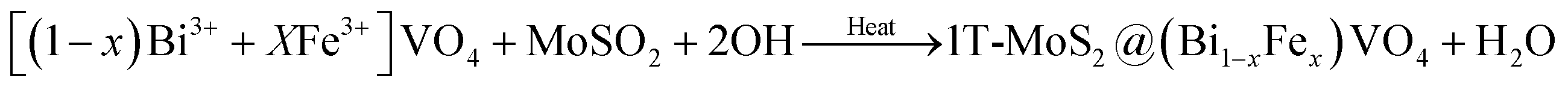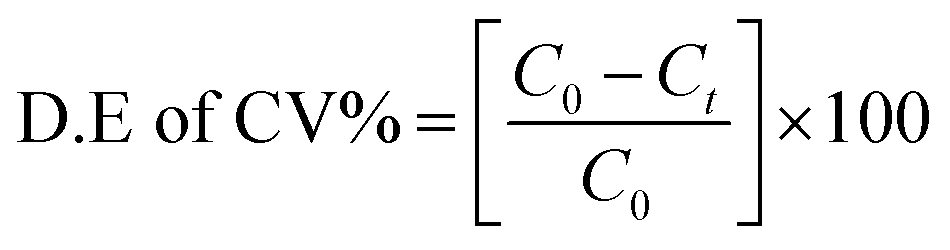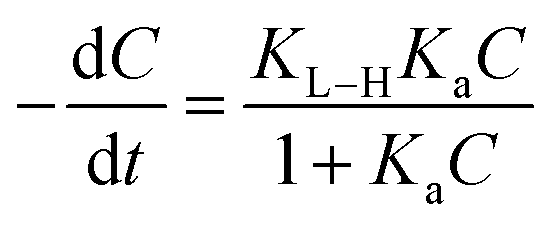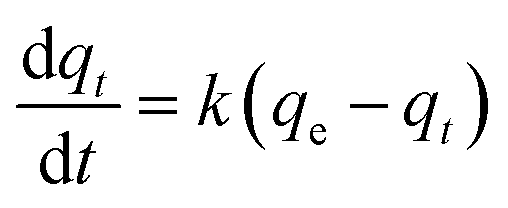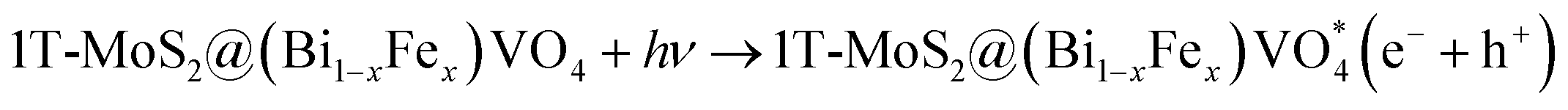DOI:
10.1039/D1RA00807B
(Paper)
RSC Adv., 2021,
11, 13105-13118
Construction of 1T-MoS2 quantum dots-interspersed (Bi1−xFex)VO4 heterostructures for electron transport and photocatalytic properties†
Received
30th January 2021
, Accepted 21st March 2021
First published on 7th April 2021
Abstract
The present study reports trigonal phase molybdenum disulfide quantum dots (MoS2/QDs)-decorated (Bi1−xFex)VO4 composite heterostructures. Initially, (Bi1−xFex)VO4 heterostructure nanophotocatalysts were synthesized through the hydrothermal method decorated with 1T-MoS2 via a sonication process. 1T-MoS2@(Bi1−xFex)VO4 heterostructures were characterized in detail for phase purity and crystallinity using XRD and Raman spectroscopy. The Raman mode evaluation indicated monoclinic, mixed monoclinic-tetragonal and tetragonal structure development with increasing Fe concentration. For physiochemical properties, SEM, EDX, XPS, PL, EPR, UV-visible and BET techniques were applied. The optical energy band gaps of 1T-MoS2@(Bi1−xFex)VO4 heterostructures were calculated using the Tauc plot method. It shows a blue shift initially within a monoclinic structure then a red shift with an increase of Fe concentration. 1T-MoS2@(Bi40Fe60)VO4 with 2 wt% of 1T-MoS2-QDs carrying a mixed phase exhibited higher photocatalytic activity. The enhanced photocatalytic activity is attributed to the higher electron transportation from (Bi1−xFex)VO4 surface onto 1T-MoS2 surface, consequently blocking the fast electron–hole recombination within (Bi1−xFex)VO4. 1T-MoS2 co-catalyst interaction with (Bi1−xFex)VO4 enhanced the light absorption in the visible region. The close contact of small 1T-MoS2-QDs with (Bi1−xFex)VO4 develops a high degree of crystallinity, with fewer defects showing mesoporous/nanoporous structures within the heterostructures which allows more active sites. Herein, the mechanism involved in the synthesis of heterostructures and optimum conditions for photocatalytic degradation of crystal violet dye are explored and discussed thoroughly.
1. Introduction
Energy shortage and water pollution are major threats worldwide owing to limited fossil fuel resources and growing consumption.1,2 Producing abundant and clean energy is the substantial solution to these two problems.3 These problems are somewhat overcome by the advanced oxidation process (AOP) with the advancement of nanotechnology.4,5 The AOP is the most environment-friendly technique and used to remove recalcitrant organic contaminants not easily treatable via traditional methods due to their chemical stability.6,7 The concept of AOPs for water and wastewater treatment was primarily discovered in 1980.8 In the AOP process, reactive oxygen species (ROS) are generated including singlet oxygen (O), ozone (O3), hydrogen peroxide (H2O2), hydroxyl radical (OH˙), and others species.7,9 Among them, OH˙ is a highly oxidizing agent with a potential of 2.8 eV and unstable as compared to other oxidizing agents such as O, O3, and H2O2, possessing oxidation potential of 1.67, 2.07 and 1.77 eV, respectively.10 Photocatalysts are materials that produce strong oxidizing agents, i.e., O, O3, and OH˙.11 In AOPs, photocatalysts or semiconductor materials can directly change solar energy into chemical energy which is a very facile method for renewable energy production and environmental remediation.12,13 Photocatalytic degradation has gained much attention in recent years as it offers a stable, clean and nontoxic direction to lessen environmental pollution.14,15 Common semiconductors used as photocatalysts (with bandgap) such as ZrO2 (3.87 eV),16 ZnS (3.6 eV),17 SnO2 (3.54 eV),18 ZnO (3.4 eV),19 TiO2 (3.3 eV),20 Fe2O3 (2.67 eV),21 CdS (2.42 eV),22 BiVO4 (2.41 eV),23,24 and PbS (0.28 eV)25 have been extensively studied, but their performance is restricted due to either broad bandgap which is functional under UV-light which is only 5% of the solar spectrum or unsuitable band edge energy.26,27 Therefore, there is an urgent need to develop such an efficient and effective photocatalyst that can harvest visible light, which is 55% of the solar spectrum, and yield optimum response for practical and commercial applications. Hence the main goal of this work is to extend the visible light absorption capability of photocatalysts.
Semiconducting material-based photocatalytic schemes are attracting a lot of attention recently because they allow the fast and effective decomposition of organic pollutants.28 In this context, transition metal oxide semiconductors have gained much interest owing to diverse physicochemical properties and applications including good stability, cost-effectiveness, high energy density, catalysts for degradation of organic pollutants, gas sensors and battery applications.27,29,30 Among these various transition metal oxides, bismuth vanadate (BiVO4) and ferric vanadate (FeVO4) are potential candidates for light-driven photocatalysts due to narrow band gaps ranging from 2.0 to 2.72 eV due to their remarkable chemical stability and catalytic activity, minimal optical damage and commercial cost-effective availability.13,24,31-33 Both BiVO4 and FeVO4 possess a suitable energy band in the visible light range indicating tremendous photocatalytic and electrochemical applications.26,33–35 From a literature survey, it is observed that in scheelite ABO4 the B site is partially filled by substituted material.36 Taking the same idea in monoclinic BiVO4, Bi3+ is 8-coordinated with ionic radius of 1.17 Å, and the ionic radius of 4-coordinated V5+ is 0.355 Å.37 FeVO4 has two different crystal structures of triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and orthorhombic (Cmcm) symmetry respectively.35,38 The ionic radius of 8-coordinated Fe3+ is 0.78 Å.39 Reviewed literature has suggested that the heterostructures of BiVO4–FeVO4 would be useful in efficient generation of electron–hole pairs and thus enhancing the photocatalytic activity of (Bi1−xFex)VO4 heterostructure nanophotocatalysts.31,33,35,40 In this research, the effect of Fe substitution for Bi in BiVO4 was investigated and the photocatalytic response was studied in detail. The (Bi1−xFex)VO4 phase transition was probed from monoclinic to tetragonal with the increase of Fe concentration at Bi sites.
) and orthorhombic (Cmcm) symmetry respectively.35,38 The ionic radius of 8-coordinated Fe3+ is 0.78 Å.39 Reviewed literature has suggested that the heterostructures of BiVO4–FeVO4 would be useful in efficient generation of electron–hole pairs and thus enhancing the photocatalytic activity of (Bi1−xFex)VO4 heterostructure nanophotocatalysts.31,33,35,40 In this research, the effect of Fe substitution for Bi in BiVO4 was investigated and the photocatalytic response was studied in detail. The (Bi1−xFex)VO4 phase transition was probed from monoclinic to tetragonal with the increase of Fe concentration at Bi sites.
Furthermore, an emerging transition metal sulfide named molybdenum disulfide (MoS2) is comparable to graphene, with a sandwich structure of three stacked atomic layers (S–Mo–S). Mo atom is covalently combined with six S atoms which form a layer of MoS2; Mo–S allows normal parabolic bonding with bond length of 2.39 Å.41–45 Recently, MoS2-based nanocomposites have emerged as the most prominent candidates owing to outstanding electronic conductivity, substitute for noble metal co-catalysts, abundant availability, and cost-effectiveness.42,46–48 MoS2 is very sensitive for photodetection, found in two-phase structures, one in trigonal phase (1T-MoS2) and the second named as hexagonal phase (2H-MoS2) with an optical energy bandgap ranging from 1.2 to 1.9 eV.42,47,49–51
Inspired by these concepts, 1T-MoS2 quantum dots-interspersed in (Bi1−xFex)VO4, hereafter 1T-MoS2@(Bi1−xFex)VO4 heterostructures, were prepared through a sonication-assisted hydrothermal method. The synthesized 1T-MoS2@(Bi1−xFex)VO4 heterostructures exhibit excellent visible light-dependent photocatalytic activity. Photoluminance study revealed excellent controlled electron–hole transfer activity. The 1T-MoS2@(Bi0.40Fe0.60)VO4 heterostructures with 2.0 wt% of 1T-MoS2 loading with mixed phase exhibited optimal enhanced photocatalytic response, as well as good stability and reusability.
