DOI:
10.1039/D0TC01389G
(Paper)
J. Mater. Chem. C, 2020,
8, 12470-12477
Spiro-type host materials with rigidified skeletons for RGB phosphorescent OLEDs†
Received
18th March 2020
, Accepted 1st August 2020
First published on 3rd August 2020
Abstract
A universal host material for red, green and blue phosphors raises challenges for the fabrication of efficient phosphorescent organic light-emitting diodes (PHOLEDs). Herein, two spiro host materials, namely 3-(4,6-diphenyl-1,3,5-triazin-2-yl)-9′,9′-dimethyl-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (QAF-TRZ) and 3′-(4,6-diphenyl-1,3,5-triazin-2-yl)-10-phenyl-10H-spiro[acridine-9,9′-fluorene] (STF-TRZ), are designed and synthesized. The designed strategy is more appropriate and rarely found among the reported spiro-based skeletons for organic light-emitting diodes (OLEDs). Both materials exhibited excellent electroluminescence performance. In particular, the RGB three-color PHOLEDs based on QAF-TRZ and STF-TRZ as hosts and FIrpic, Ir(ppy)2acac, and Ir(MDQ)2acac as emitters are fabricated. The blue PHOLEDs based on QAF-TRZ realize a maximum external quantum efficiency (EQE) of 19.4%, and remain 17.2% at 1000 cd m−2. On the other hand, green PHOLEDs show maximum EQEs of 21% and 19% for QAF-TRZ and STF-TRZ, respectively, and both devices exhibit low efficiency roll-off. The red devices obtain EQEs of 22.6% and 19.4% based on QAF-TRZ and STF-TRZ, respectively, in which the former has among the most efficient value. Consequently, these results confirm that host materials incorporating the spiro-motif with sufficiently high triplet energy are promising for obtaining high performance RGB-based PHOLEDs. The better device performance based on QAF-TRZ stems from its better hole transport capability, resulting in a superior charge recombination efficiency.
Introduction
Organic light-emitting diodes (OLEDs) have attracted widespread interest in both academia and industry, because of the fascinating applications in flat panel displays and solid-state lighting.1–5 Generally, in fluorescent OLEDs the non-radiative process limits the internal quantum efficiency (IQE) to 25% while the external quantum efficiency (EQE) hardly exceeds 5%.6 To resolve this issue, Forrest et al. reported phosphorescent organic light-emitting diodes (PHOLEDs) containing platinum(II) or iridium(III) complexes, which could successfully obtain 100% IQE via spin–orbit coupling, utilizing both singlet and triplet excitons.7–11 Since then, many efforts have been made to improve device performance and stability. On the other hand, phosphorescent OLEDs doped into a host material is also highly critical to mitigate triplet–triplet annihilation and concentration quenching phenomena.12,13 Host materials usually have two design strategies. Firstly, to prevent the reverse energy transfer from guest to host, the host materials must have a higher triplet energy level (ET) than the guest. Secondly, to reduce the turn-on voltage and facilitate effective injection of hole and electron, appropriate levels of the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) of the host to adjacent material layers are highly required.14–19
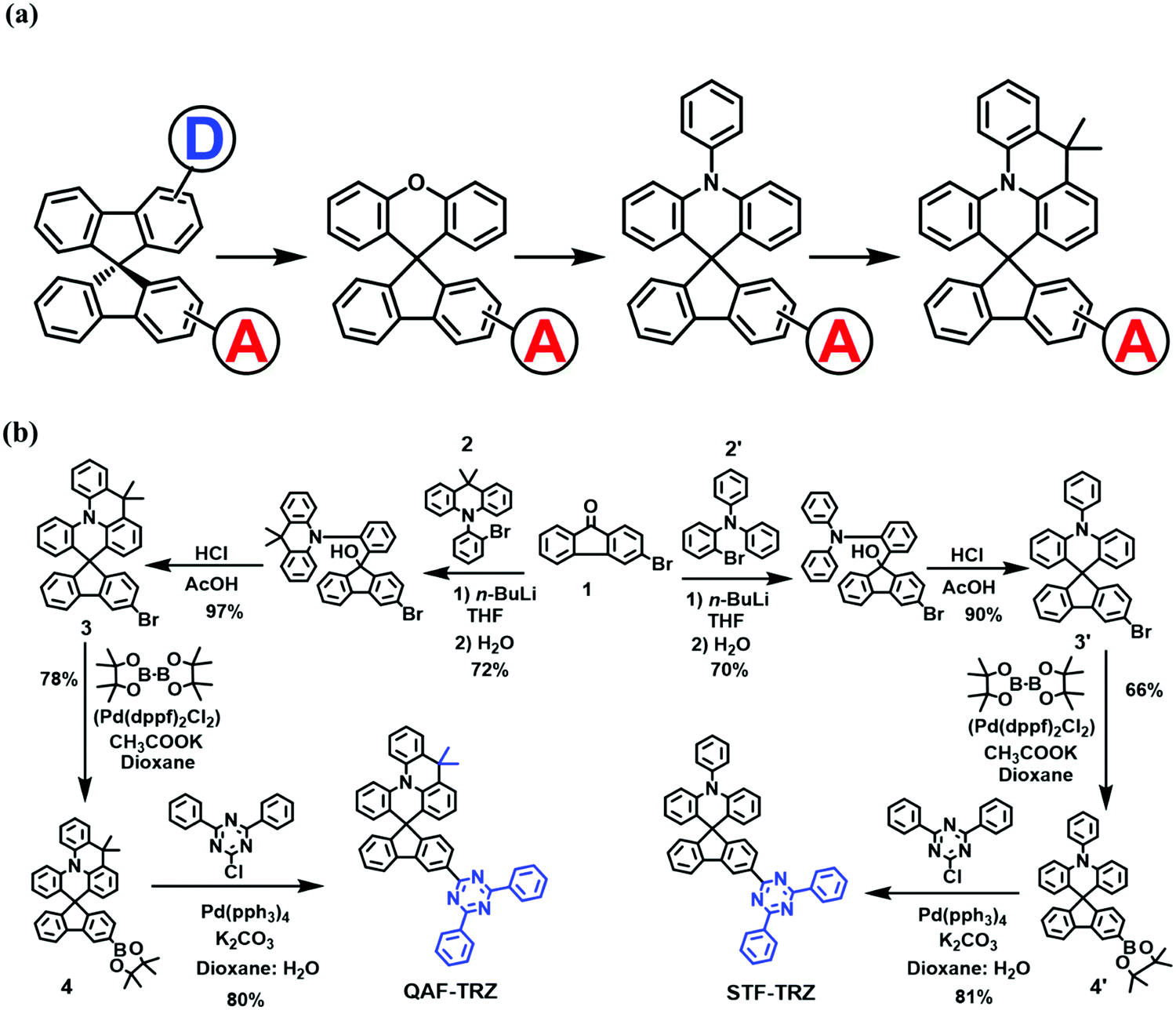
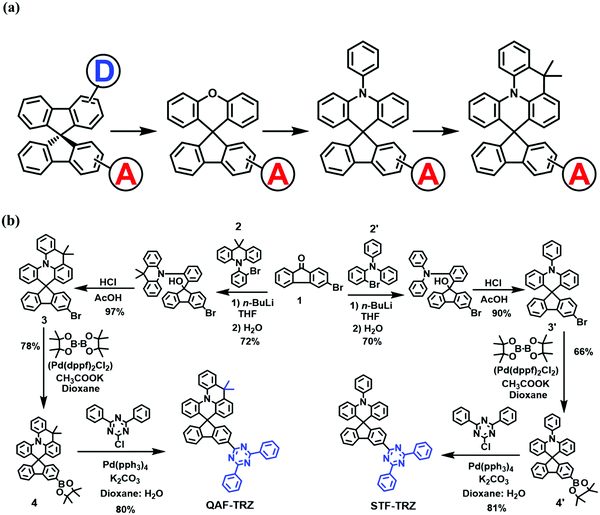
Compared with unipolar hosts,20,21 bipolar host materials attract more attention because both the hole and electron transport elements can be found in a single molecule.22–27 In this regard, many groups have strived to design bipolar host materials with good efficiency.7,22,23,28 Among them, spiro-type29–36 bipolar hosts are of particular interest due to the following features: (i) the 3-D configuration can enhance the thermal and morphological stabilities, (ii) the functionalization of spiro-hosts is flexible, i.e. chemists have the flexibility to modify not only the acceptor unit but also the donor unit of the hemisphere, (iii) this modification can balance hole/electron transport. In designing bipolar compounds in the most classic 9,9′-spirobifluorene, many electron-rich spiro backbones have been developed with an appended electron acceptor (Scheme 1a).
