
Vacancy defect engineering of BiVO4 photoanodes for photoelectrochemical water splitting
Songcan
Wang
 *a,
Xin
Wang
a,
Boyan
Liu
a,
Zhaochen
Guo
a,
Kostya (Ken)
Ostrikov
b,
Lianzhou
Wang
*c and
Wei
Huang
a
*a,
Xin
Wang
a,
Boyan
Liu
a,
Zhaochen
Guo
a,
Kostya (Ken)
Ostrikov
b,
Lianzhou
Wang
*c and
Wei
Huang
a
aFrontiers Science Center for Flexible Electronics, Xi'an Institute of Flexible Electronics (IFE) and Xi'an Institute of Biomedical Materials & Engineering, Northwestern Polytechnical University, 127 West Youyi Road, Xi'an 710072, China. E-mail: iamscwang@nwpu.edu.cn
bSchool of Chemistry and Physics and Centre for Materials Science Queensland University of Technology Brisbane, QLD 4000, Australia
cNanomaterials Centre, Australian Institute for Bioengineering and Nanotechnology and School of Chemical Engineering, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: l.wang@uq.edu.au
First published on 5th October 2021
Abstract
Photoelectrochemical (PEC) water splitting has been regarded as a promising technology for sustainable hydrogen production. The development of efficient photoelectrode materials is the key to improve the solar-to-hydrogen (STH) conversion efficiency towards practical application. Bismuth vanadate (BiVO4) is one of the most promising photoanode materials with the advantages of visible light absorption, good chemical stability, nontoxic feature, and low cost. However, the PEC performance of BiVO4 photoanodes is limited by the relatively short hole diffusion length and poor electron transport properties. The recent rapid development of vacancy defect engineering has significantly improved the PEC performance of BiVO4. In this review article, the fundamental properties of BiVO4 are presented, followed by an overview of the methods for creating different kinds of vacancy defects in BiVO4 photoanodes. Then, the roles of vacancy defects in tuning the electronic structure, promoting charge separation, and increasing surface photoreaction kinetics of BiVO4 photoanodes are critically discussed. Finally, the major challenges and some encouraging perspectives for future research on vacancy defect engineering of BiVO4 photoanodes are presented, providing guidelines for the design of efficient BiVO4 photoanodes for solar fuel production.
 Songcan Wang | Songcan Wang is currently a professor at the Institute of Flexible Electronics (IFE), Northwestern Polytechnical University (NPU), China. He received his B.Eng. (2011) and M.Eng. (2014) from Central South University (CSU), China, and PhD degree from the University of Queensland (UQ), Australia in 2018. Before joining NPU, he worked as a postdoctoral research fellow in Professor Lianzhou Wang's group at UQ for about 1.5 years. His research interests focus on the synthesis of semiconductor nanomaterials for solar energy conversion and storage, including photoelectrochemical cells, photocatalysis, and rechargeable batteries. |
 Kostya (Ken) Ostrikov | Kostya (Ken) Ostrikov is a Professor with Queensland University of Technology, Australia, a Founding Leader of the CSIRO–QUT Joint Sustainable Processes and Devices Laboratory, and Academician of the Academy of Europe and the European Academy of Sciences. His research focuses on nanoscale control of energy and matter contributing to the solution of the grand challenge of directing energy and matter at nanoscales, to develop renewable energy and energy-efficient technologies for a sustainable future. |
 Lianzhou Wang | Lianzhou Wang is currently a professor in the School of Chemical Engineering and the director of the Nanomaterials Centre at the University of Queensland, Australia. After receiving his PhD degree from the Shanghai Institute of Ceramics in 1999, he worked at NIM and AIST in Japan for 5 years. Since 2004, he has been working at the University of Queensland and his research interest focuses on the design and development of semiconducting materials toward efficient solar energy conversion and storage. |
 Wei Huang | Wei Huang received his B.Sc., M.Sc., and Ph.D. in chemistry from Peking University in 1983, 1988, and 1992, respectively. He was appointed as the Deputy President of Nanjing University of Posts and Telecommunications in 2006, the President of Nanjing Tech University in 2012, and the Provost of Northwestern Polytechnical University in 2017. He is a member of the Chinese Academy of Sciences, Foreign Academician and Honorary Doctor of the Russian Academy of Sciences, and Fellow of the Royal Society of Chemistry. His research interests include organic optoelectronics, nanomaterials, polymer chemistry, plastic electronics, and bioelectronics. |
1. Introduction
The growth of global population and the development of industry have boosted the demand for energy. However, the contemporary society is heavily dependent on fossil fuels such as petroleum, coal and natural gas. The accelerating consumption of fossil fuels not only causes the depletion of non-renewable energy, but also increases the emission of CO2 into the atmosphere.1–5 Consequently, our human community needs to confront the severe threat of the energy crisis and environmental issues. Therefore, sustainable development has become the consensus among all nations and regions throughout the world. The development of cutting-edge technologies for the production of clean and renewable energy is the pursuit for scientists. Solar energy is the most abundant renewable energy source on earth. The solar energy reaching the earth in one hour is almost equal to the global energy demand in one year.6 Nevertheless, the low energy density and intermittence make the direct utilization of solar energy unfavourable. Therefore, it is desirable to develop suitable technologies for efficient utilization of solar energy. Photoelectrochemical (PEC) water splitting that converts solar energy into hydrogen as the carbon-free energy carrier has been regarded as a promising technology for solar energy utilization.7–10 PEC water splitting can store solar energy in the chemical bonds of hydrogen, and hydrogen can be used whenever required. An ideal PEC water splitting system for practical applications should be fulfilled with a high solar-to-hydrogen (STH) conversion efficiency (>10%), long-term stability, and low cost.11 However, practical application of PEC water splitting has not been realized in the past five decades due to the very low STH efficiency achieved in the cost-effective systems.12–14The STH efficiency is mainly determined by the photoelectrode materials since the three main PEC processes take place at the photoelectrode materials: (1) light absorption to generate electron–hole pairs; (2) charge separation and transport; (3) transfer of photogenerated charge carriers from the photoelectrode material to the electrolyte for surface reactions.15–17 Amongst the above-mentioned processes, light absorption is determined by the bandgap of a semiconductor, which determines the theoretical maximum of STH efficiency under AM 1.5 G illumination. Charge separation and transport are determined by the electronic properties of the photoelectrode materials, whereas surface charge transfer properties are mainly determined by the surface catalytic activity of the photoelectrode materials. Therefore, the development of efficient photoelectrode materials is the most important task to achieve high-performance PEC water splitting. In the past five decades, numerous photoelectrode materials including oxides (e.g. TiO2, WO3, BiVO4, Cu2O, Fe2O3),18–24 nitrides (e.g. TaON, Ta3N5, C3N4),25–31 chalcogenides (e.g. MoS2, CuInS2, CZTS, CIGS),32–37 and metal halide perovskites (e.g. p-MAPbI3)38–41 have been developed as photoelectrode materials since the first discovery of PEC water splitting in 1972.42 However, ideal photoelectrode materials to drive PEC water splitting towards practical applications are still lacking.
Amongst all the available photoelectrode materials, bismuth vanadate (BiVO4) has emerged as one of the most promising photoanode materials for PEC water splitting due to its narrow bandgap for visible light absorption, appropriate band edge positions that require a relatively low onset potential, low cost, and high stability in aqueous solutions, and has been intensively studied.43–47 In the past decades, monoclinic scheelite-phase BiVO4 has been confirmed to be an excellent photoanode material for water oxidation.48 Although the photocatalytic water oxidation performance of BiVO4 has been studied since 1998,49 the PEC performance of BiVO4 photoanodes was moderate due to the lack of suitable methods for the fabrication of efficient BiVO4 films. Since 2003, with the successful synthesis of BiVO4 films by metal–organic decomposition (MOD),50 the performance of BiVO4 was boosted and an increasing number of efficient methods have been developed.51–54 Pure BiVO4 photoanodes suffer from severe charge recombination because of the relatively low carrier mobility and short hole diffusion length, leading to very poor PEC performance.55 To improve the PEC performance of BiVO4 photoanodes, various strategies such as heterojunction formation,56–61 heteroatom doping,62–65 nanostructure construction,66–68 photo-charging,69–74 crystal facet engineering,75–78 and vacancy defect engineering79–82 have been developed, which can effectively enhance light absorption, charge separation and transport, or surface charge transfer properties.
Owing to the rapid progress of BiVO4 photoanodes, several review articles discussing nanostructure engineering,83 crystal facet engineering,84 and modification strategies85 to improve the PEC performance of BiVO4 have been reviewed. In recent years, vacancy defect engineering has emerged as an important and efficient strategy to tailor the optoelectronic structure of photoelectrode materials.86–92 In particular, an increasing number of studies on vacancy defect engineering of BiVO4 photoanodes including the generation of oxygen vacancies, vanadium vacancies, and bismuth vacancies have been reported.93–99 Since vacancy defects serve as self-dopants in tailoring the conductivity, band edge positions and surface molecular adsorption properties of BiVO4 photoanodes, the PEC performance can be effectively improved.100 Considering the rapid development and increasing interest in this important field, a comprehensive and timely review solely focusing on analysing the formation of vacancy defects in BiVO4 photoanodes and their roles in promoting the PEC performance is urgently needed. In this review article, some general properties of BiVO4 such as crystal structures, optical properties, carrier transport, and photogenerated carrier lifetime are briefly introduced. This is followed by an overview of the formation of oxygen vacancies, vanadium vacancies, and bismuth vacancies in BiVO4 photoanodes. Subsequently, the roles of vacancy defects in BiVO4 photoanodes are critically discussed. Finally, a concise summary of vacancy defect engineering of BiVO4 photoanodes for PEC water splitting, the key challenges and perspectives in this attractive field are presented. We hope to shed light on the development of vacancy defect engineering for the design of efficient photoelectrodes for solar fuel production.
2. Crystal structure and optical and electronic properties
BiVO4 is a bright yellow solid that is generally applied as a novel environmentally friendly inorganic yellow pigment because of its high hiding power, weather resistance, and nontoxic feature.101,102 The bandgap of BiVO4 is 2.4–2.5 eV, which can absorb visible light up to approximately 520 nm. In addition, the valence band maximum (VBM) of BiVO4 is more positive than the water oxidation potential, making BiVO4 an attractive photoanode material for PEC water splitting. Since crystal, optical and electronic structures have a close relationship with the PEC performance of BiVO4 photoanodes, in this section, we will briefly introduce the crystal, optical and electronic properties of BiVO4 and critically discuss their effects on PEC water splitting performance, which is essential for the design of efficient BiVO4 photoanodes.2.1. Crystal structures
As shown in Fig. 1, BiVO4 has three main crystal forms: pucherite, dreyerite and clinobisvanite.48,83 More specifically, pucherite is a natural mineral of BiVO4 with an orthorhombic crystal structure, which cannot be synthesized by general laboratory routes. Dreyerite occurs in a tetragonal zircon (t-z) structure, whereas clinobisvanite is a monoclinic scheelite (m-s) structure. In addition, when the atomic positions are modified to construct a four-fold symmetry structure in the clinobisvanite, another tetragonal scheelite (t-s) structure is formed. The crystal structures and bond lengths of BiVO4 are listed in Table 1.103 Both dreyerite and clinobisvanite can be prepared in the laboratory under different conditions. Generally, BiVO4 (m-s) is achieved by a solid-state reaction at high temperatures.104 In addition, sol–gel and hydrothermal methods can also obtain BiVO4 (m-s).105 BiVO4 (t-z) is synthesized in aqueous media by low-temperature processes. For example, BiVO4 (t-z) was prepared via a coprecipitation method using a Bi(NO3)3 solution containing nitric acid and an aqueous NH4VO3 solution at room temperature.106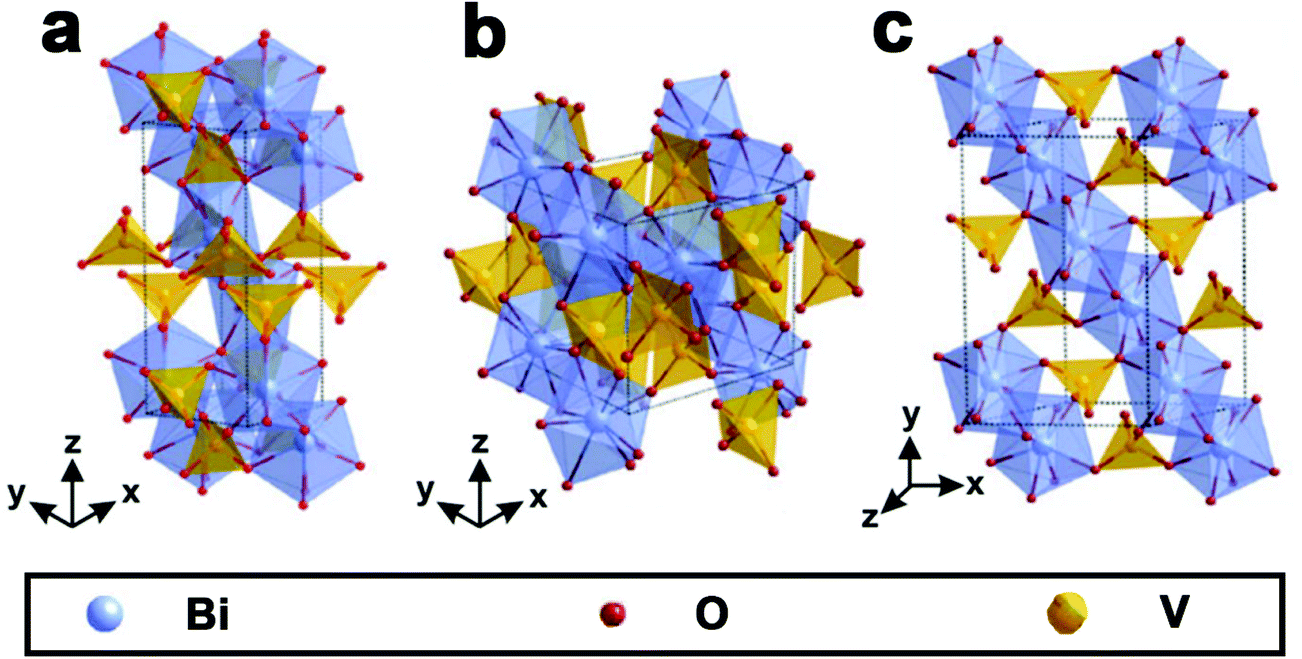 | ||
| Fig. 1 Crystal structure of BiVO4: (a) pucherite, (b) dreyerite and (c) clinobisvanite. Reproduced from ref. 107 with permission. Copyright 2016, Springer Nature. | ||
| Crystal System | Orthorhombic | t-z | m-s | t-s |
|---|---|---|---|---|
| Crystal structure (Å) | ||||
| Space group | Pnca | 141/amd | C 2/c | 141/a |
| A | 5.332 | 7.303 | 7.247 | 5.147 |
| B | 5.060 | 7.303 | 11.697 | 5.147 |
| C | 12.020 | 6.584 | 5.090 | 11.722 |
| Bond length (Å) | ||||
| Bi–O | 2.20 | 2.41 | 2.35 | 2.40 |
| 2.31 | 2.55 | 2.37 | 2.47 | |
| 2.53 | 2.52 | |||
| 2.73 | 2.63 | |||
| V–O | 1.76 | 1.7 | 1.69 | 1.73 |
| 2.73 | 1.76 | |||
BiVO4 (m-s) can be transformed from both BiVO4 (t-z) and BiVO4 (t-s) by a thermal treatment process. As illustrated in Fig. 2, by annealing BiVO4 (t-z) at 670–770 K, followed by cooling to room temperature, BiVO4 (m-s) can be obtained. However, BiVO4 (m-s) cannot be converted back to BiVO4 (t-z) by thermal treatment. BiVO4 (t-s) is a high temperature phase and the phase transition between BiVO4 (t-s) and BiVO4 (m-s) is reversible at 528 K.108 Interestingly, different crystalline structures of BiVO4 nanoparticles (t-z, m-s, and t-z/m-s heterostructure) can be synthesized by a fast microwave assisted method and annealing treatment process.109
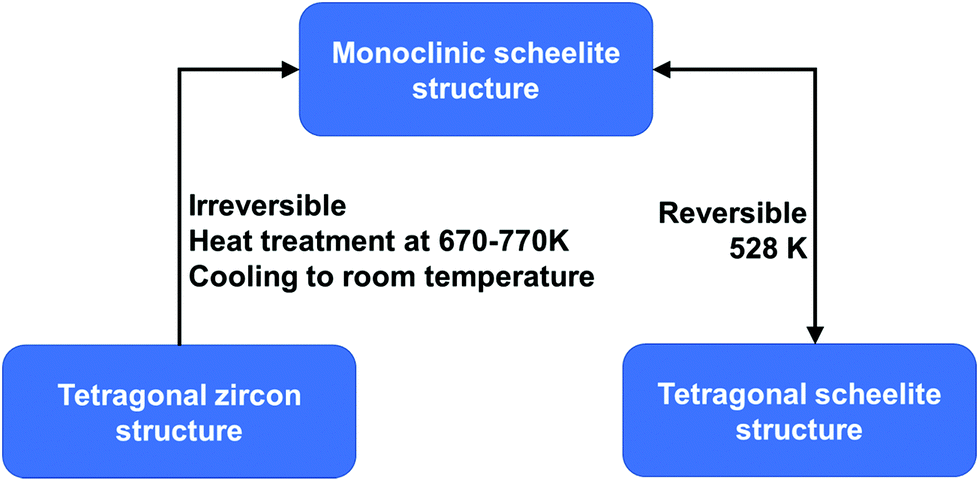 | ||
| Fig. 2 Conditions for phase transformation between different crystal structures of BiVO4. Reproduced from ref. 108 with permission. Copyright 2001, American Chemical Society. | ||
The photocatalytic activity of BiVO4 is affected by its crystal structure. For example, BiVO4 powders with m-s and t-z structures were selectively prepared by tuning the ratio of vanadium to bismuth in the starting materials.110 The bandgap of BiVO4 (t-z) is 2.9 eV while that of BiVO4 (m-s) is 2.4 eV, as demonstrated in Fig. 3a. As a result, BiVO4 (t-z) and BiVO4 (m-s) showed similar photocatalytic activities under UV light (300 < λ < 380 nm) illumination in the presence of silver nitrate as the electron sacrificial reagent. However, BiVO4 (m-s) exhibited a much higher photocatalytic activity than BiVO4 (t-z) under visible light (λ > 420 nm) illumination. Therefore, the different photocatalytic activity between BiVO4 (t-z) and BiVO4 (m-s) is mainly derived from their different bandgaps. BiVO4 powder with a scheelite structure was prepared by hydrolysing a mixture of Bi(NO3)3 and Na3VO4 solution containing nitric acid with Na2CO3 or NaHCO3 at room temperature.108 By tuning the reaction time, BiVO4 (t-s) and BiVO4 (m-s) can be selectively obtained. This pioneering work provides a reliable process for the synthesis of the high-temperature formed BiVO4 (t-s) at room temperature, enabling the possible study of its photocatalytic activity. Interestingly, even though BiVO4 (t-s) and BiVO4 (m-s) exhibit very similar crystal structures and bandgaps (Fig. 3b), their photocatalytic activities are significantly different. BiVO4 (m-s) showed a high photocatalytic activity for O2 evolution in an aqueous AgNO3 solution under visible light illumination, whereas its BiVO4 (t-s) counterpart exhibited a negligible O2 evolution activity (Fig. 3c). It was believed that the high visible light photocatalytic activity of BiVO4 (m-s) is attributed to the distortion of a Bi–O polyhedron by a 6s2 lone pair of Bi3+ that induces the local polarization, promoting the separation of photogenerated electron–hole pairs.