2. Experimental details
2.1 Preparation of catalysts
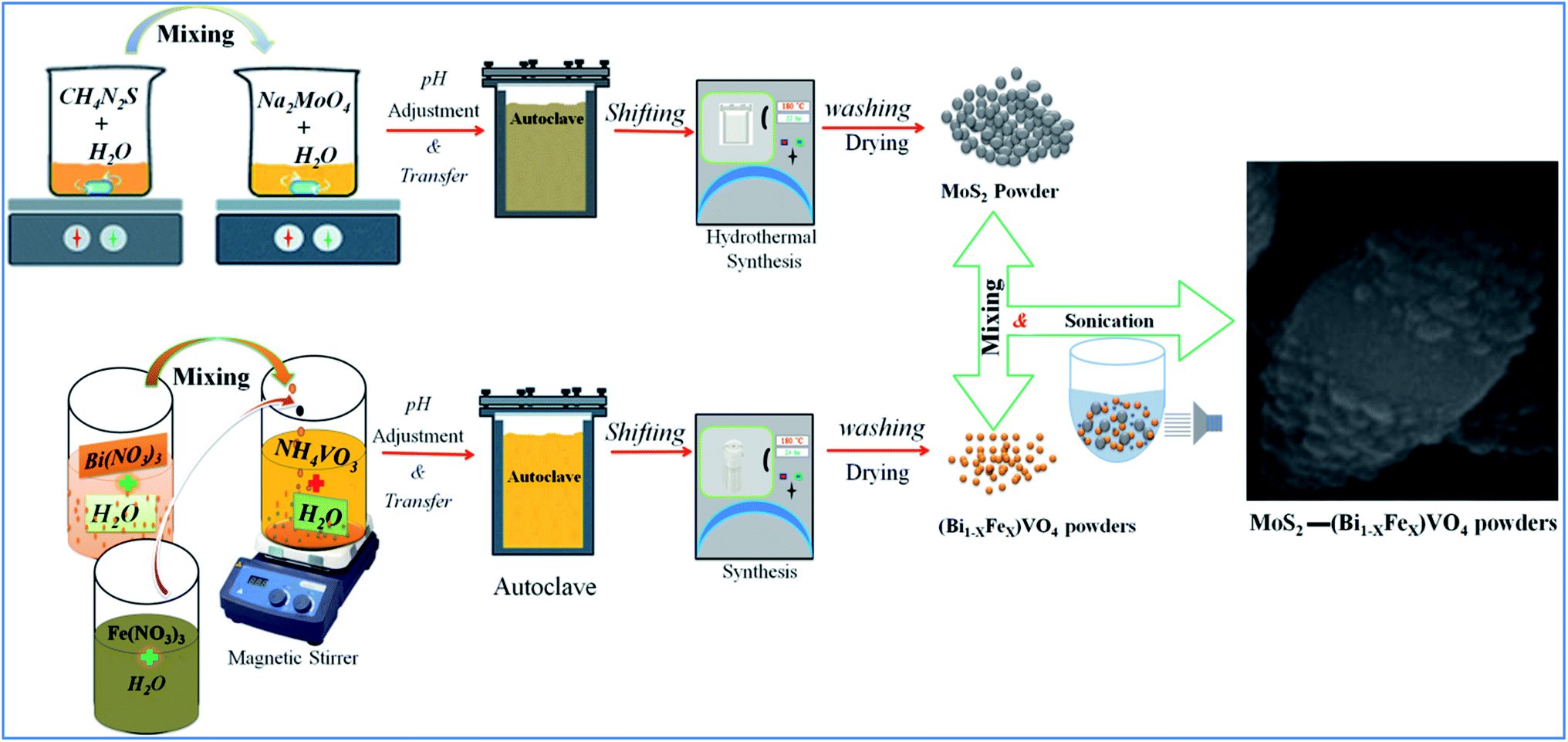
In a typical method, first, we prepared 1T-MoS2 by taking 6.24 g of thiourea (CH4N2S) and 4.84 g of sodium molybdate (NaMoO4) and making solutions of them in 30 ml deionized H2O separately named solution A and solution B. Then solution A was mixed with solution B dropwise under continuous stirring for 30 min. Ethanol (C2H5OH) was added to this mixture under a further 30 min of stirring. The as-formed mixture was transferred to a 200 ml Teflon-lined stainless steel autoclave and put into a muffle furnace for hydrothermal treatment at 180 °C for 22 h. The resultant products were centrifuged for 2 h at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to isolate the supernatant by centrifugation.49,52 The supernatant was 1T-MoS2 nanostructure. This synthesis of 1T-MoS2-QDs could be formulated as explained through the following equations:
000 rpm to isolate the supernatant by centrifugation.49,52 The supernatant was 1T-MoS2 nanostructure. This synthesis of 1T-MoS2-QDs could be formulated as explained through the following equations:| | |
CH4N2S + 2H2O → 2NH3 + H2S + CO2
| (1) |
| | |
2Na2MoO4·2H2O + 5H2S + C2H5OH → 1T-2MoS2 + Na2SO4 + NaOH + H2O + CO2 + 18H+
| (2) |
In the second phase, we prepared (Bi1−xFex)VO4 by the hydrothermal method as reported elsewhere.35,53 Then for the preparation of 1T-MoS2@(Bi1−xFex)VO4, an ultrasonic mixing method was used, for which 200 mg of (Bi1−xFex)VO4 and 0.2 mg ml−1 of MoS2 were mixed and sonicated for 2 h. The resulting solution was centrifuged and dehydrated at 80 °C to get the desired product: 2% of 1T-MoS2@(Bi1−xFex)VO4. The inclusive chemical reaction involved in the formation of the heterostructures can be written as:
| | |
Fe(NO3)3 + Bi(NO3)3 + H2O → 2HNO3 + BiONO3 + FeONO3
| (3) |
| | |
FeONO3 + BiONO3 + VO3− → [(1 − x)Bi3+ + XFe3+]VO4 + NO3−
| (4) |
| |
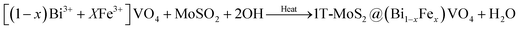 | (5) |
In the same way, a series of hybrid 1T-MoS2@(Bi1−xFex)VO4 catalysts containing different amounts of Fe (0.25%, 0.60%, and 0.80%) were achieved by regulating the 2.0% value of 1T-MoS2 solution. Optimum conditions for the construction of 1T-MoS2@(Bi1−xFex)VO4 were observed by the dosage variation effect of 1T-MoS2 on the photocatalytic activity of the photocatalysts. The 1T-MoS2@(Bi1−xFex)VO4 with different dosage of MoS2, i.e., 0.5 wt%, 1.0 wt%, 2.0 wt%, 3.0 wt% and 5.0 wt%, were each mixed with 200 mg of (Bi1−xFex)VO4. Among which the 1T-MoS2@(Bi1−xFex)VO4 with 2.0 wt% of 1T-MoS2 exhibited a remarkable photocatalytic response, which may be due to higher oxygen vacancies, large separation of charges, and the reduced rate of electron–hole recombination within the catalyst as compared to other concentration ratios. Hence, for cost-effectiveness and time saving and for brevity of description, we just describe 1T-MoS2@(Bi1−xFex)VO4 with 2.0% of 1T-MoS2 for further characterization. The proposed schematic diagram of preparation of 1T-MoS2 decorated on the surface of (Bi1−xFex)VO4 is shown in Fig. 1.
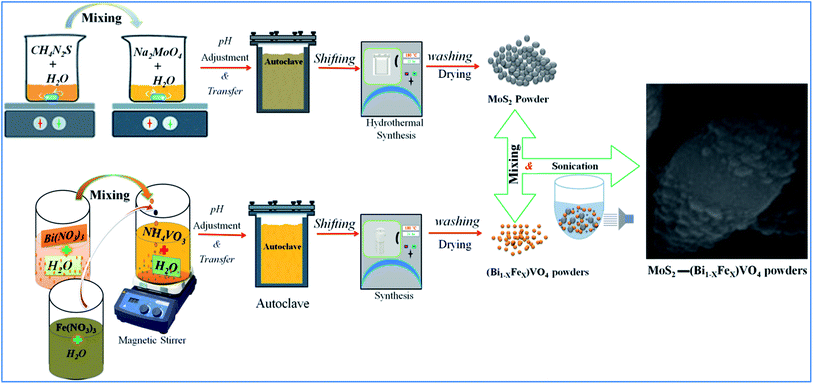 |
| | Fig. 1 Schematic representation of experimental setup for preparation of 1T-MoS2 decorated on the surface of (Bi1−xFex)VO4. | |
2.2 Characterization
X-ray diffraction (XRD) patterns were measured on a Bruker D8 diffractometer with monochromatic Cu Kα (λ = 0.15418 nm) radiation with a scanning rate of 2θ min−1 over the range of 20–60°. The morphology and topography of the materials were analyzed by high energy field emission scanning electron microscopy (SEM, JEOL-7001F), equipped with EDX for elemental analysis. The Brunauer–Emmett–Teller (BET) surface areas measurements were performed with an ASAP 2010 system. Optical properties were tested using a UV-visible spectrometer (PerkinElmer Lambda 35), equipped with an integrating sphere assembly, in a wavelength span of 300 to 800 nm at room temperature. For photoluminescence (PL), a Hitachi luminescence spectrometer (F-4500) was used with the 325 nm line of a He–Cd laser as the excitation source at ambient temperature.
2.3 Photocatalytic activity
The photocatalytic reaction response of the as-prepared samples was measured by degrading crystal violet (CV) dye solution under visible light irradiation. A xenon lamp of 300 W with a 420 nm cut-off filter was used as visible light source; the distance between the beaker and the lamp was kept at 25 cm.32,54 In detail, 10 mg of the as-prepared catalyst was added into 0.15 g L−1 (150 ml) dye solution. The suspension was stirred vigorously in the dark for 30 min to ensure adsorption–desorption stability and then put under the visible light source. At regular intervals of every 10 min, 3 ml of suspensions was taken out and the mixture was centrifuged at 10000 rpm for 2 min to eliminate the catalyst particles.55,56 The absorbance of the dye solution was quantified by a UV-visible (PerkinElmer λ-35) spectrophotometer. Nitrogen cooling system containing an ice bath was used to keep the temperature constant and to avoid thermal degradation or decomposition throughout the experimentation.34,35,57 The degradation efficiency of the dye (CV%) solutions was obtained by the following formula:32,34| |
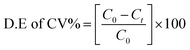 | (6) |
where C0 is the initial concentration of dye solution and C is the concentration of dye after every interval of 10 minutes.34
3. Results and discussion
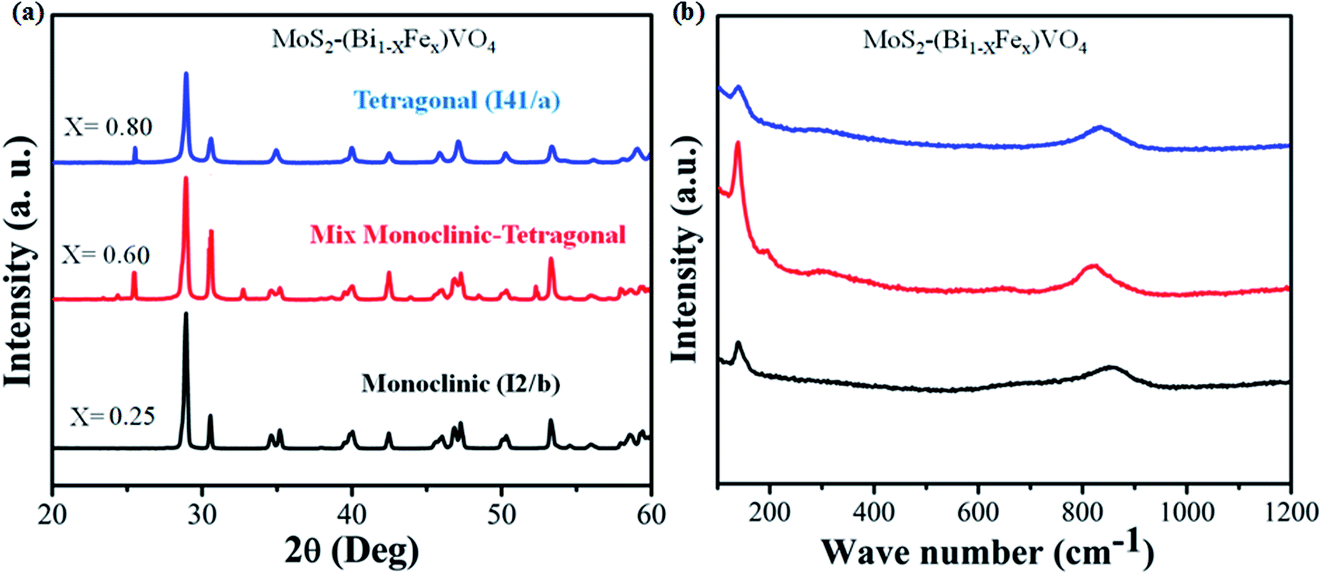
The phases of 1T-MoS2@(Bi1−xFex)VO4 with varying Fe concentrations were evaluated by XRD. It provides evidence regarding the purity and crystallinity of 1T-MoS2@(Bi1−xFex)VO4. Fig. 2(a) shows the room temperature XRD patterns of 1T-MoS2@(Bi1−xFex)VO4 corresponding to monoclinic (x = 0.25) phase indexed to JCPDS card no. 83-1699, mixed monoclinic-tetragonal (x = 0.60) phase indexed to JCPDS card no. 83-1699 and 14-0133, and tetragonal (x = 0.80) phase indexed to JCPDS card no. 75-2481. It is observed from XRD patterns that as the Fe concentration increased, monoclinic phase shifts to a mixture of the monoclinic and tetragonal phases and subsequently pure tetragonal BiVO4 phase, and a peak shift was also observed as the peaks belonging to tetragonal phase become intense while the peaks corresponding to monoclinic phases decrease.35,53,58 After 1T-MoS2 QD interspersed, all 1T-MoS2@(Bi1−xFex)VO4 samples show analogous diffraction patterns and exhibit no XRD peak shift as compared to pure (Bi1−xFex)VO4 [XRD patterns of pure BiVO4, FeVO4, 1T-MoS2 and (Bi1−xFex)VO4 are given in Fig. S1 (ESI†)]. This suggests that (Bi1−xFex)VO4 is the main structure of the prepared photocatalysts, and its structure does not change after 1T-MoS2 QD interspersing. It is clear from Fig. 2(a) that no diffraction peaks of 1T-MoS2 QDs were observed, which may be because of the low concentration (2.0 wt%) or because of the low crystallinity in the composites. The crystallite size of as-synthesized samples was evaluated from the (121) peak of XRD patterns by applying Scherrer's formula to further probe the development of the crystal structure.59 The crystallite size (D) values of the samples are given in Table 1. From Table 1, the crystallite size of Fe-doped BiVO4 increased, which implies that the presence of Fe3+ could influence the formation of crystallized particles of BiVO4. Furthermore, the strain of the nanomaterials was calculated by the Williamson–Hall method as it is expected that some Fe3+ ions may deposit onto grain boundaries owing to higher concentration and may cause lattice strain and hence reduce the lattice parameter.