 |
| | Scheme 1 (a) The derivatization of donor/acceptor host materials in the spiro-skeleton. (b) The synthetic routes for QAF-TRZ and STF-TRZ. | |
Herein, two novel spiro-type bipolar host materials, namely 3-(4,6-diphenyl-1,3,5-triazin-2-yl)-9′,9′-dimethyl-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (QAF-TRZ) and 3′-(4,6-diphenyl-1,3,5-triazin-2-yl)-10-phenyl-10H-spiro[acridine-9,9′-fluorene] (STF-TRZ), containing 9,9-dimethyl-10-phenyl-9,10-dihydroacridine and a triphenylamine donor linking with a strong triazine acceptor through the spiro bridge are prepared, respectively. In comparison, QAF-TRZ with the new rigidified donor moiety has a prominent effect on the photophysical and thermal properties and electroluminescence (EL) device performance. We systematically investigated both materials as universal hosts in blue/green/red PHOLEDs. As a result, the devices based on QAF-TRZ show superior performances and lower efficiency roll-off than STF-TRZ, demonstrating the potential of this new spiro-structure as a PHOLED host.
Experimental section
Compound 1 and 2′ were purchased while compound 2 was synthesized according to prior literature.37,38
3-Bromo-9′,9′-dimethyl-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (3)
The compound 2, (10-(2-bromophenyl)-9,9-dimethyl-9,10-dihydroacridine) (2.0 g, 5.6 mmol) was dissolved in THF (40 mL) under an inert environment. The n-butyl lithium (1.6 M solution in hexane 2.2 mL, 5.5 mmol) was added slowly at −78 °C, and the reaction mixture was stirred for 1 hour at the same condition. Then, THF solution (20 mL) of 3-bromo-9H-fluoren-9-one (1.1 g, 4.6 mmol) was added and further stirred for 2 hours at −78 °C. It was then stirred overnight at room temperature. After completion, the reaction mixture was quenched with water (5 mL). The THF was evaporated and the obtained viscous solid was extracted with dichloromethane (DCM) and washed with water (30 mL × 3). The organic portion was collected and purified through column until white solid, about 1.8 g (72%), was obtained, which was used in the next reaction after purification. The intermediate 3-bromo-9-(2-(9,9-dimethylacridin-10(9H)-yl)phenyl)-9H-fluoren-9-ol was dissolved in acetic acid (30 mL) refluxed at 80 °C for 20 minutes. Then, hydrochloric acid (8 mL) was added slowly to the reaction mixture. After 4 hours, a white solid appeared. It was filtered off and washed first with water and then petroleum ether (PE). Column chromatography was used for further purification: PE/DCM (7/3, v/v) (1.6 g, 97%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.05 (d, J = 1.8 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 1.7 Hz, 1H), 7.72 (dd, J = 12.3, 4.3 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.60 (ddd, J = 8.1, 6.1, 1.5 Hz, 2H), 7.54–7.46 (m, 1H), 7.39–7.30 (m, 2H), 7.27–7.18 (m, 2H), 7.13 (d, J = 7.7 Hz, 1H), 7.07 (dd, J = 8.2, 1.8 Hz, 1H), 7.02–6.94 (m, 1H), 6.85 (td, J = 7.6, 3.0 Hz, 2H), 6.65 (ddd, J = 9.6, 7.9, 1.4 Hz, 1H), 6.45 (ddd, J = 9.0, 7.8, 1.2 Hz, 1H), 1.99 (s, 3H), 1.39 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm) 154.60, 152.92, 150.51, 149.26, 144.90, 141.43, 139.18, 138.61, 136.98, 136.35, 135.57, 134.29, 131.71, 131.01, 129.11, 128.68, 128.42, 127.40, 126.79, 126.59, 125.41, 124.47, 124.03, 123.71, 123.61, 123.04, 122.51, 121.40, 120.37, 118.50, 116.91, 57.36, 36.93, 31.81, 23.02. MS (EI) m/z: 525.08 [M+].
3′-Bromo-10-phenyl-10H-spiro[acridine-9,9′-fluorene] (3′)
The intermediate 3′ was synthesized according to the similar procedure as explained above for compound 3 except by changing the corresponding reagents. It was further purified using column chromatography with eluents: PE/DCM (8/2, v/v) (1.3 g, 90%). 1H NMR (400 MHz, CDCl3δ (ppm) 7.97 (d, J = 1.7 Hz, 1H), 7.83–7.71 (m, 3H), 7.62 (t, J = 7.5 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.50–7.39 (m, 3H), 7.32 (dd, J = 18.3, 9.5 Hz, 2H), 7.01–6.92 (m, 2H), 6.62 (t, J = 7.5 Hz, 2H), 6.42 (dd, J = 16.2, 8.5 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ (ppm) 156.81, 155.39, 141.34, 140.96, 137.93, 131.16, 129.17, 128.56, 127.74, 127.38, 125.97, 124.01, 123.17, 121.69, 120.65, 120.17, 114.79, 56.66. MS (EI) m/z: 485.24 [M+].
9′,9′-Dimethyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (4)
A mixture of 3-bromo-9′,9′-dimethyl-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (1.3 g, 2.4 mmol), 4,4,40,40,5,5,50,50-octamethyl-2,20-bi(1,3,2-dioxaborolane) (1.2 g, 4.9 mmol), 1,1′-bis(diphenylphosphino)ferrocene-palladium(II) (Pd(dppf)2Cl2) 87.0 mg, 0.1 mmol), potassium acetate (1.1 g, 12.0 mmol), and anhydrous 1,4-dioxane (50 mL) was degassed and then refluxed under an inert atmosphere for 24 hours at 80 °C. The reaction mixture was washed three times with water (30 mL × 3) after dilution with DCM. In order to remove water completely from the organic part, anhydrous MgSO4 was added. The solvent of crude product was dried through a rotary evaporator, which was purified using column chromatography to obtain a white powder with the eluents: PE/DCM (4/6, v/v) (1.1 g, 78%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.37 (s, 1H), 8.17 (s, 1H), 7.96 (dd, J = 14.1, 7.5 Hz, 1H), 7.80–7.69 (m, 1H), 7.68–7.54 (m, 2H), 7.53–7.42 (m, 2H), 7.38–7.29 (m, 2H), 7.18 (ddd, J = 23.6, 14.6, 8.3 Hz, 3H), 6.94 (t, J = 7.5 Hz, 1H), 6.82 (dd, J = 9.2, 4.6 Hz, 2H), 6.67–6.60 (m, 1H), 6.44 (t, J = 7.2 Hz, 1H), 1.98 (s, 3H), 1.47 (s, 6H), 1.39 (s, 3H), 1.32 (s, 6H).13C NMR (100 MHz, CDCl3) δ (ppm) 153.73, 149.96, 139.28, 138.62, 136.99, 136.41, 135.24, 134.68, 134.19, 134.14, 129.23, 128.24, 128.06, 127.42, 127.13, 127.06, 126.90, 126.67, 126.46, 126.37, 124.39, 123.98, 123.85, 123.27, 123.02, 122.86, 122.11, 120.38, 120.34, 118.42, 118.38, 116.98, 116.92, 84.05, 83.72, 57.68, 36.92, 31.77, 31.66, 25.00, 24.83, 24.79, 23.02. MS (EI) m/z: 573.29 [M+].
