in cancer cells and the drug is released only at the desired target area. It is noteworthy that the dose applied in the current study was below the maximum tolerant dose of MMC. Furthermore, the ABC moiety could be easily conjugated with other types of drugs. Therefore, it could be applied to other cytotoxic drugs that cause a high rate of ADRs. In other words, this method may significantly expand the scope of anticancer therapeutic agents.Author contributions
Conceptualization: A. R. P., and K. Tanaka; formal analysis: T. I., and S. M.; funding acquisition: A. R. P., and K. Tanaka; investigation: A. R. P., P. A., K. Terashima, K. M., M. F.; visualization: A. R. P.; writing-original draft preparation: A. R. P., and K. Tanaka; writing-review and editing: A. R. P., and K. Tanaka.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (W2019-2-049). This work was financially supported by JSPS KAKENHI [Grant Numbers JP16H03287, JP18K19154, JP15H05843 (to K. Tanaka) and JP18K14341, JP18K07265, 18H05503 (to A. R. P.)]. Funding was also provided by the Russian Government Program for Competitive Growth (to Kazan Federal University). The authors are also grateful to Professor Takamitsu Hosoya and Associate Professor Suguru Yoshida of the Tokyo Medical and Dental University for their fruitful discussion on bulky aryl azide at the very beginning of this research.References
- K. Nurgali, R. T. Jagoe and R. Abalo, Front. Pharmacol., 2018, 9, 245 CrossRef PubMed.
- S. K. Sharma and K. D. Bagshawe, Adv. Drug Delivery Rev., 2017, 118, 2–7 CrossRef CAS PubMed.
- Y. Wang, S. Song, J. Liu, D. Liu and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 536–540 CAS.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767–777 CrossRef CAS PubMed.
- I. A. Müller, F. Kratz, M. Jung and A. Warnecke, Tetrahedron Lett., 2010, 51, 4371–4374 CrossRef.
- C. Li and S. Wallace, Adv. Drug Delivery Rev., 2008, 60, 886–898 CrossRef CAS PubMed.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- M. Azoulay, G. Tuffin, W. Sallem and J.-C. Florent, Bioorg. Med. Chem. Lett., 2006, 16, 3147–3149 CrossRef CAS PubMed.
- R. van Brakel, R. C. M. Vulders, R. J. Bokdam, H. Grull and M. S. Robillard, Bioconjugate Chem., 2008, 19, 714–718 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- E. M. Herrlinger, M. Hau, D. M. Redhaber, G. Greve, D. Willmann, S. Steimle, M. Muller, M. Lubbert, C. C. Miething, R. Schule and M. Jung, ChemBioChem, 2020, 21, 2329–2347 CrossRef CAS PubMed.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS PubMed.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- J. P. Kehrer and S. S. Biswal, Toxicol. Sci., 2000, 57, 6–15 CrossRef CAS PubMed.
- K. Muguruma, A. R. Pradipta, Y. Ode, K. Terashima, H. Michiba, M. Fujii and K. Tanaka, Bioorg. Med. Chem., 2020, 28, 115831 CrossRef CAS PubMed.
- G. Houen, K. Bock and A. L. Jensen, Acta Chem. Scand., 1994, 48, 52–60 CrossRef CAS PubMed.
- R. A. Alarcon, Arch. Biochem. Biophys., 1970, 137, 365–372 CrossRef CAS PubMed.
- K. Uchida, Trends Cardiovasc. Med., 1999, 9, 109–113 CrossRef CAS PubMed.
- R. Shi, T. Rickett and W. Sun, Mol. Nutr. Food Res., 2011, 55, 1320–1331 CrossRef CAS PubMed.
- A. R. Pradipta, M. Taichi, I. Nakase, E. Saigitbatalova, A. Kurbangalieva, S. Kitazume, N. Taniguchi and K. Tanaka, ACS Sens., 2016, 1, 623–632 CrossRef CAS.
