
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerobically-initiated C(sp3)–H bond amination through the use of activated azodicarboxylates†
André
Shamsabadi
,
Antoine
Maruani
,
Nehaal
Ahmed
and
Vijay
Chudasama
 *
*
Department of Chemistry, University College London, London, UK. E-mail: v.chudasama@ucl.ac.uk
First published on 30th July 2020
Abstract
Significant advancements in C–N bond formation via C–H bond functionalisation have made it a staple in the production of nitrogen-containing compounds in both industry and academia. However, transition metal-free synthesis, particularly in the case of C(sp3)–N formation, has remained a significant challenge to the synthetic community. Herein we report a procedure for α-C(sp3)–H amination of ethereal compounds through use of azodicarboxylates as the nitrogen source and freely-available atmospheric oxygen to access ethereal radical intermediates via aerobic C–H activation. The use of fluorinated alcohols as solvent is observed to greatly increase the efficiency of the reaction and we show experimentally and theoretically the key role of H-bonding between fluorinated alcohols and azodicarboxylates. Calculations of the condensed Fukui functions of a H-bonded fluorinated alcohol-azodicarboxylate complex correlates with a significantly increased susceptibility of azodicarboxylates to undergo reaction with radicals, which informs a number of recent reports in the literature.
Introduction
The drive to develop efficient and direct transformations for the construction of complex molecules is always at the forefront of research in synthetic organic chemistry.1 With growing environmental concerns around energy efficiency and waste reduction, it is clear that modern organic synthesis must continue to develop even more powerful methods for the construction of complex organic frameworks through simplification and dematerialisation.2 As such, C–H bond functionalisation reactions have emerged as a favourable route to introduce functionality and therefore increase complexity of molecules in single-step chemical transformations.3 In particular, C–H bond aminations are an extremely desirable process due to the plethora of nitrogen-containing compounds found in pharmaceuticals.4 However, there are still many challenges in this area: (i) the amination of C(sp3)–H bonds remains difficult despite significant advancements in the field of C(sp2)–N formation (e.g. Buchwald–Hartwig amination),5,6 (ii) the overreliance of directing groups, especially in the case of C(sp3)–H bond activation, resulting in synthetic routes requiring additional installation and removal steps of the protecting/directing group,7,8 (iii) the current standard of using nitrene insertion reactions as a means for C(sp3)–H amination suffers from suboptimal regioselectivity and requires the use of specialised reagents such as organic azides9–11 or azoles12 as the nitrogen source, and (iv) the prevalent use of toxic/expensive transition metal reagents in the activation of inert C–H bonds.13,14 Despite providing a direct route to C–N bond formation, the existence of these limitations and the current quantity of external additives15,16 required for efficient reaction means that there is a need to develop C–H bond activation methodologies that are more attractive from a green chemistry standpoint.17The use of radical intermediates in C–H bond activation reactions has seen major developments in the past few decades owing to improvements in indiscriminate C–H bond abstraction and reaction regioselectivity.18 Radical pathways offer fascinating alternative approaches for connecting molecular fragments with high step- and atom-economy that are often complementary to traditional two-electron processes. Ideally, said radical pathways would be initiated in a green manner and/or without the need for complex synthetic manipulation (see above). There has been some work carried out with this focus by exploiting aerobic C–H activation, especially on aldehydic C(sp2)–H bonds,19–21 but there has been a lack of a substantive breakthrough in controlling and exploiting aerobic C(sp3)–H activation, which is widely considered as more desirable. With this in mind, we noted that the mechanism for the oxygen-initiated oxidation of ethers has been extensively studied22–24 and it is widely accepted that interaction of the α-C(sp3)–H in ethers with dioxygen results in the generation of a nucleophilic radical intermediate (Scheme 1). Thus, it should be appreciated that the aerobically initiated auto-oxidation of ethers offers a unique opportunity for the clean and simple access to ethereal radical species without the use of any metal reagents or initiator species.25 Unfortunately, the majority of research exploring ether auto-oxidation has resulted in methods to suppress the process rather than utilise it as a means to achieve discriminate C(sp3)–H functionalisation.26–28
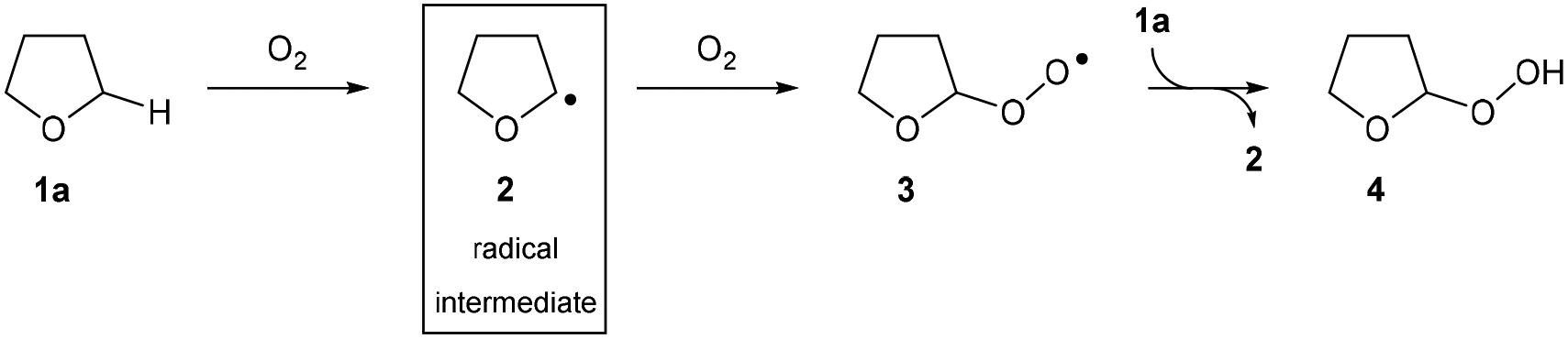 | ||
| Scheme 1 Aerobically initiated auto-oxidation of ethers generates ethereal radical species. | ||
In modern radical-based C–N bond formation, commercially available azodicarboxylates are the standout class of compounds used as radical acceptors. Their strong electrophilic nature and possession of a vacant bonding orbital renders these species excellent candidates for attack by nucleophilic radicals and this has been extensively observed when utilising acyl radical chemistry for the formation of C(sp2)–N bonds.29–33 Unfortunately, translation of these protocols to C(sp3)–N formation has been a significant challenge and has either required the substrate to be used in vast excess (ca. 90 eq.), considered suboptimal for organic synthesis, or required use of a specialised initiator/polarity reversal catalyst.34,35 We desired a protocol that would substantially increase the efficiency of the process; omitting the requirement of any additional additive whilst utilising molecular oxygen in air to initiate α-C(sp3)–H activation. In consideration of this, we looked towards the use of fluorinated alcohols such as 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE) as reaction solvents. These species exhibit higher relative acidity and substantial H-bond donating ability when compared to traditional solvents.36 We postulated that H-bonding of fluorinated alcohols to azodicarboxylates would result in a decrease in the energy of the LUMO, thus increasing susceptibility to nucleophilic attack from ethereal radicals and resulting in an increased efficiency of reaction.