10-Phenyl-3′-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10H-spiro[acridine-9,9′ fluorene] (4′)
The compound 4′ was synthesized according to the similar procedure mentioned above for compound 4 only by changing the corresponding reagents. In order to obtain pure product, it was passed through column chromatography with the eluents: PE/DCM (4/6, v/v) (0.8 g, 66%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.30 (s, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.81–7.69 (m, 3H), 7.60 (t, J = 8.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 2H), 7.46 (dd, J = 14.3, 7.5 Hz, 2H), 7.39 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 7.4 Hz, 1H), 6.97–6.89 (m, 2H), 6.57 (t, J = 7.5 Hz, 2H), 6.39 (dd, J = 13.7, 7.3 Hz, 4H), 1.42 (s, 12H). 13C NMR (100 MHz, CDCl3) δ (ppm) 159.53, 156.68, 141.17, 138.95, 135.10, 131.15, 128.40, 127.87, 127.58, 127.21, 126.33, 125.66, 125.35, 124.54, 120.54, 120.18, 114.63, 83.90, 56.97, 24.94. MS (EI) m/z: 533.35 [M+].
3-(4,6-Diphenyl-1,3,5-triazin-2-yl)-9′,9′-dimethyl-9′H-spiro[fluorene-9,5′-quinolino[3,2,1-de]acridine] (QAF-TRZ)
The compounds 9′,9′-dimethyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9′H-spiro[fluorene-9,5′quinolino[3,2,1-de]acridine] (0.8 g,1.5 mmol), 2-chloro-4,6-diphenyl-1,3,5 triazine (0.5 g, 1.8 mmol), tetrakis(triphenylphosphine)palladium(0) (50.0 mg, 0.05 mmol) and potassium carbonate (0.5 g, 3.7 mmol) were dissolved in 1,4-dioxane/H2O (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), and refluxed for 24 hours at 85 °C. The mixture was diluted with DCM and washed thrice with distilled water (100 mL). The organic layer was dried and purified through column chromatography using PE/DCM (7/3, v/v) to obtain a pale white powder (0.8 g, 80%). 1H NMR (600 MHz, CDCl3) δ (ppm) 9.25 (s, 1H), 9.02 (s, 1H), 8.87 (dd, J = 14.1, 7.3 Hz, 2H), 8.73 (d, J = 7.1 Hz, 2H), 8.36 (d, J = 8.1 Hz, 1H), 8.14 (d, J = 7.6 Hz, 1H), 7.93 (d, J = 7.5 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.70–7.48 (m, 8H), 7.36 (s, 1H), 7.31 (t, J = 7.2 Hz, 1H), 7.20 (t, J = 8.9 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.83 (dd, J = 13.1, 7.2 Hz, 2H), 6.69 (dd, J = 14.3, 7.9 Hz, 1H), 6.49 (dd, J = 18.7, 7.8 Hz, 1H), 1.97 (s, 3H), 1.39 (s, 3H). 13C NMR (150 MHz, CDCl3) δ (ppm) 171.73, 158.31, 155.01, 154.51, 150.38, 143.51, 142.24, 139.26, 138.69, 137.70, 137.03, 136.67, 136.16, 135.75, 134.32, 132.58, 131.96, 129.64, 129.42, 127.81, 127.04, 126.63, 124.51, 124.31, 123.39, 123.08, 122.39, 120.95, 118.56, 117.02, 57.79, 36.98, 31.84, 23.08. MALDI-TOF (m/z) calculated for C49H34N4 [M+]: 678.27, found: 678.22. Anal. calcd for C49H34N4 C, 86.70; H, 5.05: N, 8.25 found C, 85.92; H, 4.97: N, 8.07.
:4), and refluxed for 24 hours at 85 °C. The mixture was diluted with DCM and washed thrice with distilled water (100 mL). The organic layer was dried and purified through column chromatography using PE/DCM (7/3, v/v) to obtain a pale white powder (0.8 g, 80%). 1H NMR (600 MHz, CDCl3) δ (ppm) 9.25 (s, 1H), 9.02 (s, 1H), 8.87 (dd, J = 14.1, 7.3 Hz, 2H), 8.73 (d, J = 7.1 Hz, 2H), 8.36 (d, J = 8.1 Hz, 1H), 8.14 (d, J = 7.6 Hz, 1H), 7.93 (d, J = 7.5 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.70–7.48 (m, 8H), 7.36 (s, 1H), 7.31 (t, J = 7.2 Hz, 1H), 7.20 (t, J = 8.9 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.83 (dd, J = 13.1, 7.2 Hz, 2H), 6.69 (dd, J = 14.3, 7.9 Hz, 1H), 6.49 (dd, J = 18.7, 7.8 Hz, 1H), 1.97 (s, 3H), 1.39 (s, 3H). 13C NMR (150 MHz, CDCl3) δ (ppm) 171.73, 158.31, 155.01, 154.51, 150.38, 143.51, 142.24, 139.26, 138.69, 137.70, 137.03, 136.67, 136.16, 135.75, 134.32, 132.58, 131.96, 129.64, 129.42, 127.81, 127.04, 126.63, 124.51, 124.31, 123.39, 123.08, 122.39, 120.95, 118.56, 117.02, 57.79, 36.98, 31.84, 23.08. MALDI-TOF (m/z) calculated for C49H34N4 [M+]: 678.27, found: 678.22. Anal. calcd for C49H34N4 C, 86.70; H, 5.05: N, 8.25 found C, 85.92; H, 4.97: N, 8.07.