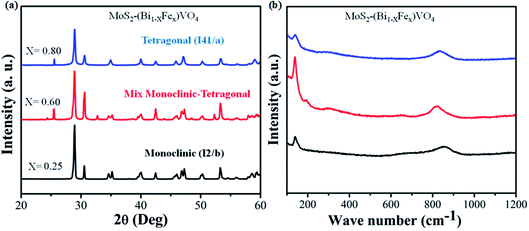 |
| | Fig. 2 (a) XRD patterns and (b) Raman spectra for the 1T-MoS2@(Bi1−xFex)VO4 nanoparticles synthesized at 180 °C. | |
Table 1 Physical and structural properties of the samples obtained from XRD analysis
| Sample |
Dcryst (nm) |
Lattice constant (Å) |
Cell volume (Å) |
Lattice strain, ε (%) |
| a |
b |
c |
| BiVO4 |
19.06 |
5.19 |
5.19 |
11.75 |
309.52 |
0.206 |
| FeVO4 |
23.55 |
6.71 |
8.06 |
9.25 |
462.46 |
0.416 |
| (Bi0.5Fe0.5)VO4 |
26.76 |
5.19 |
5.19 |
11.69 |
301.93 |
0.377 |
| 1T-MoS2@(Bi0.75Fe0.25)VO4 |
30.62 |
5.19 |
5.19 |
11.75 |
309.74 |
0.275 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
20.91 |
5.19 |
5.19 |
11.70 |
309.20 |
0.315 |
| 1T-MoS2@(Bi0.20Fe0.80)VO4 |
34.30 |
5.14 |
5.14 |
11.72 |
310.52 |
0.617 |
Raman spectroscopy is a versatile characterization technique generally used for monitoring the local molecular bonding and structures of all types of catalytic materials.13,34 Therefore, Raman analysis was conducted for structural information and to find the crystal local structure and electronic dimensions of materials. In Fig. 2(b), seven vibrational modes are located at around 198, 210, 326, 369, 639, 708 and 835 cm−1 that belong to BiVO4, while the vibrational modes at 139, 313, 359, 644, 823 and 893 cm−1 correspond to FeVO4 in the samples indicating the replacement of Bi3+ with Fe3+.35 It was observed that the number of vibrational modes decreased gradually to increase crystal symmetry and resulted peak shift. Contrary to this, when Fe concentration is increased, some new peaks emerge and become broader dominating some BiVO4 peaks. No vibrational peak of 1T-MoS2 QDs (2.0 wt%) was detected in the spectra of 1T-MoS2@(Bi1−xFex)VO4, due to the low amount of MoS2 in 1T-MoS2@(Bi1−xFex)VO4 heterostructures, which also confirmed our previous result.
The morphologies of prepared powder samples were examined via high-resolution SEM. This gives information regarding the structure of 1T-MoS2@(Bi1−xFex)VO4 possessing different compositions. The SEM images show that nearly all the (Bi1−xFex)VO4 samples exhibit a ball-shaped spherical configuration, as depicted in Fig. 3. The particle edge is more or less ball-shaped owing to some clusters and agglomeration of the particles. From SEM images, the particle size of the samples is found around 20–60 nm range. The SEM images indicate that BiVO4 nanoparticles have spherical symmetry and uniformity. Yet, an increase in Fe concentration causes the structural transformation of the particles slightly from spherical to non-uniform hexagonal and spherical. At higher Fe concentration, fewer globular and more corners, randomized particles and agglomerations were observed as exhibited in Fig. 3. So, the addition of Fe dopant gradually affects the structure of (Bi1−xFex)VO4, and observable transformation occurs from homogenous spherical morphology to irregular spheres and agglomerations of nanostructures. Moreover, oxygen adsorption also influences the morphology. Subsequently, the surface decoration of (Bi1−xFex)VO4 with 1T-MoS2 QDs was accomplished by sonication of (Bi1−xFex)VO4 in an aqueous solution of 1T-MoS2 QDs for 2 h. During the sonication process, 1T-MoS2 QDs are adsorbed on the (Bi1−xFex)VO4 surface, leading to the fabrication of 1T-MoS2 QDs on (Bi1−xFex)VO4. The obtained 1T-MoS2@(Bi1−xFex)VO4 composites, however, retained the globular spherical morphology as indicated in Fig. 3. EDX analysis (Fig. 3(g)) of 1T-MoS2@(Bi0.40Fe0.60)VO4 heterostructures verifies the homogenous distribution and presence of 1T-MoS2 QDs on the (Bi1−xFex)VO4 surface confirming that the heterostructures consist of only Mo, S, Bi, Fe, V and O elements, while Cu presence is due to the copper holding grid, with no other impurities found within the detection limit as shown in Fig. 3(g).
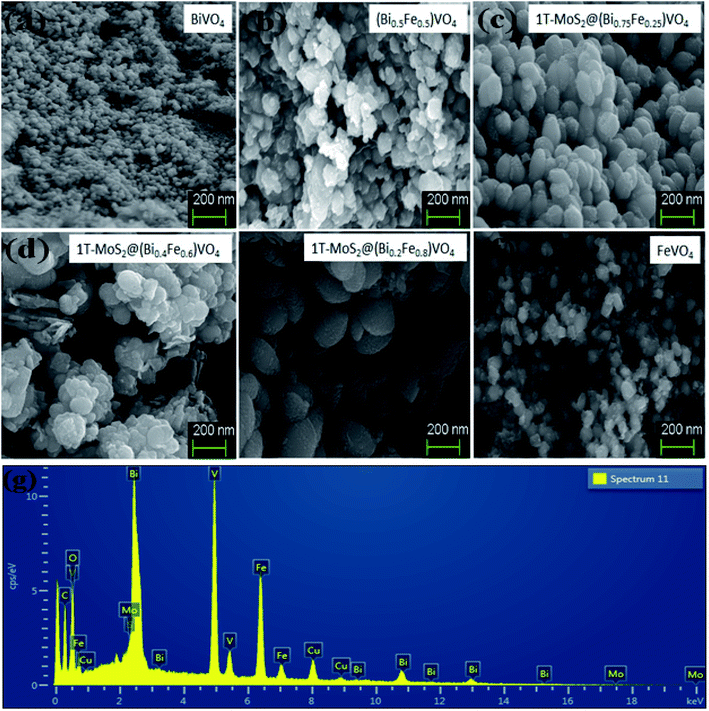 |
| | Fig. 3 SEM analysis for (a) pure BiVO4, (b) (Bi0.5Fe0.5)VO4, (c) 1T-MoS2@(Bi0.75Fe0.25)VO4, (d) 1T-MoS2@(Bi0.20Fe0.80)VO4, (e) 1T-MoS2@(Bi0.40Fe0.60)VO4, and (f) pure FeVO4. (g) EDX analysis of 1T-MoS2@(Bi0.40Fe0.60)VO4 heterostructures. | |
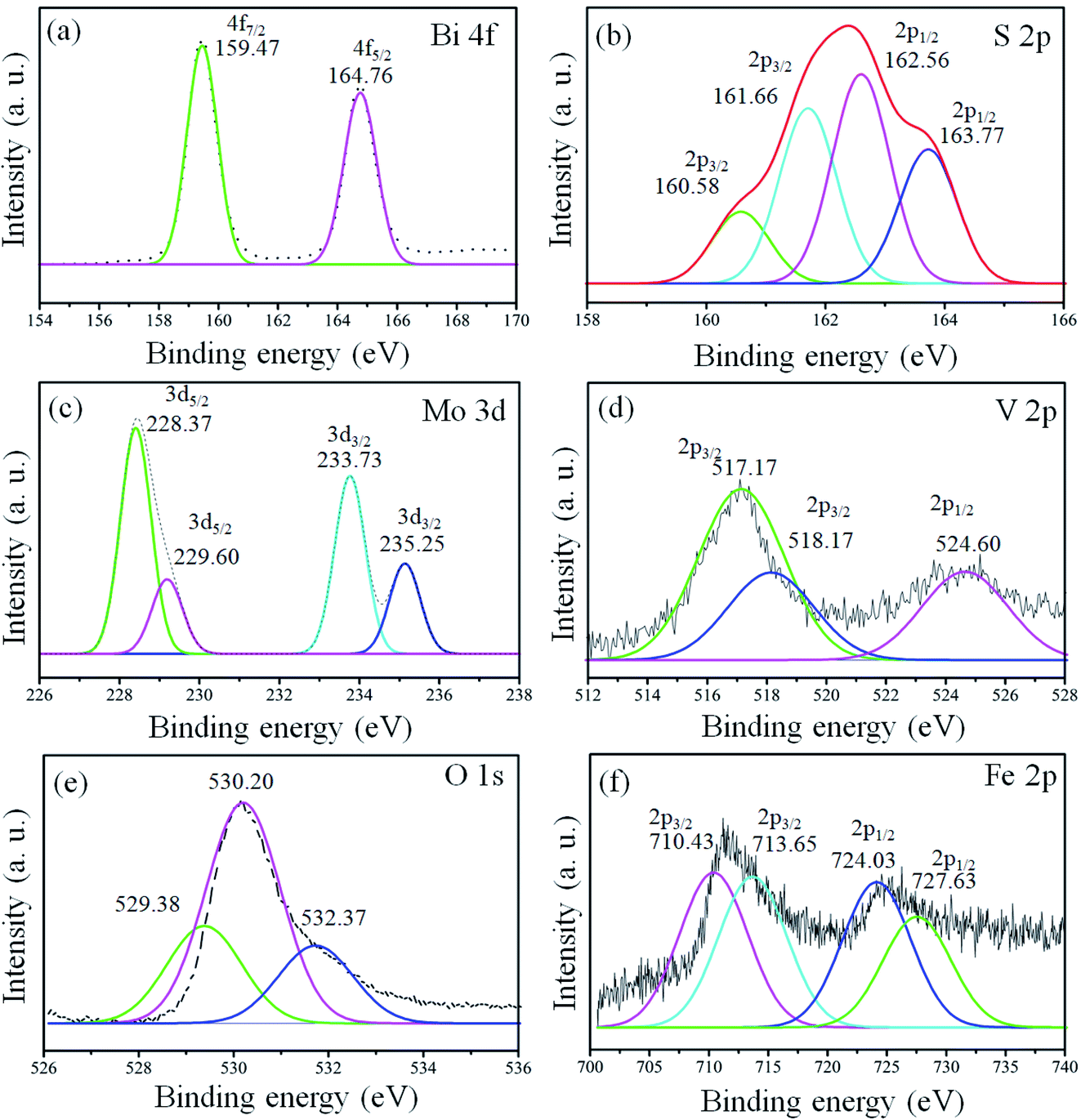
X-ray photoelectron spectroscopy (XPS) analysis was conducted for the confirmation of the elemental, chemical composition and surface bond analysis.14,26 XPS survey scan (Fig. S2, ESI†) was performed to confirm the presence of essential components present in the samples. The survey spectrum indicates that binding energy peaks of essential elements (Bi, S, Mo, O, V, and Fe) in the 1T-MoS2@(Bi1−xFex)VO4 samples were observed as shown in Fig. S2, ESI.