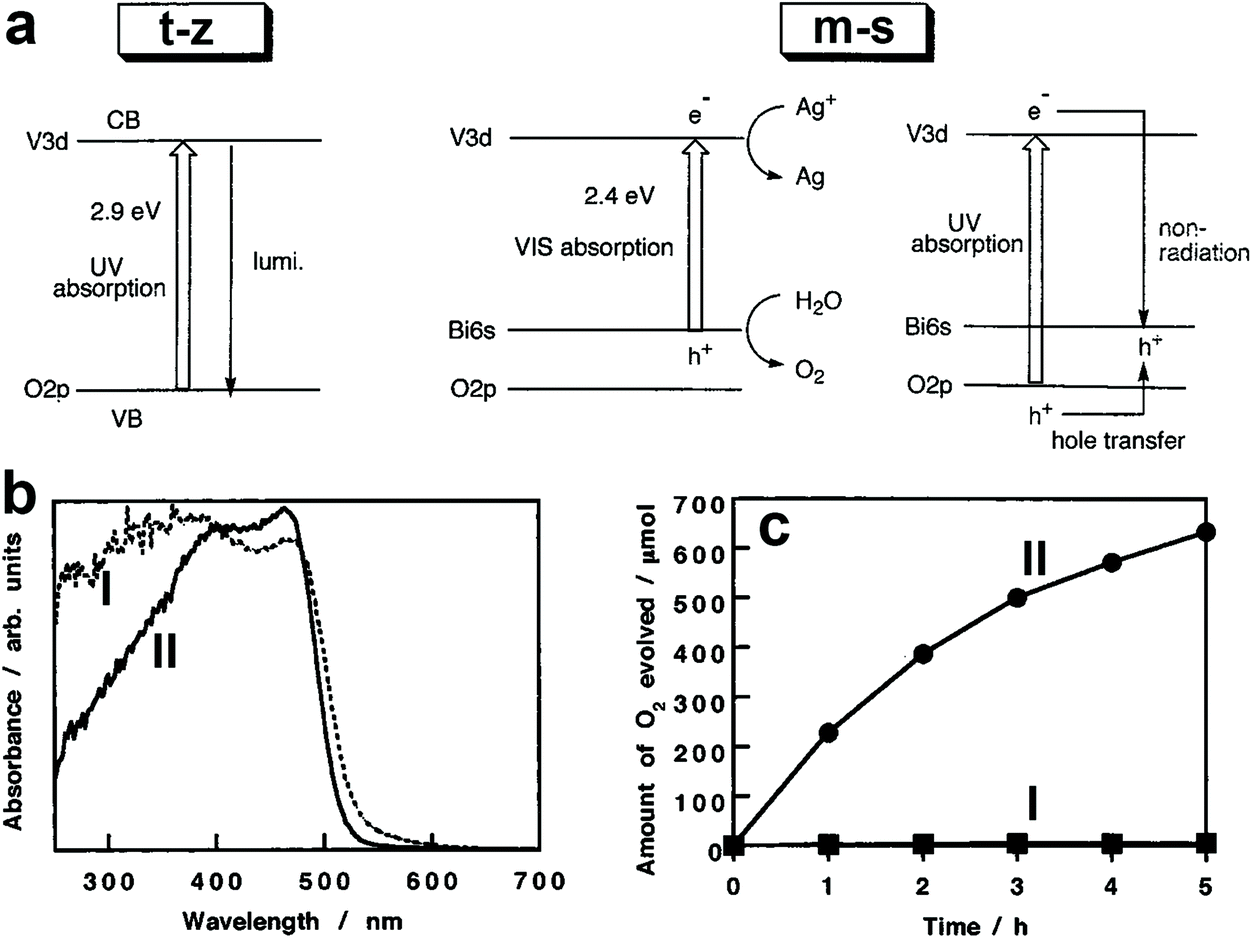 | ||
| Fig. 3 (a) Band structures of the t-z and m-s BiVO4. Reproduced from ref. 110 with permission. Copyright 1999, American Chemical Society. (b) UV-vis light absorption curves and (c) oxygen evolution curves in an aqueous AgNO3 solution under visible light illumination (λ > 420 nm) of the BiVO4 samples I: BiVO4 (t-s) and II: BiVO4 (m-s). Reproduced from ref. 108 with permission. Copyright 2001, American Chemical Society. | ||
2.2. Optical properties
According to the discussion in section 2.1, BiVO4 (m-s) exhibits the best photocatalytic activity, and has been widely studied as an excellent water oxidation material for both photocatalytic and PEC water splitting.111–115 Therefore, the optical properties discussed in this section are focused on BiVO4 (m-s). Based on the density functional theory (DFT) calculations, the minimum bandgap of BiVO4 is 2.174 eV, and the two k-points at the VBM and conduction band minimum (CBM) are located at different positions, indicating the indirect bandgap characteristics (Fig. 4a).116 The calculated bandgap is underestimated compared to the experimental values (2.4–2.5 eV depending on the preparation methods), due to the well-known drawback of generalized gradient approximation (GGA). The band structure of BiVO4 is shown in Fig. 4b, where the conduction band of BiVO4 mainly originates from the V 3d orbitals, while the valence band is composed of the hybridization of the O 2p and Bi 6s orbitals,117 which is consistent with the band structure proposed by Kudo et al.110 Owing to the relatively narrow bandgap and the positive VBM, BiVO4 exhibits excellent photocatalytic oxygen evolution and organic compound decomposition under visible light illumination.118–120 However, since the CBM of BiVO4 is close to but a little more positive than the hydrogen evolution potential, spontaneous photocatalytic hydrogen evolution cannot take place. Thus, BiVO4 can only achieve the half water oxidation reaction. As shown in Fig. 4c, the absorption coefficient (α) of pure BiVO4 increases significantly at the bandgap energy, with a value of 1 × 104 cm−1 at the band edge absorption onset of approximately 500 nm, reaching 1 × 105 cm−1 at around 460 nm.121 Moreover, a plateau can be observed between 430 and 390 nm. When the wavelength is shorter than 390 nm, the absorption coefficient increases gradually. The strong absorption of green to ultraviolet photons in the visible region and transmission of yellow to infrared photons results in the yellow colour of BiVO4.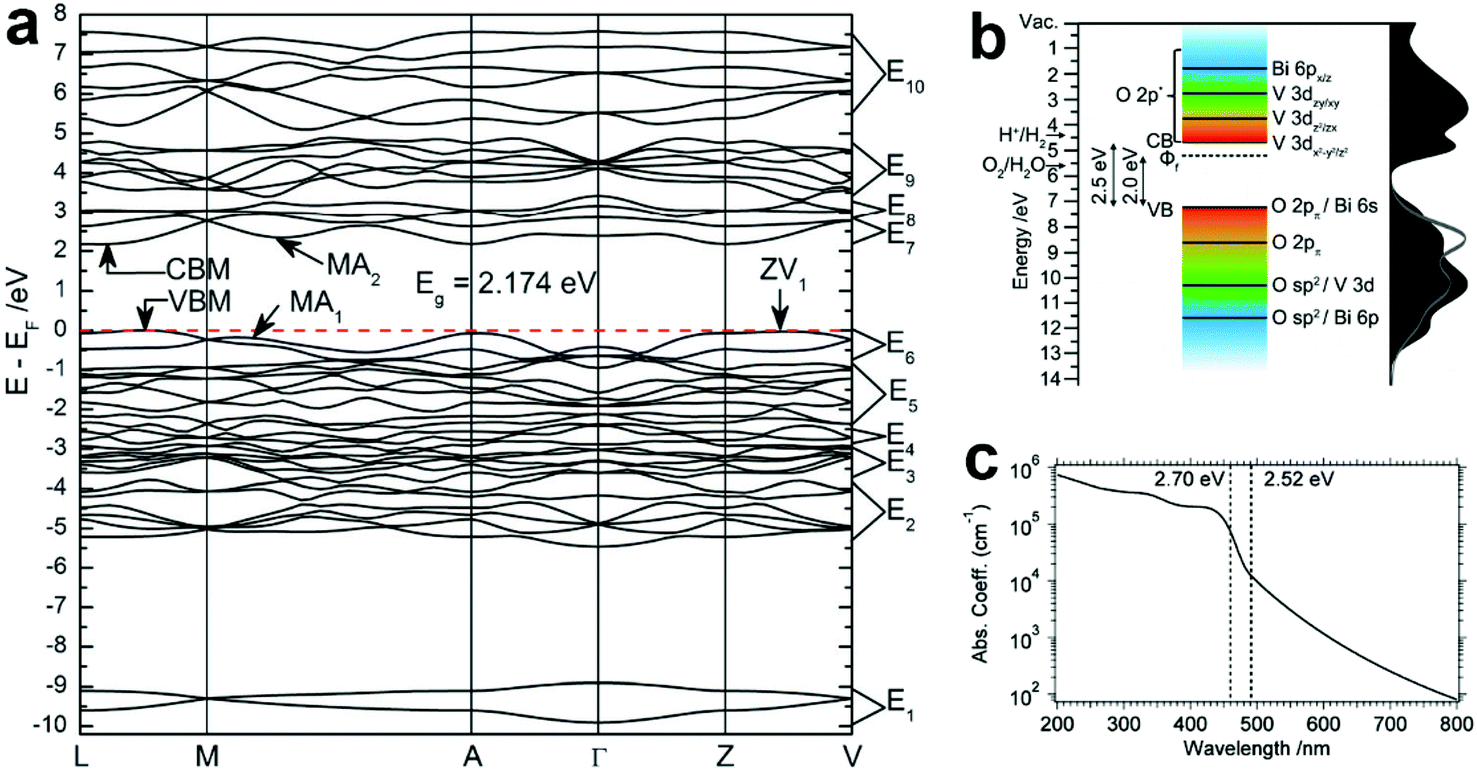 | ||
| Fig. 4 (a) The calculated band structure of BiVO4 (m-s). Reproduced from ref. 116 with permission. Copyright 2011, Royal Society of Chemistry. (b) Schematic illustration of the band structure of BiVO4 (m-s). Reproduced from ref. 117 with permission. Copyright 2014, American Chemical Society. (c) The absorption coefficient curve of BiVO4 (m-s) between 200 and 800 nm. Reproduced from ref. 121 with permission. Copyright 2015, American Chemical Society. | ||
2.3. Carrier transport
The efficient transport of majority charge carriers in BiVO4 is essential to achieve a high charge separation efficiency when used as a photoanode. Previous experimental studies revealed that the carrier transport in BiVO4 photoanodes is poor, leading to severe charge recombination. However, the underlying mechanism was unclear due to the lack of cutting-edge time-resolved technology to characterize the dynamic charge separation and transport process. In 2013, Roel van de Krol's group applied a time-resolved microwave conductivity (TRMC) technology to investigate BiVO4 under one sun illumination, and found that the carrier mobility of undoped BiVO4 is ∼0.04 cm2 V−1 s−1, which is at least 1–2 orders of magnitude lower than that of other typical semiconductor photoelectrode materials (e.g. the electron mobility of ZnO is 100–200 cm2 V−1 s−1) for PEC water splitting.122 DFT calculations revealed that the low carrier mobility is attributed to the relatively localized charge carriers due to the weakly dispersive valence band and conduction band in the band structure curve. By using THz spectroscopy, the temporal evolution of polaron formation leading to a build-up of a polaron population in parallel to initial carrier trapping was observed in BiVO4.123 In addition, the self-trapped carriers have a mobility of 0.02 cm2 V−1 s−1, leading to a thermal hopping activation energy of ∼90 meV. Interestingly, BiVO4 showed facet-dependent charge carrier transport properties.124 The effective hole and electron masses of the (010) facet are estimated to be approximately 0.16m0 and 0.11m0, respectively. In comparison, the effective hole and electron masses of the (011) facet are around 0.40m0 and 0.24m0, respectively. Since the drift velocity of electrons or holes is proportional to the reciprocal of the effective mass, lower effective masses indicate more efficient charge carrier transport. Therefore, charge carrier transport properties in the (010) facet are better than those in the (011) facet for BiVO4. In addition, O 2p and Bi 6s contributions in the valence bands are more localized in the (011) facet compared to those of the (010) facet. Thus, the (011) facet exhibits slower charge mobility than the (010) facet.On the other hand, the relatively low electronic conductivity also causes severe charge recombination in BiVO4 photoanodes. Doping BiVO4 with either Mo6+ or W6+ or simultaneous doping with Mo6+ and W6+ at the V5+ site is effective to increase the electronic conductivity,125–128 thereby promoting the PEC water splitting performance. Generally, the increased carrier density after doping can be demonstrated from Mott–Schottky plots. Interestingly, DFT calculations revealed a marginal decrease in the value of carrier mobility with an increase in the doping level (singly and co-doped samples), which is consistent with the experimental results that the intrinsic mobility of electrons in doped BiVO4 is smaller than that in un-doped BiVO4.129 However, W/Mo doping is efficient to increase the electron carrier concentration, leading to higher conductivity of the BiVO4 photoanode.