Herein, we report the appraisal of this idea and demonstrate the exploitation of the aerobic C–H bond activation of ethers as a means to achieve site-selective radical generation. The use of fluorinated alcohols as both the reaction solvent and to ‘activate’ azodicarboxylates through a H-bonding interaction results in efficient, regioselective and metal-free amination of ethereal α-C–H bonds that requires no additional initiator species, additives or directing groups.
Results and discussion
To the best of our knowledge, the closest related example to what we wanted to achieve was the pioneering work of Askani who reported an unexpected reaction of dimethyl azodicarboxylate 5a and reaction solvent THF 1a under the action of UV-light to afford adduct species 6aa.34 Askani confirmed a radical pathway for the reaction by repeating the process using benzoyl peroxide in place of UV-light as the radical initiator (Scheme 2).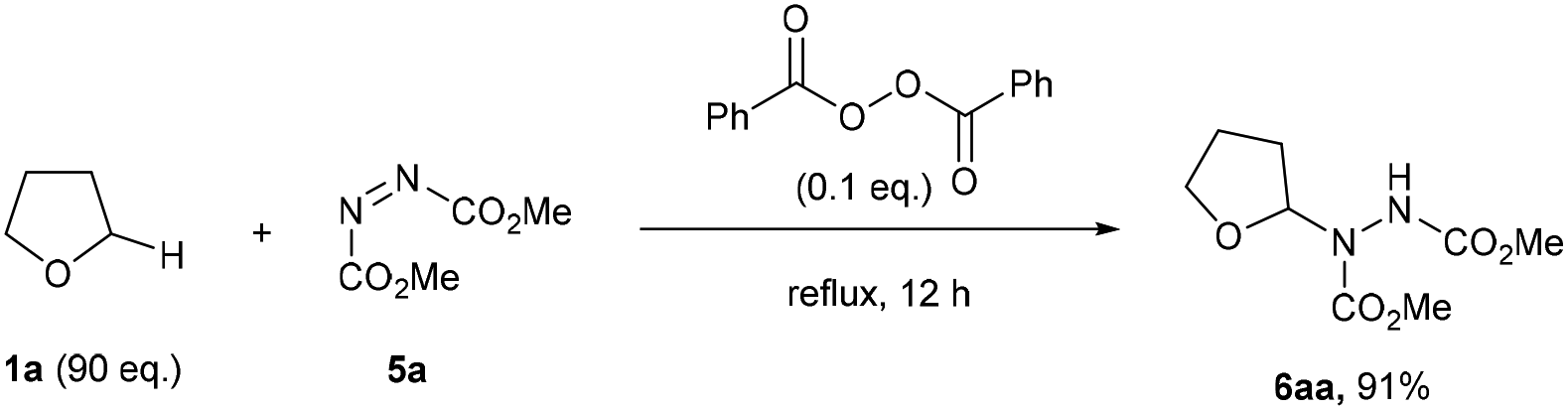 | ||
| Scheme 2 Previously reported peroxide-initiated synthesis of adduct 6aa requiring a vast excess of THF 1a. | ||
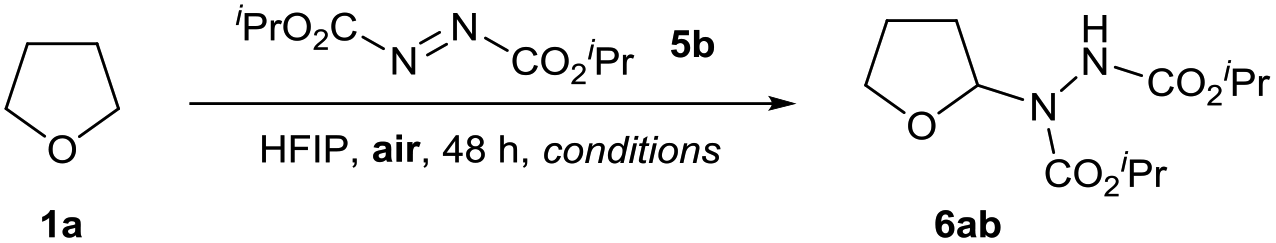
We postulated an aerobic protocol could be developed through the formation of the integral ethereal radical intermediate by exposure of the reaction mixture to air. Our investigations began with optimisation of the reaction between tetrahydrofuran (THF) 1a and commercially available diisopropyl azodicarboxylate (DIAD) 5b to form desired adduct 6ab. As previous reports for the reaction between ethers and azodicarboxylates utilised the ethereal substrate in vast excess,34,37 we were motivated to develop a protocol that employed a more desirable quantity of substrate 1a.
An initial reaction, using 5 equivalents of THF 1a at 21 °C, a stirring rate of 700 rpm and the reaction mixture exposed to air, resulted in a trace amount of desired product (2% yield, Table 1, entry 1). We were pleased to observe that the same reaction conducted with use of 1 mL of HFIP resulted in a four-fold increase in yield (8% yield, Table 1, entry 2). This suggested that use of fluorinated alcohols as solvent for the transformation could improve the reaction. We observed that when utilising HFIP, increasing the reaction temperature also had a large impact on the efficiency of the reaction (Table 1, entries 2–4); a reaction temperature of 80 °C afforded a 64% yield of desired product 6ab (Table 1, entry 4). As it is well understood that stirring rate can have a significant effect on the rate of aerobic oxidation,30 we investigated whether modification of the stirring rate affected reaction efficiency (Table 1, entries 5–7). Gratifyingly, we observed that increasing the stirring rate to 1050 rpm resulted in a far more desirable yield of 6ab (83%, Table 1, entry 5), whereas decreasing stirring rate resulted in systematic reductions in reaction yield (Table 1, entries 6 and 7).