† We used O 1s (530.3 eV) for referencing the binding energy values rather than to C 1s (284.6 eV). To further analyze the 1T-MoS2@(Bi1−xFex)VO4 composites, the high-resolution spectra for Bi, S, Mo, O, V, and Fe are depicted in Fig. 4. The XPS peaks of Bi doublets (4f7/2 and 4f5/2) were found at 159.38 eV and 164.83 eV as indicated in Fig. 4(a). Fig. 4(b) shows the doublets of S 2p (S 2p3/2 and S 2p1/2), in which the main two peaks at 161.66 eV and 162.56 eV represent the 1T-phase of MoS2, and the two small peaks at 160.58 eV and 163.77 eV correspond to the 2H-phase of MoS2. However, the concentration of the 1T-phase is higher and is found to be 80% which indicates the predominance of the 1T-phase over the 2H-phase. Mo 3d doublets (Mo 3d5/2 and Mo 3d5/2) appeared at 229.1 eV and 232.5 eV. Mo 3d peaks were more de-convoluted to obtain further information about the existence of valence state of Mo ions. The peak at 235.25 eV is developed due to oxidation of 1T-MoS2@(Bi0.40Fe0.60)VO4 by exposure to air and attributed to Mo6+. V 2p doublets (V 2p3/2 and V 2p1/2) appeared at approximately 517.23 and 524.60 eV. In the case of V, the value of binding energy of the V 2p3/2 core level is responsive to the oxidation state of the V cation. Earlier reports demonstrated curve fitting of the V 2p3/2 core peak, so we also fitted the V 2p3/2 core peak in our samples.14 The V 2p3/2 core peak was de-convoluted with the fitting of two peaks arising at binding energy values of 517.17 and 518.17 eV. The peak fitting suggests the valence state of V5+ is in bulk with the presence of V4+ ions in the 1T-MoS2@(Bi0.40Fe0.60)VO4 heterojunction composite. Fig. 4(e) depicts the O 1s spectrum also showing asymmetrical behavior and can be fitted with three Gaussians peaks for 1T-MoS2@(Bi0.40Fe0.60)VO4 heterojunction composite samples. The de-convoluted peaks appeared at around 529.38, 530.20 and 532.37 eV. The peak at 529.38 eV is due to lattice constant of V–O, Fe–O or Bi–O bonding in (Bi1−xFex)VO4, as the binding energies of V–O and Bi–O are approximately the same. The peak at 530.20 eV is due to OH group while that at 532.37 eV is attributed to molecular water adsorbed on the surface.13 The XPS spectrum of Fe 2p core peaks is shown in Fig. 4(f). Fe 2p doublet (Fe 2p3/2 and 2p1/2) evidenced a binding energy transformation of 13.6 eV which established spin-orbit coupling of Fe with host lattices. Fe 2p peaks were more de-convoluted to get further accurate information on the presence of the valence state of the Fe ions. The de-convolution of Fe 2p3/2 reveals two peaks at 710.43 and 713.65 eV corresponding to Fe3+ and Fe2+ valence states.13,26 It is observed that there is a slight shift in these peaks towards higher binding energy during the Fe doping in BiVO4 and formation of Fe–O–Bi bond, indicating that Fe and 1T-MoS2 co-doped further increase the oxygen vacancies and cause higher enhanced photocatalytic activity, as discussed below. These results confirm the existence of Fe in BiVO4, successful constriction of (Bi1−xFex)VO4 and decoration of 1T-MoS2 on (Bi1−xFex)VO4 sample surfaces.
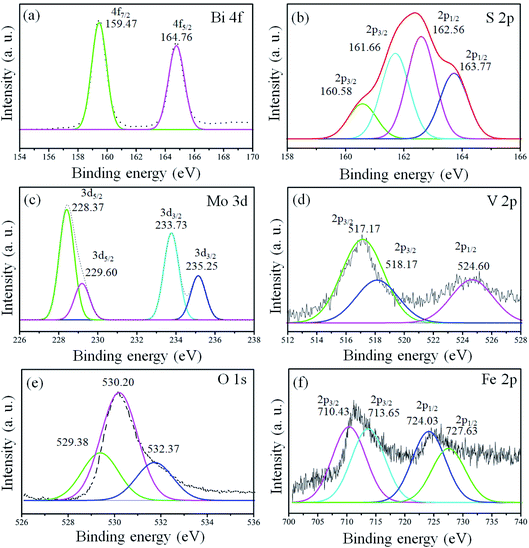 |
| | Fig. 4 High-resolution peak-fitting XPS spectra of (a) Bi 4f, (b) S 2p, (c) Mo 3d, (d) V 2p, (e) O 1s and (f) Fe 2p of 1T-MoS2@(Bi0.40Fe0.60)VO4 heterostructures. | |
3.1 Photocatalytic activity
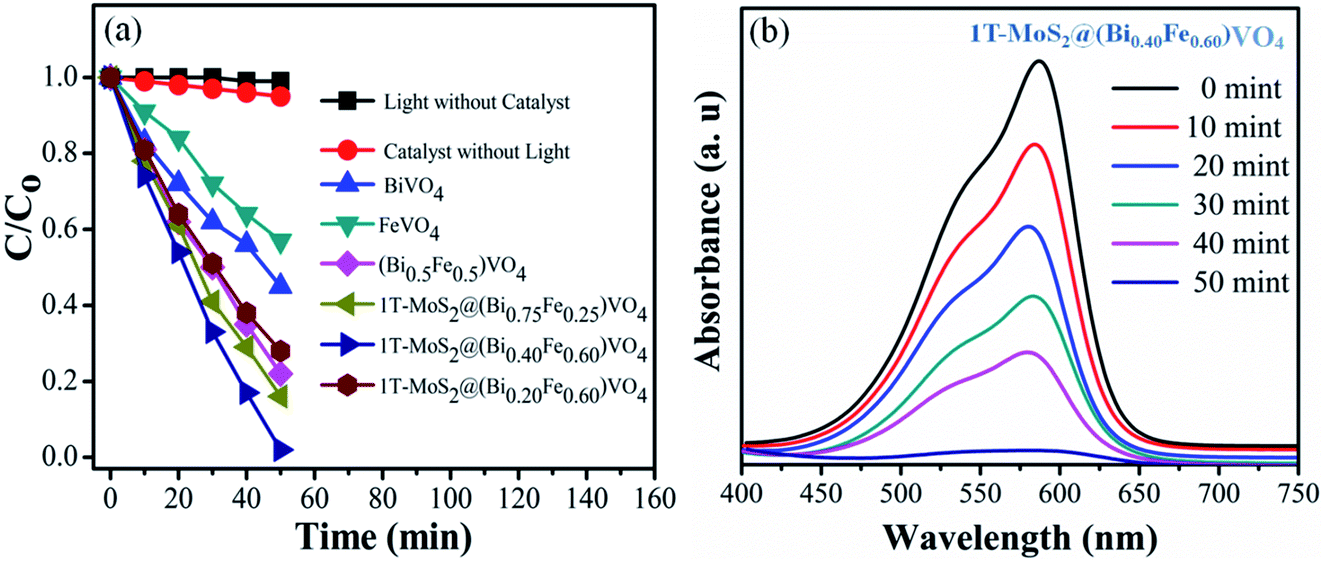
The photocatalytic conversion of CV over various samples under visible light irradiation (λ = 420 nm) is shown in Fig. 5. For reference, the CV dye solution degradation/decomposition without photocatalyst (photolysis) and with photocatalyst but without light was performed. The absorptions of CV dye solution reduce gradually in the presence of both photocatalyst and visible light irradiation, while no or insignificant photocatalytic response was observed either without photocatalysts or without light irradiation, i.e., 1% and 5% respectively in 50 min (Fig. 5(a)). From these results, it is obvious that the photocatalytic activity for CV over 1T-MoS2@(Bi1−xFex)VO4 is light-dependent.35,57 The photocatalytic activity is primarily related to the production of superoxide anions (O2−) and hydroxyl radicals (˙OH).13,27 Therefore, in the presence of 1T-MoS2@(Bi1−xFex)VO4, light photons (hν) of energy equal to or greater than the bandgap generate electron and hole pairs which transfer to the 1T-MoS2@(Bi1−xFex)VO4 surface, where they oxidize the water and CV molecules and cause advanced photocatalytic efficiency under visible light irradiation. From Fig. 5(a) the photodegradation of CV with pure BiVO4 is 55%, that for FeVO4 is 43%, (Bi0.5Fe0.5)VO4 is 78%, 1T-MoS2@(Bi0.75Fe0.25)VO4 is 86%, and 1T-MoS2@(Bi0.20Fe0.80)VO4 is 76%, while for 1T-MoS2@(Bi0.40Fe0.60)VO4 it reaches up to 99.98% in 50 min of irradiation time under similar conditions (Table 2). To get more rigorous results, a comparison between (Bi0.40Fe0.60)VO4 and 1T-MoS2@(Bi0.40Fe0.60)VO4 was conducted (Fig. S3, ESI†), where it can be seen that (Bi0.40Fe0.60)VO4 gives 89.80% degradation efficiency whereas 1T-MoS2@(Bi0.40Fe0.60)VO4 exhibited 99.98% degradation in 50 minutes. During the photoirradiation experiment, the discoloring of the solution suggests the decomposition of CV dye fragments. From absorption peaks a red shift was noticed, which may be due to benzene ring breakup within the dye molecules.13,60 From experimental outcomes and the above discussion, it is suggested that in the 1T-MoS2@(Bi0.40Fe0.60)VO4 heterostructures, the separation of photogenerated charge carriers is well controlled owing to 1T-MoS2 helping the transport of electrons (electron capture) compared with pure BiVO4, (Bi1−xFex)VO4, FeVO4 and for other 1T-MoS2@(Bi1−xFex)VO4 photocatalysts, thus improving the electron–hole pair separation lifetime involving an enhanced photoconversion efficiency.