2.4. Photogenerated carrier lifetime
In addition to the excellent conductivity for majority charge carriers, a promising photoanode should also efficiently transport photogenerated minority charge carriers to the semiconductor/electrolyte interfaces for water oxidation. Therefore, the lifetime of the photogenerated carriers is another important factor. The minority carrier diffusion length (L) is defined by L = (Dτ)1/2,130 where D is the diffusion constant, which is proportional to the carrier mobility, and τ is the minority carrier lifetime, which is highly sensitive to the crystal structure, crystallinity, facet and recombination channels in the semiconductor. According to this equation, a higher carrier mobility and a longer minority carrier lifetime can achieve a longer minority carrier diffusion length, which means that the photogenerated charge carriers are easier to be separated and transferred to the semiconductor/electrolyte interface for the subsequent photocatalytic reaction.Since BiVO4 has an indirect bandgap, the band edge photoluminescence curve cannot be observed and the direct measurement of the minority carrier lifetime through time-resolved photoluminescence cannot be achieved. Thus, transient absorption pump–probe spectroscopy was applied to determine the characteristic relaxation time.121 By using excitation pulses at 350 nm with a duration of 100 fs and a repetition rate of 1 kHz, and a white light continuum pulse of 360–700 nm, the decay time was estimated to be around 20 ns. In another study, TRMC measurements confirmed that the carrier lifetime of BiVO4 is exponentially long, reaching ∼40 ns.122 Although the carrier lifetime of BiVO4 is much longer than that of other metal oxide semiconductors such as α-Fe2O3 (3 ps), WO3 (1–9 ns), and Cu2O (40 ps), the minority carrier diffusion length of BiVO4 is calculated to be only ∼70 nm, due to the relatively low carrier mobility. It was believed that the relatively long carrier lifetime is attributed to the indirect bandgap characteristics of BiVO4 that prevent a direct recombination of the photogenerated hole–electron pairs.131
Interestingly, BiVO4 particles exhibit particle size dependent carrier dynamics and reactivity under visible light illumination, as evidenced by single-particle transient absorption microscopy.132 Upon the illumination of a 527 nm light, it was found that the well-faceted nano-aggregated BiVO4 crystals exhibit fast hole decay and little reactivity for Fe3+ reduction. In comparison, other aggregated particles with grain boundaries between small primary crystals exhibit slower hole decay and higher reactivity for Fe3+ reduction than their nano-aggregated crystal counterparts. When the secondary particle size of the aggregated crystals increases, the hole decay becomes slower and the reactivity becomes higher. Thus, the grain boundaries in aggregated particles do not function as recombination centers but play an essential role in elongating the carrier lifetime.
3. Vacancy defect engineering of BiVO4 photoanodes
In the conventional point of view, vacancy defects in semiconductor materials would induce detrimental effects on their photocatalytic activities. In fact, excessive vacancy defects in semiconductor materials lead to enhanced charge recombination due to the generation of more charge recombination centers.133,134 Nevertheless, an optimized number of vacancy defects can effectively tailor the electronic structure of a semiconductor material, and increase the conductivity and mobility, thereby promoting photocatalytic activity. In recent years, an increasing number of publications confirmed the benefit of vacancy defects in photoelectrode materials for PEC water splitting.135–140 In particular, oxygen vacancies function as shallow donors in BiVO4 photoanode materials, which can improve the electronic conductivity and promote the separation and transfer of photogenerated charge carriers.141 As a result, a much higher photocurrent density can be observed compared to pristine BiVO4 photoanodes without vacancy defects.In addition to tuning the electronic structure, oxygen vacancies can also cause distortions in the crystal structure of BiVO4 at room temperature, and the degree of distortion is determined by the vacancy concentration.142 Moreover, defects on the surface of BiVO4 can obviously increase the hydrophilicity, promoting the surface water oxidation reaction.143 Generally, most of the oxide semiconductors show n-type conductivity due to the formation of oxygen vacancies, whereas the formation of cationic vacancies or interstitial anions could increase the p-type conductivity.144,145 This is very interesting, because the switchable n–p conductivity of a metal oxide semiconductor can be designed by deliberately creating oxygen vacancies or cationic vacancies. With the increasing interest of vacancy defect engineering in PEC water splitting, other kinds of vacancy defects such as vanadium vacancies and bismuth vacancies have also been prepared. In this section, we will concisely discuss the methods to generate different kinds of vacancy defects in BiVO4 photoanodes, and critically analyse the roles of vacancy defects in BiVO4 photoanodes in terms of improving the PEC properties.
3.1. Formation of vacancy defects in BiVO4 photoanodes
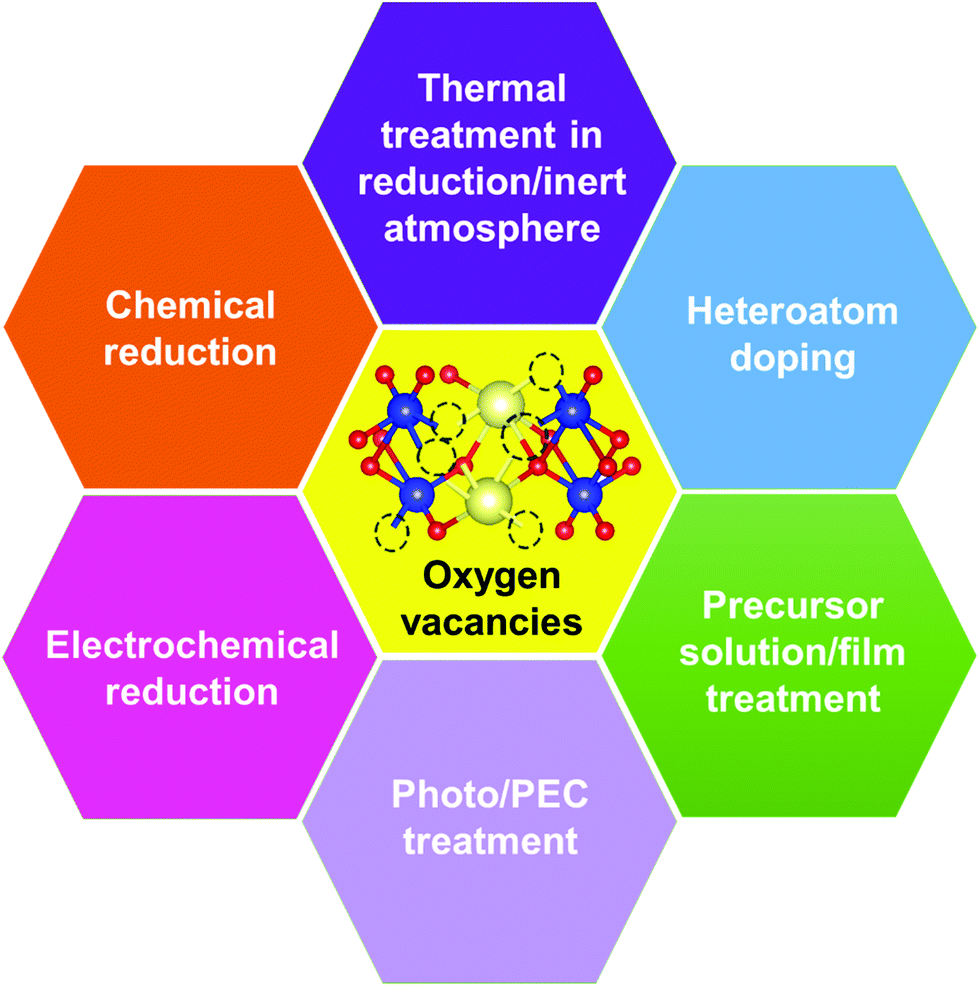 | ||
| Fig. 5 Strategies for generating oxygen vacancies in BiVO4 photoanodes. | ||
Generally, thermally treating a metal oxide semiconductor under a reduction or inert atmosphere can generate oxygen vacancies. The number of generated oxygen vacancies can be tailored by controlling the thermal treatment temperature. The formation of oxygen vacancies can be expressed using the Kröger–Vink notation:146
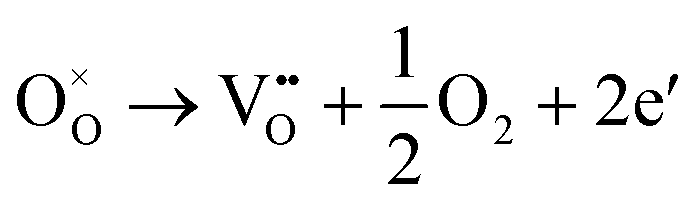 | (1) |
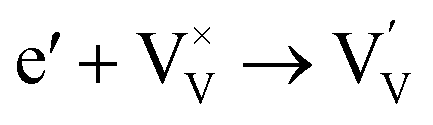 | (2) |
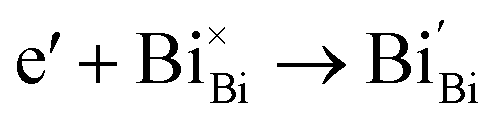 | (3) |
According to eqn (1)–(3), the formation of one oxygen vacancy 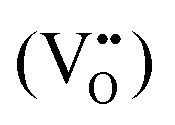 is accompanied by the reduction of one V5+ (V×V) and one Bi3+ (Bi×Bi) to generate V4+
is accompanied by the reduction of one V5+ (V×V) and one Bi3+ (Bi×Bi) to generate V4+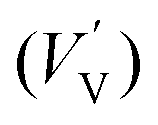 and Bi2+
and Bi2+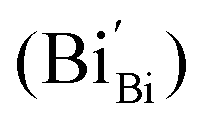 , thus maintaining electric neutrality in the BiVO4 crystal.
, thus maintaining electric neutrality in the BiVO4 crystal.
Planar BiVO4 films were annealed under a hydrogen atmosphere at elevated temperatures between 200 and 400 °C.147 Not only oxygen vacancies but also hydrogen impurities were incorporated into the BiVO4 films. With the annealing temperature increased from 200 to 400 °C, the bright yellow BiVO4 films turned yellowish green and eventually dark green. Similarly, nanoporous BiVO4 films were prepared by a typical electrodeposition-thermal method.66 To create oxygen vacancies, the obtained nanoporous BiVO4 films were annealed in a flow of 6% H2/Ar for 10 min at 300 °C.82 In addition to hydrogen treatment at elevated temperatures, hydrogen plasma treatment of BiVO4 photoanodes at room temperature can also generate oxygen vacancies.148 Likewise, the number of oxygen vacancies in three-dimensional (3D) nanoporous BiVO4 can be controllably generated by an ionized argon plasma technology.149 Moreover, anoxic annealing can also generate oxygen vacancies in BiVO4 photoanodes, which was effective to increase the mobility, lifetime, and concentration of the photogenerated carriers.150 In addition, thermally treating BiVO4 under an argon atmosphere at elevated temperature can also generate oxygen vacancies.151 Unlike under the hydrogen atmosphere, a higher temperature (e.g. 500 and 700 °C) is required to generate oxygen vacancies under an argon atmosphere, which can form new sub-gap states in the electronic structure of BiVO4.
Chemical reduction of BiVO4 photoanodes with different reducing agents is another common strategy to generate oxygen vacancies. By chemically treating BiVO4 films in a NaBH4 solution, B–O bonding at the surface of BiVO4 photoanodes was destroyed, leading to the removal of oxygen atoms and the formation of oxygen vacancies.152 In addition, Bi3+ ions were reduced to metallic Bi nanoparticles that were decorated homogeneously on the surfaces of the BiVO4 photoanode. Similarly, an effective surface-engineered sulfite treatment process was developed to generate surface oxygen vacancies in BiVO4 photoanodes without illumination.153 Interestingly, a novel pyramid-BiVO4 with sufficient oxygen vacancies was prepared by a low-temperature solvothermal process,154 which can effectively improve the separation and transfer efficiencies of the photogenerated charge carriers.
Electrochemical reduction can also generate oxygen vacancies in BiVO4 photoanodes. The number of oxygen vacancies generated can be tuned by controlling the reduction potential and the electrochemical reduction time. For example, an electrochemical reduction process was applied to treat the Mo-doped BiVO4 films.155 As shown in Fig. 6a, when the BiVO4 film was electrochemically treated at −0.8 V (versus Ag/AgCl), a quasi-oxygen vacancy was formed on the (020) facet (only Bi–O bonds crack), which can significantly increase the electron mobility and photocurrent density. Nevertheless, the reduction potential of −1.2 V induced the formation of oxygen vacancies on the surface of the (020) facet (both Bi–O and V–O bonds break simultaneously), leading to the obvious decrease of photocurrent densities. It was believed that the formation of a quasi-oxygen vacancy is essential to promote the PEC water splitting performance of the Mo-doped BiVO4 photoanodes, as evidenced by the much weaker photoluminescence signal of the −0.8-BiMoVO compared to that of BiMoVO (Fig. 6b). Similarly, a hydrothermally synthesized BiVO4 film with the (040) facet grown vertically on the fluorine doped SnO2 (FTO) glass substrate (Fig. 6c–e) was electrochemically treated at −0.1 V versus the reversible hydrogen electrode (RHE) for only 150 s, which generated an appropriate amount of oxygen vacancies.79 Surprisingly, the photocurrent density is significantly enhanced by 10 times, reaching 2.5 mA cm−2 under AM 1.5 G illumination (Fig. 6f). Upon the surface modification with cobalt borate (CoBi) as the oxygen evolution cocatalyst, the photocurrent density can be further improved, exhibiting an excellent applied bias photon-to-current efficiency (ABPE) of 1.1%.