|
|
||||
|---|---|---|---|---|
| Entry | T (°C) | Stirring rate (rpm) | HFIP (mL) | Yield b (%) |
| a Reaction Conditions: 1a (5 mmol), 5b (1 mmol), air, 48 h. b Isolated yield, conversion of 5b given in parenthesis based on amount recovered. c Use of TFE instead of HFIP. d Use of AcOH instead of HFIP. e Use of 10 mol% BHT. f Substrates and solvent degassed and reaction conducted under an argon atmosphere. | ||||
| 1 | 21 | 700 | 0 | 2 (5%) |
| 2 | 21 | 700 | 1 | 8 (16%) |
| 3 | 60 | 700 | 1 | 52 (64%) |
| 4 | 80 | 700 | 1 | 64 (77%) |
| 5 | 80 | 1050 | 1 | 83 (96%) |
| 6 | 80 | 350 | 1 | 60 (66%) |
| 7 | 80 | 0 | 1 | 54 (58%) |
| 8 | 80 | 1050 | 0.5 | 92 (100%) |
| 9 | 80 | 1050 | 2 | 78 (92%) |
| 10 | 80 | 1050 | 0 | 55 (59%) |
| 11c | 80 | 1050 | 0.5 | 82 (89%) |
| 12d | 80 | 1050 | 0.5 | 68 (75%) |
| 13e | 80 | 1050 | 0.5 | 0 (1%) |
| 14f | 80 | 1050 | 0.5 | 2 (4%) |
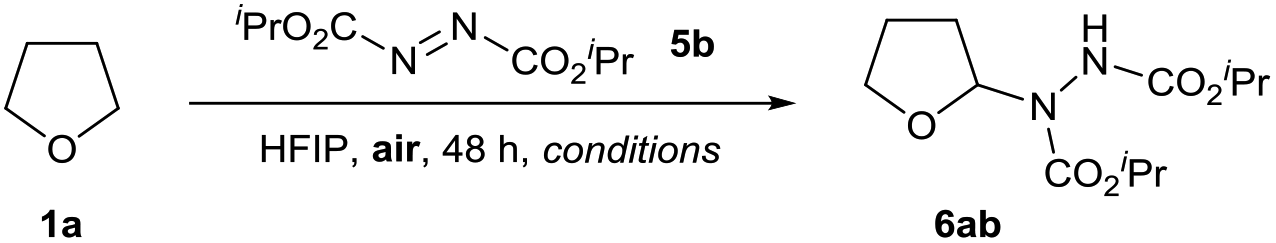
Finally, a decrease in the amount of HFIP solvent to 0.5 mL resulted in a highly desirable 92% yield of 6aa (Table 1, entry 8), whereas increasing the amount of HFIP resulted in a reduction in yield to 78%. With an efficient reaction using HFIP in hand, we investigated the effect of alternative solvents on reaction efficiency. Conducting the reaction in the absence of solvent resulted in only 55% yield (Table 1, entry 10). Substitution of HFIP to alternative fluorinated alcohol TFE resulted in only a small reduction in yield to 82% (Table 1, entry 11), whilst use of an alternative acidic solvent AcOH (despite displaying stronger acidity, it is an inferior H-bond donor compared to HFIP38) resulted in a significant decrease in yield of 6ab (68%, Table 1, entry 12). This provided some evidence that the greater the H-bonding ability of the solvent, the more efficient the reaction. To confirm the presence of a radical pathway, we conducted the reaction of 1a and 5b under our optimised conditions with 10 mol% of radical inhibitor BHT (2,6-di-tert-butyl-4-methylphenol) and observed minimal conversion of starting material 5b (Table 1, entry 13). Finally, to demonstrate that the reaction proceeds via a dioxygen-initiated pathway, the optimised reaction of 1a and 5b was conducted under an inert atmosphere (utilising degassed reagents) resulted in a modest 2% yield of 6ab (Table 1, entry 14).
With optimised conditions in hand, the generality of the procedure was then appraised (Scheme 3). Besides DIAD, all azodicarboxylates trialled gave their respective desired products in good to excellent yields (72–94% yield), resulting in ether-azodicarboxylate adducts with carbamate esters that could be removed either under basic (methyl-, ethyl-carbamate), acidic (isopropyl-carbamate) or hydrogenation (benzyl-carbamate) conditions. A gram-scale synthesis of THF-DIAD adduct 6ab was performed, which provided the desired product in an 81% yield. Use of 6- and 7-membered ring cyclic ethers also proved to be compatible under the reaction conditions with desired products isolated in good yields (62–71%). However, when employing α-substituted cyclic ether 2-methyltetrahydrofuran, only 20% of the sole regioisomer 6eb was observed, with a significant amount reduced azodicarboxylate 7 recovered. This was intriguing as we expected appearance of the tetra-substituted regioisomer 6fb, and we hypothesised that acid-mediated cleavage of the C–N bond in 6fb brought about through the presence of HFIP (pKa = 9.3) led to the formation of 7. In an attempt to reduce this pathway, we utilised comparatively less acidic fluorinated alcohol TFE (pKa = 12.5). Unfortunately, this did not seem to improve the reaction with regards to yield or reducing formation of by-product 7. When the reaction was conducted under neat conditions (without the use of solvent), a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 ratio of 6eb to 6fb in 68% yield was observed. To provide evidence that tetra-substituted α-aminated ethers are unstable under acidic conditions, subjection of this regioisomeric mixture to HFIP then led to decomposition of the tetra-substituted 6fb regioisomer When utilising cyclic acetals, it was observed that the use of HFIP as the reaction solvent resulted in the formation of a large amount of reduced-azodicarboxylate 7, thereby resulting in suboptimal reaction yields (0–21%). We again hypothesised that this was due to acid-mediated cleavage of the electron-rich C–N bond. To our delight, use of the less acidic fluorinated alcohol TFE, in this case, proved optimal with minimal quantities of reduced-azodicarboxylate 7 observed and full consumption of azodicarboxylate, resulting in far more efficient reactions (59–70% yield). Fascinatingly, when utilising 1,3,5-trioxane as the ether substrate and TFE as the reaction solvent, we observed 62% of product 6ib (i.e. formylation of azodicarboxylate species). We suspect this is due to 1,3,5-trioxane being a formaldehyde trimer which is prone to acidic decomposition. Indeed, we observed that use of HFIP resulted in only 15% yield of 6ib. We suspect this is due to rapid decomposition of 1,3,5-trioxane under the action of the more acidic HFIP prior to significant C–N formation. The scope of acyclic ethers was then appraised. Disappointingly, diethyl ether did not appear to not undergo any transformation under the reaction conditions. We suspect that this is due to the comparatively larger bond dissociation energy (BDE) of the α-C(sp3)–H in diethyl ether (ca. 400 kJ mol−1) compared to cyclic ethers (i.e. 380–390 kJ mol−1).39 To provide evidence for this, acyclic dibenzyl ether (which exhibits a BDE of 359 kJ mol−1) was trialled and the reaction proceeded efficiently with a 63% of desired adduct 6kb observed. Owing to literature describing the oxygen-induced auto-oxidation of organic sulfides,40 tetrahydrothiophene (sulfur-analogue of THF) was also trialled in the reaction conditions; this resulted in the isolation of the desired product in 49% yield. Finally, we demonstrate the potential of the reaction procedure with regards to nitrogen-based heterocycles through the use of N-Boc pyrrolidine affording an 81% yield of desired adduct 6mb.