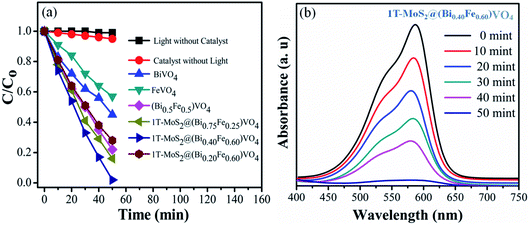 |
| | Fig. 5 (a) Degradation of CV dye solution under different conditions; (b) UV-visible absorption spectra of CV dye solution during photocatalytic degradation over 1T-MoS2@(Bi0.40Fe0.60)VO4 heterojunction photocatalysts at different time intervals. | |
Table 2 Effect of Fe concentration variation and 1T-MoS2 loading on CV dye degradation using 1T-MoS2@(Bi1−xFex)VO4 photocatalysts in 50 min
| Ratio (Bi1−xFex)VO4 |
Degradation efficiency (%) |
| BiVO4 |
55 |
| FeVO4 |
43 |
| (Bi0.5Fe0.5)VO4 |
78 |
| 1T-MoS2@(Bi0.75Fe0.25)VO4 |
86 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
99.98 |
| 1T-MoS2@(Bi0.20Fe0.80)VO4 |
76 |
A kinetics study for degradation of CV under visible light irradiation was also conducted. The Langmuir–Hinshelwood model is generally considered a suitable model to examine the reaction kinetics of heterogeneous catalysts.61,62 The applied mathematical expression is given as in eqn (7):61
| |
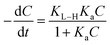 | (7) |
Here,
Ka,
C, and
KL–H correspond to the absorption coefficient, concentration and reaction rate constant. When the initial pollutant concentration is low as in this study, Langmuir's formula is moderated into the pseudo-first-order model and generally used for photocatalytic degradation activity. A pseudo-first-order kinetic model is used to fit the degradation data using the following equation:
32,61| |
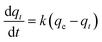 | (8) |
where
k (min
−1) is the photocatalytic reaction rate constant,
qe is the adsorbed dye at the equilibrium of CV after the adsorption–desorption equilibrium before illumination, and
qt is the adsorbed CV dye at different illumination times (min) (
Fig. 5(a)). The degradation rate constants
k for CV over BiVO
4, FeVO
4, (Bi
1−xFe
x)VO
4, and 1T-MoS
2@(Bi
1−xFe
x)VO
4 photocatalysts through pseudo-first-order equation were evaluated from the nonlinear regression of
t versus qt using the integration of the equation and are given in
Table 3. It was observed that CV dye solution over 1T-MoS
2@(Bi
0.40Fe
0.60)VO
4 presented a superior photodegradation efficiency as shown in
Fig. 5(a).
Table 3 Kinetics data for the adsorption of CV on catalysts obtained using kinetics models
| Sample |
C/C0 exp. (mg g−1) |
Pseudo-first order |
| K (min−1) visible light |
R2 |
| BiVO4 |
0.454 |
9 × 10−3 |
0.9848 |
| FeVO4 |
0.571 |
1.14 × 10−2 |
0.9975 |
| (Bi0.5Fe0.5)VO4 |
0.223 |
4.46 × 10−3 |
0.9907 |
| 1T-MoS2@(Bi0.75Fe0.25)VO4 |
0.163 |
3.26 × 10−3 |
0.9823 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
0.021 |
4 × 10−4 |
0.9863 |
| 1T-MoS2@(Bi0.20Fe0.80)VO4 |
0.285 |
5.7 × 10−3 |
0.9825 |
The optical band gap is calculated by the Tauc plot method as presented in Table 4.63 The reduction in optical bandgap results in an increase in the mobility of charge carriers, refractive index and electrical conductivity.64,65 The results obtained are in accordance with those of previous work;13,24,32,35 therefore, it is suggested that the prepared semiconductor material will also be very helpful for electronic device applications. The overall results are presented in Table 4.
Table 4 Determination of optical band gap, mobility of charge carriers, refractive index and conductivity
| Sample |
Optical band gap (eV) |
Mobility of charge carriers (cm2 V−1 s−1) |
Refractive index |
Conductivity (S cm−1) |
| BiVO4 |
2.41 |
3.18 × 103 |
2.54 |
4.19 × 104 |
| (Bi0.5Fe0.5)VO4 |
2.38 |
3.24 × 103 |
2.55 |
4.30 × 104 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
1.98 |
4.27 × 103 |
2.71 |
6.19 × 104 |
3.2 Surface chemistry
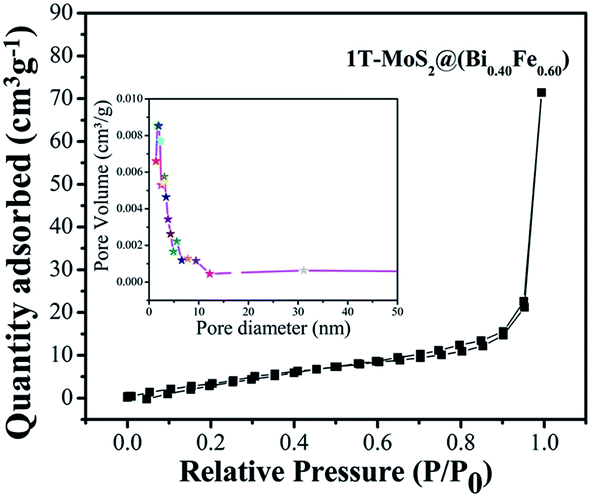
The photocatalytic activity is correlated to the adsorption–desorption-releasing process. The appropriate adsorption of pollutant molecules on the surface of photocatalysts is the major requirement for an excellent photoinduced response. In addition, in the function of photocatalytic activity, the rate of reaction is proportional to the number of photons (hν) adsorbed on the surface of a photocatalyst.12,15,29 Adsorptions of nonreactive gas at the atomic level have a significant role in determining the surface area, as well as surface roughness and pore interior. BET specific surface area is evaluated by N2 adsorption–degradation curve.29 The nitrogen adsorption–desorption curve for surface area measurement of 1T-MoS2@(Bi0.40Fe0.60)VO4 is shown in Fig. 6. For pore size, the Barrett–Joyner–Halenda (BJH) method was applied using desorption isotherms.29 1T-MoS2@(Bi0.40Fe0.60)VO4 exhibits sufficient large surface area of 40 m2 g−1 and calculated pore volume and average pore diameter are 0.122 cc g−1 and 1.685 nm respectively as shown in the inset of Fig. 6. As is well known, higher surface area means a high ability to adsorb large quantity of dye pollutants from solution; moreover, pore size and pore volume also have a major effect on the photocatalytic activity. Thus, the surface area, pore volume, and pore size of the as-synthesized sample nanopowders were measured as shown in Table 5. It is observed that with the increase of Fe concentration the surface area increases as compared to pure BiVO4 and the optimum increment is at x = 60 as depicted in Table 5. This may be owing to Fe3+ having small ionic radius of 0.78 Å; secondly, the surface area may be large owing to increase in adsorption in the presence of Fe3+; thirdly, interspersing of 1T-MoS2 quantum dots inside the (Bi0.40Fe0.60)VO4 nanoparticles allows an increase in dye adsorption on the catalyst surface; and lastly, it may be due to the arrangement of metal ions.15,29 The adsorption curve of 1T-MoS2@(Bi0.40Fe0.60)VO4 is a type IV curve indicating the presence of mesopores/nanopores, and a type H3 hysteresis loop. The enhanced surface area of 1T-MoS2@(Bi0.40Fe0.60)VO4 reveals the existence of a multichannel structure inside the composite which allows facile and easy mass transport, light-capturing due to multiple light reactions and gas diffusion within the pores of the material. The presence of mesopores and nanopores within the pore structures helped to adsorb extra dye fragments on the photocatalyst surface, hence the combined influence of multiple light reflections and adsorption consequently resulting in enhancing the photocatalytic activity and increasing the dye degradation.12,14,15,26
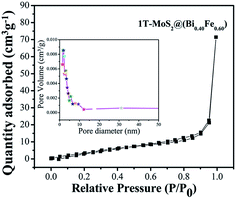 |
| | Fig. 6 Nitrogen gas adsorption–desorption isotherm distribution curve for 1T-MoS2@(Bi0.40Fe0.60)VO4 at 77 K. Inset: pore volume vs. pore diameter curve from BJH method. | |
Table 5 Summary of the physical properties obtained, BET surface area, pore volume, and pore size of sample nanopowders
| Sample |
BET surface area (m2 g−1) |
Vp (cc g−1) |
Dp (nm) |
| BiVO4 |
14 |
0.025 |
2.181 |
| FeVO4 |
26 |
0.052 |
2.183 |
| (Bi0.5Fe0.5)VO4 |
26 |
0.117 |
1.681 |
| 1T-MoS2@(Bi0.75Fe0.25)VO4 |
13 |
0.059 |
1.931 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
40 |
0.122 |
1.685 |
| 1T-MoS2@(Bi0.20Fe0.80)VO4 |
21 |
0.107 |
2.192 |
3.3 Photoluminescence and electrochemical impedance spectroscopy analysis study
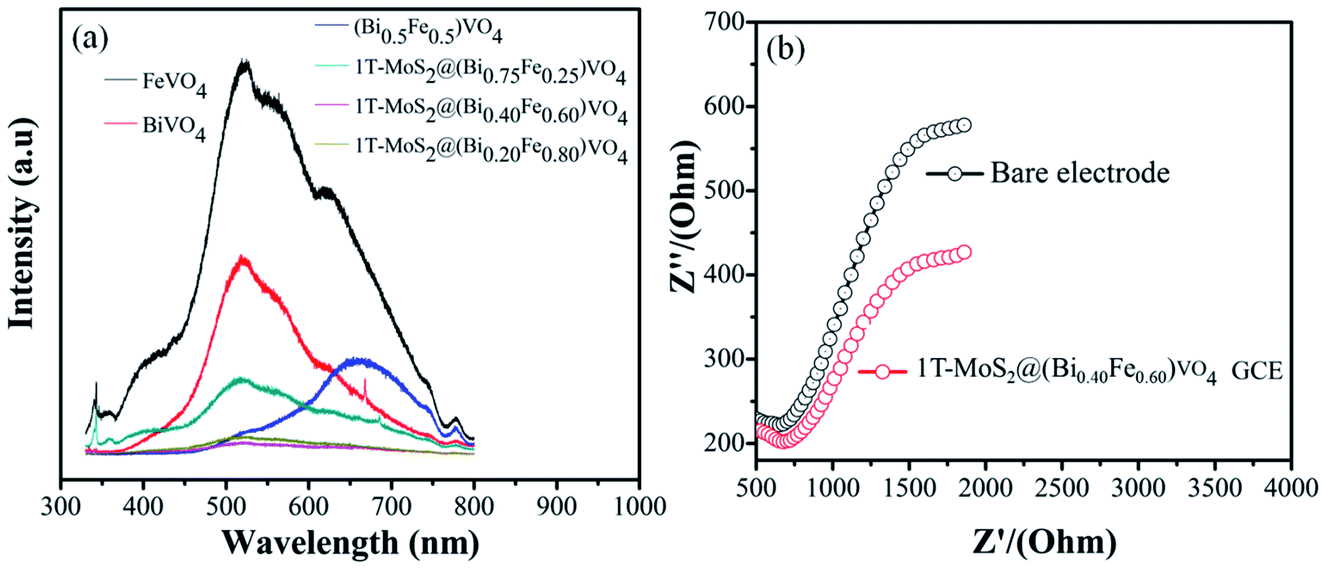