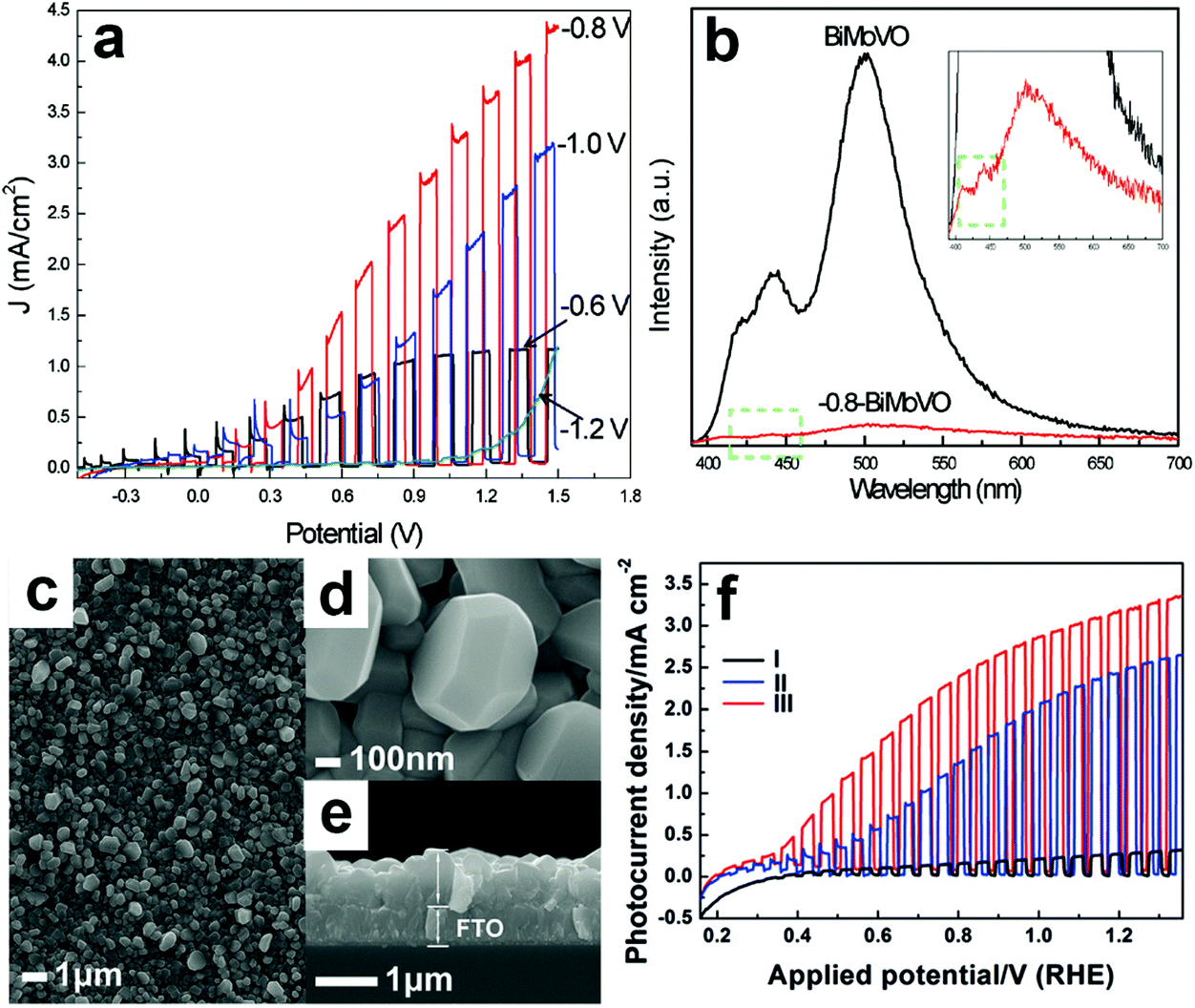 | ||
| Fig. 6 (a) Photocurrent density versus applied potential curves of the Mo-doped BiVO4 films electrochemically treated at −0.6, −0.8, −1.0, and −1.2 V versus Ag/AgCl, respectively. (b) Photoluminescence spectra of Mo-doped BiVO4 without electrochemical reduction (BiMoVO) and electrochemically treated at −0.8 V (−0.8-BiMoVO). Reproduction from ref. 155 with permission. Copyright 2017, Wiley-VCH. (c) Low-magnification, (d) high-magnification, and (e) cross-sectional SEM images of BiVO4 films with (040) facet grown vertically on the FTO substrate. (f) Photocurrent density versus applied potential curves of samples I: BVO4 film, II: electrochemically-treated BiVO4 film (E-BVO), and III: CoBi/E-BVO. Reproduced from ref. 79 with permission. Copyright 2017, Wiley-VCH. | ||
Interestingly, photo or PEC treatment of BiVO4 photoanodes in the presence of Na2SO3 solution can also generate oxygen vacancies. The formation of oxygen vacancies is attributed to the redox reaction capacity of the photogenerated electrons and holes, as demonstrated below:95,156
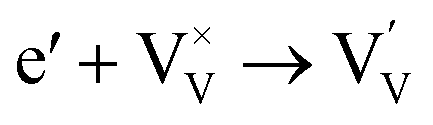 | (4) |
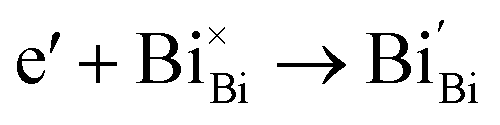 | (5) |
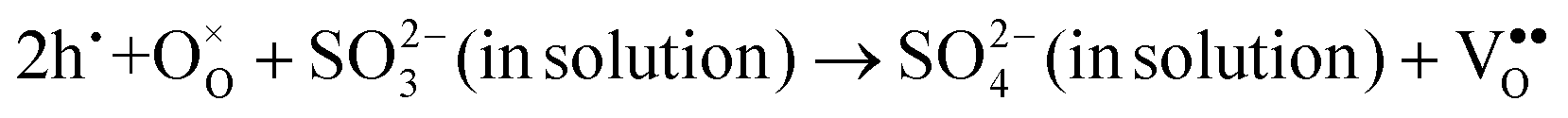 | (6) |
For example, a bare BiVO4 photoanode was placed in a Na2SO3 solution under light illumination.156 Owing to the presence of a hole sacrificial agent (Na2SO3), the photogenerated holes are consumed while the photogenerated electrons can reduce BiVO4 itself, forming oxygen vacancies. Similarly, a facile photoetching process was applied to generate enriched oxygen vacancies at the surface of BiVO4 photoanodes, while avoiding the formation of excessive bulk defects.95 The surface oxygen vacancies increased the carrier density to enhance band bending, leading to a 2.3-fold higher charge separation efficiency. Recently, a PEC activation process was developed to generate oxygen vacancies at the surface and passivated the surface states for BiVO4 photoanodes, thus boosting charge transfer at the BiVO4/electrolyte interface, suppressing surface recombination, and leading to an enhanced PEC performance.94
Suitable treatment on the precursor solutions or films can also generate oxygen vacancies in the obtained BiVO4 photoanodes. For example, BiVO4 photoanodes were prepared by a spray pyrolysis process.157 By treating the precursor solution with ultraviolet light and ultrasonication (Fig. 7a), the nanostructure as well as the number of oxygen vacancies can be tuned in the final obtained BiVO4 films. Systematic studies revealed that the nanoporous structure was mainly attributed to the ultrasonic treatment, while the formation of oxygen vacancies was primarily derived from ultraviolet treatment. Bismuth precursor films were prepared by an electrodeposition method.158 By thermally treating the bismuth precursor films with vanadyl acetylacetonate (VO(C5H7O2)2) vapors (Fig. 7b), BiVO4 films with abundant oxygen vacancies can be achieved. Interestingly, the number of oxygen vacancies in the BiVO4 film can be tuned by changing the pH value in the precursor electrolyte. BiVO4 films obtained by converting from the bismuth precursor films electrodeposited in an acidic electrolyte contain much more oxygen vacancies compared to their counterparts prepared by electrodeposition in an alkaline electrolyte (Fig. 7c). A bismuth oxide precursor film was prepared by an electrodeposition method, which was converted to a transparent BiVO4 photoanode with well-controlled oxygen vacancies through a mild thermal treatment process in the presence of VO(C5H7O2)2.80 The formation of a bismuth oxide precursor film is the key to generate oxygen vacancies in the obtained BiVO4 photoanode. By carefully tuning the electrodeposition and the thermal treatment processes, an appropriate amount of oxygen vacancies was formed in the BiVO4 photoanodes, leading to a significantly enhanced charge separation efficiency. As a result, a high photocurrent density of 5.87 mA cm−2 was achieved at 1.23 V versus RHE under AM 1.5 G illumination (Fig. 7d and e). Very recently, a new sulfur oxidation strategy was developed to prepare planar BiVO4 photoanodes with in situ formed oxygen vacancies.81 The oxidation of sulfur during the calcination process can create an oxygen deficient atmosphere, leading to the formation of oxygen vacancies in the BiVO4 photoanodes.
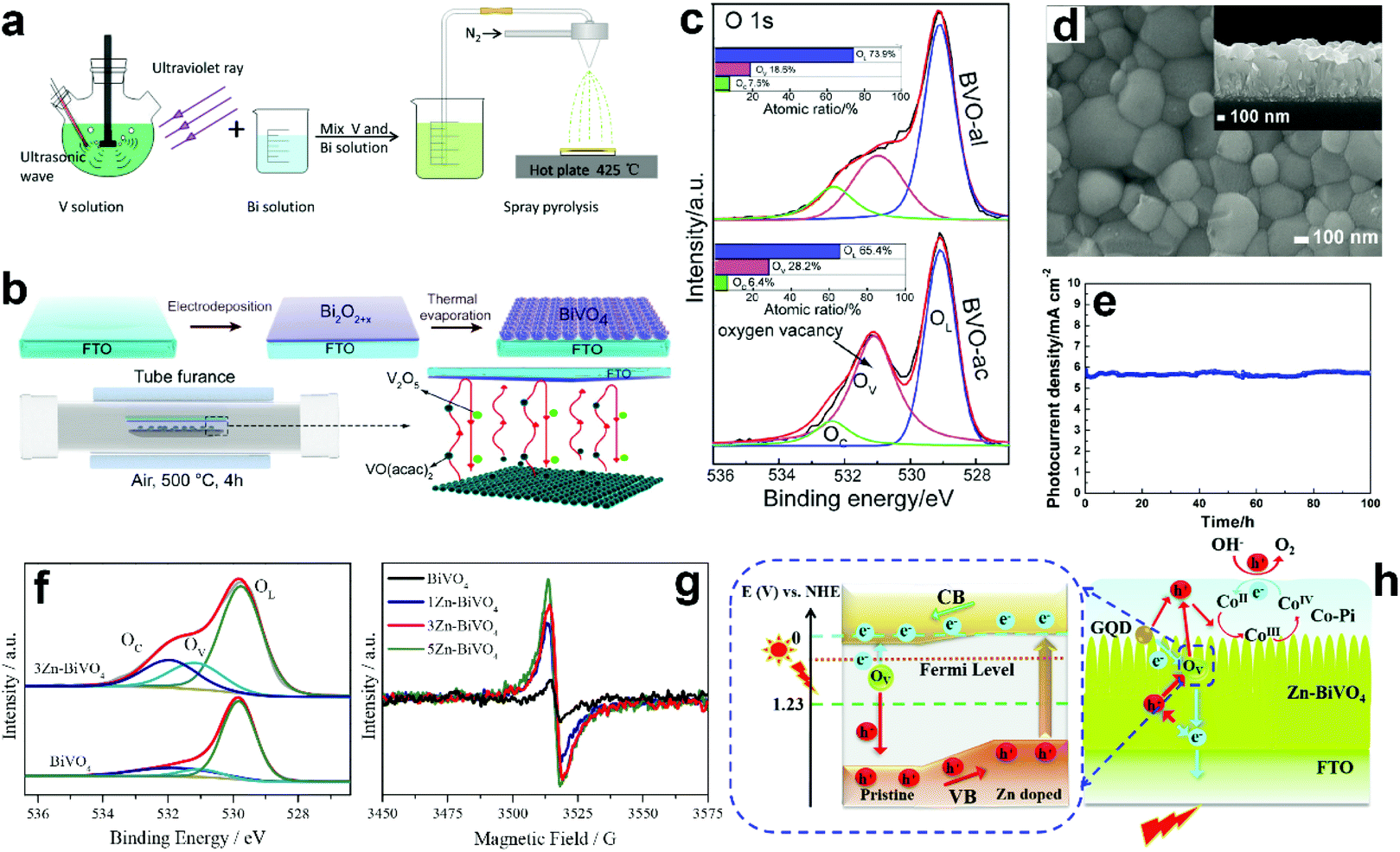 | ||
| Fig. 7 (a) Schematic illustration of the preparation of nanoporous BiVO4 photoanodes with abundant oxygen vacancies. Reproduced from ref. 157 with permission. Copyright 2020 Author(s). (b) Schematic illustration of the BiVO4 films prepared by a thermal evaporation process. (c) High resolution XPS O 1s spectra of BiVO4 films converted from bismuth precursor films prepared in an acidic electrolyte (BVO-ac) and alkaline electrolyte (BVO-al) (Inset: atomic ratios of OL, OV, and OC calculated from the XPS spectrum of O 1s). Reproduced from ref. 158 with permission. Copyright 2019, Wiley-VCH. (d) SEM image of the BiVO4 film with oxygen vacancies, inset: the corresponding cross-sectional view. (e) Photocurrent density versus time of the dual BiVO4 photoanodes with abundant oxygen vacancies coated with FeOOH/NiOOH oxygen evolution cocatalysts at 1.23 V versus RHE under AM 1.5 G illumination. Reproduced from ref. 80 with permission. Copyright 2018, Wiley-VCH. (f) High resolution O 1s XPS spectra of BiVO4 and 3 at% Zn doped BiVO4 (3Zn-BiVO4). (g) EPR curves of BiVO4 with different Zn-dopant contents. (h) Schematic of the proposed band gap for the pristine BiVO4/Zn-doped BiVO4 homojunction photoanode. Reproduced from ref. 159 with permission. Copyright 2019, Elsevier. | ||
Doping BiVO4 photoanodes with other elements can induce the formation of oxygen vacancies. Both theoretical and experimental results revealed that Zn doping can replace the Bi-sites, thus inducing a controllable number of oxygen vacancies in the BiVO4 films (Fig. 7f).159 Electron paramagnetic resonance (EPR) results demonstrated that with the increasing content of Zn dopant in BiVO4, more oxygen vacancies are formed (Fig. 7g). As illustrated in Fig. 7h, the synergistic effect of Zn doping and oxygen vacancies not only change the conduction and valence band positions, forming a local built-in electric field, but also increase the charge carrier density, promoting charge separation and transfer. In addition, water adsorption on Bi-sites was also activated, which is also beneficial for water splitting. Interestingly, a BiVO4 homojunction with abundant oxygen vacancies was prepared by a surface crystal orientation reconstruction induced by a one-step Mo doping method.160 In addition to the generation of oxygen vacancies, Mo doping also promoted the formation of a BiVO4 nanolayer with crystal orientation of (121) on the (110) facet. Compared to the pristine BiVO4 photoanode, the photocurrent density of the Mo doped one is obviously improved. Similarly, Gd doping of BiVO4 can also generate surface oxygen vacancies.161
Protecting oxygen vacancies from being oxidized during PEC water splitting is also very important to maintain the high activity and stability. Although hydrogenation treatment is a facile and efficient process to create oxygen vacancies in BiVO4 photoanodes, PEC activity of the obtained oxygen deficient BiVO4 photoanode would decrease due to the gradual healing of oxygen vacancies in the BiVO4 photoanode. It was found that coating an ultrathin amorphous TiO2 layer on a hydrogenated nanoporous BiVO4 (H-BiVO4) photoanode is effective to protect the oxygen vacancies from being oxidized during PEC water splitting.162 Without TiO2 coating, the photocurrent density of the H-BiVO4 photoanode is decreased to the value of the pristine one after consecutive light illumination for 3 h with external bias. However, the TiO2 coated H-BiVO4 photoanode only exhibits 5% decay of the photocurrent density in the same period of time. In addition, oxygen vacancies can also be stabilized in the BiVO4 Scheelite structure by incorporating Sr2+ to replace Bi3+, resulting in the formation of corner-sharing V2O7 tetrahedral dimers.163 The migration of oxygen vacancies took place through a cooperative mechanism involving V2O7-dimer breaking and reforming assisted by synergic rotation and deformation of the neighboring VO4 tetrahedra.
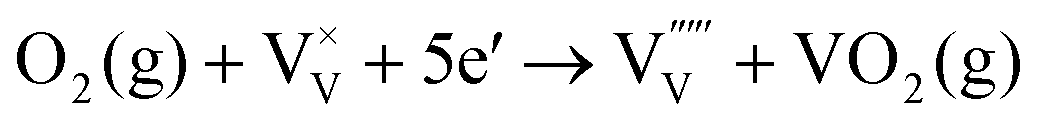 | (7) |
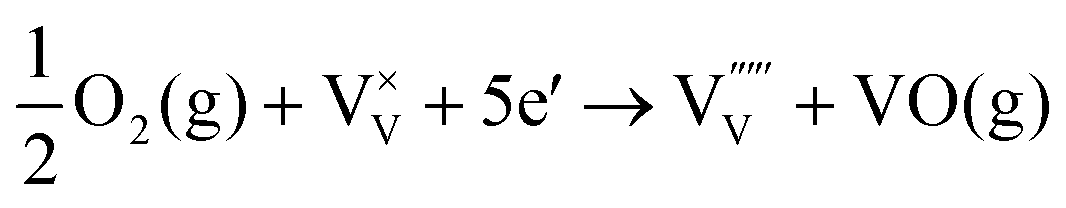 | (8) |
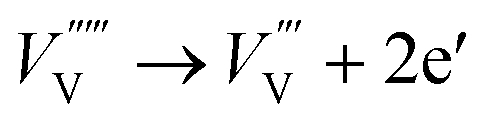 | (9) |
| 2VO(g) → VO2(g) + V(g) | (10) |
The energy level of vanadium vacancies is located at around 0.3 eV above the valence band edge of BiVO4,128 which would be deep enough in the bandgap to form trap states, rendering charge recombination centers for the photogenerated electron–hole pairs. Unlike oxygen vacancies that have positive effects on the PEC performance of BiVO4 photoanodes, vanadium vacancies generally reduce the photocurrent densities. Fortunately, vanadium loss in BiVO4 photoanodes can be avoided by supplying excess vanadium in the gas phase during the calcination process.133
Very recently, the formation of vanadium vacancies and their impact on PEC water splitting performance of the BiVO4 photoanodes were systematically studied.99 BiVO4 films with a high intrinsic level of vanadium vacancies (denoted as VV-BiVO4) were prepared by high-temperature self-assembly of flame-made aerosols, followed by calcination at 450 °C for 2 h in air without the addition of any vanadium source. X-ray photoelectron spectroscopy (XPS) confirmed the presence of vanadium vacancies on the surfaces of the obtained BiVO4 photoanodes, whereas photoluminescence spectroscopy revealed an increased photogenerated charge recombination efficiency. The underlying mechanism was further studied by DFT calculations, which revealed that vanadium vacancies formed a new sub-band gap level near the Fermi level of BiVO4, acting as recombination centers. Thus, many more photogenerated charge carriers were recombined in the bulk of BiVO4 before reaching the surfaces for the subsequent oxygen evolution reaction, leading to a very low photocurrent density.
To further confirm the detrimental effect of vanadium vacancies on the PEC performance of BiVO4 photoanodes, the vanadium deficient BiVO4 photoanodes were annealed in the presence of an additional vanadium source to compensate for the vanadium loss (denoted as V-treated BiVO4).99 As expected, the photocurrent density was increased by 2 times compared to its vanadium deficient counterpart in the presence of Na2SO3 as the hole scavenger (Fig. 8a). In addition, by surface loading the V-treated BiVO4 photoanode with FeOOH/NiOOH dual cocatalysts (Fig. 8b–d), the photocurrent densities of V-treated BiVO4/FeOOH/NiOOH are also much higher than those of its VV-BiVO4/FeOOH/NiOOH counterpart for PEC water splitting (Fig. 8e), which is due to the enhanced charge separation and transfer efficiencies in the V-treated BiVO4 photoanode (Fig. 8f).