:70 ratio of 6eb to 6fb in 68% yield was observed. To provide evidence that tetra-substituted α-aminated ethers are unstable under acidic conditions, subjection of this regioisomeric mixture to HFIP then led to decomposition of the tetra-substituted 6fb regioisomer When utilising cyclic acetals, it was observed that the use of HFIP as the reaction solvent resulted in the formation of a large amount of reduced-azodicarboxylate 7, thereby resulting in suboptimal reaction yields (0–21%). We again hypothesised that this was due to acid-mediated cleavage of the electron-rich C–N bond. To our delight, use of the less acidic fluorinated alcohol TFE, in this case, proved optimal with minimal quantities of reduced-azodicarboxylate 7 observed and full consumption of azodicarboxylate, resulting in far more efficient reactions (59–70% yield). Fascinatingly, when utilising 1,3,5-trioxane as the ether substrate and TFE as the reaction solvent, we observed 62% of product 6ib (i.e. formylation of azodicarboxylate species). We suspect this is due to 1,3,5-trioxane being a formaldehyde trimer which is prone to acidic decomposition. Indeed, we observed that use of HFIP resulted in only 15% yield of 6ib. We suspect this is due to rapid decomposition of 1,3,5-trioxane under the action of the more acidic HFIP prior to significant C–N formation. The scope of acyclic ethers was then appraised. Disappointingly, diethyl ether did not appear to not undergo any transformation under the reaction conditions. We suspect that this is due to the comparatively larger bond dissociation energy (BDE) of the α-C(sp3)–H in diethyl ether (ca. 400 kJ mol−1) compared to cyclic ethers (i.e. 380–390 kJ mol−1).39 To provide evidence for this, acyclic dibenzyl ether (which exhibits a BDE of 359 kJ mol−1) was trialled and the reaction proceeded efficiently with a 63% of desired adduct 6kb observed. Owing to literature describing the oxygen-induced auto-oxidation of organic sulfides,40 tetrahydrothiophene (sulfur-analogue of THF) was also trialled in the reaction conditions; this resulted in the isolation of the desired product in 49% yield. Finally, we demonstrate the potential of the reaction procedure with regards to nitrogen-based heterocycles through the use of N-Boc pyrrolidine affording an 81% yield of desired adduct 6mb.
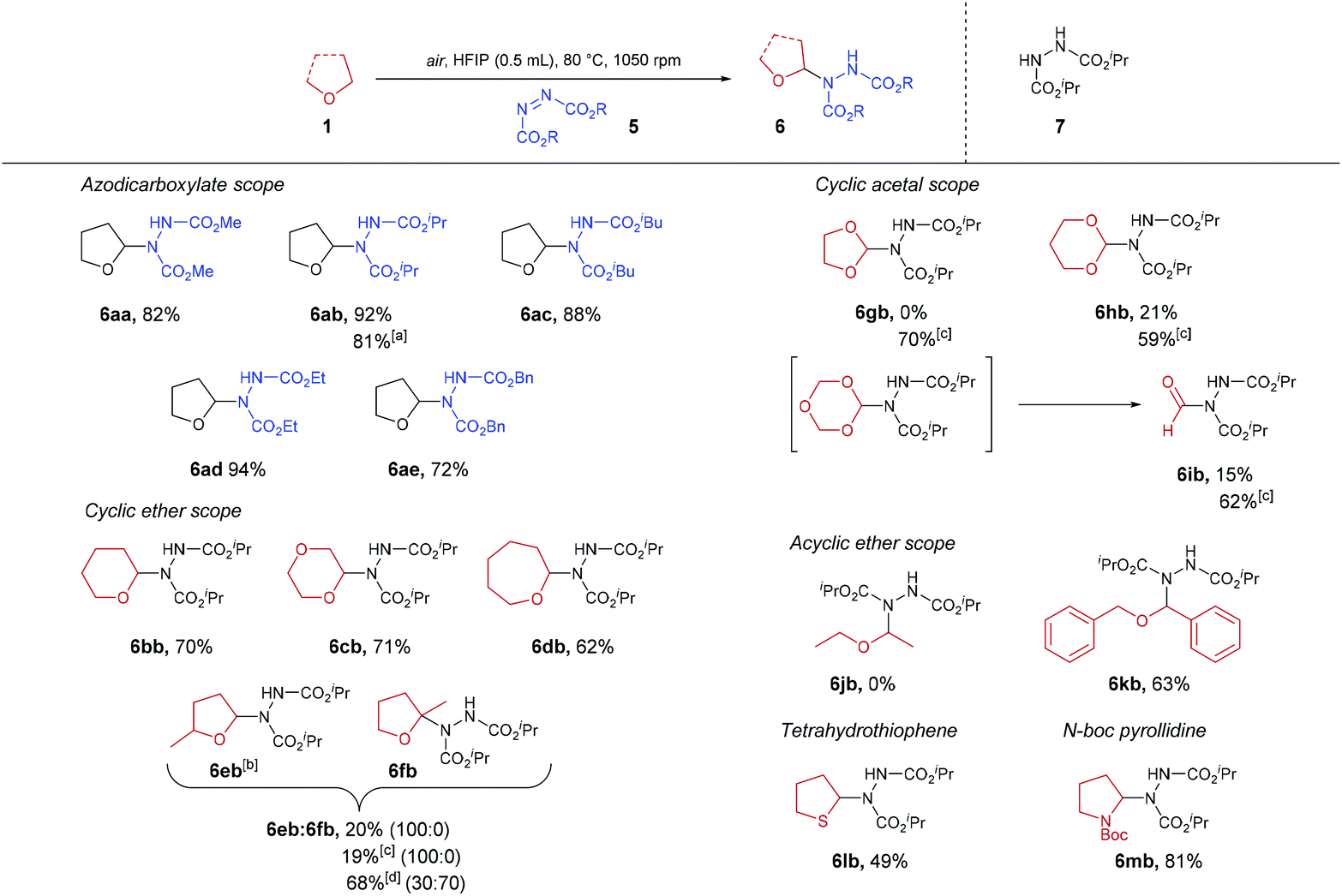 | ||
| Scheme 3 α-C(sp3)–H amination of ethers 1 using azodicarboxylates 5. aGram scale synthesis. b1:1 Ratio of diastereomers. cUse of TFE as solvent. dNeat conditions. | ||
For our initial investigations of the reaction mechanism, a control experiment using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger was conducted under the optimised conditions (Scheme 4). As expected, a complete retardation of the reaction was observed and the THF-TEMPO adduct was detected by LC-MS analysis (MS = 228.2 Da), suggesting the mechanism proceeds through the initial formation of a THF radical.