Several studies show that PL emission spectra can be stimulated through the recombination of photoexcited electrons and holes.66 A lower PL emission peak intensity suggests less recombination of photogenerated charge carriers. Consequently, the electron–hole transfer and recombination operations in the photoaided degradation experimentation could be tested by the PL technique.15,35,57 Fig. 7(a) exhibits the PL spectra of pure BiVO4, FeVO4, (Bi1−xFex)VO4 and different 1T-MoS2@(Bi1−xFex)VO4 composites in the range of 330–800 nm for an excitation source of 325 nm. The peaks increase owing to the recombination of photoexcited electrons and holes. It is noticed that the emission intensities of FeVO4 and BiVO4 are higher. However, after the coupling of (Bi1−xFex)VO4 with 1T-MoS2 quantum dots, the emission intensity is reduced, suggesting that the charge separation rate in 1T-MoS2@(Bi1−xFex)VO4 composites is more efficient as compared to BiVO4, FeVO4, and (Bi1−xFex)VO4. It is observed that the 1T-MoS2@(Bi0.40Fe0.60)VO4 composite exhibits the lowest PL emission intensity, indicating a large separation efficiency of photogenerated charge carriers.34 To further understand the charge transfer in 1T-MoS2@(Bi1−xFex)VO4 photocatalysts, the electrochemical impedance spectroscopy (EIS) technique was used. The measurement was also carried out for a bare glassy carbon electrode (GCE) and 1T-MoS2@(Bi0.40Fe0.60)VO4 composite. As shown in Fig. 7(b), it is experimentally evident that the 1T-MoS2@(Bi0.40Fe0.60)VO4 composite has a small-scale semicircle radius as compared to bare GCE. It is well recognized that the radius of curvature is an indicator of charge transport impedance, and a smaller semicircle radius signifies greater charge transportation efficiency.26,34,35
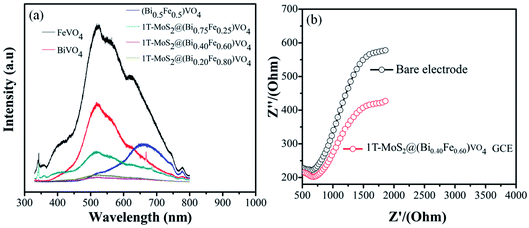 |
| | Fig. 7 (a) PL spectra (excited at 325 nm) of composites at different concentrations and (b) EIS Nyquist plots of the bare and as-modified 1T-MoS2@(Bi0.40Fe0.60)VO4 nanocomposite GCE samples. | |
3.4 Radical scavenger and electron paramagnetic resonance study for active species
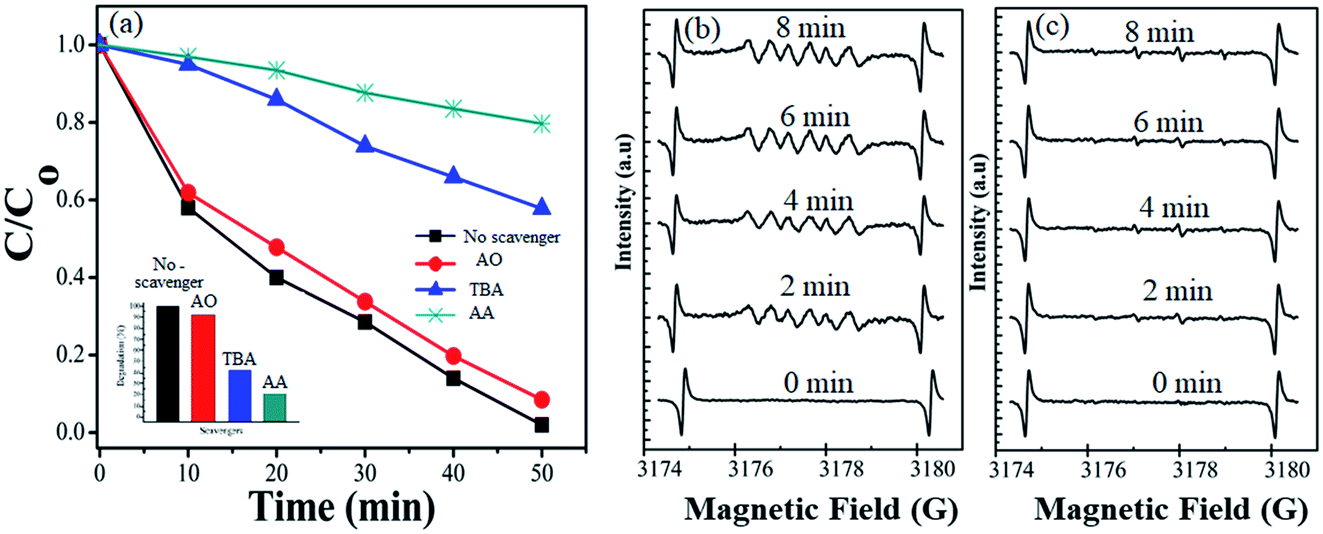
During photocatalytic degradation, different active species are generated, including hydroxyl radical (˙HO), singlet oxygen (1O2), superoxide radical (˙O2−) and photogenerated hole (h+).67–69 Radical scavenger analyses were performed to check out the agents involved in the photodegradation of the dye. The results of effective species detection are indicated in Fig. 8(a). The result showed that CV solution degradation over 1T-MoS2@(Bi0.40Fe0.60)VO4 was highly depressed when ascorbic acid (˙O2− radical) is added into the system. It is seen that in the presence of t-butanol (˙OH radical) and diammonium oxalate monohydrate (h+ scavenger), the concentration slightly decreases, which reveals that ˙OH and h+ had small roles as main reactive species in the photocatalytic degradation. Thus ˙O2− plays a major role as reactive species in the degradation of CV dye solution. To further strengthen our results, electron paramagnetic resonance (EPR) analysis was performed which also indicates that once an electron entered the conduction band of (Bi1−xFex)VO4, it brings oxygen, which makes centers for degradation of CV dye. Hydroxyl radicals (˙OH) were also generated when ˙O2− interacted with holes (h+) and H+ ions in H2O.14,34 No EPR signals were observed when the reaction was performed in the dark. DMPO-˙O2− and DMPO OH˙ adducts were observed when the experiment was executed under visible light illumination. The signal intensity increases progressively while prolonging the reaction time as shown in Fig. 8(b), while no significant enhancement in DMPO OH˙ adducts was noticed when the 1T-MoS2@(Bi0.40Fe0.60)VO4 heterojunction nanocomposite solution was irradiated under visible light as shown in Fig. 8(c). These results again confirm that ˙O2− radicals were the major active species during CV photocatalytic degradation over 1T-MoS2@(Bi0.40Fe0.60)VO4 in this study.
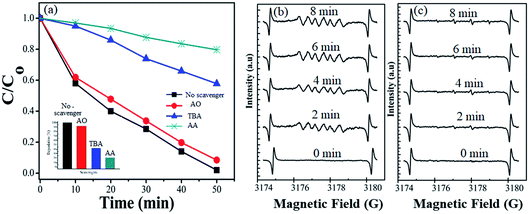 |
| | Fig. 8 (a) Effects of different radical scavengers on CV degradation over 1T-MoS2@(Bi0.40Fe0.60)VO4 under visible light irradiation. (b) DMPO spin trapping EPR spectra for DMPO-˙O2− and (c) DMPO-˙OH− under visible light over 1T-MoS2@(Bi0.40Fe0.60)VO4 photocatalyst. | |
3.5 Photocatalytic mechanism
The comprehensive mechanism of 1T-MoS2@(Bi0.40Fe0.60)VO4 photocatalyzed reactions is explored to understand the procedure of photocatalysis. The complete mechanism for photocatalysis over the 1T-MoS2@(Bi0.40Fe0.60)VO4 catalyst was proposed based on prior investigations as illustrated in Fig. 9.
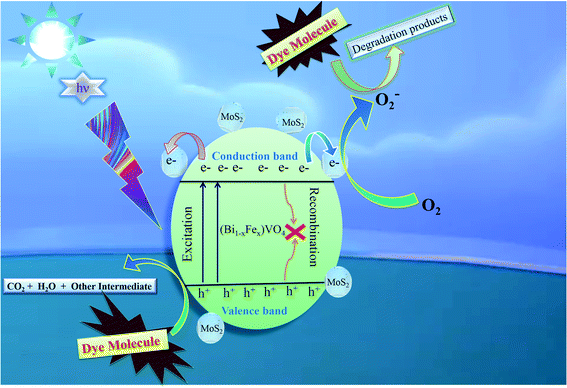 |
| | Fig. 9 Proposed basic mechanism of photocatalysis of 1T-MoS2@(Bi1−xFex)VO4 heterostructures. | |
The overall process includes first the production of photoexcited electrons (e−) from the valence band (VB) into the conduction band (CB) of (Bi0.40Fe0.60)VO4 when light photons fall on it. Secondly, holes (h+) are generated in the VB owing to the migration of electrons from VB to CB. In the third step the photoexcited electrons of the CB are captured or trapped to the 1T-MoS2 surface from where the electrons rapidly take part in the photocatalytic activity. The close contact level between tiny 1T-MoS2 particles and (Bi0.40Fe0.60)VO4 provides more opportunity to transport photogenerated electrons from the CB of (Bi0.40Fe0.60)VO4 to 1T-MoS2 surface from where they react with surface adsorbed oxygen (O2) molecules and convert them into superoxide anion radicals (˙O2−). In the fourth and final step, the holes in VB are transmitted to the surface to oxidize the water and hydroxyl ions into hydroxyl radical (˙OH) species.70,71 In comparison with the pure BiVO4, FeVO4 and (Bi1−xFex)VO4 samples, the fabrication of 1T-MoS2 doped (Bi1−xFex)VO4 catalyst could boost the efficient separation and transport of photoproduced charges for degradation of CV dye. Consequently, the photocatalytic response of BiVO4 was extensively enhanced.15 Transfer of the electron–hole pairs within and on the surface of the 1T-MoS2 doped (Bi1−xFex)VO4 photocatalyst and photocatalyzed reactions may be expressed by the following equations:
| |
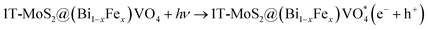 | (9) |
The electrons from the 1T-MoS2 surface can reduce O2 to superoxide anions:
These radicals interact with the pollutant CV dye molecules and decompose them into harmless fragments:
| | |
OH˙ + CV → CO2 + H2O
| (13) |
| | |
˙O2− + CV → CO2 + H2O
| (14) |
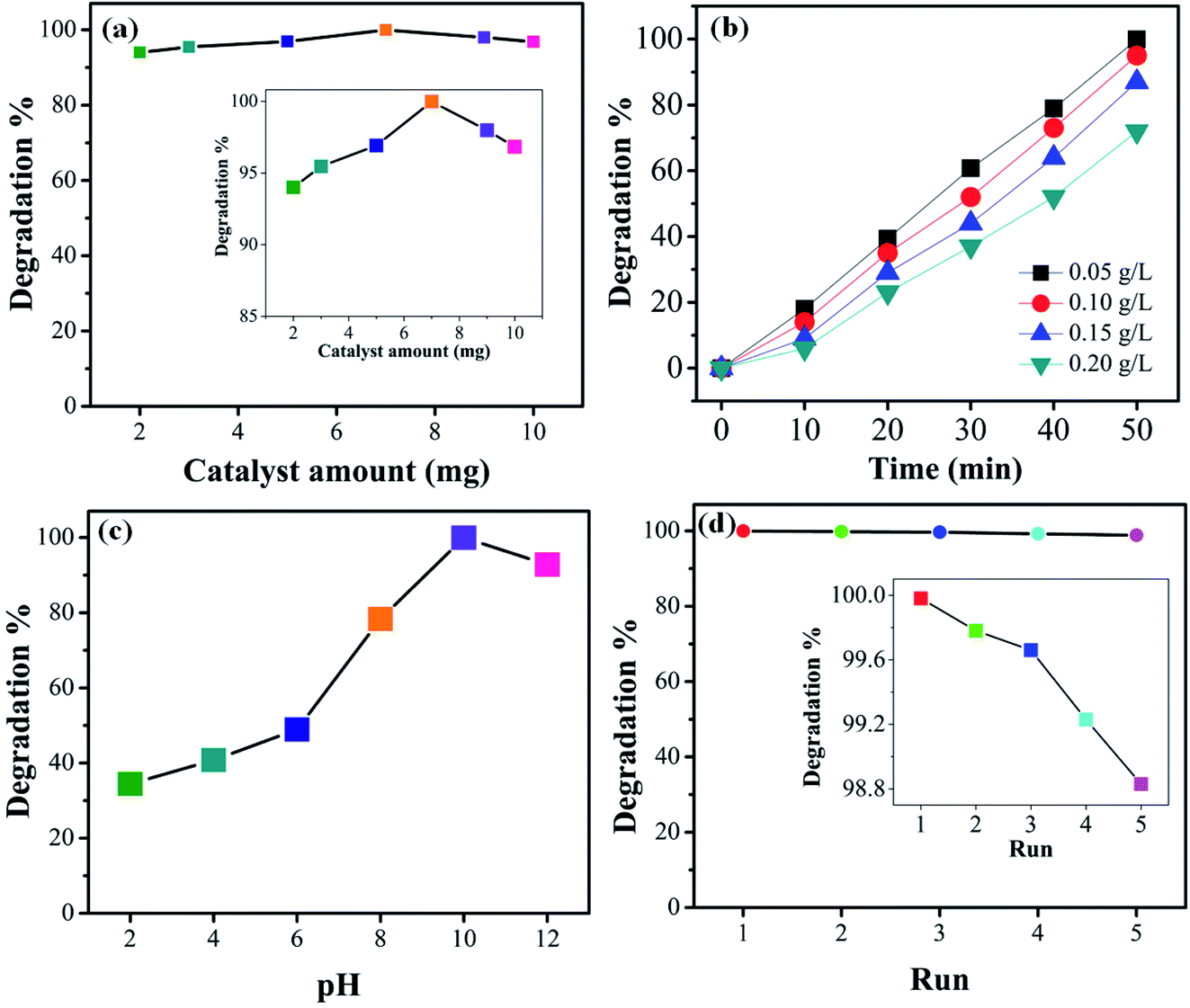
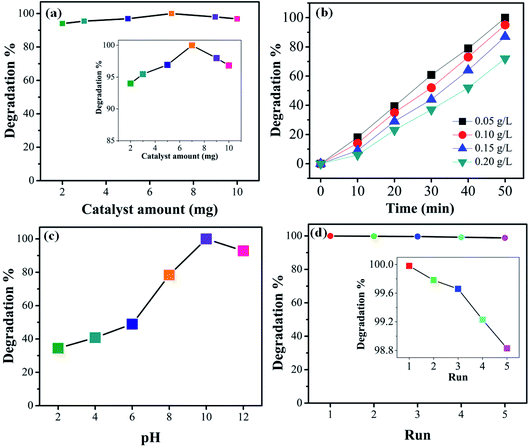
3.6 Effect of catalyst dosage and initial dye concentration
For practical and economic purposes the effect of different catalyst dosages was evaluated by varying the quantity in the range 2–10 mg in the presence of visible light. From experimental results it was noted that the degradation is almost independent of the catalyst concentration. The optimal catalyst amount (Fig. 10(a)) is 7 mg. It can be seen that for higher amount the photocatalytic activity slightly decreases. This may be due to the agglomeration of the catalyst particles or, beside this, at a higher concentration of photocatalyst, the recombination between photogenerated electron hole pairs increases which may decrease the degradation efficiency. The photodegradation efficiency of the 1T-MoS2@(Bi1−xFex)VO4 catalyst was also evaluated against the removal of CV dye with different (0.05, 0.10, 0.15, and 0.20 g L−1) concentrations. Fig. 10(b) indicates the dye removal efficiency decreases as the dye concentration increases. This decrease in the removal efficiency may be due to the large amount of dye molecules on the photocatalyst surface while the numbers of hydroxyl radicals were reduced; meanwhile, another reason for the lower efficiency is limitation in light transmutation and as a result the number of photons reaching the photocatalyst decreases, consequently resulting in lower degradation efficiency.