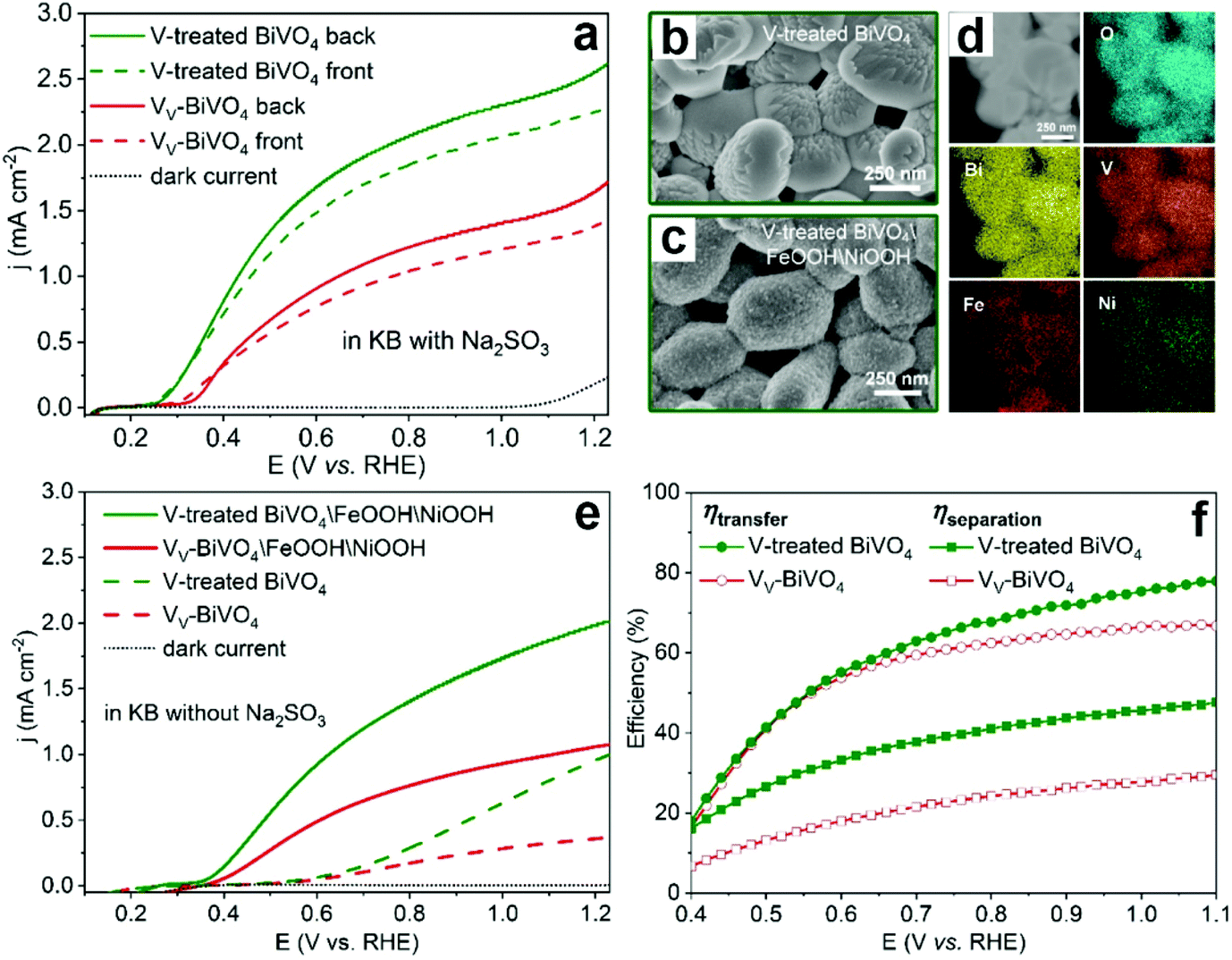 | ||
| Fig. 8 (a) Photocurrent density versus applied potential curves of the BiVO4 with vanadium vacancies (VV-BiVO4) and V-treated BiVO4 photoanodes. SEM images of V-treated BiVO4 (b) before and (c) after the deposition of FeOOH/NiOOH cocatalysts. (d) STEM image and elemental mapping of O, Bi, V, Fe and Ni for V-treated BiVO4. (e) Photocurrent density versus applied potential curves of VV-BiVO4 and V-treated BiVO4 with and without FeOOH/NiOOH cocatalysts. (f) Charge-transfer and charge-separation efficiencies versus applied potential curves of VV-BiVO4 and V-treated BiVO4 photoanodes. Reproduced from ref. 99 with permission. Copyright 2021, American Chemical Society. | ||
Based on the current understanding of vanadium vacancies, it is necessary to add excess vanadium for BiVO4 films during the calcination process to achieve the highly active m-s phase while avoiding the formation of vanadium vacancies. It should be mentioned that only very few reports studied the PEC performance of BiVO4 photoanodes with vanadium vacancies. Thus, more experimental and theoretical studies are still required to gain more insights into the real function of vanadium vacancies during PEC water splitting. It may be too early to conclude that vanadium vacancies are detrimental to the PEC performance of BiVO4 photoanodes. Like oxygen vacancies, the location (e.g. in the surface or in the bulk) and quantity of vanadium vacancies may also affect the PEC performance of BiVO4 photoanodes. Therefore, the development of suitable strategies to accurately control the generation of vanadium vacancies in BiVO4 photoanodes should be essential to promote the further development of this field.
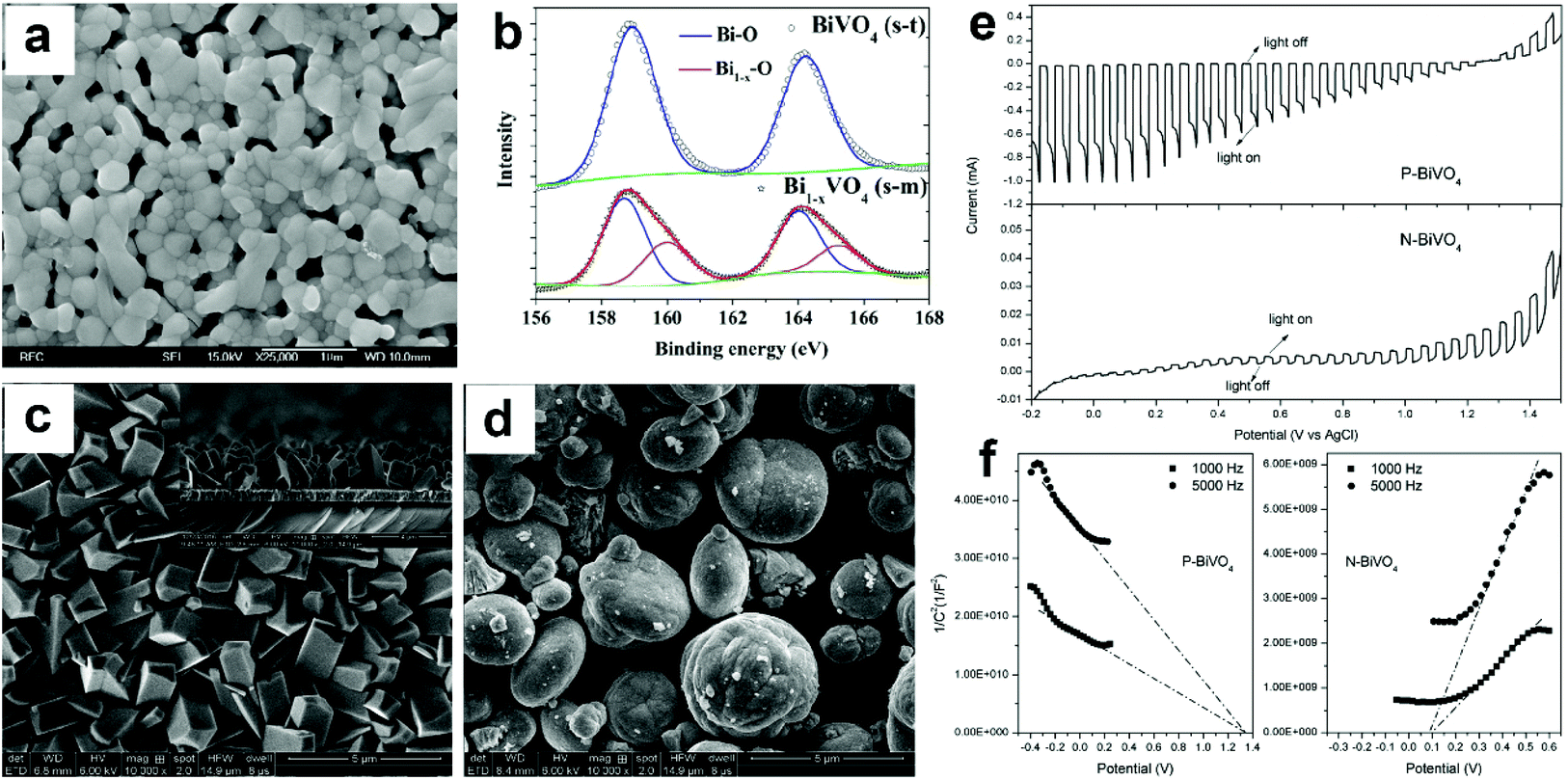 | ||
| Fig. 9 (a) SEM image of Bi1−xVO4 (m-s). (b) High resolution Bi 4f XPS spectra of BiVO4 (s-t) and Bi1−xVO4 (m-s). Reproduced from ref. 96 with permission. Copyright 2017, Elsevier. SEM images of (c) P-BiVO4 and (d) N-BiVO4. (e) Photocurrent density versus applied potential curves and (f) Mott–Schottky plots of the P-BiVO4 and N-BiVO4 photoelectrodes. Reproduced from ref. 97 with permission. Copyright 2019, Elsevier. | ||
Compared to oxygen vacancies that have been intensively studied in BiVO4 photoanodes, only a few reports mentioned bismuth vacancies in BiVO4. Different from vanadium vacancies that are reported to have a detrimental effect on the PEC performance of BiVO4, surface bismuth vacancies exhibit a positive effect on tuning the optoelectronic structure of BiVO4, leading to an enhanced photocatalytic activity. Moreover, bismuth vacancies can also change the conductivity of BiVO4 from n-type to p-type, enabling the design of an unbiased PEC water splitting system composed of only BiVO4 as both the photoanode and photocathode.98 However, the underlying mechanism of how bismuth vacancies affect the electronic structure and PEC performance of BiVO4 is still unclear, which requires more efforts in the research of this field. The development of cutting-edge characterization technology and the combination of DFT calculations may gain more insights into understanding the real role of bismuth vacancies in BiVO4 photoanodes.
3.2. Roles of vacancy defects in BiVO4 photoanodes
During PEC water splitting, the BiVO4 photoanode will absorb photons with energy higher than its bandgap under light illumination, and electrons and holes will be generated. Then, electrons will migrate to the counter electrode for the hydrogen evolution reaction (HER), while holes will transfer to the surface of BiVO4 for the oxygen evolution reaction (OER). During this process, charge recombination also occurs. Vacancy defects will affect the crystal structure and electronic and optical properties of BiVO4 photoanodes,165 inducing profound impact on their PEC performances. For example, DFT calculations revealed that oxygen vacancies can change the bandgap of BiVO4 from an indirect one into a direct one, enhance the hybridization of O 2p, V 3d and Bi 6s orbitals, and shift the band edges to lower energies.166 Considering the positive effect of vacancy defects on the design of efficient BiVO4 photoanodes for PEC water splitting, it is necessary to deeply understand the roles of vacancy defects in tuning the electronic structure, promoting charge separation, and increasing surface photoreaction kinetics. In this section, we will critically discuss the main roles of vacancy defects in BiVO4 photoanodes during PEC water splitting, and hope to provide new insights for the design of efficient BiVO4 photoanodes for solar fuel production.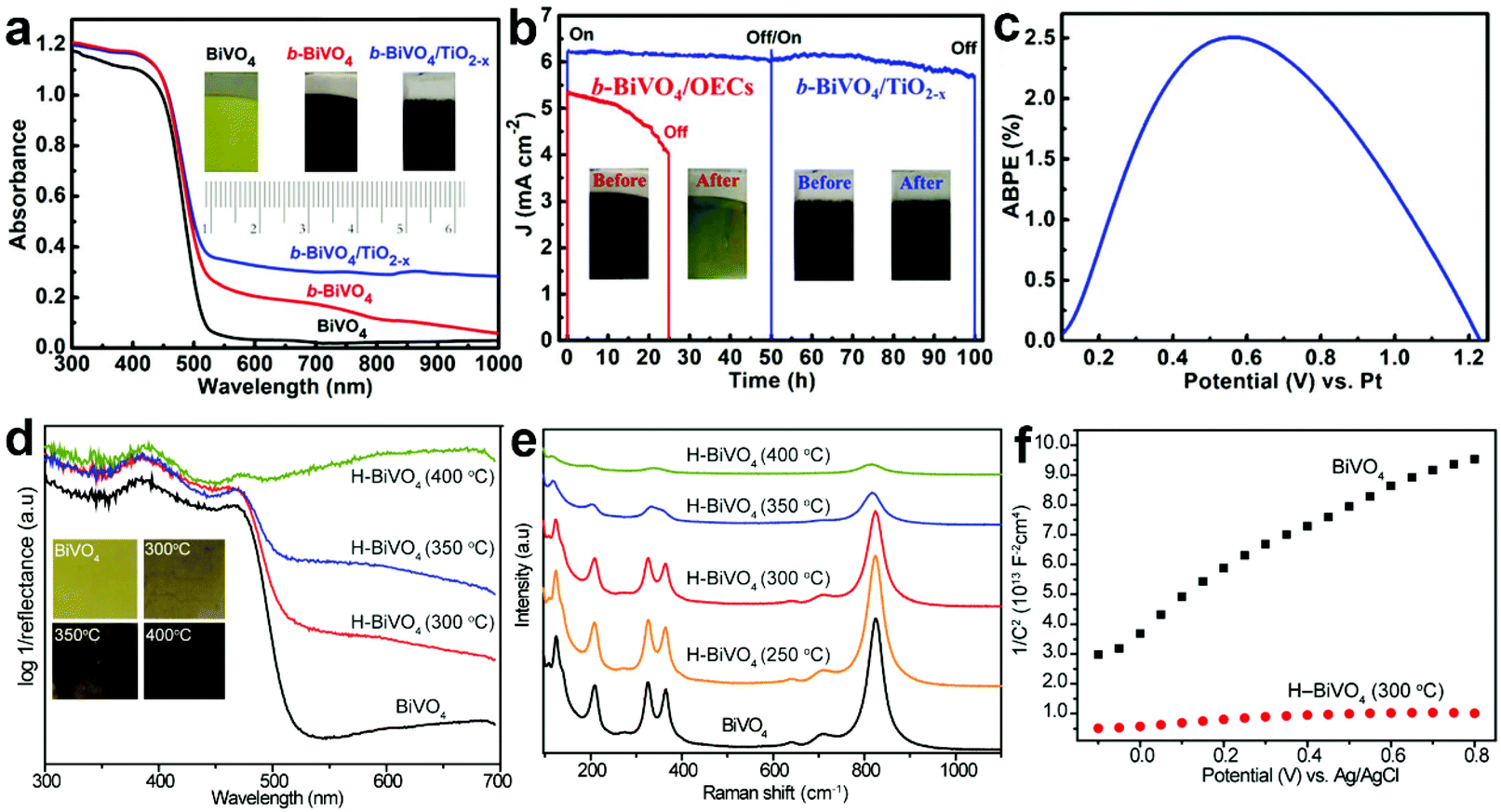 | ||
| Fig. 10 (a) UV-vis absorption spectra of the BiVO4, b-BiVO4, and b-BiVO4/TiO2−x photoanodes. Inset: digital images of the BiVO4, b-BiVO4, and b-BiVO4/TiO2−x photoanodes. (b) Photocurrent density versus time plots of the b-BiVO4/TiO2−x and b-BiVO4/FeOOH/NiOOH photoanodes for PEC water splitting at 1.23 V versus RHE. (c) ABPE curve of the b-BiVO4/TiO2−x photoanode obtained in a two-electrode cell for PEC water splitting. Reproduced from ref. 148 with permission. Copyright 2019, Wiley-VCH. (d) UV-vis absorption spectra of the air-annealed BiVO4 film and H-BiVO4 films hydrogen treated at 300, 350, and 400 °C. Inset: digital images of the air-annealed BiVO4 film and H-BiVO4 films. (e) Raman spectra of the air-annealed BiVO4 film and H-BiVO4 films hydrogen treated at 300, 350, and 400 °C. (f) Mott–Schottky plots of BiVO4 and H-BiVO4 (300 °C) in the dark with a frequency of 10 kHz. Reproduced from ref. 147 with permission. Copyright 2013, American Chemical Society. | ||
In addition to light absorption, the conductivity of semiconductor films is another important factor for efficient PEC water splitting, because it significantly affects the photogenerated charge separation and transfer properties in the photoanode. As for BiVO4, oxygen vacancies can increase the charge carrier density, thus improving the conductivity. For example, DFT calculations revealed that both oxygen vacancies and hydrogen impurities in the hydrogen-treated BiVO4 (H-BiVO4) photoanodes are shallow donors with low formation energies, which can increase the donor densities without introducing deep trap states.147 In addition, the light absorption efficiency is improved with the increase of the hydrogen treatment temperature (Fig. 10d), which is consistent with the formation of more oxygen vacancies and hydrogen impurities, as evidenced by the considerably broader Raman peaks (Fig. 10e). Compared to pristine BiVO4, the H-BiVO4 exhibits a significantly smaller slope in the Mott–Schottky plot (Fig. 10f), indicating the dramatic increase of the donor density, which effectively increases the conductivity within the film. Moreover, electrochemical impedance measurements also confirm that the donor densities of BiVO4 films are dramatically increased by hydrogenation. In addition, oxygen vacancies can be introduced into BiVO4 photoanodes upon electrochemical reduction, which is accompanied by the formation of partial reduction of Bi3+ and V5+.79 The carrier concentration was improved by two times, which significantly improves the charge separation and transfer efficiency. Compared to pristine BiVO4, hydrogen-treated BiVO4 with oxygen vacancies (H-BiVO4−x) exhibits a much higher photocurrent density, due to the enhanced carrier density and conductivity.169 DFT calculations demonstrated that the carrier concentration and mobility of BiVO4 may be significantly enhanced by doping with oxygen vacancies and nitrogen impurities.170 The underlying mechanism is that the localized electrons provided by an oxygen vacancy may be easily ionized, thus contributing to polaron carriers and n-type properties of BiVO4.