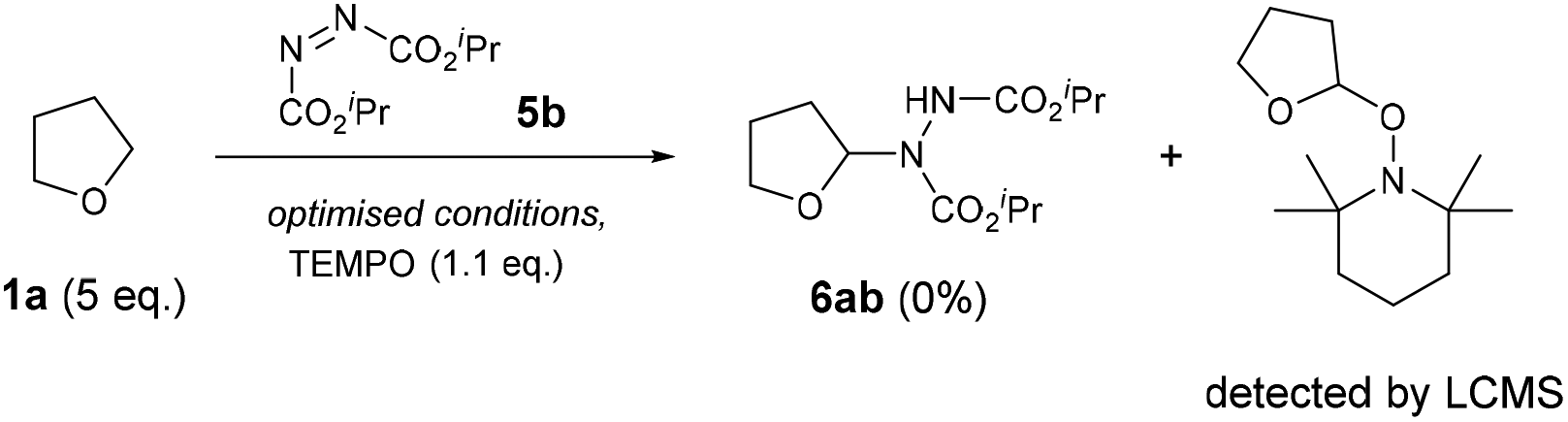 | ||
| Scheme 4 TEMPO trapping experiment. | ||
It is well understood that fluorinated alcohols such as HFIP and TFE are excellent hydrogen-bond donors and this has been extensively studied in the interaction with hydrogen-bond accepting ethers.36 We proposed that fluorinated alcohols can also participate in efficient H-bonding to azodicarboxylates. We therefore experimentally examined, through 1H NMR titration, the formation of H-bond complexes of HFIP with DIAD. The titration was conducted through successive additions of DIAD to an NMR sample of a fixed quantity of HFIP in CDCl3 and the change in the chemical shift (δ) of the hydroxyl proton was recorded. A significant downfield shift of the proton was observed in the 1H NMR with increasing quantity of DIAD, clearly indicating the formation of a hydrogen-bonded complex (Fig. 1). The presence of only one hydroxyl proton signal indicates a fast equilibrium on the NMR timescale.
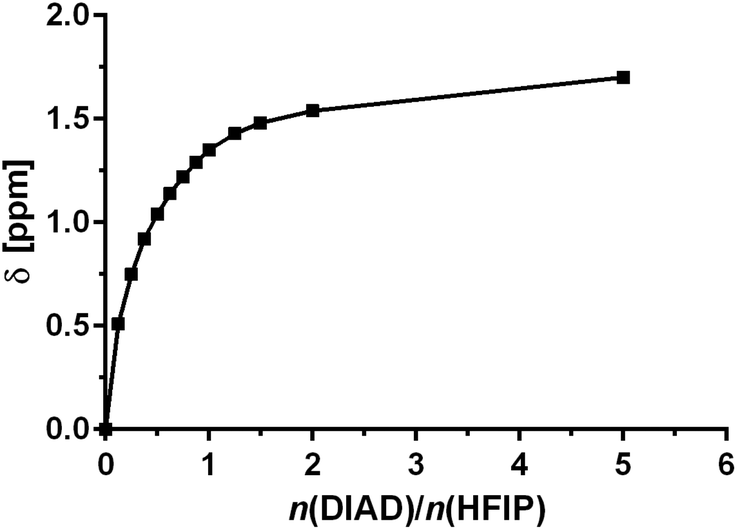 | ||
| Fig. 1 NMR titration curve of HFIP with DIAD 5b. | ||
The binding constant values were calculated from 1H-NMR titration data using Bindfit41,42 for 1:2 (host:guest) non-cooperative binding stoichiometry. The following equation was used for calculating the binding constant from titration experiments via non-linear fitting method (Nelder–Mead):
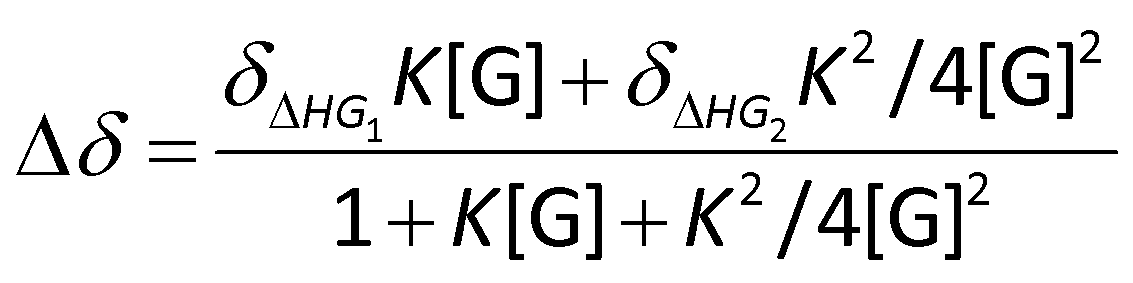
NMR data points for DIAD fitted well into this model and yielded a binding constant K of 4.4 ± 0.5 M−1 (initial H-bonding between HFIP and DIAD), indicating a significant interaction.