3′-(4,6-Diphenyl-1,3,5-triazin-2-yl)-10-phenyl-10H-spiro[acridine-9,9′-fluorene] (STF-TRZ)
A mixture of 10-phenyl-3′-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10H-spiro[acridine-9,9′-fluorene] (0.7 g, 1.3 mmol), 2-chloro-4,6-diphenyl-1,3,5 triazine (0.4 g, 1.6 mmol), tetrakis (triphenylphosphine) palladium (0) (50.0 mg, 0.04 mmol) and potassium carbonate (0.4 g, 3.2 mmol) was dissolved in 1,4-dioxane/H2O (40:4) refluxed over night at 85 °C. The crude organic layer was subjected to column chromatography for further purification using PE/DCM (7/3, v/v) (0.68 g, 81%). 1H NMR (600 MHz, CDCl3) δ (ppm) 9.15 (s, 1H), 8.81 (d, J = 6.9 Hz, 4H), 8.69 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 7.5 Hz, 1H), 7.73 (t, J = 7.6 Hz, 2H), 7.68–7.56 (m, 8H), 7.54 (d, J = 7.8 Hz, 2H), 7.47 (dd, J = 17.7, 10.3 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 6.94 (t, J = 7.7 Hz, 2H), 6.58 (t, J = 7.4 Hz, 2H), 6.47 (d, J = 6.9 Hz, 2H), 6.39 (d, J = 8.4 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ (ppm) 171.83, 171.64, 160.63, 156.52, 141.28, 140.99, 139.87, 138.70, 136.25, 135.95, 132.51, 131.17, 131.08, 129.54, 128.97, 128.84, 128.65, 128.49, 127.77, 127.74, 127.39, 125.96, 125.87, 124.25, 120.63, 120.53, 120.40, 114.73, 57.03. MALDI-TOF (m/z) calculated for C46H30N4 [M+]: 638.24, found: 638.18. Anal. calcd for C46H30N4 C, 86.49; H, 4.73: N, 8.77 found C, 85.58; H, 4.69: N, 8.39.
Results and discussion
Synthesis and characterization
The synthetic routes for two compounds are depicted in Scheme 1b. The intermediate 3 and 3′ were synthesized through a three-step consecutive reaction under the same conditions. Similarly, the intermediate 4 and 4′ were synthesized through the Miyaura borylation reaction.39 Finally, QAF-TRZ and STF-TRZ were successfully obtained in good yields through the Suzuki–Miyaura coupling reaction with 2-chloro-4,6-diphenyl-1,3,5 triazine and corresponding reagents 4 and 4′. To make it highly pure, the products were further sublimated under vacuum after column chromatography before being used in OLED devices. The chemical structures of QAF-TRZ and STF-TRZ were fully characterized through 1H and 13C NMR spectroscopy, mass spectrometry, and elemental analysis.
Thermal properties
The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under inert atmosphere are shown in Fig. S1a and b (ESI†) for both compounds. The measured thermal properties and values are depicted in Table 1. The spiro annulated structure reveals high decomposition temperatures (Td) with 5% weight loss observed at 430 and 424 °C, while the glass transition temperatures (Tg) are 195 and 158 °C for QAF-TRZ and STF-TRZ, respectively. Comparably, QAF-TRZ has more favorable Td and Tg over the STF-TRZ for OLED preparation in vacuum deposition due to the rigidification in the spiro-skeleton.
Table 1 Summary of the physical properties of QAF-TRZ and STF-TRZ
| Compounds |
Abs
(nm) |
Fl
(nm) |
Phos
(nm) |
E
g
(eV) |
E
T
(eV) |
HOMO/LUMOf (eV) |
T
d/Tgg (°C) |
|
Calculated from the UV-vis absorption measured in toluene.
Determined from the room temperature fluorescence in toluene.
Calculated from the 77 K phosphorescence in toluene.
Calculated from the UV-vis absorption in toluene.
Calculated from the phosphorescence in toluene.
HOMO is calculated from the oxidation potential with respect to reference ferrocene; LUMO is calculated from the energy gap plus HOMO.
T
d: decomposition temperature, Tg: glass transition temperature.
|
|
QAF-TRZ
|
305 |
447 |
447 |
3.44 |
2.88 |
−5.67/−2.23 |
430/195 |
|
STF-TRZ
|
305 |
450 |
446 |
3.44 |
2.86 |
−5.36/−1.91 |
424/158 |
Theoretical calculations
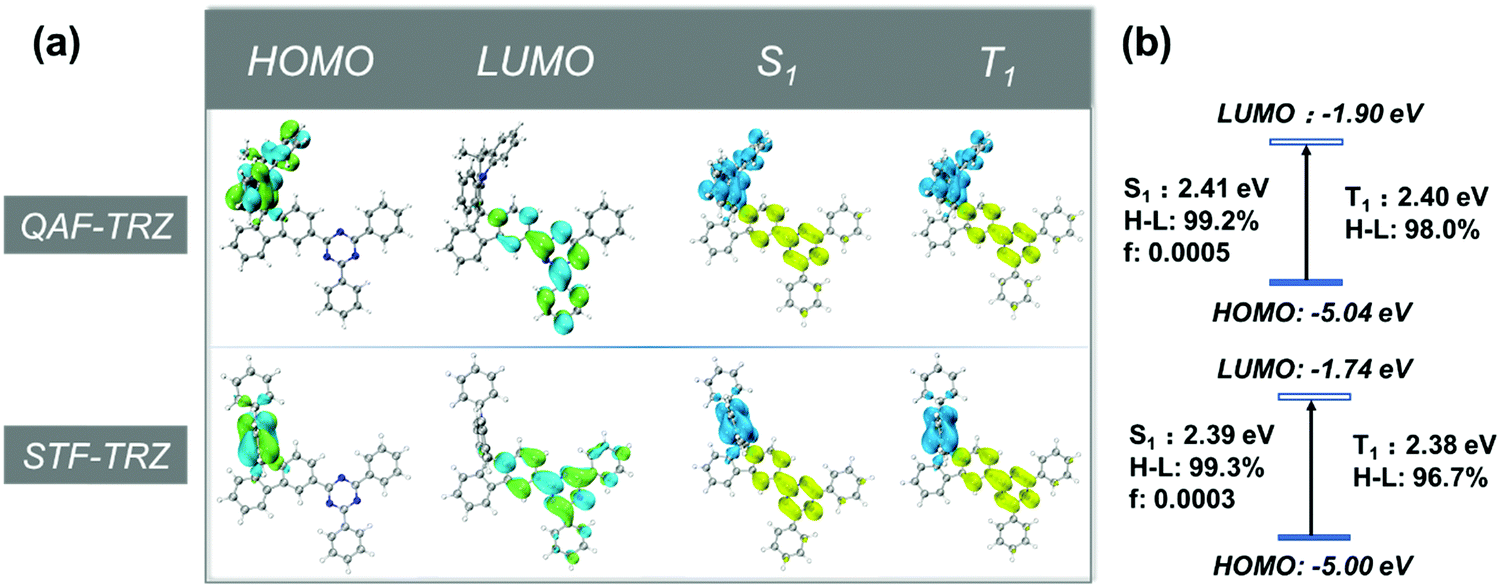
Theoretical calculations were performed to better understand the electronic properties of the two compounds by using density functional theory (DFT) and time-dependent DFT (TD-DFT). Ground-state geometries and excited state geometries were optimized at the B3LYP/6-31G level.40 The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of QAF-TRZ and STF-TRZ are mainly located at the donor and acceptor segment, respectively (Fig. 1a). The energy level of the HOMO and LUMO of QAF-TRZ and STF-TRZ are −5.04/−1.90 eV and −5.00/−1.74 eV, respectively (Fig. 1b); the fused strategy on the donor leads to a deeper HOMO and higher singlet/triplet energies as unveiled from the photophysical measurements (vide infra). Furthermore, the energy differences between the S1 and T1 states for QAF-TRZ and STF-TRZ are almost the same. Based on the electron–hole distribution analysis,41 99.2% and 99.3% of the HOMO-to-LUMO transitions contributed to the S0 → S1 excitation for QAF-TRZ and STF-TRZ, respectively, while contributions to the S0 → T1 excitation was 98.0% and 96.7%, respectively, for QAF-TRZ and STF-TRZ.