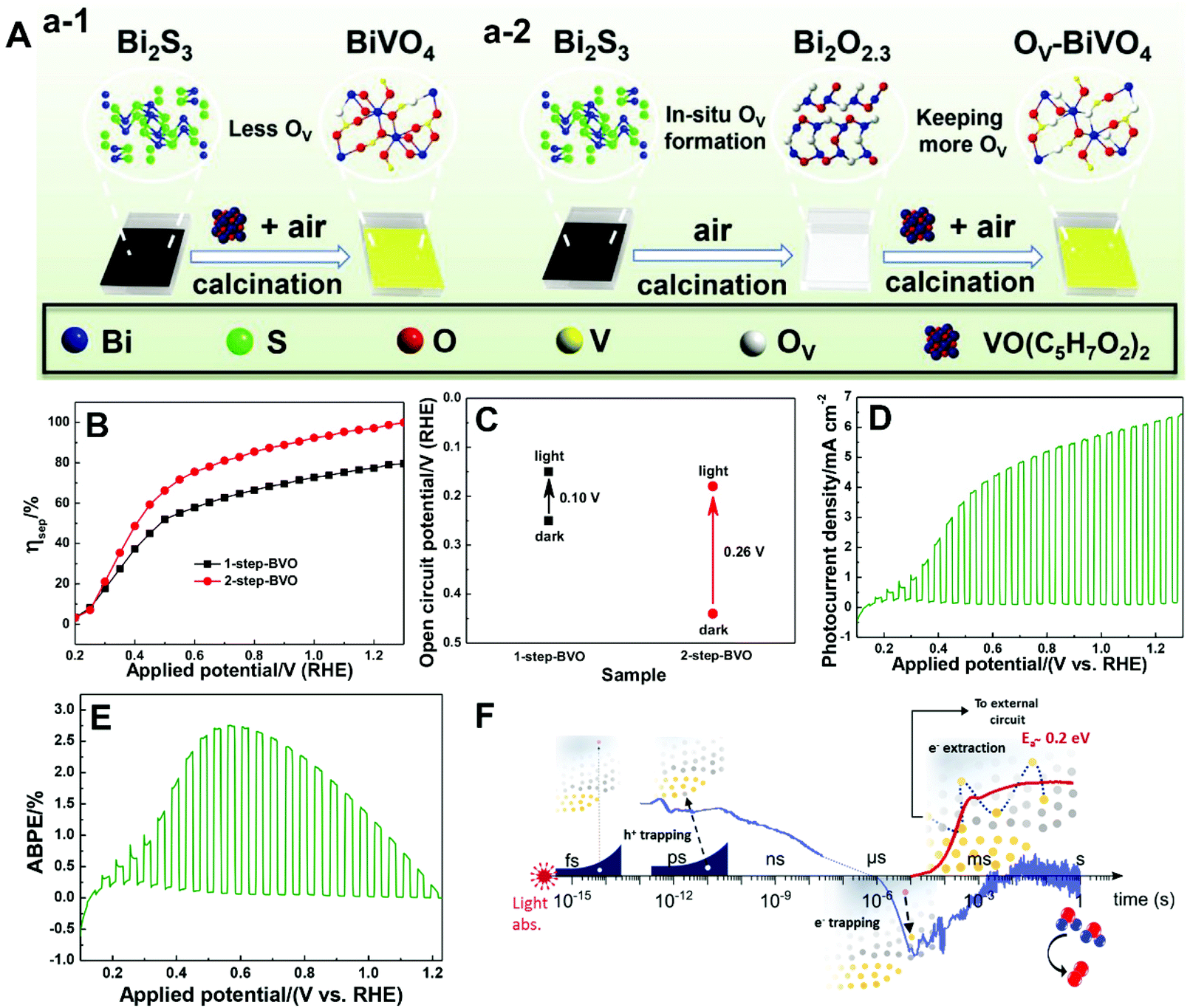 | ||
| Fig. 11 (A) Schematic illustration of the fabrication of BiVO4 films with (a-1) less oxygen vacancies (1-step-BVO) and (a-2) more oxygen vacancies (2-step-BVO). (B) Charge separation efficiencies and (C) open-circuit potential under AM 1.5 G illumination of one-step-BVO and two-step-BVO photoanodes. (D) Photocurrent density versus potential and (E) ABPE curve of dual 2-step-BVO/NiFeOx photoanodes. Reproduced from ref. 81 with permission. Copyright 2020, Wiley-VCH. (F) Schematic illustration of the role of oxygen vacancies in charge carrier trapping and electron transport in BiVO4 from the time scale of light absorption to water oxidation. Reproduced from ref. 173 with permission. Copyright 2019, American Chemical Society. | ||
As illustrated in Fig. 11F, oxygen vacancies play an important role in space-charge layer formation and n-type doping of bulk BiVO4 to reduce resistance losses during charge transport, promoting charge separation.173 Moreover, oxygen vacancies also participate in the trapping of photogenerated electrons and holes. In the bulk of BiVO4, oxygen vacancies can trap the photogenerated holes that are energetically unable to drive water oxidation, leading to the loss of photogenerated holes. On the other hand, electron trapping is reversible, and the de-trapping activation energy is approximately 0.2 eV. Generally, electron trapping takes place in the space-charge layer, in which most oxygen vacancies are ionized in the dark, leading to a thermally activated electron extraction into the external circuit. As a result, a higher PEC water splitting performance is achieved at higher temperatures, which is essential for technological application of BiVO4 photoanodes with oxygen vacancies under one sun or concentrated sunlight illumination.
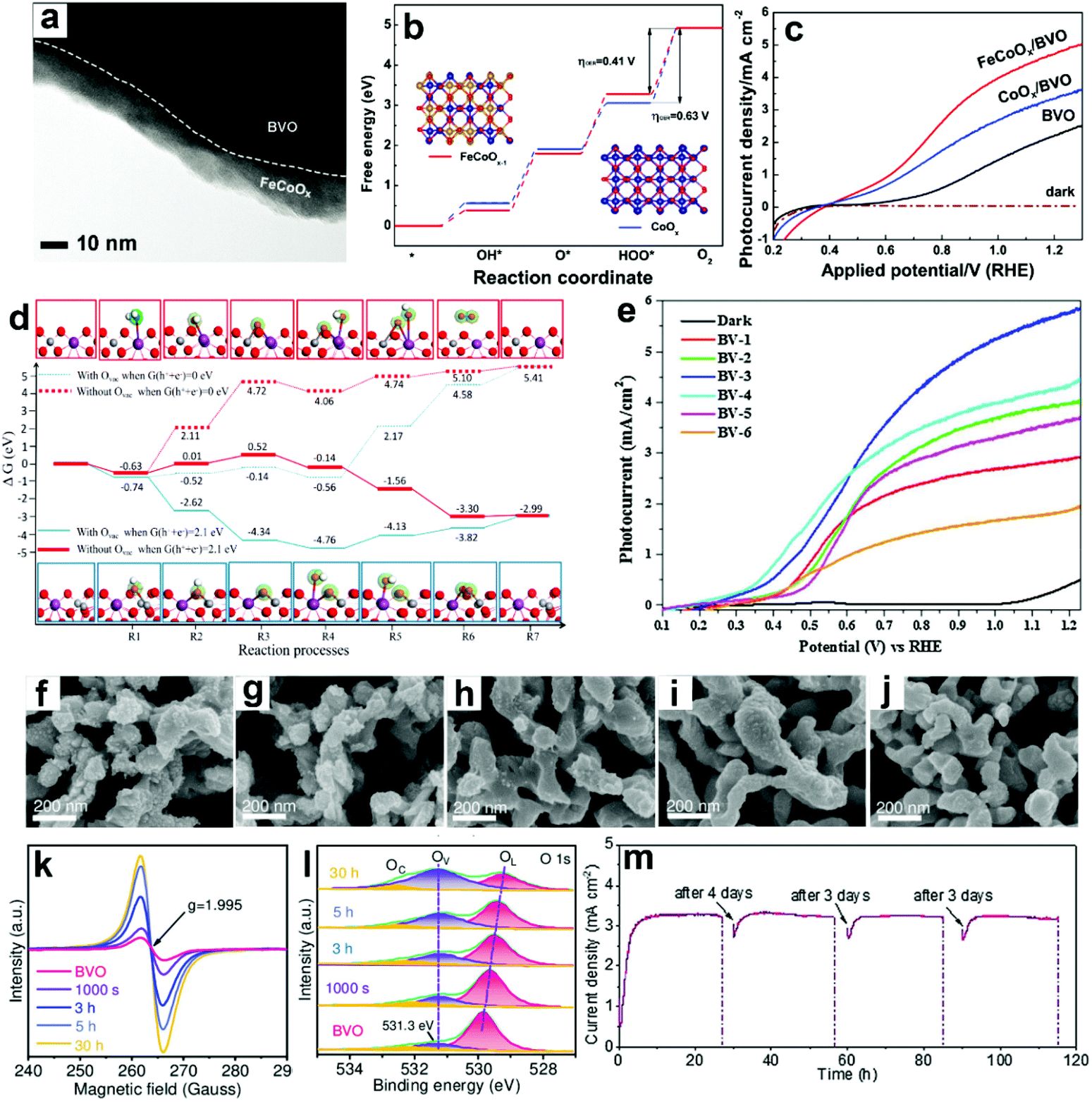 | ||
| Fig. 12 (a) TEM image of BiVO4 coated with FeCoOx as the oxygen evolution cocatalyst (FeCoOx/BVO). (b) Free energy diagram of CoOx and FeCoOx−1 (i.e., FeCoOx with oxygen vacancies) for the OER pathway. Insets: the models of FeCoOx−1 (left) and CoOx (right): blue spheres: Co atoms, gold spheres: Fe atoms, and red spheres: O atoms. (c) Photocurrent density versus applied potential curves. Reproduced from ref. 174 with permission. Copyright 2018, Wiley-VCH. (d) Free energy diagram of the OER on BiVO4 (010) facets and BiVO4 (010) facets with oxygen vacancies. Reproduced from ref. 177 with permission. Copyright 2017, American Chemical Society. (e) Photocurrent density versus applied potential curves for sulfite oxidation under light illumination (100 mW cm−2). Reproduced from ref. 179 with permission. Copyright 2017, Elsevier. Characteristics of BiVO4 photoanodes potentiostatically photopolarized at 0.8 V versus RHE as a function of time (0 s, 1000 s, 3 h, 5 h, and 30 h) under AM 1.5 G (100 mA cm−2) illumination: SEM images of samples for (f) 0 s, (g) 1000 s, (h) 3 h, (i) 5 h, (j) 30 h. (k) EPR spectra. (l) High-resolution O 1s XPS peak. (m) Cycling photostability of BiVO4 photoanodes potentiostatically photopolarized at 0.8 V versus RHE in 1 M KBi (pH 9.5). Reproduced from ref. 94 with permission. Copyright 2020, Wiley-VCH. | ||
DFT calculations also suggested that the V site is the active site for PEC water splitting in BiVO4 photoanodes, and the number of active sites is significantly increased by the formation of oxygen vacancies.177 Compared to pristine BiVO4 photoanodes, the adsorption energies for H2Oads, OHads, and Oads are higher in the oxygen vacancy enriched one (Fig. 12d), indicating the enhanced hole transfer efficiency from the photoanode surface to the electrolyte for the OER. These DFT calculation results are consistent with the experimental results.178 A multi-cycle electrodeposition process followed by high-temperature annealing was developed to prepare multi-layer BiVO4 photoanodes with abundant oxygen vacancies and V4+ in the crystal lattice, which promotes mass and charge transfer, leading to a photocurrent density of 5.80 mA cm−2 in sulfite oxidation at 1.23 V versus RHE under AM 1.5 G illumination (Fig. 12e).179 By incorporating nitrogen into BiVO4 photoanodes, oxygen vacancies and V5+/V4+ redox channels were generated, which diminished surface recombination, promoting the injection of photogenerated holes into the electrolyte for the OER.180
It was found that oxygen vacancies can directly enhance OER reactivity by tailoring coordination of surface metal sites in BiVO4 photoanodes.149 In the presence of oxygen vacancies, the optimized surface catalytic kinetics significantly promote the surface reaching holes for the OER on the photoanode surface without any oxygen evolution cocatalysts, exhibiting a high photocurrent density of 4.32 mA cm−2 at 1.23 V versus RHE under AM 1.5 G illumination. Similarly, a potentiostatically photopolarized process generated surface oxygen vacancies in BiVO4 photoanodes.94 By tuning the potentiostatic photopolarization time, porous BiVO4 photoanodes with different numbers of oxygen vacancies were obtained (Fig. 12f–l). Without any oxygen evolution cocatalysts, the obtained BiVO4 photoanode exhibited a record-high photocurrent density of 4.60 mA cm−2 at 1.23 V versus RHE with a low onset potential of 0.23 V versus RHE in a borate buffer electrolyte without any sacrificial agent under AM 1.5 G illumination. Impressively, an excellent photostability of over 100 h was achieved under the intermittent test (Fig. 12m). These very encouraging works demonstrate the excellent cocatalyst-like function of oxygen vacancies, which may inspire the design of efficient cocatalyst-free BiVO4 photoanodes for PEC water splitting.
4. Conclusion and outlook
BiVO4 is one of the most promising photoanode materials for PEC water splitting, and has attracted great attention in recent years. With the in-depth theoretical understanding on the material design and the development of cutting-edge time-resolve technologies, great breakthroughs have been achieved. The record photocurrent density of the BiVO4 single junction photoanode is 6.24 mA cm−2 at 1.23 V versus RHE under AM 1.5 G illumination in the presence of oxygen vacancies and the NiFeOx cocatalyst,81 whereas the WO3/BiVO4/Co–Pi heterojunction photoanode can achieve a record photocurrent density of 6.72 mA cm−2 at 1.23 V versus RHE under AM 1.5 G illumination.181 Since the theoretical photocurrent density of BiVO4 photoanodes is 7.5 mA cm−2 under AM 1.5 G illumination, the experimental values achieved indicate the superior PEC performance of BiVO4 compared to other photoanode materials. Recently, vacancy defect engineering has stood out to be an efficient strategy to tailor the optoelectronic properties of BiVO4 photoanodes, which has attracted increasing attention. This review article briefly introduces the fundamental properties of BiVO4 including crystal structure, optical properties, carrier transport and photogenerated carrier lifetime. Then, emerging strategies to generate vacancy defects including oxygen vacancies, vanadium vacancies and bismuth vacancies in BiVO4 photoanodes are systematically presented. Finally, the roles of vacancy defects in BiVO4 photoanodes in terms of tuning the electronic structure, promoting charge separation, and increasing surface photoreaction kinetics are critically discussed.Although great achievements have been made for vacancy defect engineering of BiVO4 photoanodes, some emerging challenges should not be ignored. For example, most of the reports confirmed the positive effect of oxygen vacancies for BiVO4 photoanodes. However, recent DFT calculations revealed that neutral oxygen vacancies not only apparently decrease the bandgap but also cause localized lattice distortions by forming stable V–O–V bonds and localize electrons in V5+ atoms by forming V4+ small polarons, which act as recombination centers and dramatically accelerate the nonradiative electron recombination by a factor of approximately 1100 compared to that of the pristine one.182 Therefore, in-depth understanding of the formation of oxygen vacancies and their effect on the optoelectronic properties of BiVO4 photoanodes is still required. In addition, BiVO4 exhibits facet-dependent PEC water splitting performance, so oxygen vacancies on different facets of BiVO4 may show different PEC performance. How to selectively generate oxygen vacancies on different facets of BiVO4 is challenging while worth investigating. Many reports have shown the different roles of oxygen vacancies at the surface and in the bulk of BiVO4 photoanodes. How to control the oxygen vacancies in different locations of the BiVO4 film is another challenging task. On the other hand, excessive oxygen vacancies generally show a detrimental effect on the PEC performance of BiVO4; is it possible to quantitatively control the oxygen vacancies? The development of cutting-edge technologies may provide a reliable way for the quantitative analysis of oxygen vacancies. With the development of knowledge to deeply understand the formation of oxygen vacancies, maybe we can develop a reliable approach to quantitatively control the oxygen vacancies in BiVO4 photoanodes. Compared to oxygen vacancies, very few reports investigated other defects such as vanadium or bismuth defects in BiVO4 photoanodes. How to control the type, location and quantity of these kinds of defects is another question. In addition, the multiple-effect roles of vacancy defects in the three main PEC processes are difficult to be decoupled for better analysis, which is challenging for the further development of vacancy defect engineering of BiVO4 photoanodes. Considering that the photocurrent densities of BiVO4 photoanodes are already very close to the theoretical maximum, to further improve the PEC performance of BiVO4, the next step should be the effective reduction of its bandgap. How to reduce the bandgap of BiVO4 while keeping the high charge separation and transfer properties through vacancy defect engineering is also very challenging.