Having experimental support for the H-bonding effect of HFIP to DIAD 5b, we decided to conduct theoretical studies on the nature and effect such an interaction would have on the mechanism of the overall chemical transformation. First, we were interested in whether DIAD was likely to be accepting a H-bond through a nitrogen atom on the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, or the carbonyl oxygen on the carboxylate esters adjacent to the NN bond (Fig. 2). Thus, the energy of DIAD 5b, DIAD-HFIP complex H-bonded through a carbonyl oxygen atom (denoted as 5bO) and DIAD-HFIP complex H-bonded through a nitrogen atom (denoted as 5bN) were calculated.
N bond, or the carbonyl oxygen on the carboxylate esters adjacent to the NN bond (Fig. 2). Thus, the energy of DIAD 5b, DIAD-HFIP complex H-bonded through a carbonyl oxygen atom (denoted as 5bO) and DIAD-HFIP complex H-bonded through a nitrogen atom (denoted as 5bN) were calculated.
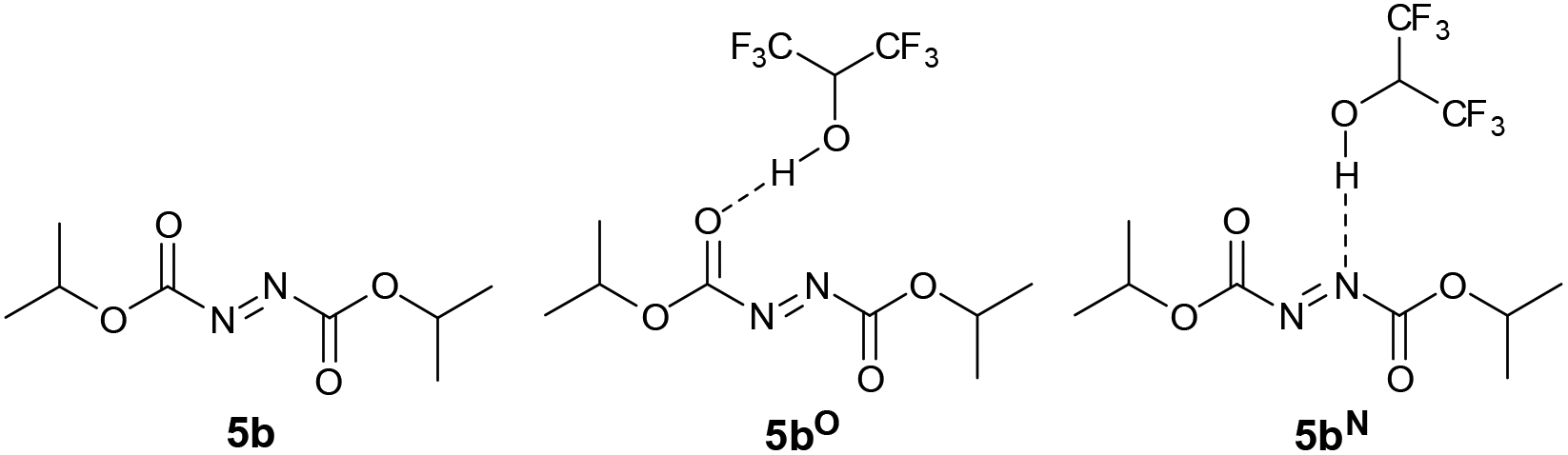 | ||
| Fig. 2 DIAD 5b, DIAD-HFIP complex H-bonded through a carbonyl oxygen atom 5bO and DIAD-HFIP complex H-bonded through a nitrogen atom 5bN. | ||
The molecules were subjected to geometry optimisation at B3LYP/6-311++G(d,p) level; the resulting optimised structure are given in Fig. 3. Following this, calculations in vacuo were performed at M06-2X/6-311++G(d,p) level of theory and the energy difference between 5bO and 5bN was found to be 1.8 kcal mol−1, 5bN being the lowest in energy. The energy difference being relatively small, both complexes were further investigated to understand the role of HFIP in the reaction.
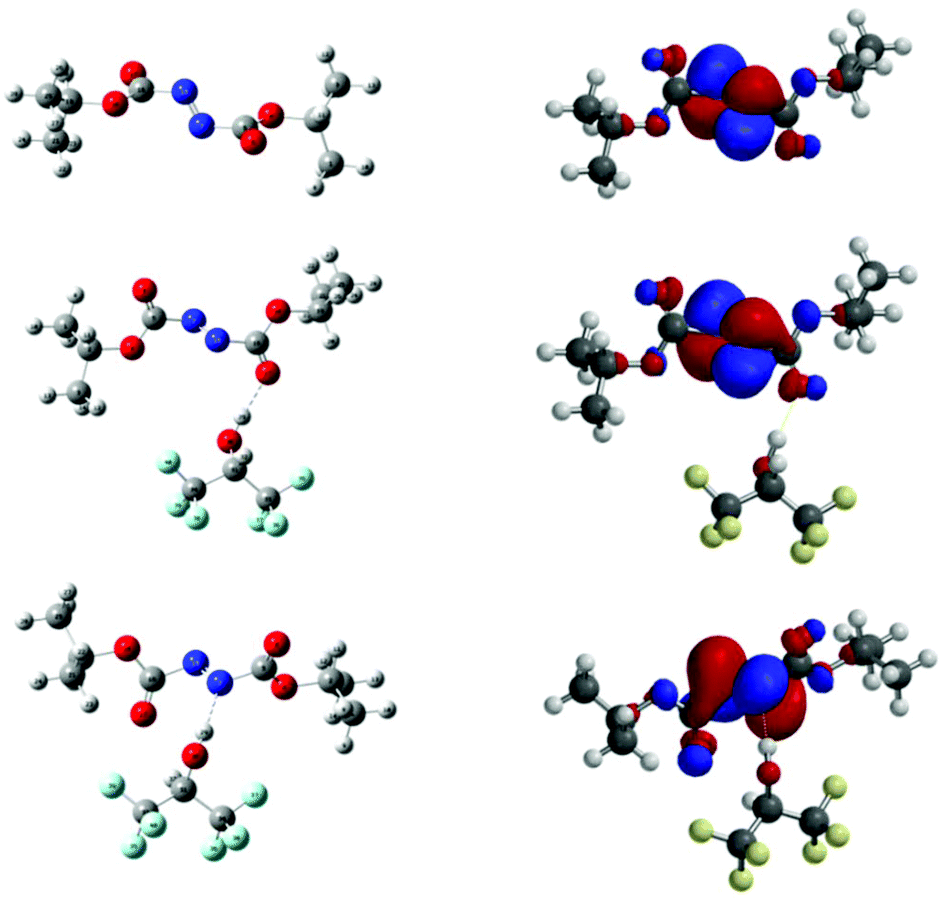 | ||
| Fig. 3 (Left) Optimised geometry of 5b, 5bO and 5bN at M06–2X/6-311++G(d,p) level. (Right) Corresponding calculated LUMO. | ||
As α-ethereal radicals are nucleophilic in nature they will interact favourably with electrophilic entities such as azodicarboxylates; as such, the LUMO energies of 5b and 5bO and 5bN were calculated. They were found to be −2.42 eV for DIAD 5b, −2.93 eV for DIAD-HFIP complex 5bO and −3.25 eV for DIAD-HFIP complex 5bN. This suggests that H-bonding to azodicarboxylates does indeed lower the energy of the LUMO, with a significant increase observed upon H-bonding through a nitrogen atom compared to a carbonyl oxygen atom. The spin density maps of nitrogen-centred radical intermediates of the form 8 following THF radical attack on 5b, 5bO and 5bN respectively have also been calculated and are presented in ESI.†
In computational chemistry, Fukui functions are the reactivity descriptors that enable characterising the relative susceptibility of sites to electrophilic, nucleophilic and radical attack and hence are commonly used for the establishment of the regio- and chemoselectivity observed in reactions. The condensed Fukui function represent the same underlying idea but applied to an atom within a molecule rather than a point in three-dimensional space. The atoms of a molecule, which have the largest Fukui values, are the most feasible sites for the attack. For radical attack, the condensed Fukui function of an atom A is defined as