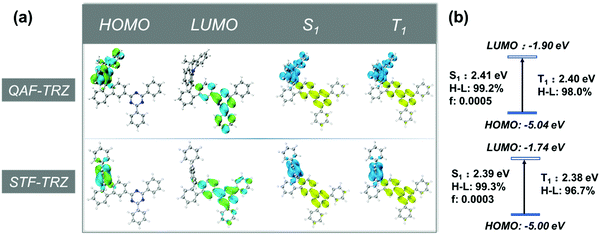 |
| | Fig. 1 (a) The distribution of HOMOs and LUMOs and the analysis for distribution of hole (blue), electron (yellow) for S1 and T1 and (b) energy level diagram of HOMOs, LUMOs, S1 and T1. | |
Photophysical properties
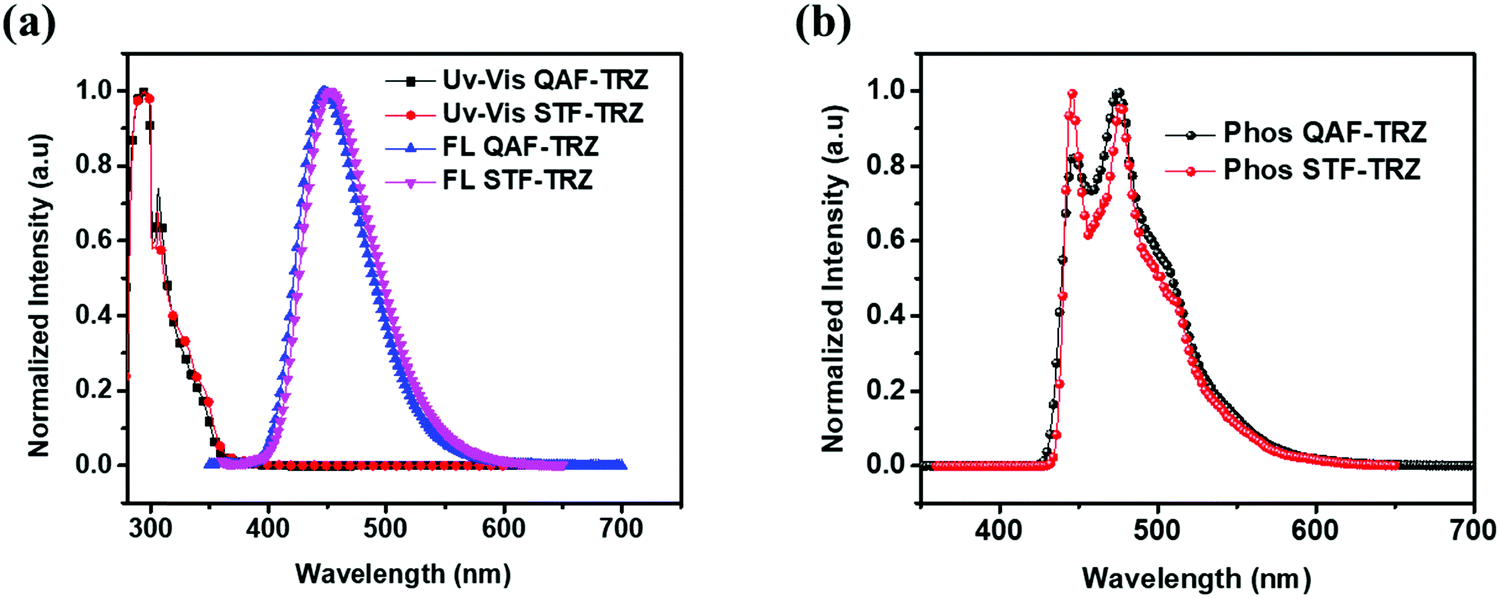
The photophysical behavior of QAF-TRZ and STF-TRZ was recorded using ultraviolet-visible (UV-vis) and fluorescence spectroscopy in toluene at room temperature. Phosphorescence spectra were measured at frozen temperature (77 K) using the same solvent and are depicted in Fig. 2a and b, respectively. QAF-TRZ and STF-TRZ show very similar absorption peaks at 305 nm, which is attributed to the π–π* transitions. The HOMO–LUMO energy gaps were calculated from the UV absorption onset, and both are 3.44 eV. The fluorescence peaks as observed in the photoluminescence spectra (PL) for QAF-TRZ and STF-TRZ were 447 and 450 nm, respectively. Apart from that, the photophysical properties of QAF-TRZ and STF-TRZ were also measured in neat film and their spectra are illustrated in Fig. S2 (ESI†). Notably, the absoption peak for QAF-TRZ and STF-TRZ is blue-shifted as compared to that of the solution results; the triplet-energy levels for QAF-TRZ and STF-TRZ are quite similar as well. Moreover, the triplet energies calculated from the vibronic band of the phosphorescence spectra at 77 K are 2.88 eV for QAF-TRZ, and 2.86 eV for STF-TRZ. Therefore, such triplet energies are high enough to prevent back energy transfer from emitters.
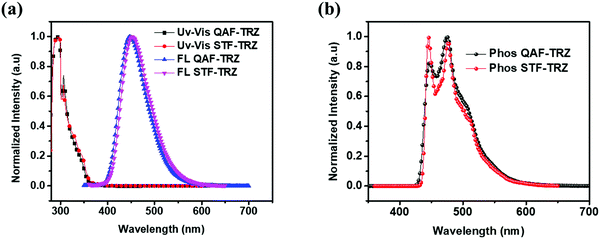 |
| | Fig. 2 (a) Normalized UV-vis absorption and fluorescence emission spectra in toluene at room temperature. (b) Phosphorescence spectra in frozen toluene at 77 K for compounds QAF-TRZ and STF-TRZ. | |
Electrochemical properties
To investigate the electrochemical behavior of the two host materials, QAF-TRZ and STF-TRZ, cyclic voltammetry analysis was performed in anhydrous DCM using ferrocene as the internal standard and the parameters are summarized in Table 1. The oxidation potential values for the compounds QAF-TRZ and STF-TRZ are +1.37 V and +1.05 V, respectively. The obtained results indicate that QAF-TRZ is constructed with a rigid donor structure as compared to STF-TRZ. The HOMO energy levels were estimated from the onset of the oxidation potential curves to be −5.67/−5.36 eV, respectively, while the LUMO energy levels were calculated from Eg (onset of UV-vis spectra) and HOMO values, which were −2.23/−1.91 eV for QAF-TRZ and STF-TRZ, respectively (Fig. S3, ESI†).