 |
| | Fig. 10 (a) Effect of 1T-MoS2@(Bi0.40Fe0.60)VO4 catalyst dosage (inset shows zoomed view). (b) Effect of initial dye concentration on the degradation percentage. (c) Effect of pH on the degradation efficiency of CV dye. (d) Reusability of the as-prepared 1T-MoS2@(Bi0.40Fe0.60)VO4 in terms of the degradation efficiency (inset shows zoomed view). | |
3.7 Effect of pH and reusability
To study the influence of pH on the removal of CV dye, a series of experiments were performed at different pH (2–12) values. It is well known that pH strongly affects the photocatalytic efficiency during the wastewater decolorization process. This may be owing to pH affecting the agglomeration of the catalyst particles, thus changing the surface area for dye adsorption and photon absorption. The formation of hydroxyl radicals is different for acidified solutions than for alkaline media. Variations in pH can also affect the adsorption of dye molecules on the photocatalyst surface. Therefore, pH of dye solutions was adjusted by HCl and NaOH, and the results are illustrated in Fig. 10(c). CV is a cationic dye so low pH values are not favorable; higher pH value is more appropriate for its degradation. It can be seen that low pH cannot produce the required number of hydroxyl groups to produce hydroxyl radicals on the photocatalyst surface; moreover, the positively charge surface of the catalyst repels the cationic dye adsorption on the 1T-MoS2@(Bi1−xFex)VO4 catalyst surface. Initially when the hydroxyl radicals increase they cause an increase in degradation efficiency; however, at higher alkaline pH value the degradation efficiency is reduced owing to the recombination of the charge carriers. Besides, at high pH hydroxyl ions might be able to compete with the dye molecules in the incorporation process on the photocatalyst surface. From the experimental results it is observed that a pH value of 10 gives the optimum degradation efficiency, as illustrated in Fig. 10(c). For economic, practical use and purposes of reusability, the 1T-MoS2@(Bi1−xFex)VO4 catalyst was tested by running five cycles of activity as exhibited in Fig. 10(d). According to the results, it was observed that the 1T-MoS2@(Bi0.40Fe0.60)VO4 catalyst can be reused and has acceptable stability after five cycles of dye degradation.
3.8 Comparative study (Table 6)
Table 6 Comparative study of the degradation efficiency of CV dye using the 1T-MoS2@(Bi0.40Fe0.60)VO4 nanocomposite with other nanomaterial photocatalysts
| Catalyst |
Efficiency (%) |
Light source |
Time (min) |
Ref. |
| Ag doped TiO2 |
>99 |
UV light |
90 |
72 |
| Cr doped ZnO |
95 |
UV light |
60 |
73 |
| TG capped ZnS nanoparticles |
∼96 |
UV irradiation |
180 |
74 |
| Mn doped and PVP capped ZnO nanoparticles |
<100 |
UV irradiation |
180 |
75 |
| Bi7O9I3/GO |
96 |
UV light |
144 |
68 |
| CdS/CdTiO3–TiO2 |
61 |
UV light |
180 |
76 |
| Ag3VO4/g-C3N4 |
34 |
UV light |
150 |
77 |
| BaWO4 |
∼98 |
Visible light |
60 |
67 |
| Zn3(VO4)2 nanoplate |
100 |
Visible light |
60 |
27 |
| CeO2–TiO2 |
98 |
Visible light |
60 |
78 |
| BiVO4/FeVO4 nanocomposite |
99.1 |
Visible light |
60 |
35 |
| 1T-MoS2@(Bi0.40Fe0.60)VO4 |
∼99.98 |
Visible light |
50 |
Present work |
4. Conclusions
In summary, we prepared 1T-MoS2 QDs-decorated (Bi1−xFex)VO4 heterostructures by a sonication-assisted hydrothermal method for photocatalytic degradation of organic pollutant CV dye and confirmed the electron transfer process from (Bi1−xFex)VO4 onto 1T-MoS2. The 1T-MoS2 QD-decorated (Bi1−xFex)VO4 heterostructures exhibit excellent visible light-dependent response and light absorption range as well as charge transfer activity. The (Bi1−xFex)VO4 heterostructures with optimal 1T-MoS2 QDs loading of 2 wt% show a significantly enhanced photocatalytic activity. The results indicate the easy separation of electron hole pairs, where electrons were transferred to MoS2 from (Bi1−xFex)VO4 surface by irradiation of visible light. Another factor for the superior photocatalytic activity of the Ti–MoS2@(Bi0.40Fe0.60)VO4 heterostructures is the high degree of crystallinity, involving fewer defects and causing less recombination of photogenerated electrons and holes. The Ti–MoS2@(Bi0.40Fe0.60)VO4 photocatalyst was successfully recycled for five cycles providing more stability and reusability in the photocatalytic experiments without a significant loss of catalytic activity. The active species trapping experiments and EPR results allowed us to propose a possible photocatalytic mechanism of Ti–MoS2@(Bi0.40Fe0.60)VO4 and revealed that ˙O2− radicals are the major active species for CV dye degradation. This study shows that 1T-MoS2 QDs-decorated (Bi1−xFex)VO4 heterostructures prepared by this route could be of great potential for practical use of surface modification and give some new ideas for developing promising efficient photocatalysts for removal of and/or degradation of other environmental pollutants in a reasonable reaction time.
Funding detail
This work was financially supported by the State Scholarship Fund of China Scholarship Council (no. 201808410144), the National Natural Science Foundation of China (no. 51202107), and Foundation of Henan Educational Committee (no. 20A480003).
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgements
Dr Muhammad Munir Sajid is grateful to Prof. Zhengjun Zhang, Tsinghua University, Beijing, China, for support with characterization techniques and applications.
References
- K. Zhang, J. Deng, Y. Liu, S. Xie and H. Dai, in Semiconductor Photocatalysis-Materials, Mechanisms and Applications, IntechOpen, 2016 Search PubMed.
- M. Zalfani, Z.-Y. Hu, W.-B. Yu, M. Mahdouani, R. Bourguiga, M. Wu, Y. Li, G. Van Tendeloo, Y. Djaoued and B.-L. Su, Appl. Catal., B, 2017, 205, 121–132 CrossRef CAS.
- S. J. Phang, V.-L. Wong, L.-L. Tan and S.-P. Chai, Appl. Mater. Today, 2020, 20, 100741 CrossRef.
- I. A. Ike, K. G. Linden, J. D. Orbell and M. Duke, Chem. Eng. J., 2018, 338, 651–669 CrossRef CAS.
- M. Rafique, M. B. Tahir and I. Sadaf, in Advanced Research in Nanosciences for Water Technology, Springer, 2019, pp. 95–131 Search PubMed.
- W. Wu, Z.-H. Huang and T.-T. Lim, Appl. Catal., A, 2014, 480, 58–78 CrossRef CAS.
- C. Singh, R. Chaudhary and R. S. Thakur, Int. J. Energy Environ., 2011, 2, 337–350 CAS.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229–230 CrossRef CAS.
- M. Pirhashemi, A. Habibi-Yangjeh and S. R. Pouran, J. Ind. Eng. Chem., 2018, 62, 1–25 CrossRef CAS.
- X. Meng and Z. Zhang, J. Mol. Catal. A: Chem., 2016, 423, 533–549 CrossRef CAS.
- H. Al-Kandari, A. Abdullah, S. Al-Kandari and A. Mohamed, Sci. Adv. Mater., 2017, 9, 739–746 CrossRef CAS.
- S. B. Khan, M. Hou, S. Shuang and Z. Zhang, Appl. Surf. Sci., 2017, 400, 184–193 CrossRef CAS.