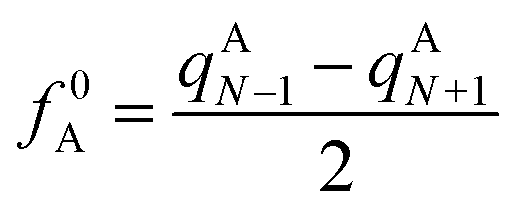
|
|
|||
|---|---|---|---|
| 5b | 5bO | 5bN | |
| N(7) | 0.22 | 0.25 | 0.11 |
| N(15) | 0.21 | 0.23 | 0.33 |
These results highlight the impact of H-bonding through the nitrogen atom where susceptibility of the non-H-bonded nitrogen N(15) on the NN bond to radical attack is significantly increased. By contrast, H-bonding through a carbonyl oxygen atom did not have a significant effect on the susceptibility of DIAD to radical attack. This fits with the theoretically optimised geometry of azodicarboxylates where the carbonyls of the carboxylate esters adjacent to the NN bond are shown to be practically orthogonal to the NN bond,44 indicating that the molecule is not conjugated and therefore hydrogen-bonding to the carbonyl oxygen atom should have minimal effect on the reactivity of azodicarboxylates.
We therefore propose that the mechanism for radical attack of ethereal radical species 2 on an azodicarboxylate-HFIP complex 5bN proceeds via H-bonding to the adjacent nitrogen on the NN bond (Scheme 5). The radical adduct species 8 is then expected to abstract a H-atom from the starting material 1a, forming desired product 6ab and reforming ethereal radical 2.
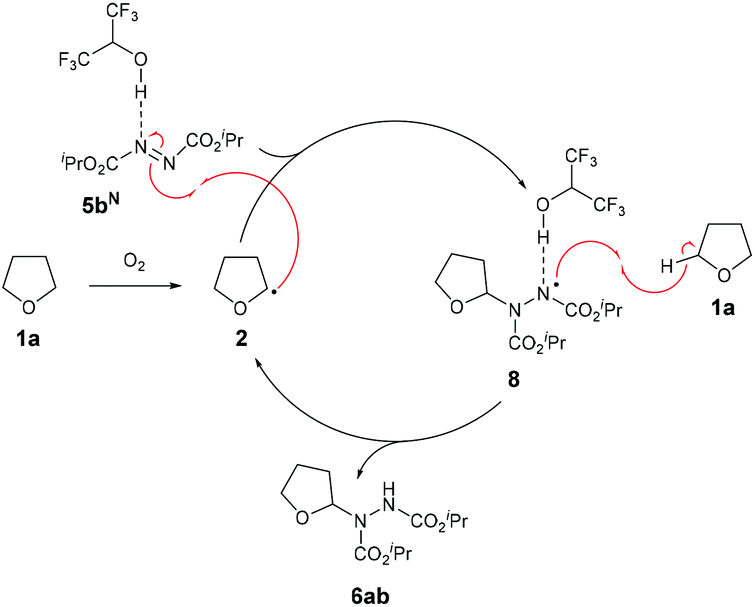 | ||
| Scheme 5 Proposed mechanism. | ||
The described formation of ethereal α-C–N bonds via aerobic C–H activation represents a fundamental advancement that allows for the increase in functionality and complexity of ether substrates in a minimalistic fashion and therefore obviates the need for any undesirable metals, initiators or additives. The use of atmospheric dioxygen represents a green, sustainable and a freely accessible form of oxidant to be utilised in the synthesis. Furthermore, the formation of the C–N bond at the α-site in ethers results in the hemiaminal ether skeleton (HES) and this functionality is commonplace in organic synthesis45 and bioactive molecules such as Tegafur (chemotherapeutic prodrug) and Crambescin B (voltage-gated sodium channel inhibitor) and are also present in a variety of HIV/AIDS medications (i.e. didanosine, zalcitabine). The formed ether-azodicarboxylates also offer platforms for further synthetic manipulation. As an example, access to (protected) α-amino species is highly desirable and we demonstrate the formation of these structures through cleavage of the N–N bond (Scheme 6).46 A one-pot alkylation-elimination protocol was carried out on compound 6ab utilising tert-butyl bromoacetate as the alkylating agent and sodium hydride to facilitate base-mediated alkylation and E1cB elimination gave desired product 9 in a 75% yield. This result demonstrates that the amination procedure reported in the study is a powerful tool for the metal-free formation of synthetically useful α-hydrazo and α-amino ethers, as well as being a key step forward in the field of C–H activation.