- C.-H. Yang, L.-T. Lee, J.-H. Yang, Y. Wang and G.-H. Lee, Tetrahedron, 1994, 50, 12133–12142 CrossRef CAS.
- G. T. Anderson, J. R. Henry and S. M. Weinreb, J. Org. Chem., 1991, 56, 6946–6948 CrossRef CAS.
- W. Broeckx, N. Overbergh, C. Samyn, G. Smets and G. L’abbé, Tetrahedron, 1971, 27, 3527–3534 CrossRef CAS.
- K. R. Henery-Logan and R. A. Clark, Tetrahedron Lett., 1968, 9, 801–806 CrossRef.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1966, 99, 475–490 CrossRef CAS.
- A. R. Pradipta, M. Fujii, T. Tanei, K. Morimoto, K. Shimazu, S. Noguchi and K. Tanaka, Bioorg. Med. Chem., 2019, 27, 2228–2234 CrossRef CAS PubMed.
- T. Tanei, A. R. Pradipta, K. Morimoto, M. Fujii, M. Arata, A. Ito, M. Yoshida, E. Saigitbatalova, A. Kurbangalieva, J.-I. Ikeda, E. Morii, S. Noguchi and K. Tanaka, Adv. Sci., 2019, 6, 1801479 CrossRef PubMed.
- A. R. Pradipta, T. Tanei, K. Morimoto, K. Shimazu, S. Noguchi and K. Tanaka, Adv. Sci., 2020, 7, 1901519 CrossRef CAS PubMed.
- M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184 CrossRef CAS PubMed.
- S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- K. N. Houk, J. Sims, C. R. Watts and L. J. Luskus, J. Am. Chem. Soc., 1973, 95, 7301–7315 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- R. Laplaza, F. Peccati, R. A. Boto, C. Quan, A. Carbone, J. P. Piquemal, Y. Maday and J. Contreras-García, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, e1497 Search PubMed.
- J. F. Stevens and C. S. Maier, Mol. Nutr. Food Res., 2008, 52, 7–25 CrossRef CAS PubMed.
- S. T. Crooke and W. T. Bradner, Cancer Treat. Rev., 1976, 3, 121–139 CrossRef CAS PubMed.
- W. E. Derek, N. Rashmi, C. Simon, M.-B. Kyle, P. J. M. Jonathan and M. P. Amadeo, Curr. Drug Metab., 2015, 16, 412–426 CrossRef PubMed.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253–7269 CrossRef CAS PubMed.
- Y. Amitay, H. Shmeeda, Y. Patil, J. Gorin, D. Tzemach, L. Mak, P. Ohana and A. Gabizon, Pharm. Res., 2016, 33, 686–700 CrossRef CAS PubMed.
- T. Sun, A. Morger, B. Castagner and J. C. Leroux, Chem. Commun., 2015, 51, 5721–5724 RSC.
- P. D. Bass, D. A. Gubler, T. C. Judd and R. M. Williams, Chem. Rev., 2013, 113, 6816–6863 CrossRef CAS PubMed.
- M. He, P. J. Sheldon and D. H. Sherman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 926 CrossRef CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- C. Carvalho, R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS PubMed.
- S. M. Swain, F. S. Whaley and M. S. Ewer, Cancer, 2003, 97, 2869–2879 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- B. A. Weaver, Mol. Biol. Cell, 2014, 25, 2677–2681 CrossRef PubMed.
- S. B. Horwitz, Ann. Oncol., 1994, 5, S3–S6 CrossRef PubMed.
- M. Inaba, T. Kobayashi, T. Tashiro and Y. Sakurai, Japanese Journal of Cancer Research GANN, 1988, 79, 509–516 CrossRef CAS PubMed.
- T. Bohnert and L.-S. Gan, J. Pharm. Sci., 2013, 102, 2953–2994 CrossRef CAS PubMed.
- I. Petitpas, A. A. Bhattacharya, S. Twine, M. East and S. Curry, J. Biol. Chem., 2001, 276, 22804–22809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06083f |
| This journal is © The Royal Society of Chemistry 2021 |