- M. M. Sajid, N. A. Shad, Y. Javed, S. B. Khan, Z. Zhang, N. Amin and H. Zhai, Nano-Struct. Nano-Objects, 2020, 22, 100431 CrossRef CAS.
- M. M. Sajid, N. A. Shad, Y. Javed, S. B. Khan, Z. Zhang, N. Amin and H. Zhai, Surf. Interfaces, 2020, 100502 CrossRef CAS.
- S. Irfan, L. Li, A. S. Saleemi and C.-W. Nan, J. Mater. Chem. A, 2017, 5, 11143–11151 RSC.
- C. F. Braganza and A. Salker, Chin. J. Catal., 2016, 37, 1991–1996 CrossRef CAS.
- A. Phuruangrat, K. Karthik, B. Kuntalue, P. Dumrongrojthanath, S. Thongtem and T. Thongtem, Chalcogenide Lett., 2019, 16, 387–393 CAS.
- M. Sathishkumar and S. Geethalakshmi, Mater. Today: Proc., 2020, 20, 54–63 CAS.
- H. Razavi-Khosroshahi, K. Edalati, J. Wu, Y. Nakashima, M. Arita, Y. Ikoma, M. Sadakiyo, Y. Inagaki, A. Staykov and M. Yamauchi, J. Mater. Chem. A, 2017, 5, 20298–20303 RSC.
- G. Takaoka, T. Nose and M. Kawashita, Vacuum, 2008, 83, 679–682 CrossRef CAS.
- P. Mallick, Mater. Sci.-Pol., 2014, 32, 193–197 CrossRef CAS.
- A. Ashour, Turk. J. Phys., 2004, 27, 551–558 Search PubMed.
- F.-y. Chen, X. Zhang, Y.-b. Tang, X.-g. Wang and K.-k. Shu, RSC Adv., 2020, 10, 5234–5240 RSC.
- M. M. Sajid, N. Amin, N. A. Shad, Y. Javed and Z. Zhang, J. Mater. Sci. Eng. B, 2019, 242, 83–89 CrossRef CAS.
- L. Maskaeva, E. Mostovshchikova, V. Voronin, E. Lekomtseva, P. Bogatova and V. Markov, Semiconductors, 2020, 54, 1230–1240 CrossRef CAS.
- M. M. Sajid, N. A. Shad, Y. Javed, S. B. Khan, N. Amin, Z. Zhang, Z. Imran and M. I. Yousuf, Appl. Nanosci., 2020, 10, 421–433 CrossRef CAS.
- M. M. Sajid, N. A. Shad, S. B. Khan, Z. Zhang and N. Amin, J. Alloys Compd., 2019, 775, 281–289 CrossRef CAS.
- R. A. Senthil, S. Osman, J. Pan, Y. Sun, T. R. Kumar and A. Manikandan, Ceram. Int., 2019, 45, 18683–18690 CrossRef CAS.
- S. Irfan, Y. Shen, S. Rizwan, H. C. Wang, S. B. Khan and C. W. Nan, J. Am. Ceram. Soc., 2017, 100, 31–40 CrossRef CAS.
- D. T. Tran, T. Kshetri, N. D. Chuong, J. Gautam, H. Van Hien, N. H. Kim and J. H. Lee, Nano Today, 2018, 22, 100–131 CrossRef.
- M. Kryjewski, T. Goslinski and J. Mielcarek, Coord. Chem. Rev., 2015, 300, 101–120 CrossRef CAS.
- M. M. Sajid, N. A. Shad, Y. Javed, S. B. Khan, Z. Zhang and N. Amin, Res. Chem. Intermed., 2020, 46, 1201–1215 CrossRef CAS.
- N. Li, X. Wu, M. Wang, K. Huang, J. He, W. Ma, H. Chen, Y. Li and S. Feng, Int. J. Hydrogen Energy, 2019, 44, 23046–23053 CrossRef CAS.
- M. M. Sajid, S. B. Khan, N. A. Shad and N. Amin, RSC Adv., 2018, 8, 35403–35412 RSC.
- M. M. Sajid, S. B. Khan, N. A. Shad, N. Amin and Z. Zhang, RSC Adv., 2018, 8, 23489–23498 RSC.
- B. Xiao and M. Schmidt, Inorg. Chem., 2017, 56, 14948–14959 CrossRef CAS PubMed.
- X. Zhang, Z. Zhu, Z. Guo, F. Mo and Z.-c. Wu, Dyes Pigm., 2018, 156, 67–73 CrossRef CAS.
- G. Bera, V. Reddy, P. Rambabu, P. Mal, P. Das, N. Mohapatra, G. Padmaja and G. Turpu, J. Appl. Phys., 2017, 122, 115101 CrossRef.
- M. Stachowicz, B. Bagiński, M. D. Welch, P. M. Kartashov, R. Macdonald, J. Balcerzak, J. Tyczkowski and K. Woźniak, Am. Mineral., 2019, 104, 595–602 CrossRef.
- G. Rajeshkhanna, S. Kandula, K. R. Shrestha, N. H. Kim and J. H. Lee, Small, 2018, 14, 1803638 CrossRef PubMed.
- Z. Wang and B. Mi, Environ. Sci. Technol., 2017, 51, 8229–8244 CrossRef CAS PubMed.
- J.-W. Jiang, Front. Phys., 2015, 10, 287–302 CrossRef.
- B. Sun, Z. Liang, Y. Qian, X. Xu, Y. Han and J. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7257–7269 CrossRef CAS PubMed.
- X. Xu, X. Tian, B. Sun, Z. Liang, H. Cui, J. Tian and M. Shao, Appl. Catal., B, 2020, 272, 118984 CrossRef CAS.
- Q. Tang and D.-e. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- H. Zhai, J. Qi, Y. Tan, L. Yang, H. Li, Y. Kang, H. Liu, J. Shang and H. S. Park, Appl. Mater. Today, 2020, 18, 100536 CrossRef.
- Z. Liang, Y. Guo, Y. Xue, H. Cui and J. Tian, Mater. Chem. Front., 2019, 3, 2032–2040 RSC.
- J. Xiong, J. Li, J. Shi, X. Zhang, W. Cai, Z. Yang and H. Cheng, Appl. Catal., B, 2019, 243, 614–620 CrossRef CAS.
- M. Maqsood, S. Afzal, A. Shakoor, N. A. Niaz, A. Majid, N. Hassan and H. Kanwal, J. Mater. Sci.: Mater. Electron., 2018, 29, 16080–16087 CrossRef CAS.
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan and N. Williams, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- Y. Li, L. Wang, S. Zhang, X. Dong, Y. Song, T. Cai and Y. Liu, Catal. Sci. Technol., 2017, 7, 718–724 RSC.
- S. Das and A. K. Nandi, 2019.
- D. Zhou, H. Bin, J. Guo, L. X. Pang, Z. M. Qi, T. Shao, Q. P. Wang, Z. X. Yue and X. Yao, J. Am. Ceram. Soc., 2014, 97, 2915–2920 CrossRef CAS.
- R. Jaiswal, N. Patel, D. Kothari and A. Miotello, Appl. Catal., B, 2012, 126, 47–54 CrossRef CAS.
- Z. Zhang, W. Wang, M. Shang and W. Yin, Catal. Commun., 2010, 11, 982–986 CrossRef CAS.
- S. Rizwan and S. Fatima, Bismuth Ferrites/Graphene Nanoplatelets Nanohybrids for Efficient Organic Dye Removal, in Bismuth-Advanced Applications and Defects Characterization, IntechOpen, 2018 Search PubMed.
- M. M. Sajid, N. A. Shad, A. M. Afzal, Y. Javed, S. B. Khan, N. Amin, A. Shah, I. Yousaf and H. Zhai, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 314 CrossRef CAS.
- N. Van Landschoot, E. Kelder and J. Schoonman, J. Solid State Electrochem., 2003, 8, 28–33 CrossRef CAS.
- S. B. Khan, S. Irfan and S.-L. Lee, Nanomaterials, 2019, 9, 1024 CrossRef CAS PubMed.
- V. Venkatraman, A. E. Yemene and J. de Mello, Sci. Rep., 2019, 9, 1–13 CAS.
- N. A. Shad, M. M. Sajid, N. Amin, Y. Javed, K. Akhtar, G. Ahmad, S. Hassan and M. Ikram, Ceram. Int., 2019, 45, 19015–19021 CrossRef CAS.
- S. Ahmadi, A. Rahdar, C. A. Igwegbe, S. Mortazavi-Derazkola, A. M. Banach, S. Rahdar, A. K. Singh, S. Rodriguez-Couto and G. Z. Kyzas, Polyhedron, 2020, 190, 114792 CrossRef CAS.
- M. Mehta, S. Krishnamurthy, S. Basu, T. P. Nixon and A. P. Singh, Mater. Today Chem., 2020, 17, 100283 CrossRef CAS.
- N. A. Mohamed, H. Ullah, J. Safaei, A. F. Ismail, M. F. Mohamad Noh, M. F. Soh, M. A. Ibrahim, N. A. Ludin and M. A. Mat Teridi, Journal of Physical Chemistry C, 2019, 123, 9013–9026 CrossRef CAS.
- X. Zhang, Y. Huang, F. Ma, Z. Zhang and X. Wei, J. Phys. Chem. Solids, 2018, 121, 85–92 CrossRef CAS.
- V. Rumyantsev, M. Fadeev, V. Aleshkin, N. Kulikov, V. Utochkin, N. Mikhailov, S. Dvoretskii, S. Pavlov, H. W. Hübers and V. Gavrilenko, Phys. Status Solidi B, 2019, 256, 1800546 CrossRef.
- M. Y. A. Khan, M. Zahoor, A. Shaheen, N. Jamil, M. I. Arshad, S. Z. Bajwa, N. A. Shad, R. Butt, I. Ali and M. Z. Iqbal, Mater. Res. Bull., 2018, 104, 38–43 CrossRef.
- S.-Y. Chou, W.-H. Chung, L.-W. Chen, Y.-M. Dai, W.-Y. Lin, J.-H. Lin and C.-C. Chen, RSC Adv., 2016, 6, 82743–82758 RSC.
- X. Xu, Y. Sun, Z. Fan, D. Zhao, S. Xiong, B. Zhang, S. Zhou and G. Liu, Front. Chem., 2018, 6, 64 CrossRef PubMed.
- Y. Nosaka and A. Y. Nosaka, Identification and roles of the active species generated on various photocatalysts, in Photocatalysis and Water Purification, ed. P. Pichat, Wiley-VCH, Weinheim, Germany, 2013, pp. 3–24 Search PubMed.
- V. I. Avdeev and A. F. Bedilo, J. Phys. Chem. C, 2013, 117, 14701–14709 CrossRef CAS.
- A. Gupta, A. Pal and C. Sahoo, Dyes Pigm., 2006, 69, 224–232 CrossRef CAS.
- I. Zammit, V. Vaiano, G. Iervolino and L. Rizzo, RSC Adv., 2018, 8, 26124–26132 RSC.
- M. Sharma, T. Jain, S. Singh and O. Pandey, Sol. Energy, 2012, 86, 626–633 CrossRef CAS.
- M. Mittal, M. Sharma and O. Pandey, J. Nanosci. Nanotechnol., 2014, 14, 2725–2733 CrossRef CAS PubMed.
- Y. Li, W. Zhang, L. Li, C. Yi, H. Lv and Q. Song, RSC Adv., 2016, 6, 51374–51386 RSC.
- S. Wang, D. Li, C. Sun, S. Yang, Y. Guan and H. He, Appl. Catal., B, 2014, 144, 885–892 CrossRef CAS.
- A. Morlando, J. McNamara, Y. Rehman, V. Sencadas, P. J. Barker and K. Konstantinov, J. Mater. Sci., 2020, 55, 8095–8108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00807b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Haifa Zhai
*a,
Haifa Zhai