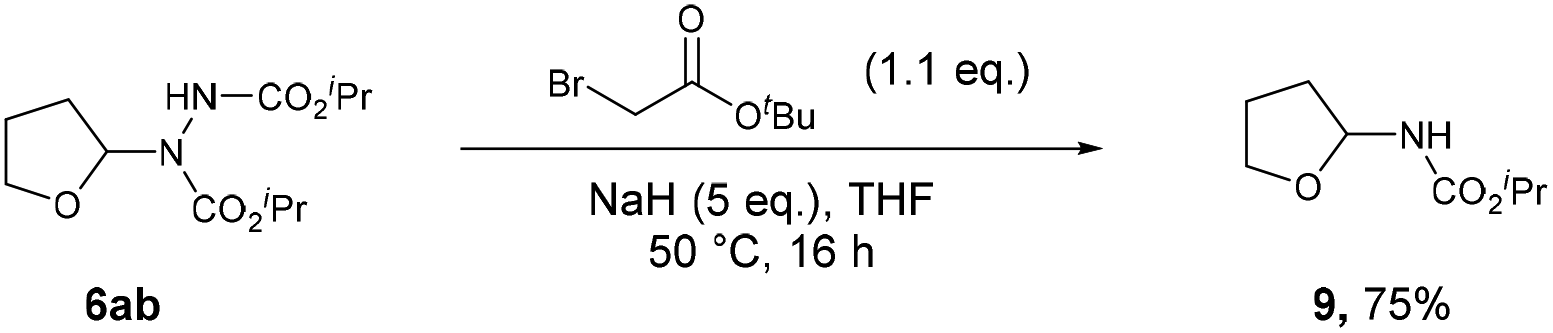 | ||
| Scheme 6 Metal-free rupture of the N–N bond in 6ab to form 9. | ||
Conclusions
In summary, an aerobic approach for α-C(sp3)–H amination of ethereal substrates utilising azodicarboxylates as nitrogen source has been enabled through the use of fluorinated alcohols. The use of atmospheric oxygen to generate radical species and the dual function of fluorinated alcohols as both solvent and activating agent feeds into the sought-after goal of simplification and dematerialisation of C–H activation methods. A broad selection of ether- and acetal-azodicarboxylate adducts are efficiently prepared by the disclosed methodology. We provide experimental evidence for the H-bonding interaction between HFIP and DIAD and explore theoretical studies suggesting that the H–bonding of HFIP specifically to the nitrogen atom in DIAD results in a substantial lowering of the LUMO energy and increases its susceptibility to radical attack. This finding not only informs the reactions disclosed in this article but also a number of recent reports in the literature that utilise this combination.47–50 We also anticipate that the interaction we detail between fluorinated alcohols and azodicarboxylates will provide new opportunities for X–N bond formations, particularly in radical-based synthesis. Moreover, the formed ether-azodicarboxylate adducts offer opportunity for further synthetic manipulation, and in particular, to gain access to α-amino ethers. To the best of our knowledge, the use of aerobic ethereal C–H bond activation to access reactive radical species without the use of any additional reagents is largely unexplored and thus the methods we describe in this manuscript represent a fundamentally novel contribution to the field of C–H activation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the UCL Graduate School for funding A. S. and N. A. The authors also acknowledge the UCL Chemistry Mass Spectrometry Facility (Dr K. Karu) and the EPSRC U.K. National MS Facility (Swansea).Notes and references
- R. A. Sheldon, Green Chem., 2008, 10, 359–360 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. De Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- D. Hazelard, P. A. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500–2521 RSC.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., 2019, 58, 17118–17129 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS.
- D. Mu, X. Wang, G. Chen and G. He, J. Org. Chem., 2017, 82, 4497–4503 CrossRef CAS.
- J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702–4705 CrossRef CAS.
- L. Zhang, H. Yi, J. Wang and A. Lei, J. Org. Chem., 2017, 82, 10704–10709 CrossRef CAS.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- D. Basu, S. Kumar, S. Sudhir and R. Bandichhor, J. Chem. Sci., 2018, 130, 71 CrossRef.
- K. N. Betz, N. D. Chiappini and J. Du Bois, Org. Lett., 2020, 22, 1687–1691 CrossRef CAS.
- J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645–1657 CAS.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS.
- A. Maity, S. M. Hyun and D. C. Powers, Nat. Chem., 2018, 10, 200–204 CrossRef CAS.
- K. Miyamoto, J. Yamashita, S. Narita, Y. Sakai, K. Hirano, T. Saito, C. Wang, M. Ochiai and M. Uchiyama, Chem. Commun., 2017, 53, 9781–9784 RSC.
- S. Di Tommaso, P. Rotureau, B. Sirjean, R. Fournet, W. Benaissa, P. Gruez and C. Adamo, Process Saf. Prog., 2014, 33, 64–69 CrossRef CAS.
- S. Di Tommaso, P. Rotureau, O. Crescenzi and C. Adamo, Phys. Chem. Chem. Phys., 2011, 13, 14636–14645 RSC.
- H. Matsubara, S. Suzuki and S. Hirano, Org. Biomol. Chem., 2015, 13, 4686–4692 RSC.
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2019, 17, 2865–2872 RSC.
- H. L. Jackson, W. B. McCormack, C. S. Rondestvedt, K. C. Smeltz and I. E. Viele, J. Chem. Educ., 1970, 47, A175 CrossRef CAS.
- R. J. Kelly, Chem. Health Saf., 1996, 3, 28–36 CAS.
- D. E. Clark, Chem. Health Saf., 2001, 8, 12–21 CrossRef CAS.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 RSC.
- G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS.
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 RSC.
- A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC.
- R. Askani, Chem. Ber., 1965, 98, 2551–2555 CrossRef CAS.
- Y. Amaoka, S. Kamijo, T. Hoshikawa and M. Inoue, J. Org. Chem., 2012, 77, 9959–9969 CrossRef CAS.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- G. Ahlgren, Tetrahedron Lett., 1974, 15, 2779–2782 CrossRef.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- V. Tumanov, Pet. Chem., 2005, 45, 350–363 Search PubMed.
- J. A. Howard and S. Korcek, Can. J. Chem., 1971, 49, 2178–2182 CrossRef CAS.
- http://supramolecular.org .
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- M. S. Goh, L. Rintoul, M. C. Pfrunder, J. C. McMurtrie and D. P. Arnold, J. Mol. Struct., 2015, 1098, 298–305 CrossRef CAS.
- L. Dian, Q. Xing, D. Zhang-Negrerie and Y. Du, Org. Biomol. Chem., 2018, 16, 4384–4398 RSC.
- A. Shamsabadi, J. Ren and V. Chudasama, RSC Adv., 2017, 7, 27608–27611 RSC.
- K. Y. Lee, Y. J. Im, T. H. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2001, 22, 131–132 CAS.
- S. M. Inamdar, V. K. More and S. K. Mandal, Tetrahedron Lett., 2013, 54, 530–532 CrossRef CAS.
- R. J. Tang, T. Milcent and B. Crousse, Eur. J. Org. Chem., 2017, 2017, 4753–4757 CrossRef CAS.
- R. J. Tang, P. Retailleau, T. Milcent and B. Crousse, ACS Omega, 2019, 4, 8960–8966 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1H and 13C), IR, LRMS (ESI) and HRMS (ESI) of all featured compounds. See DOI: 10.1039/d0ob01562h |
| This journal is © The Royal Society of Chemistry 2020 |