Electroluminescence (EL) properties
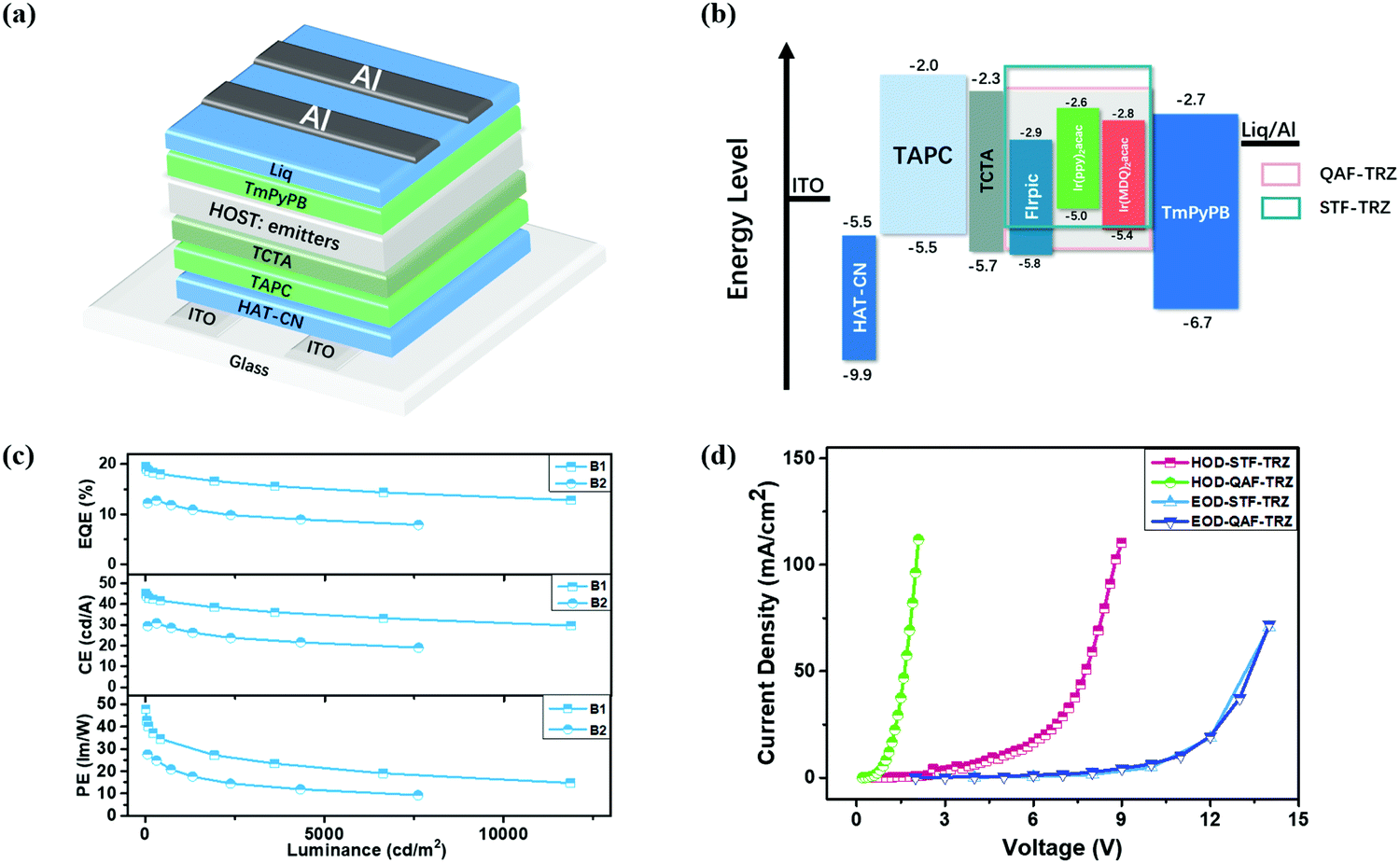
To further evaluate the EL properties of these two host materials, we first fabricated blue PHOLED devices based on FIrpic. As shown in Fig. 3a, the blue device structure is ITO/1,4,5,8,9,11-hexaazatriphenylene-hexacarbonitrile (HAT-CN) (10 nm)/1,1-bis[4-[N,N-di(p-tolyl)amino]phenyl]cyclohexane (TAPC) (40 nm)/tris(4-(9H-carbazol-9-yl)phenyl)amine (TCTA) (10 nm)/QAF-TRZ or STF-TRZ:FIrpic 15 wt% (20 nm)/3,3′-(5′-(3-(pyridin-3-yl)phenyl)-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)dipyridine (TmPyPB) (45 nm)/(8-quinolinolato lithium) Liq (2 nm)/Al (100 nm) (B1 for QAF-TRZ and B2 for STF-TRZ). The energy level diagrams of all the blue, green and red devices fabricated in the present studies are displayed in Fig. 3b. ITO acts as the anode while Al acts as the cathode. HAT-CN and Liq are the hole and electron-injection layers, respectively. TAPC is employed as the hole transport layer because of its remarkable hole mobility and TCTA plays the role of the electron blocking layer. Furthermore, TmPyPB is chosen as the electron transport layer for better energy level alignment. The chemical structures of these commercial materials are shown in Fig. S4 (ESI†). We all know that the ET of the PHOLED host is higher than that of the emitters for effective exciton confinement, better energy transfer from the host to the guest and reducing the non-radiative transition. In this work, the ET of QAF-TRZ and STF-TRZ are 2.88 and 2.86 eV, respectively, which indicates that all are appropriate as the host for FIrpic (ET = 2.62 eV). We first fabricated the blue devices by varying the concentration of FIrpic from 10 to 20% (Table S1, ESI†), and the optimized concentration was 15%. The current density–voltage–luminance (J–V–L) characteristics for both B1 and B2 are shown in Fig. S5a (ESI†) and their device performance is summarized in Table 2. Device B1 exhibits a maximum EQE, current efficiency (CE), and power efficiency (PE) of 19.4%, 45 cd A−1 and 47.8 lm W−1, respectively. The EQE can be maintained as 17.2% at 1000 cd m−2. However, device B2 shows a maximum EQE, CE, and PE of 12.7%, 30.8 cd A−1 and 27.5 lm W−1, which reveals an apparently lower device performance as compared with B1 (Fig. 3c). Conventional hosts such as 1,3-bis(N-carbazolyl)benzene (mCP) and 2,6-bis(3-(carbazol-9-yl)phenyl)pyridine (26DCzppy) were also used for comparison. The device results are summarized in Table S2 (ESI†). Among them, QAF-TRZ shows the best device performance, making it a good candidate as a host material for PhOLED.
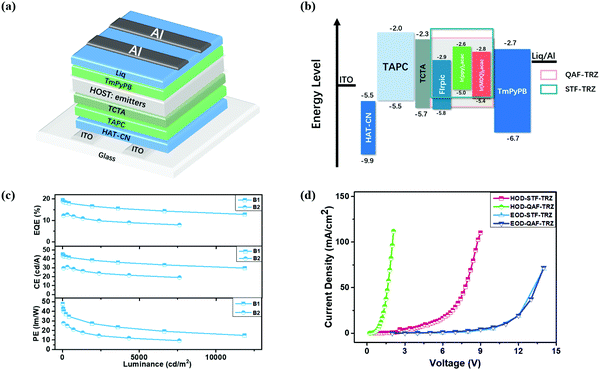 |
| | Fig. 3 (a) Device structure, (b) energy level diagrams of devices, (c) blue PHOLED performance and (d) hole-only and electron-only devices based on QAF-TRZ and STF-TRZ. | |
Table 2 Electroluminescence characteristics of the devices
| Device |
V
(V) |
CEb (cd A−1) |
PEb (lm W−1) |
EQEc (%) |
CIEd (x,y) |
|
Driving voltage at 100 cd m−2.
Maximum value of current efficiency and power efficiency.
EQEs of maximum value, 100 cd m−2 and 1000 cd m−2.