Owing to the suitable band edge positions and the relatively narrow bandgap for visible light absorption, BiVO4 photoanodes are very promising for unbiased PEC water splitting when coupled with another photocathode or photovoltaic (PV) device. For example, connecting the WO3/BiVO4/Co–Pi heterojunction photoanode with a double-junction GaAs/InGaAsP photovoltaic cell can achieve a STH efficiency of 8.1%.181 In spite of the high STH efficiency, the utilization of the double-junction GaAs/InGaAsP photovoltaic cell is extremely expensive, which should be taken into account for possible practical applications. With the rapid development of low-cost metal halide perovskite solar cells, coupling BiVO4 photoanodes with perovskite solar cells may be a good choice to keep the high STH efficiency while reducing the cost. Our group developed a wireless device composed of the oxygen vacancy enriched BiVO4/FeOOH/NiOOH dual photoanodes and a single sealed perovskite solar cell, leading to a STH efficiency of 6.5%.80 The long-term stability of this device is mainly determined by the perovskite solar cell. Compared to the BiVO4-PV system, the BiVO4-photocathode system is more desirable for possible practical applications due to the much lower cost. By coupling the BiVO4 photoanode with a Cu2O photocathode, a STH efficiency of 3% is achieved.183 In addition, a standalone overall solar water splitting system composed of a BiVO4 photoanode and a Cu2ZnSnS4 photocathode exhibited a higher STH efficiency of 3.17%.184 Very recently, an integrated system composed of a BiVO4 photoanode and an organic polymer-based photocathode (PBDB-T:ITIC:PC71BM) made a new breakthrough of the STH efficiency to 4.3%.185 Therefore, with the development of a more efficient photocathode, the STH efficiency of the BiVO4 photoanode coupled with another photocathode to construct an unbiased PEC water splitting system can be further improved. Possibly, the combination of vacancy defect engineering with other state-of-the-art strategies can develop both an efficient BiVO4 photoanode and photocathode, leading to a new breakthrough of the STH efficiency, thus promoting the PEC water splitting technology a step closer to practical applications. We wish to see more exciting development of efficient BiVO4 photoanodes upon vacancy defect engineering in the near future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the financial support from National Natural Science Foundation of China (No. 52002328), the Fundamental Research Funds for the Central Universities, the Joint Research Funds of Department of Science & Technology of Shaanxi Province and Northwestern Polytechnical University (2020GXLH-Z-018), and Australian Research Council through its Discovery Projects (DPs) and Laureate Fellowship.References
- Z.-j. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016–8035 CrossRef CAS PubMed.
- Z.-H. Zhou, K.-H. Chen, S. Gao, Z.-W. Yang and L.-N. He, Research, 2020, 2020, 9398285 CAS.
- M.-q. Cao, K. Liu, H.-m. Zhou, H.-m. Li, X.-h. Gao, X.-q. Qiu and M. Liu, J. Cent. South Univ., 2019, 26, 1503–1509 CrossRef CAS.
- P. Wang, S. Wang, H. Wang, Z. Wu and L. Wang, Part. Part. Syst. Charact., 2018, 35, 1700371 CrossRef.
- Z. Sun and Z.-q. Sun, J. Cent. South Univ., 2020, 27, 1074–1103 CrossRef CAS.
- M. Z. Jacobson, M. A. Delucchi, Z. A. F. Bauer, S. C. Goodman, W. E. Chapman, M. A. Cameron, C. Bozonnat, L. Chobadi, H. A. Clonts, P. Enevoldsen, J. R. Erwin, S. N. Fobi, O. K. Goldstrom, E. M. Hennessy, J. Liu, J. Lo, C. B. Meyer, S. B. Morris, K. R. Moy, P. L. O'Neill, I. Petkov, S. Redfern, R. Schucker, M. A. Sontag, J. Wang, E. Weiner and A. S. Yachanin, Joule, 2017, 1, 108–121 CrossRef.
- Y. Qiu, Z. Pan, H. Chen, D. Ye, L. Guo, Z. Fan and S. Yang, Sci. Bull., 2019, 64, 1348–1380 CrossRef CAS.
- Z. Luo, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 2158–2181 RSC.
- D. K. Lee, D. Lee, M. A. Lumley and K. S. Choi, Chem. Soc. Rev., 2019, 48, 2126–2157 RSC.
- Y. He, T. Hamann and D. Wang, Chem. Soc. Rev., 2019, 48, 2182–2215 RSC.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- B. Liu, S. Feng, L. Yang, C. Li, Z. Luo, T. Wang and J. Gong, Energy Environ. Sci., 2020, 13, 221–228 RSC.
- S. Wang and L. Wang, Tungsten, 2019, 1, 19–45 CrossRef.
- W. Wang, M. Xu, X. Xu, W. Zhou and Z. Shao, Angew. Chem., Int. Ed., 2020, 59, 136–152 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- T. Yao, X. An, H. Han, J. Q. Chen and C. Li, Adv. Energy Mater., 2018, 8, 1800210 CrossRef.
- A. Riapanitra, Y. Asakura and S. Yin, Tungsten, 2019, 1, 306–317 CrossRef.
- S. Wang, H. Chen, G. Gao, T. Butburee, M. Lyu, S. Thaweesak, J.-H. Yun, A. Du, G. Liu and L. Wang, Nano Energy, 2016, 24, 94–102 CrossRef CAS.
- T. Butburee, Y. Bai, H. Wang, H. Chen, Z. Wang, G. Liu, J. Zou, P. Khemthong, G. Q. Lu and L. Wang, Adv. Mater., 2018, 30, 1705666 CrossRef PubMed.
- P. Peerakiatkhajohn, J.-H. Yun, H. Chen, M. Lyu, T. Butburee and L. Wang, Adv. Mater., 2016, 28, 6405–6410 CrossRef CAS PubMed.
- S. Wang, L. Wang and W. Huang, J. Mater. Chem. A, 2020, 8, 24307–24352 RSC.
- P. Liu, C. Wang, L. Wang, X. Wu, L. Zheng and H. G. Yang, Research, 2020, 2020, 5473217 CAS.
- S. Wang, F. Tang and L. Wang, J. Inorg. Mater., 2018, 33, 173–197 CrossRef.
- X. Zou, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 21474–21502 RSC.
- Y. Yang, S. Wang, Y. Jiao, Z. Wang, M. Xiao, A. Du, Y. Li, J. Wang and L. Wang, Adv. Funct. Mater., 2018, 28, 1805698 CrossRef.
- R. Chen, C. Zhen, Y. Yang, X. Sun, J. T. S. Irvine, L. Wang, G. Liu and H.-M. Cheng, Nano Energy, 2019, 59, 683–688 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
- G. Liu, S. Ye, P. Yan, F. Xiong, P. Fu, Z. Wang, Z. Chen, J. Shi and C. Li, Energy Environ. Sci., 2016, 9, 1327–1334 RSC.
- X. Wang, B. Liu, X. Xiao, S. Wang and W. Huang, J. Mater. Chem. C, 2021 10.1039/D1TC04142H.
- S. Wang, Y. Li, X. Wang, G. Zi, C. Zhou, B. Liu, G. Liu, L. Wang and W. Huang, J. Mater. Sci. Technol., 2022, 104, 155–162 CrossRef.
- R. Liu, H.-L. Fei and G.-L. Ye, Tungsten, 2020, 2, 147–161 CrossRef.
- K. Matoba, M. Takahashi, Y. Matsuda and S. Higashimoto, J. Electroanal. Chem., 2021, 895, 115489 CrossRef CAS.
- C.-B. Sun, Y.-W. Zhong, W.-J. Fu, Z.-Q. Zhao, J. Liu, J. Ding, X.-P. Han, Y.-D. Deng, W.-B. Hu and C. Zhong, Tungsten, 2020, 2, 109–133 CrossRef.
- K. Feng, D. Huang, L. Li, K. Wang, J. Li, T. Harada, S. Ikeda and F. Jiang, Appl. Catal., B, 2020, 268, 118438 CrossRef CAS.
- C. T. Altaf, N. S. Sahsuvar, N. Abdullayeva, O. Coskun, A. Kumtepe, E. Karagoz, M. Sankir and N. D. Sankir, ACS Sustainable Chem. Eng., 2020, 8, 15209–15222 CrossRef.
- J. Ke, F. He, H. Wu, S. Lyu, J. Liu, B. Yang, Z. Li, Q. Zhang, J. Chen, L. Lei, Y. Hou and K. Ostrikov, Nano-Micro Lett., 2021, 13, 24 CrossRef PubMed.
- F. Zhang, Z. Ma, Z. Shi, X. Chen, D. Wu, X. Li and C. Shan, Energy Mater. Adv., 2021, 2021, 5198145 Search PubMed.
- J. Chen, C. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Energy Material Advances, 2020, 10, 1902433 CrossRef CAS.
- J. Yuan, H. Liu, S. Wang and X. Li, Nanoscale, 2021, 13, 10281–10304 RSC.
- M. Crespo-Quesada, L. M. Pazos-Outón, J. Warnan, M. F. Kuehnel, R. H. Friend and E. Reisner, Nat. Commun., 2016, 7, 12555 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 2001 CrossRef PubMed.
- K. H. Ye, H. Li, D. Huang, S. Xiao, W. Qiu, M. Li, Y. Hu, W. Mai, H. Ji and S. Yang, Nat. Commun., 2019, 10, 3687 CrossRef PubMed.
- W. T. Qiu, S. Xiao, J. W. Ke, Z. Wang, S. T. Tang, K. Zhang, W. Qian, Y. C. Huang, D. Huang, Y. X. Tong and S. H. Yang, Angew. Chem., Int. Ed., 2019, 58, 19087–19095 CrossRef CAS PubMed.
- X. Ning, B. Lu, Z. Zhang, P. Du, H. Ren, D. Shan, J. Chen, Y. Gao and X. Lu, Angew. Chem., Int. Ed., 2019, 58, 16800–16805 CrossRef CAS PubMed.
- Q. Meng, B. Zhang, L. Fan, H. Liu, M. Valvo, K. Edstrom, M. Cuartero, R. De Marco, G. A. Crespo and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 19027–19033 CrossRef CAS PubMed.
- Y. Park, K. J. McDonald and K. S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229–230 CrossRef CAS.
- K. Sayama, A. Nomura, Z. Zou, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2003, 2908–2909 RSC.
- S. K. Pilli, T. E. Furtak, L. D. Brown, T. G. Deutsch, J. A. Turner and A. M. Herring, Energy Environ. Sci., 2011, 4, 5028–5034 RSC.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- K. J. McDonald and K.-S. Choi, Energy Environ. Sci., 2012, 5, 8553 RSC.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed.
- X. Xu, S. Jin, C. Yang, J. Pan, W. Du, J. Hu, H. Zeng and Y. Zhou, Sol. RRL, 2019, 3, 1900115 CrossRef CAS.
- S. Wang, J.-H. Yun, B. Luo, T. Butburee, P. Peerakiatkhajohn, S. Thaweesak, M. Xiao and L. Wang, J. Mater. Sci. Technol., 2017, 33, 1–22 CrossRef.
- H. Ren, T. Dittrich, H. Ma, J. N. Hart, S. Fengler, S. Chen, Y. Li, Y. Wang, F. Cao, M. Schieda, Y. H. Ng, Z. Xie, X. Bo, P. Koshy, L. R. Sheppard, C. Zhao and C. C. Sorrell, Adv. Mater., 2019, 31, 1807204 CrossRef PubMed.
- Y. Zhou, L. Zhang, L. Lin, B. R. Wygant, Y. Liu, Y. Zhu, Y. Zheng, C. B. Mullins, Y. Zhao, X. Zhang and G. Yu, Nano Lett., 2017, 17, 8012–8017 CrossRef CAS PubMed.
- M. Zhong, T. Hisatomi, Y. Kuang, J. Zhao, M. Liu, A. Iwase, Q. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
- Y. Kuang, Q. Jia, G. Ma, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Nakabayashi, N. Shibata, T. Yamada, A. Kudo and K. Domen, Nat. Energy, 2016, 2, 16191 CrossRef.
- K.-H. Ye, Z. Wang, J. Gu, S. Xiao, Y. Yuan, Y. Zhu, Y. Zhang, W. Mai and S. Yang, Energy Environ. Sci., 2017, 10, 772–779 RSC.
- Y. Shi, Y. Yu, Y. Yu, Y. Huang, B. Zhao and B. Zhang, ACS Energy Lett., 2018, 3, 1648–1654 CrossRef CAS.
- Q. Shi, S. Murcia-López, P. Tang, C. Flox, J. R. Morante, Z. Bian, H. Wang and T. Andreu, ACS Catal., 2018, 8, 3331–3342 CrossRef CAS.
- W. J. Jo, J.-W. Jang, K.-j. Kong, H. J. Kang, J. Y. Kim, H. Jun, K. P. S. Parmar and J. S. Lee, Angew. Chem., Int. Ed., 2012, 51, 3147–3151 CrossRef CAS PubMed.
- T. W. Kim, Y. Ping, G. A. Galli and K.-S. Choi, Nat. Commun., 2015, 6, 8769 CrossRef CAS PubMed.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- Y. Kuang, Q. Jia, H. Nishiyama, T. Yamada, A. Kudo and K. Domen, Adv. Energy Mater., 2016, 6, 1501645 CrossRef.
- Y. Qiu, W. Liu, W. Chen, W. Chen, G. Zhou, P.-C. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, Sci. Adv., 2016, 2, e1501764 CrossRef PubMed.
- T. Li, J. He, B. Peña and C. P. Berlinguette, Angew. Chem., Int. Ed., 2016, 55, 1769–1772 CrossRef CAS PubMed.
- B. J. Trześniewski and W. A. Smith, J. Mater. Chem. A, 2016, 4, 2919–2926 RSC.
- E. Y. Liu, J. E. Thorne, Y. He and D. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 22083–22087 CrossRef CAS PubMed.
- B. J. Trześniewski, I. A. Digdaya, T. Nagaki, S. Ravishankar, I. Herraiz-Cardona, D. A. Vermaas, A. Longo, S. Gimenez and W. A. Smith, Energy Environ. Sci., 2017, 10, 1517–1529 RSC.
- B. Lamm, B. J. Trześniewski, H. Döscher, W. A. Smith and M. Stefik, ACS Energy Lett., 2018, 3, 112–124 CrossRef CAS.
- M. Tayebi, A. Tayyebi and B.-K. Lee, Sol. Energy, 2019, 191, 427–434 CrossRef CAS.
- H. S. Han, S. Shin, D. H. Kim, I. J. Park, J. S. Kim, P.-S. Huang, J.-K. Lee, I. S. Cho and X. Zheng, Energy Environ. Sci., 2018, 11, 1299–1306 RSC.
- P. Li, X. Chen, H. He, X. Zhou, Y. Zhou and Z. Zou, Adv. Mater., 2018, 30, 1703119 CrossRef PubMed.
- J. Song, M. J. Seo, T. H. Lee, Y.-R. Jo, J. Lee, T. L. Kim, S.-Y. Kim, S.-M. Kim, S. Y. Jeong, H. An, S. Kim, B. H. Lee, D. Lee, H. W. Jang, B.-J. Kim and S. Lee, ACS Catal., 2018, 8, 5952–5962 CrossRef CAS.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- S. Wang, P. Chen, J. H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed.
- S. Wang, P. Chen, Y. Bai, J.-H. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef PubMed.
- S. Wang, T. He, P. Chen, A. Du, K. Ostrikov, W. Huang and L. Wang, Adv. Mater., 2020, 32, 2001385 CrossRef CAS PubMed.