CIE (Commission International de l'Eclairge); measured at a driving current density at 5 mA cm−2.
|
|
B1
|
3.4 |
45.0 |
47.8 |
19.4/18.3/17.2 |
(0.16,0.35) |
|
B2
|
3.5 |
30.8 |
27.5 |
12.7/12.3/11.3 |
(0.17,0.37) |
|
G1
|
2.8 |
80.0 |
91.0 |
21.0/20.4/20.0 |
(0.34,0.63) |
|
G2
|
2.9 |
72.0 |
76.0 |
19.0/18.9/18.8 |
(0.34,0.61) |
|
R1
|
3.8 |
26.5 |
24.1 |
22.6/21.8/17.1 |
(0.63,0.36) |
|
R2
|
3.8 |
29.0 |
25.3 |
19.4/19.0/15.5 |
(0.62,0.37) |
To further investigate the difference in the blue PHOLED performance under the same device structure, we fabricated electron- and hole-only devices (EOD and HOD) to estimate their charge mobilities. The device structures are as follows: ITO/TmPyPB (20 nm)/QAF-TRZ or STF-TRZ (100 nm)/TmPyPB (20 nm)/Liq (2 nm)/Al (100 nm) (as EOD) and ITO/MoO3 (10 nm)/QAF-TRZ or STF-TRZ (100 nm)/MoO3 (10 nm)/Al (100 nm) (as HOD). As shown in Fig. 3d, the J–V curves of QAF-TRZ and STF-TRZ almost overlap under the same driving voltages in the EOD device, indicating that the two host materials have similar electron mobility. On the contrary, QAF-TRZ shows a higher current density than STF-TRZ at a low driving voltage in the HOD. Therefore, the better hole mobility of QAF-TRZ may favor the hole-electron combination for forming excitons in device B1.
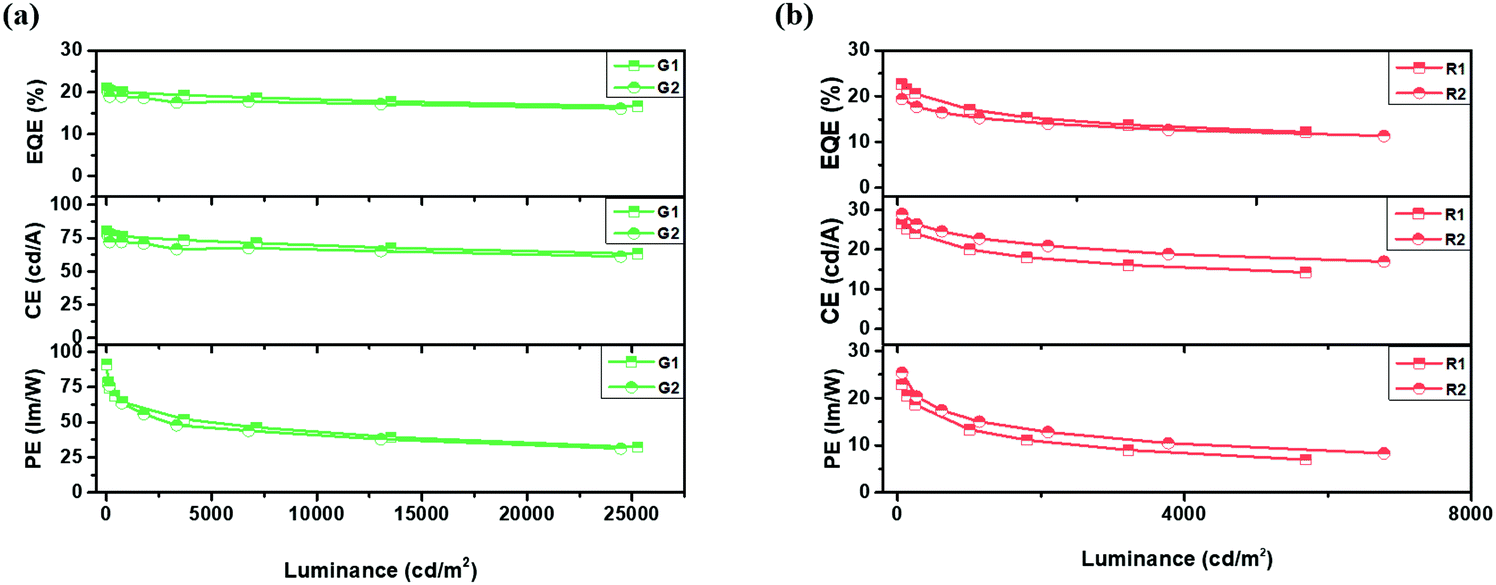
Furthermore, green and red PHOLEDs were also fabricated with device structures of ITO/HAT-CN (10 nm)/TAPC (40 nm)/TCTA (10 nm)/QAF-TRZ or STF-TRZ:Ir(ppy)2acac 10 wt% (20 nm)/TmPyPB (45 nm)/Liq (2 nm)/Al (100 nm) and ITO/HAT-CN (10 nm)/TAPC (40 nm)/TCTA (10 nm)/QAF-TRZ or STF-TRZ:Ir(MDQ)2acac 6 wt% (20 nm)/TmPyPB (50 nm)/Liq (2 nm)/Al (100 nm) (G1, R1 for QAF-TRZ and G2, R2 for STF-TRZ). The ETs of green (Ir(ppy)2acac) and red (Ir(MDQ)2acac) emitters are 2.4 eV and 2.0 eV, respectively, which are much lower than those of the hosts, and therefore can realize effective energy transfer. The device performance is shown in Fig. 4 and also summarized in Table 2. It can be seen that the maximum EQE, CE, and PE of R1 and R2 are 22.6%, 26.5 cd A−1, and 24.1 lm W−1 and 19.4%, 29.0 cd A−1, and 25.3 lm W−1, respectively. The EQE of R1 based on the QAF-TRZ is among the highest value in red PHOLEDs. Also, the EQE can be kept as high as 17.1% at 1000 cd m−2. Moreover, the green devices (G1 and G2) based on our two host materials exhibit maximum EQE, CE, and PE of 21.0%, 80 cd A−1, and 91 lm W−1 and 19%, 72 cd A−1, and 76 lm W−1, respectively. Notably, G1 and G2 have quite similar performance and low-efficiency roll-off. At a luminance of 1000 cd m−2, the EQE of devices G1 and G2 remain 20% and 18.8%, and merely decrease by 4% and 1% from their maximum values, respectively. This low roll-off property is mainly attributed to the well-balanced charge injection and better recombination arisen from the high ET of the hosts. The J–V–L characteristics of green and red devices are exhibited in Fig. S5b and c (ESI†), respectively. In addition, both hosts almost lead to identical RGB EL emissions (Fig. S5d, ESI†), i.e., 472 nm for B1 and B2, 524 nm for G1 and G2, and 612 nm for R1 and R2. Thus, there is no other emission peak in the EL spectra, substantiating an efficient energy transfer from the hosts to the emitters.
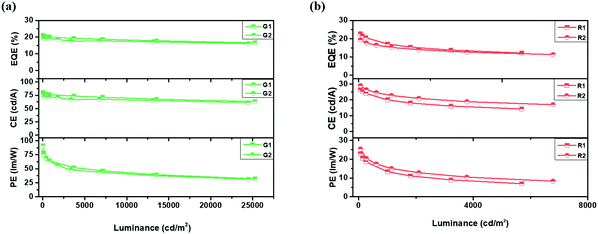 |
| | Fig. 4 (a) Green and (b) red PHOLEDs device performance for QAF-TRZ and STF-TRZ. | |
Conclusion
We have designed and synthesized two host materials, QAF-TRZ and STF-TRZ, for red, green and blue PHOLEDs. Both materials have excellent thermal properties with a high Td of over 420 °C. The EQE of the blue PHOLED reached nearly 20% when using QAF-TRZ as the host. The maximum EQEs for the green PHOLEDs QAF-TRZ and STF-TRZ as the hosts were 21% and 19%, respectively. Both the green PHOLEDs exhibited a low-efficiency roll-off, in which the EQEs merely decreased by 4% and 1%, respectively, from their maximum values when the devices were driven to a luminance of 1000 cd m−2. Moreover, the red PHOLEDs exhibited maximum EQEs of 22.6% and 19.4% using QAF-TRZ and STF-TRZ as the hosts, respectively, in which the former has the best EQE among all the red PHOLEDs. The superior device performance is attributed to the new rigidified donor moiety with the spiro motif incorporating triazine (at para position) type acceptors. The device based on QAF-TRZ exhibits even higher efficiencies because it has a higher hole mobility with better charge recombination. We believe our simple spiro-skeleton strategy can facilitate the development of efficient and stable hosts for RGB-based PHOLEDs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (No. 2016YFB0400700), the National Natural Science Foundation of China (No. 51773141, 51873139 and 21572152) and the Natural Science Foundation of Jiangsu Province of China (BK20181442). This project is also funded by Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 111 Project of The State Administration of Foreign Experts Affairs of China. We greatly thank Yang-Kun Qu for analyzing the molecular simulations.
References
- X.-D. Zhu, Y.-L. Zhang, Y. Yuan, Q. Zheng, Y.-J. Yu, Y. Li, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2019, 7, 6714–6720 RSC.
- S. J. Kim, Y. J. Kim, Y. H. Son, J. A. Hur, H. A. Um, J. Shin, T. W. Lee, M. J. Cho, J. K. Kim and S. Joo, Chem. Commun., 2013, 49, 6788–6790 RSC.
- M. Hu, Y. Liu, Y. Chen, W. Song, L. Gao, H. Mu, J. Huang and J. Su, RSC Adv., 2017, 7, 7287–7292 RSC.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- W. Jiang, L. Duan, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, J. Mater. Chem., 2010, 20, 6131–6137 RSC.
- L.-S. Cui, S.-B. Ruan, F. Bencheikh, R. Nagata, L. Zhang, K. Inada, H. Nakanotani, L.-S. Liao and C. Adachi, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- R. K. Konidena, K. H. Lee and J. Y. Lee, Chem. – Asian J., 2019, 14, 313–321 CrossRef CAS PubMed.
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2015, 54, 1176–1180 CrossRef CAS PubMed.
- Y. Zhang, J. Lee and S. R. Forrest, Nat. Commun., 2014, 5, 1–7 CAS.
- C. Adachi, M. A. Baldo, S. R. Forrest and M. E. Thompson, Appl. Phys. Lett., 2000, 77, 904–906 CrossRef CAS.
- S. Ye, Y. Liu, K. Lu, W. Wu, C. Du, Y. Liu, H. Liu, T. Wu and G. Yu, Adv. Funct. Mater., 2010, 20, 3125–3135 CrossRef CAS.
- P. Wu, W. Song, Z. Xia, Y. Chen, G. Tian, J. Huang and J. Su, Dyes Pigm., 2019, 162, 153–159 CrossRef CAS.
- Q. Dong, F. Tai, H. Lian, Z. Chen, M. Hu, J. Huang and W.-Y. Wong, Dyes Pigm., 2017, 143, 470–478 CrossRef CAS.
- M. A. Baldo, D. O’brien, M. Thompson and S. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422 CrossRef CAS.
- M. A. Baldo, D. O’brien, Y. You, A. Shoustikov, S. Sibley, M. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. H. Tsai, H. W. Lin, H. C. Su, T. H. Ke, C. C. Wu, F. C. Fang, Y. L. Liao, K. T. Wong and C. I. Wu, Adv. Mater., 2006, 18, 1216–1220 CrossRef CAS.
- D. Zhang, C. Zhao, Y. Zhang, X. Song, P. Wei, M. Cai and L. Duan, ACS Appl. Mater. Interfaces, 2017, 9, 4769–4777 CrossRef CAS PubMed.
- S.-R. Park, S.-M. Kim, J.-H. Kang, J.-H. Lee and M. C. Suh, Dyes Pigm., 2017, 141, 217–224 CrossRef CAS.
- X. Zhan, Z. Wu, Y. Lin, Y. Xie, Q. Peng, Q. Li, D. Ma and Z. Li, Chem. Sci., 2016, 7, 4355–4363 RSC.
- Z. Hu, W. Fu, L. Yan, J. Miao, H. Yu, Y. He, O. Goto, H. Meng, H. Chen and W. Huang, Chem. Sci., 2016, 7, 5007–5012 RSC.
- V. Govindan, K.-C. Yang, Y.-S. Fu and C.-G. Wu, New J. Org. Chem., 2018, 42, 7332–7339 RSC.
- A. Chaskar, H. F. Chen and K. T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- K. S. Yook and J. Y. Lee, Chem. Rec., 2016, 16, 159–172 CrossRef CAS PubMed.
- T. Chatterjee, W.-Y. Hung, W.-F. Tang, H.-F. Chen and K.-T. Wong, Org. Electron., 2017, 50, 204–212 CrossRef CAS.
- S. Kumar, D. Li, Y.-K. Wang, Y. Yuan, A. Khan, Z.-Q. Jiang and L.-S. Liao, Synth. Metods, 2019, 254, 42–48 CrossRef CAS.
- D. Thirion, J. Rault-Berthelot, L. Vignau and C. Poriel, Org. Lett., 2011, 13, 4418–4421 CrossRef CAS PubMed.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- M. Romain, D. Tondelier, J. C. Vanel, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2013, 52, 14147–14151 CrossRef CAS PubMed.
- C. Poriel, N. Cocherel, J. Rault-Berthelot, L. Vignau and O. Jeannin, Chem. – Eur. J., 2011, 17, 12631–12645 CrossRef CAS PubMed.
- C. Poriel, J. Rault-Berthelot, F. Barriere and A. M. Slawin, Org. Lett., 2008, 10, 373–376 CrossRef CAS PubMed.
- L. Sicard, C. Quinton, J. D. Peltier, D. Tondelier, B. Geffroy, U. Biapo, R. Métivier, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2017, 23, 7719–7727 CrossRef CAS PubMed.
- J. D. Peltier, B. Heinrich, B. Donnio, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2017, 23, 17290–17303 CrossRef CAS PubMed.
- I. Bulut, P. Chávez, S. Fall, S. Méry, B. Heinrich, J. Rault-Berthelot, C. Poriel, P. Lévêque and N. Leclerc, RSC Adv., 2016, 6, 25952–25959 RSC.
- C. Poriel, J. J. Liang, J. Rault-Berthelot, F. Barrière, N. Cocherel, A. M. Slawin, D. Horhant, M. Virboul, G. Alcaraz and N. Audebrand, Chem. – Eur. J., 2007, 13, 10055–10069 CrossRef CAS PubMed.
- F. Moreau, N. Audebrand, C. Poriel, V. Moizan-Baslé and J. Ouvry, J. Mater. Chem., 2011, 21, 18715–18722 RSC.
- Y. K. Wang, Q. Sun, S. F. Wu, Y. Yuan, Q. Li, Z. Q. Jiang, M. K. Fung and L. S. Liao, Adv. Funct. Mater., 2016, 26, 7929–7936 CrossRef CAS.
- A. Khan, Y.-K. Wang, C.-C. Huang, S. Kumar, M.-K. Fung, Z.-Q. Jiang and L.-S. Liao, Org. Electron., 2019, 105520 Search PubMed.
- T. Liu, H. Sun, C. Fan, D. Ma, C. Zhong and C. Yang, Org. Electron., 2014, 15, 3568–3576 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frish, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- L. Tian and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc01389g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Man-Keung
Fung
*,
Man-Keung
Fung