- J.-B. Pan, B.-H. Wang, J.-B. Wang, H.-Z. Ding, W. Zhou, X. Liu, J.-R. Zhang, S. Shen, J.-K. Guo, L. Chen, C.-T. Au, L.-L. Jiang and S.-F. Yin, Angew. Chem., Int. Ed., 2021, 60, 1433–1440 CrossRef CAS PubMed.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- S. Chen, D. Huang, P. Xu, X. Gong, W. Xue, L. Lei, R. Deng, J. Li and Z. Li, ACS Catal., 2020, 10, 1024–1059 CrossRef CAS.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- S. Bai, N. Zhang, C. Gao and Y. Xiong, Nano Energy, 2018, 53, 296–336 CrossRef CAS.
- D. Maarisetty and S. S. Baral, J. Mater. Chem. A, 2020, 8, 18560–18604 RSC.
- L. Ran, J. Hou, S. Cao, Z. Li, Y. Zhang, Y. Wu, B. Zhang, P. Zhai and L. Sun, Sol. RRL, 2020, 4, 1900487 CrossRef CAS.
- P. Raizada, V. Soni, A. Kumar, P. Singh, A. A. P. Khan, A. M. Asiri, V. K. Thakur and V.-H. Nguyen, J. Materiomics, 2021, 7, 388–418 CrossRef.
- S. A. Monny, Z. Wang, T. Lin, P. Chen, B. Luo and L. Wang, Chem. Commun., 2020, 56, 9376–9379 RSC.
- Z. Wang, X. Mao, P. Chen, M. Xiao, S. A. Monny, S. Wang, M. Konarova, A. Du and L. Wang, Angew. Chem., Int. Ed., 2019, 58, 1030–1034 CrossRef CAS PubMed.
- Z. Wang, L. Zhang, T. U. Schülli, Y. Bai, S. A. Monny, A. Du and L. Wang, Angew. Chem., Int. Ed., 2019, 58, 17604–17609 CrossRef CAS PubMed.
- K. R. Tolod, S. Hernández, M. Castellino, F. A. Deorsola, E. Davarpanah and N. Russo, Int. J. Hydrogen Energy, 2020, 45, 605–618 CrossRef CAS.
- R.-T. Gao and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 23094–23099 CrossRef CAS PubMed.
- S. Feng, T. Wang, B. Liu, C. Hu, L. Li, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 2044–2048 CrossRef CAS PubMed.
- A. Tayyebi, T. Soltani, H. Hong and B.-K. Lee, J. Colloid Interface Sci., 2018, 514, 565–575 CrossRef CAS PubMed.
- J. Wang, Y. Song, J. Hu, Y. Li, Z. Wang, P. Yang, G. Wang, Q. Ma, Q. Che, Y. Dai and B. Huang, Appl. Catal., B, 2019, 251, 94–101 CrossRef CAS.
- X. Liang, P. Wang, F. Tong, X. Liu, C. Wang, M. Wang, Q. Zhang, Z. Wang, Y. Liu, Z. Zheng, Y. Dai and B. Huang, Adv. Funct. Mater., 2021, 31, 2008656 CrossRef CAS.
- T. Tran-Phu, Z. Fusco, I. D. Bernardo, J. Lipton-Duffin, C. Y. Toe, R. Daiyan, T. Gengenbach, C.-H. Lin, R. Bo, H. T. Nguyen, G. M. J. Barca, T. Wu, H. Chen, R. Amal and A. Tricoli, Chem. Mater., 2021, 33, 3553–3565 CrossRef CAS.
- L. Hao, H. Huang, Y. Zhang and T. Ma, Adv. Funct. Mater., 2021, 31, 2100919 CrossRef CAS.
- Wendusu, T. Honda, T. Masui and N. Imanaka, RSC Adv., 2013, 3, 24941–24945 RSC.
- X. Zhang, T. Chen, Y. Xu, W. Jiang, J. Liu and Z. Xie, J. Sol-Gel Sci. Technol., 2019, 91, 127–137 CrossRef CAS.
- D. T. T. Trinh, W. Khanitchaidecha, D. Channei and A. Nakaruk, Res. Chem. Intermed., 2019, 45, 5217–5259 CrossRef CAS.
- J. Jian, Y. Xu, X. Yang, W. Liu, M. Fu, H. Yu, F. Xu, F. Feng, L. Jia, D. Friedrich, R. van de Krol and H. Wang, Nat. Commun., 2019, 10, 2609 CrossRef PubMed.
- M. Q. Pham, T. M. Ngo, V. H. Nguyen, L. X. Nong, D.-V. N. Vo, T. V. Tran, T.-D. Nguyen, X.-T. Bui and T. D. Nguyen, Arabian J. Chem., 2020, 13, 8388–8394 CrossRef CAS.
- X. Zhang, Z. Ai, F. Jia, L. Zhang, X. Fan and Z. Zou, Mater. Chem. Phys., 2007, 103, 162–167 CrossRef CAS.
- F. F. Abdi, S. P. Berglund and R. van de Krol, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, 2016, pp. 355–391 Search PubMed.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- N. D. Phu, L. H. Hoang, P. K. Vu, M.-H. Kong, X.-B. Chen, H. C. Wen and W. C. Chou, J. Mater. Sci.: Mater. Electron., 2016, 27, 6452–6456 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed., 2018, 57, 2248–2252 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. Liu, J. Pan, X. Li, X. Meng, H. Yuan, Y. Li, Y. Zhao, D. Wang, J. Ma, S. Zhu and L. Kong, Nanoscale, 2020, 12, 14853–14862 RSC.
- Z. Zhao, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2011, 13, 4746–4753 RSC.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, P.-A. Glans, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, Chem. Mater., 2014, 26, 5365–5373 CrossRef CAS.
- Y.-X. Ma, B. Gao, J. He, J.-F. Ma and Y. Zhao, Chem. Eng. J., 2021, 422, 130092 CrossRef CAS.
- H. Chen, S. Wang, J. Wu, X. Zhang, J. Zhang, M. Lyu, B. Luo, G. Qian and L. Wang, J. Mater. Chem. A, 2020, 8, 13231–13240 RSC.
- H. Tian, H. Wu, Y. Fang, R. Li and Y. Huang, J. Hazard. Mater., 2020, 399, 123159 CrossRef CAS PubMed.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, Y.-S. Liu, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, J. Phys. Chem. C, 2015, 119, 2969–2974 CrossRef CAS.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS.
- M. Ziwritsch, S. Müller, H. Hempel, T. Unold, F. F. Abdi, R. van de Krol, D. Friedrich and R. Eichberger, ACS Energy Lett., 2016, 1, 888–894 CrossRef CAS.
- J. Yang, D. Wang, X. Zhou and C. Li, Chem. – Eur. J., 2013, 19, 1320–1326 CrossRef CAS PubMed.
- L. Chen, F. M. Toma, J. K. Cooper, A. Lyon, Y. Lin, I. D. Sharp and J. W. Ager, ChemSusChem, 2015, 8, 1066–1071 CrossRef CAS PubMed.
- M. Li, L. Zhao and L. Guo, Int. J. Hydrogen Energy, 2010, 35, 7127–7133 CrossRef CAS.
- M. Tayebi and B.-K. Lee, Catal. Today, 2021, 361, 183–190 CrossRef CAS.
- W.-J. Yin, S.-H. Wei, M. M. Al-Jassim, J. Turner and Y. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155102 CrossRef.
- V. Pasumarthi, T. Liu, M. Dupuis and C. Li, J. Mater. Chem. A, 2019, 7, 3054–3065 RSC.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H.-M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef PubMed.
- T. Das, X. Rocquefelte, R. Laskowski, L. Lajaunie, S. Jobic, P. Blaha and K. Schwarz, Chem. Mater., 2017, 29, 3380–3386 CrossRef CAS.
- M. Yabuta, A. Takeda, T. Sugimoto, K. Watanabe, A. Kudo and Y. Matsumoto, J. Phys. Chem. C, 2017, 121, 22060–22066 CrossRef CAS.
- M. Lamers, S. Fiechter, D. Friedrich, F. F. Abdi and R. van de Krol, J. Mater. Chem. A, 2018, 6, 18694–18700 RSC.
- Z. Chen, Z. Liu, J. Zhan, Y. She, P. Zhang, W. Wei, C. Peng, W. Li and J. Tang, Chem. Phys. Lett., 2021, 766, 138342 CrossRef CAS.
- Z. Ma, H. Hou, K. Song, Z. Fang, L. Wang, F. Gao, W. Yang, B. Tang and Y. Kuang, Chem. Eng. J., 2020, 379, 122266 CrossRef CAS.
- L. Grad, Z. Novotny, M. Hengsberger and J. Osterwalder, Sci. Rep., 2020, 10, 10686 CrossRef CAS PubMed.
- S. Corby, L. Francàs, A. Kafizas and J. R. Durrant, Chem. Sci., 2020, 11, 2907–2914 RSC.
- M. Kim, B. Lee, H. Ju, J. Y. Kim, J. Kim and S. W. Lee, Adv. Mater., 2019, 31, 1903316 CrossRef PubMed.
- C. Zhu, C. Li, M. Zheng and J.-J. Delaunay, ACS Appl. Mater. Interfaces, 2015, 7, 22355–22363 CrossRef CAS PubMed.
- Z. Wang and L. Wang, EcoMat, 2021, 3, e12075 CAS.
- G. Wang, Y. Yang, Y. Ling, H. Wang, X. Lu, Y.-C. Pu, J. Z. Zhang, Y. Tong and Y. Li, J. Mater. Chem. A, 2016, 4, 2849–2855 RSC.
- N. Österbacka and J. Wiktor, J. Phys. Chem. C, 2021, 125, 1200–1207 CrossRef.
- Y. Zhang, Y. Bu, F. Jiang, H. Li, X. Chen and J.-P. Ao, Electrochim. Acta, 2021, 366, 137288 CrossRef CAS.
- Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Adv. Mater., 2016, 28, 3831–3892 CrossRef CAS PubMed.
- M. D. Bhatt and J. Y. Lee, J. Electroanal. Chem., 2018, 828, 97–101 CrossRef CAS.
- F. A. Kröger and H. J. Vink, J. Phys. Chem. Solids, 1958, 5, 208–223 CrossRef.
- G. Wang, Y. Ling, X. Lu, F. Qian, Y. Tong, J. Z. Zhang, V. Lordi, C. R. Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957–10964 CrossRef CAS.
- Z. Tian, P. Zhang, P. Qin, D. Sun, S. Zhang, X. Guo, W. Zhao, D. Zhao and F. Huang, Adv. Energy Mater., 2019, 9, 1901287 CrossRef.
- S. Jin, X. Ma, J. Pan, C. Zhu, S. E. Saji, J. Hu, X. Xu, L. Sun and Z. Yin, Appl. Catal., B, 2021, 281, 119477 CrossRef CAS.
- Y. Zhang, X. Chen, F. Jiang, Y. Bu and J.-P. Ao, ACS Sustainable Chem. Eng., 2020, 8, 9184–9194 CrossRef CAS.
- H. L. Tan, A. Suyanto, A. T. D. Denko, W. H. Saputera, R. Amal, F. E. Osterloh and Y. H. Ng, Part. Part. Syst. Charact., 2017, 34, 1600290 CrossRef.
- J. Li, L. Guo, N. Lei, Q. Song and Z. Liang, ChemElectroChem, 2017, 4, 2852–2861 CrossRef CAS.
- Y. Peng, H. Wu, M. Yuan, F.-F. Li, X. Zou, Y. H. Ng and H.-Y. Hsu, Sustainable Energy Fuels, 2021, 5, 2284–2293 RSC.
- D. Xu, Y. Liu, Y. Zhang, Z. Shi, M. Yang, C. Zhang and B. Liu, Chem. Eng. J., 2020, 393, 124693 CrossRef CAS.
- Y. Bu, J. Tian, Z. Chen, Q. Zhang, W. Li, F. Tian and J.-P. Ao, Adv. Mater. Interfaces, 2017, 4, 1601235 CrossRef.
- X. Yin, J. Li, L. Du, F. Zhan, K. Kawashima, W. Li, W. Qiu, Y. Liu, X. Yang, K. Wang, Y. Ning and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 4403–4410 CrossRef CAS.
- X. Chen, D. Wang, Y. Huang, Y. Zhang, C. Li, S. Wang, Y. Liu and X. Zhang, APL Mater., 2020, 8, 031112 CrossRef CAS.
- Q. Qin, Q. Cai, J. Li, C. Jian, W. Hong and W. Liu, Sol. RRL, 2019, 3, 1900301 CrossRef CAS.
- Q. Pan, K. Yang, G. Wang, D. Li, J. Sun, B. Yang, Z. Zou, W. Hu, K. Wen and H. Yang, Chem. Eng. J., 2019, 372, 399–407 CrossRef CAS.
- M. Fang, Q. Cai, Q. Qin, W. Hong and W. Liu, Chem. Eng. J., 2021, 421, 127796 CrossRef CAS.
- Y. Luo, G. Tan, G. Dong, H. Ren and A. Xia, Appl. Surf. Sci., 2016, 364, 156–165 CrossRef CAS.
- Y. Zhang, X. Zhang, D. Wang, F. Wan and Y. Liu, Appl. Surf. Sci., 2017, 403, 389–395 CrossRef CAS.
- X. Yang, A. J. Fernández-Carrión, J. Wang, F. Porcher, F. Fayon, M. Allix and X. Kuang, Nat. Commun., 2018, 9, 4484 CrossRef PubMed.
- B. Anke, M. Rohloff, M. G. Willinger, W. Hetaba, A. Fischer and M. Lerch, Solid State Sci., 2017, 63, 1–8 CrossRef CAS.
- F. S. Hegner, D. Forrer, J. R. Galán-Mascarós, N. López and A. Selloni, J. Phys. Chem. Lett., 2019, 10, 6672–6678 CrossRef CAS PubMed.
- Y. Yuan, Y. Huang, F. Ma, Z. Zhang and X. Wei, J. Mater. Sci., 2017, 52, 8546–8555 CrossRef CAS.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H.-M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Gan, X. Lu, B. B. Rajeeva, R. Menz, Y. Tong and Y. Zheng, ChemElectroChem, 2015, 2, 1385–1395 CrossRef CAS.
- H. Seo, Y. Ping and G. Galli, Chem. Mater., 2018, 30, 7793–7802 CrossRef CAS.
- X. Gu, Y. Luo, Q. Li, R. Wang, S. Fu, X. Lv, Q. He, Y. Zhang, Q. Yan, X. Xu, F. Ji and Y. Qiu, Front. Chem., 2020, 8, 601983 CrossRef CAS PubMed.
- Y. Zhang, J. Bai, J. Wang, S. Chen, H. Zhu, J. Li, L. Li, T. Zhou and B. Zhou, Chem. Eng. J., 2020, 401, 126134 CrossRef CAS.
- S. Selim, E. Pastor, M. Garcia-Tecedor, M. R. Morris, L. Francas, M. Sachs, B. Moss, S. Corby, C. A. Mesa, S. Gimenez, A. Kafizas, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 18791–18798 CrossRef CAS PubMed.
- S. Wang, T. He, J.-H. Yun, Y. Hu, M. Xiao, A. Du and L. Wang, Adv. Funct. Mater., 2018, 28, 1802685 CrossRef.
- W. Wang, P. J. Strohbeen, D. Lee, C. Zhou, J. K. Kawasaki, K.-S. Choi, M. Liu and G. Galli, Chem. Mater., 2020, 32, 2899–2909 CrossRef CAS.
- Y. Zhang, Y. Guo, H. Duan, H. Li, C. Sun and H. Liu, Phys. Chem. Chem. Phys., 2014, 16, 24519–24526 RSC.
- J. Hu, X. Zhao, W. Chen, H. Su and Z. Chen, J. Phys. Chem. C, 2017, 121, 18702–18709 CrossRef CAS.
- X. Zhao, J. Hu, X. Yao, S. Chen and Z. Chen, ACS Appl. Energy Mater., 2018, 1, 3410–3419 CrossRef CAS.
- J.-M. Wu, Y. Chen, L. Pan, P. Wang, Y. Cui, D. Kong, L. Wang, X. Zhang and J.-J. Zou, Appl. Catal., B, 2018, 221, 187–195 CrossRef CAS.
- A. Kahraman, M. B. Vishlaghi, I. Baylam, H. Ogasawara, A. Sennaroğlu and S. Kaya, J. Phys. Chem. Lett., 2020, 11, 8758–8764 CrossRef CAS PubMed.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- C. Cheng, Q. Fang, S. Fernandez-Alberti and R. Long, J. Phys. Chem. Lett., 2021, 12, 3514–3521 CrossRef CAS PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- D. Huang, K. Wang, L. Li, K. Feng, N. An, S. Ikeda, Y. Kuang, Y. Ng and F. Jiang, Energy Environ. Sci., 2021, 14, 1480–1489 RSC.
- S. Ye, W. Shi, Y. Liu, D. Li, H. Yin, H. Chi, Y. Luo, N. Ta, F. Fan, X. Wang and C. Li, J. Am. Chem. Soc., 2021, 143, 12499–12508 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |