
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of protein glycation using vibrational spectroscopy†
Bethan S.
McAvan
 a,
Aidan P.
France
a,
Bruno
Bellina
a,
Perdita E.
Barran
a,
Royston
Goodacre
b and
Andrew J.
Doig
*c
a,
Aidan P.
France
a,
Bruno
Bellina
a,
Perdita E.
Barran
a,
Royston
Goodacre
b and
Andrew J.
Doig
*c
aSchool of Chemistry, Manchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: andrew.doig@manchester.ac.uk
bDepartment of Biochemistry, Institute of Integrative Biology, University of Liverpool, Biosciences Building, Crown Street, Liverpool L69 7ZB, UK
cManchester Institute of Biotechnology and Division of Neuroscience and Experimental Psychology, Michael Smith Building, School of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester M13 9PT, UK
First published on 22nd April 2020
Abstract
Glycation is a protein modification prevalent in the progression of diseases such as Diabetes and Alzheimer's, as well as a byproduct of therapeutic protein expression, notably for monoclonal antibodies (mAbs). Quantification of glycated protein is thus advantageous in both assessing the advancement of disease diagnosis and for quality control of protein therapeutics. Vibrational spectroscopy has been highlighted as a technique that can easily be modified for rapid analysis of the glycation state of proteins, and requires minimal sample preparation. Glycated samples of lysozyme and albumin were synthesised by incubation with 0.5 M glucose for 30 days. Here we show that both FTIR-ATR and Raman spectroscopy are able to distinguish between glycated and non-glycated proteins. Principal component analysis (PCA) was used to show separation between control and glycated samples. Loadings plots found specific peaks that accounted for the variation – notably a peak at 1027 cm−1 for FTIR-ATR. In Raman spectroscopy, PCA emphasised peaks at 1040 cm−1 and 1121 cm−1. Therefore, both FTIR-ATR and Raman spectroscopy found changes in peak intensities and wavenumbers within the sugar C–O/C–C/C–N region (1200–800 cm−1). For quantification of the level of glycation of lysozyme, partial least squares regression (PLSR), with statistical validation, was employed to analyse Raman spectra from solution samples containing 0–100% glycated lysozyme, generating a robust model with R2 of 0.99. We therefore show the scope and potential of Raman spectroscopy as a high throughput quantification method for glycated proteins in solution that could be applied in disease diagnostics, as well as therapeutic protein quality control.
Introduction
Glycation describes a process by which glucose, or a similar sugar molecule, covalently binds to a protein in a non-enzymatic reaction. This reaction, first described by Maillard in 1912, is known to result in irreversible products causing biophysical and structural changes in proteins.1–6 Reducing sugars, such as glucose, fructose and galactose, drive the glycation reaction between their free aldehyde and a protein amine, typically in a Lys side chain (Fig. 1).7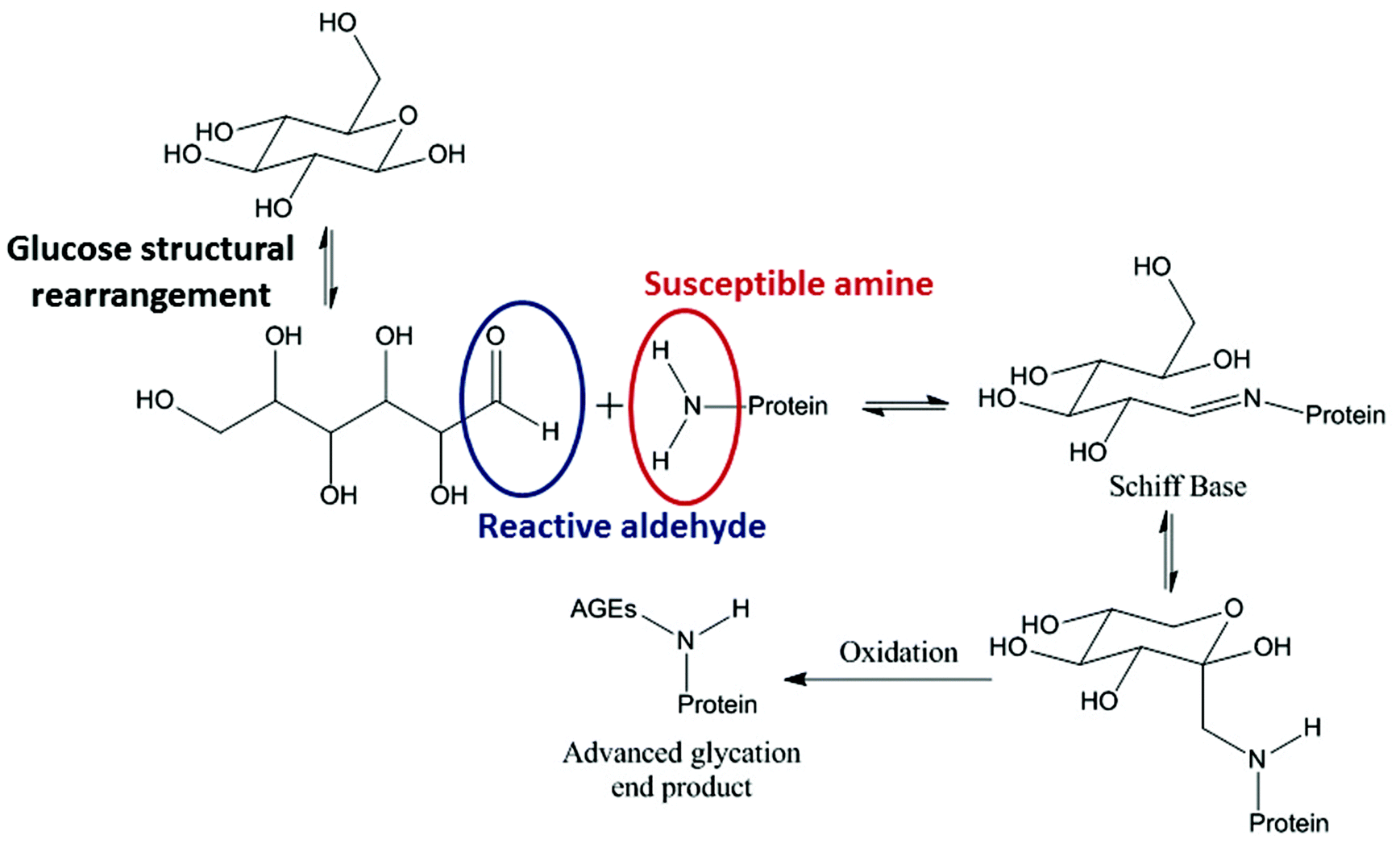 | ||
| Fig. 1 Reaction scheme showing the general process of glycation in proteins. | ||
Reducing sugars are able to act as reducing agents when they tautomerise to form an open chain molecule with a terminal aldehyde group. This free aldehyde group is then able to react with susceptible amine groups of amino acids resulting in the formation of a Schiff base. The Schiff base then undergoes spontaneous rearrangements to form the Amadori product. The Amadori product is capable of degrading through many complex pathways.8
In vivo the most problematic resultant structures are known as advanced glycation end products (AGEs) which are formed due to reaction of the glycated proteins and glucose in the blood. High levels of AGEs have been linked with many degenerative diseases and can cause vascular problems.9 In diabetic patients, abnormally high blood glucose concentrations cause glycation of plasma proteins, such as hemoglobin, giving HbA1c as the most common form. HbA1c is now used to diagnose diabetes, as the amount of glycation reflects the long term concentration of glucose in the blood. Many diabetes studies on treatment and diagnosis now also look at other plasma proteins, such as albumin and IgG, which can also be glycated.8,10 Patients with Alzheimer's disease have high accumulation of AGEs on top of the increased levels of AGEs that occur with natural aging. Research for Alzheimer's diagnostics now focuses on glycated products in the brain tissues and cerebrospinal fluid (CSF). Therefore glycation products are have a wide scope as biomarkers for diagnostic purposes.11
The pharmaceutical industry has been increasingly turning to biologically based drugs in the treatment of autoimmune and difficult to treat diseases. Monoclonal antibodies (mAbs) are one of the most common types of biotherapeutic. Glycation has been proven to be problematic during therapeutic expression. mAbs are often expressed using Chinese hamster ovary (CHO) systems in which glucose or other sugars are used as the primary feed source to drive production of the proteins. For example, the mAb Rhumab manufactured by Genentech and Roche, has been found to have between 40–60% glycation when expressed in specific CHO cell lines.12 This high glycation level is thought to be due to a modified Lys in the mAb light chain. Previous literature into the effects that glycation has on both the structure and function of proteins, specifically mAbs, have mixed outcomes. It has been reported that glycation in mAbs is often at ∼5% or less and seems to have minimal effects on binding efficacy and producing immunogenic responses from T cells in vitro.13 However, others report that minor structural deviations in sugar patterns can lead to diminished antibody functionality and batch heterogeneity, especially if the glycation is prevalent in the antigen binding domain of the mAb.5,14,15 Irrespective of the effects on structure and function, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), European Medicines Agency (EMA) and The Food and Drug Administration (FDA) still require glycation levels to be reported as standard quality control for therapeutic proteins.16–18
Analysis and quantification of glycation currently relies on laborious and expertise intensive methods such as mass spectrometry (MS) alone or combined with peptide mapping that includes targeted enzymatic digestion of protein and MS to determine the mass of fragments.19 Alternatively, boronate columns (BAC) with affinity for glycated proteins can be used in native conditions. These are limited, however, as they have similar affinities no matter how many times the protein is glycated.20–22 In this paper, we present vibrational spectroscopy as an analytical method for the quantification of glycation in a model protein, lysozyme. Lysozyme has 6 Lys residues and the N-terminus, resulting in 7 potential glycation sites. To our knowledge, this is the first reported quantification of protein glycation in solution using Raman spectroscopy.23–26 Raman spectroscopy is inherently easier to incorporate into bio-manufacturing than other widely used analytical methods such as MS, peptide mapping, or the use of columns. Furthermore, Raman spectroscopy has minimal contribution of water bands and spectroscopic information can be obtained without the requirement for a sample to be taken from a fermentation. It can therefore be used in situ.27–29 This paper therefore provides proof-of-concept for using Raman spectroscopy as a method for rapid quantification of glycation and for future development of in-line quality control of protein therapeutic manufacture. It has wider applications in disease diagnosis using glycated proteins implicated in diabetes and Alzheimer's.
Experimental section
Full experimental details are provided in the ESI.†Materials and chemicals
Recombinant human lysozyme, ≥100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg protein (Lysobac) was obtained from Sigma Aldrich (Missouri, US) as a lyophilised powder. D-(+)-Glucose ≥99.5% anhydrous was purchased from Thermo Fischer. Recombinant human albumin, was purchased from Sigma Aldrich as a lyophilised powder. Amicon Ultra 0.5 mL centrifugal filter units, 10 kDa cut off were used for buffer exchange. For the quantification, lysozyme and albumin were buffer exchanged to remove free glucose using Centripure P100 columns (Emp Biotech GmbH) before incubation. Sartorius Stedim Biotech ministart 0.2 μm syringe filters were used to filter the incubation buffers.
000 units per mg protein (Lysobac) was obtained from Sigma Aldrich (Missouri, US) as a lyophilised powder. D-(+)-Glucose ≥99.5% anhydrous was purchased from Thermo Fischer. Recombinant human albumin, was purchased from Sigma Aldrich as a lyophilised powder. Amicon Ultra 0.5 mL centrifugal filter units, 10 kDa cut off were used for buffer exchange. For the quantification, lysozyme and albumin were buffer exchanged to remove free glucose using Centripure P100 columns (Emp Biotech GmbH) before incubation. Sartorius Stedim Biotech ministart 0.2 μm syringe filters were used to filter the incubation buffers.
Sample preparation
:40 ratio into a water/formic acid solution (99.9:0.1, v:v%) at which the final protein concentration was ∼5 μM prior to analysis. For consistency, the non-glycated form of albumin was also treated in the same manner. All experiments were performed using nano-electrospray ionisation (nESI) in positive ionisation mode. Experiments were performed on a Q Exactive UHMR hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher scientific). A full list of the Q Exactive settings utilised are outlined in Table S3.† Mass spectra were recorded at a resolving power of 200000 (lysozyme) and 3125 (albumin). Full quantification and cations are shown in the ESI† and within Fig. S3–S4 and Tables S4–S6.†
Results and discussion
Lysozyme has 6 Lys residues and an N-terminus that can be glycated by glucose via a condensation reaction (Fig. 1). The project used two experimental groups. The first group was 5 mg mL−1 lysozyme with the addition of 0.5 M glucose in incubation buffer. The control group was 5 mg mL−1 lysozyme in incubation buffer without glucose. Each of the conditions had 7 vials that were incubated at 37 °C. One vial from each condition was collected at day 1, 2, 5, 7, 14, 21 and day 30 to study the structure of lysozyme as glycation increases. The glycation method was adapted from previous literature.6 The time length of 30 days was chosen to maximise the amount of glycation in an incubation time frame that avoided the use of preservatives such as sodium azide to prevent bacterial growth.Mass spectrometry determination of glycation
To determine the extent of glycation, MS was employed to determine the mass changes in the protein as a function of incubation time. The MS analysis was carried out using the parameters stated in the methods section whilst the results are shown in Fig. S1.†Table 1 summarises the results of the glucose incubation and monitors its covalent binding to lysozyme with incubation time. In order to check that glucose was covalently bound, the mass spectrometry analysis was carried out at two different collisional energies of 4 V and 20 V with the assumption that at 20 V only covalently bound glucose should be seen and any that is non-covalently associating to the protein would be removed. At both voltages, the populations (%) of glycated species were similar, as shown from the SE in Table 1, showing that no glucose was bound non-covalently. The table summarises the integrated mass peaks averaged over 20 V and 4 V, using the 8+ and 7+ charge states for both voltages. The full quantitative analysis is described in the ESI (Time course mass spectrometry†). The protein is 100% non-glycated at day 0. Lysozyme becomes increasingly glycated the longer it is incubated with glucose and thus the glycated population increases with time. After 30 days the most populated state of lysozyme is that with 1 or 2 bound glucose molecules. In this analysis, as far as we can tell, we never saw more than 4 Lys glycated or any significant change in protein glycation status after 21 days, suggesting that 3 of the Lys side chains are not susceptible to being glycated. The difference in susceptibility to being glycated is a result that has been widely published and investigated using peptide mapping. It is thought to arise from the surrounding environment and solvent accessibility with Lys residues less open to the solvent or having amine groups hydrogen bonded to other protein groups being less likely to be glycated.22,30,31| Population % of lysozyme with bound glucose as a function of time | ||||||||
|---|---|---|---|---|---|---|---|---|
| Number of glucose bound | Day 0 (Lys standard) | Day 1 | Day 2 | Day 5 | Day 7 | Day 14 | Day 21 | Day 30 |
| 0 Glucose | 100 ± 0 | 61 ± 4 | 58 ± 3 | 46 ± 2 | 40 ± 3 | 19 ± 1 | 13 ± 1 | 12 ± 1 |
| 1 Glucose | 0 | 27 ± 2 | 27 ± 1 | 41 ± 0 | 41 ± 1 | 40 ± 1 | 35 ± 2 | 36 ± 1 |
| 2 Glucose | 0 | 13 ± 2 | 11 ± 1 | 18 ± 1 | 18 ± 2 | 30 ± 1 | 33 ± 1 | 35 ± 1 |
| 3 Glucose | 0 | 0 | 4 ± 1 | 5 ± 1 | 5 ± 1 | 11 ± 1 | 14 ± 2 | 14 ± 1 |
| 4 Glucose | 0 | 0 | 0 | 0 | 0 | 0 | 5 ± 1 | 4 ± 0 |
FTIR-ATR analysis of glycated lysozyme
FTIR-ATR was carried out on all the controls and glycated lysozyme from days 1 to 30 of the incubation using lyophilised samples outlined in the methods. The full FTIR spectra of all of the samples are shown in Fig. 2a, where the amide region of the spectrum stretches from 1700 cm−1 to 1200 cm−1 and contains amide regions I, II and III. The overlap between sugar peaks and amide peaks becomes increasingly complicated as increasing sugar peaks can stretch from 1500 cm−1 to 1000 cm−1.32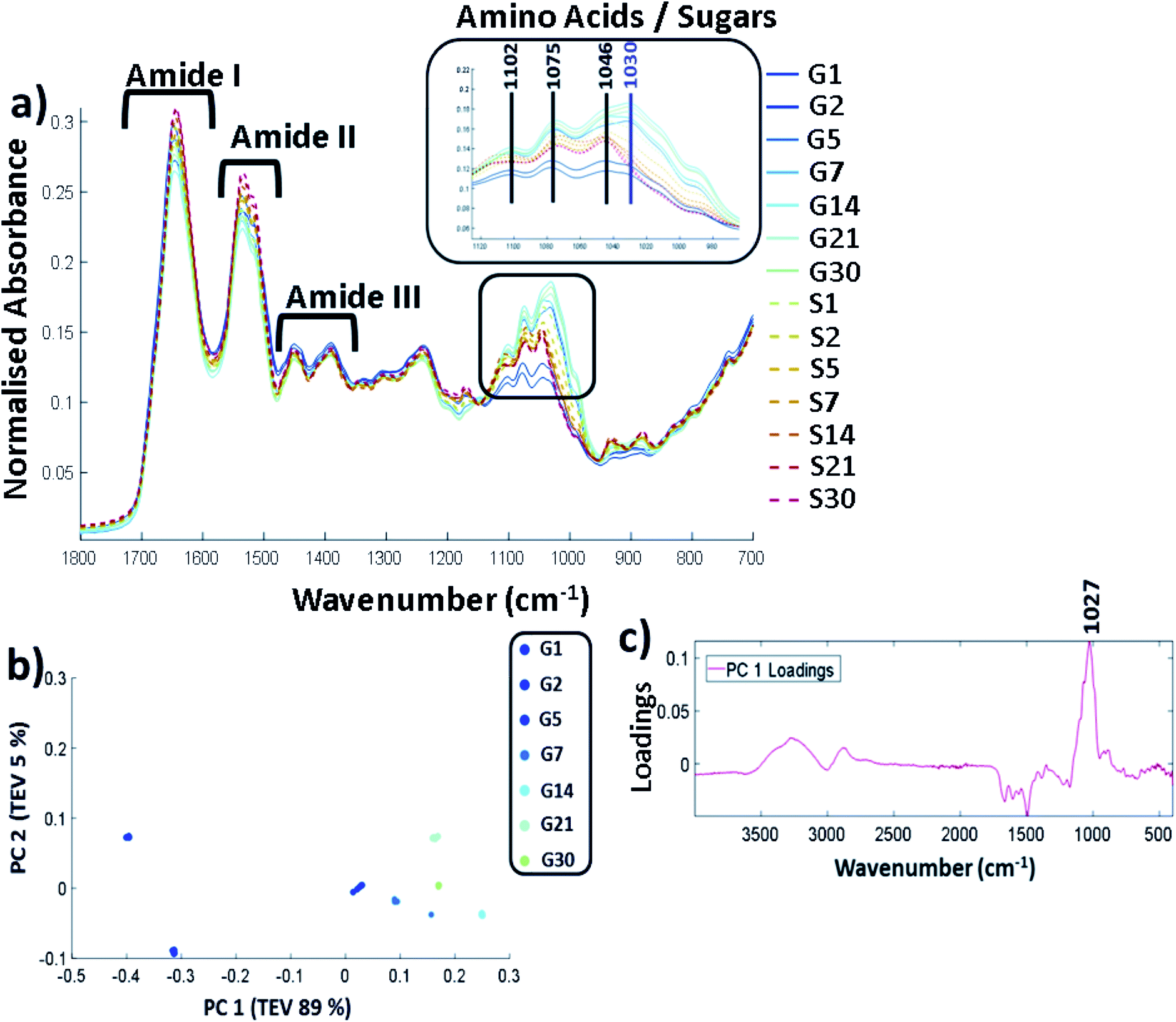 | ||
| Fig. 2 (a) FTIR-ATR spectrum showing the controls (samples incubated with no glucose labelled S and glycated lysozyme G from incubation day 1 through until day 30. Spectra are cut to only show between 1800 cm−1 to 700 cm−1, b) PCA scores plot carried out across full spectra (400–4000 cm−1) and (c) the corresponding PCA loadings plot. Samples were filtered, buffer exchanged to H2O and stored at −20 °C until the analysis was undertaken. The FTIR was carried on solid samples at 25 °C using the parameters stated in the methods. All FTIR data is an average of 5 replicates per sample. The data were scaled to allow a more accurate comparison between the samples as described in ESI (Data pre-processing and chemometrics†). TEV = total explained variance. | ||
Amide I is represented by the peak at 1649 cm−1 and amide II at 1537 cm−1. Amide III is 1452 cm−1 and 1390 cm−1 with some possible overlap with the sugar peaks. The predominant changes are in at the sugar region around 1000 cm−1–1200 cm−1 and are highlighted in Fig. 2a where a unique peak in the spectra of the glycated samples can be seen at 1030 cm−1 (highlighted in purple) which is not present in the control samples. A full spectral assignment is given in Table S2.†
In order to determine the specific peak changes, PCA was carried out on the glycated samples. PCA scores plots are used to highlight patterns within a dataset, in this case spectral variation between the peaks from the samples. It can be seen from Fig. 2b that the samples separated out on PC1 that accounts for 89% of the total explained variance (TEV) with a general trend from G1 to G30. The corresponding loadings plot (Fig. 2c) can be used to highlight the most important peaks in terms of identifying the variability between samples. A peak at 1027 cm−1 has been identified as the main peak causing the separation across PC1. We therefore deduce that the unique peak in the FTIR-ATR spectra in Fig. 2a at 1030/1027 cm−1, due to C–O or C–N bonds, is caused by glycation of lysozyme.
Fig. 3 shows three FTIR-ATR spectra to determine the difference between free glucose and bound glucose peaks. Lysozyme mixed with free glucose in solution and then freeze dried has a main peak at 1022 cm−1. The S30 sample of lysozyme incubated with no glucose for 30 days has a peak at 1046 cm−1 therefore further validating that in G30 the peak at 1030 cm−1 is only found in glycated samples and could be attributed to covalently bound glucose only.
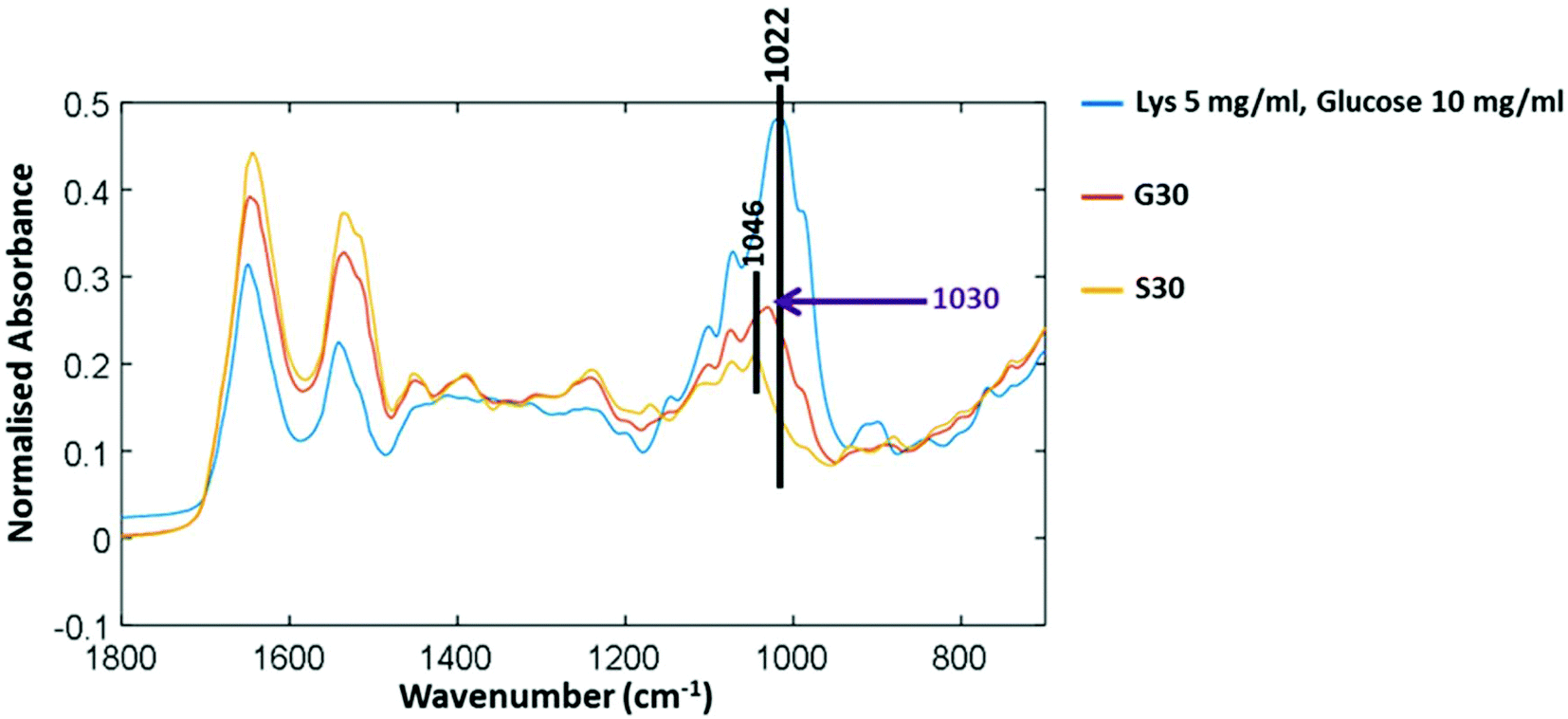 | ||
| Fig. 3 FTIR-ATR spectra of control lysozyme (S30) after 30 days of incubation with no glucose; glycated lysozyme after 30 day (G30); and lysozyme mixed with free glucose. Samples are lyophilized and analysed under the same conditions to compare bound and free glucose peaks. | ||
Analysis of glycation using Raman spectroscopy
Raman spectroscopy is a complementary vibrational analysis technique to FTIR spectroscopy with the benefit of having little spectral contribution from water. A larger scale glycation set-up was used in order to create two sets of samples: lysozyme was incubated for 30 days, with and without glucose. Raman spectroscopy requires a minimum concentration of >10 mg mL−1 and the 96 well plate set-up needs 360 μL per well. We therefore simplified the incubation set-up in order to create larger volumes so we could create a concentration gradient. Glycated samples were buffer exchanged to remove free glucose (full details are described in sample preparation). The samples were anlaysed using MS, which is summarised in Table 2 below. In comparison to the first glycation (Table 1) this method shows that although the most abundant species is lysozyme with 2 glucose bound, there are fewer lysozymes with 3 glucose bound, suggesting that the altered reaction conditions may affect the glycation rate.| Population of lysozyme with bound glucose | Control (%) 30 day incubation (no glucose) | Glycated (%) 30 day glucose incubation |
|---|---|---|
| 0 | 100 | 24 |
| 1 | 0 | 47 |
| 2 | 0 | 23 |
| 3 | 0 | 6 |
The samples were concentrated to 12 mg mL−1 and a concentration gradient was created by spiking glycated lysozyme into the control samples to create a series of 0% to 100% glycated lysozyme samples. The full concentration gradient is shown in Table S7.† Using a 96 well plate set-up, described in the Experimental section, the samples were run in a randomised order so as to avoid bias from laser power variation between the wells, or for any time-dependent changes. The samples were measured in triplicate and the average of each sample is shown in Fig. 4a. The 39%, 78% and 94% glycated samples were outliers and were removed (PCA including these data points are shown in Fig. S6†).
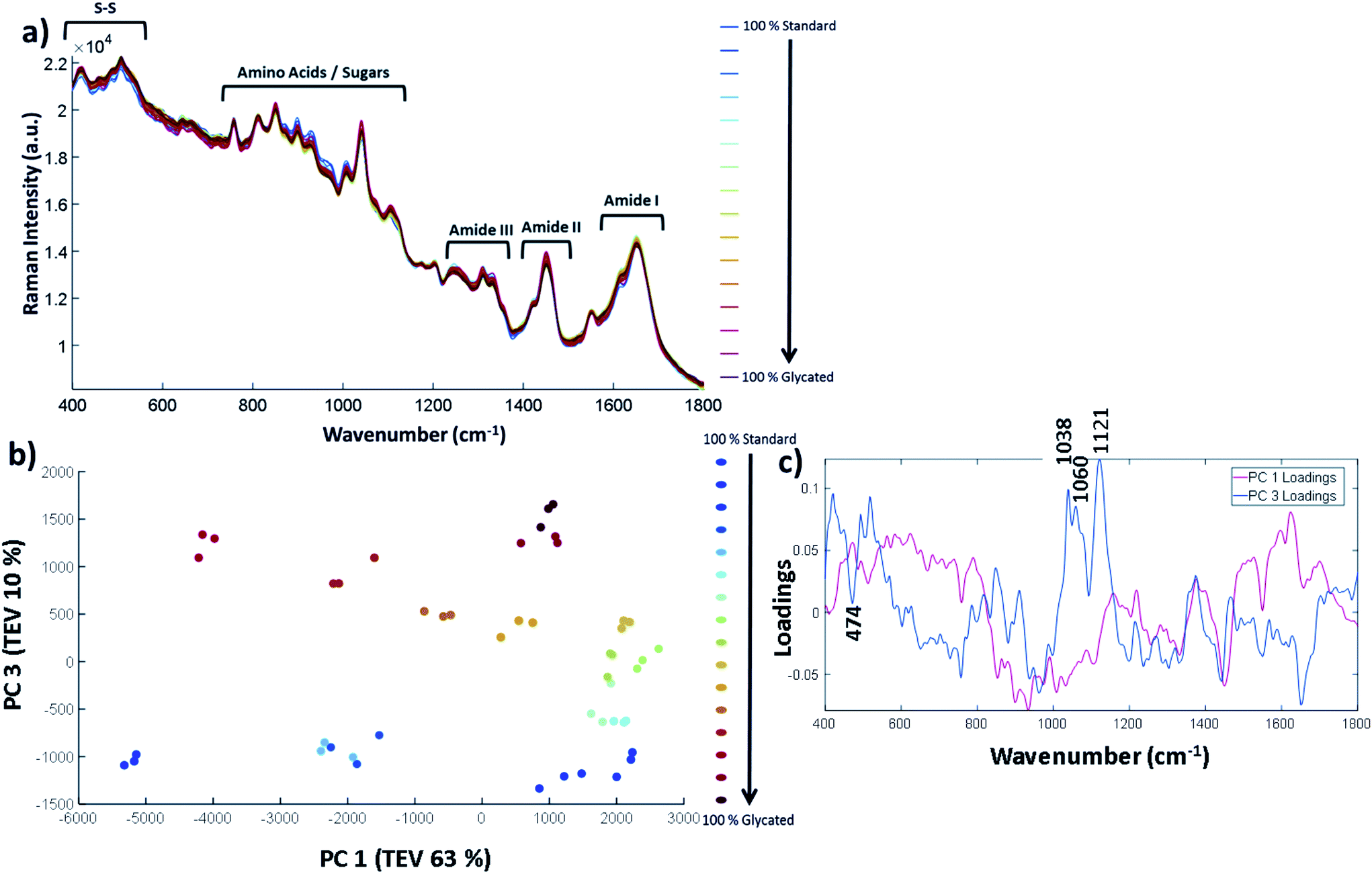 | ||
| Fig. 4 Raman spectra and PCA of glycation concentration with 9%, 78% and 94% glycated sample outliers removed. (a) Pre-processed Raman spectra with three repeats per sample, (b) PCA scores plot of samples excluding outliers showing PC 1 has a total explained variance (TEV) of 63% and PC 3 has a TEV of 10%, and (c) the corresponding loadings plot to highlight peaks that contributed to the separation of samples in the PCA. | ||
The full Raman spectra are shown in Fig. 4a. The vibrational frequency regions can be split up similarly to that of FTIR-ATR where 1600–1800 cm−1 is the Amide I, 1400–1500 cm−1 is the Amide II and 1200–1400 cm−1 is the Amide III region. At lower wavenumbers, such as between 750–1100 cm−1, the amino acid and sugar molecules vibrational frequencies result from C–O bonds vibrations and others. A full spectral assignment can be found in Table S8.† In general, it is difficult to pull out spectral differences between the % glycation of samples in Fig. 4a. Therefore, in order to highlight smaller spectral differences and to look at correlations within the data, we have used PCA with the three spectral repeats for each sample plotted separately. PC 1 vs. PC 2 showed no clear separation between glycated and non-glycated samples; by contrast, PC 1 plotted against PC 3 (Fig. 4b) separated the spectra on amount of lysozyme glycation. The three repeats from each sample show good reproducibility as they can be seen to cluster together for each set. Fig. 4c shows the corresponding loadings plot. PC 1 loadings are difficult to interpret but separation does depend on a peak at 1452 cm−1 which is the CH/CH2/CH3 vibrational frequency. The peaks in PC 3 were found to correspond to glucose peaks. PC 3 loadings highlight important spectral bands within the sugar regions at 1038 cm−1, 1060 cm−1 and 1121 cm−1, reflecting the level of glycation of lysozyme.
In order to help distinguish glycated protein from protein with free glucose we again performed a glucose spike in experiment with lysozyme (Fig. 5). Furthermore, we glycated albumin, a protein that is known to be glycated in the blood of diabetic patients.33 Human albumin has an expected mass of ∼66.5 kDa, congruent with our MS findings (shown in Table S6†). Albumin has 59 Lys, 23 Arg and the N-terminal amine that can potentially be glycated. Lys side chains are the most susceptible to be glycated.34 Peptide mapping is often used to quantify and identify the glycation sites with the number of glycation sites dependent upon the method of glycation used. Albumin is usually reported to have higher levels of glycation than lysozyme. Bovine serum albumin incubated with glucose in a similar manner showed that up to 48 glucose molecules had bound to the protein.35 Increasing the temperature can push this number up to ∼57.36 Quantification of human serum albumin (HSA) from diabetic patients has found up to 15 Lys residues with glycation.37 Therefore albumin serves as model protein that is able to achieve higher levels of glycation than that of lysozyme. Glycation levels of albumin are dependent on the conditions used to induce glucose binding. From our MS analysis we can estimate that 9–24 glucose molecules bound to albumin with the most prevalent number of glycations being 17 under our experimental conditions (Fig. S4 & Tables S5–S6†).
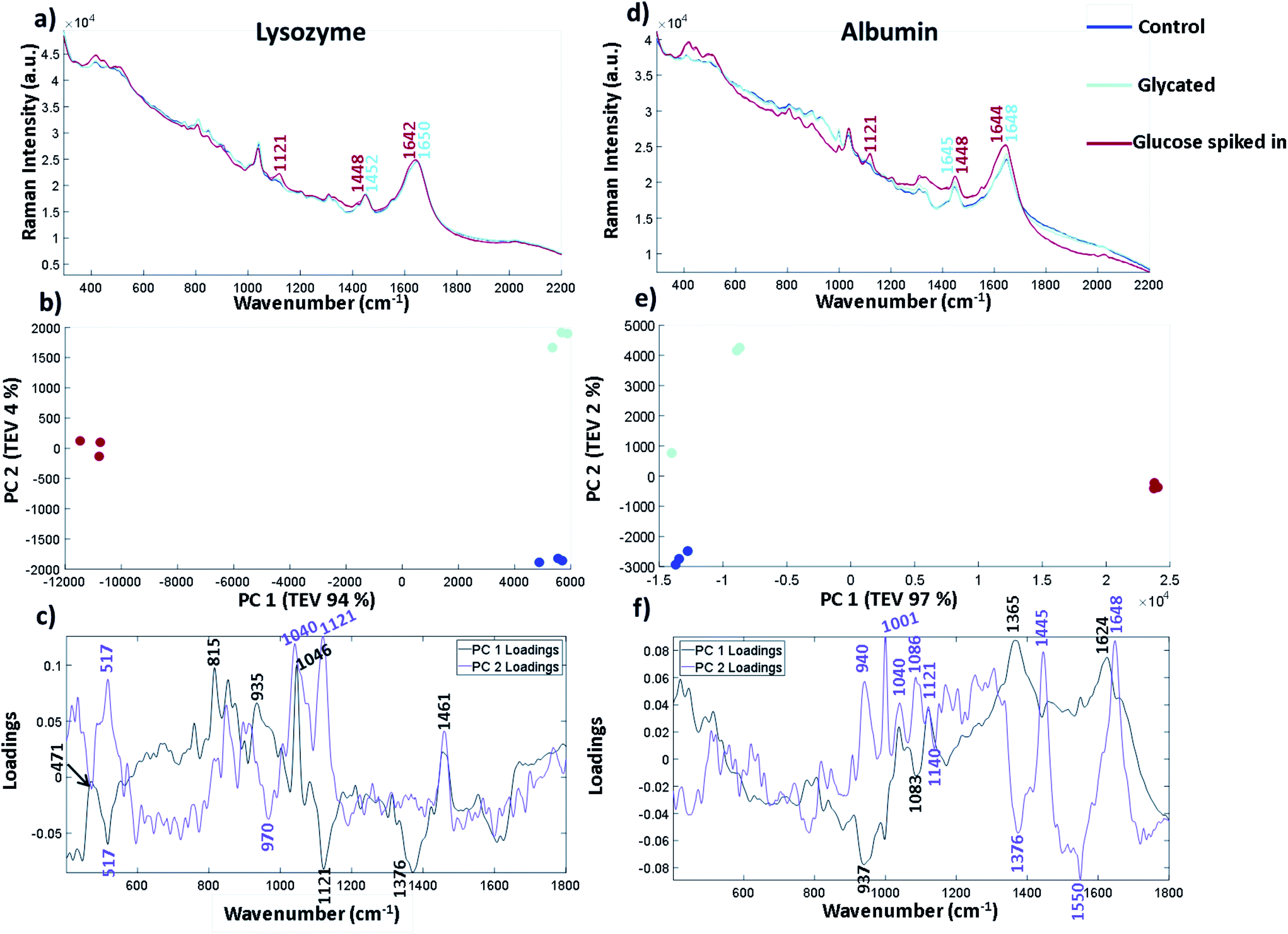 | ||
| Fig. 5 Raman spectra of lysozyme and albumin all shown as an average of three replicates. (a) and (d) show lysozyme and albumin at 12 mg mL−1 in 25 mM MOPS and 115 mM NaCl at pH 7.4 glycated, control and with 10 mg mL−1 glucose added. (b) and (e) PCA scores plots of the albumin and lysozyme samples showing the three separate replicates per condition. (c) and (e) show the corresponding PCA loadings plot for lysozyme and albumin. Both loadings plots highlight peaks at 1040 cm−1 as being significant to the separation in PC 2. | ||
Glycated and non-glycated albumin were produced using identical methods to that of lysozyme detailed in the Experimental section. The inclusion of a further protein that can become glycated and is of medical importance was used to determine whether the vibrational bands identified originated from bound glucose and could be translated to other proteins.
The spectra in Fig. 5 are plotted without buffer subtraction due to the addition of spiked in glucose changing the baseline slope and intensity. The spectra have been normalised and baselined using EMSC to allow comparison. Fig. 5a shows the three repeated spectra of lysozyme control, lysozyme glycated and lysozyme with glucose spike. We have also included a spectrum of glucose in the same buffer (25 mM MOPS and 115 mM NaCl at pH 7.4) in Fig. S8† for comparison. The labelled peaks are ones which showed a spectral shift between the glycated samples and the spiked in glucose at peak positions already attributed to different forms of glucose. In the sample with spiked in glucose the intensity of the peak at 1121 cm−1 arising from the C–OH vibration increases. This peak is very weak and broad in both the control and glycated lysozyme. In the CH2/CH3 region there is a peak shift from 1448 cm−1 in the spiked glucose sample to 1452 cm−1 in the glycated and control sample, suggesting that vibrations arising from C–H stretching are different in glucose than in the protein. The Amide I region also shows a slight shift in the band at 1650 cm−1 in the control and glycated protein to 1642 cm−1 in the spiked sample suggesting a change in the α-helical content of lysozyme, but this could be obscured by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the aldehyde group in the open form of glucose. Similar spectral differences can be picked out in Fig. 5d for albumin, where 1121 cm−1 is the main peak shown in the glucose spike and subtle spectral shifts again are seen for CH2/CH3 and the amide I region. We again used PCA to pull out smaller differences in the spectra for both lysozyme and albumin. Fig. 5b and e show the three spectral repeats of each sample. In both PCA plots, PC1 accounts for by far the largest total explained variance. In general the PCA plots are very similar, showing a mirror image of one another along the y-axis suggesting the spectral differences in the samples follow a similar pattern of separation. For lysozyme, the most significant peaks that led to the separation of the data points in lysozyme across PC 1 are 1046 cm−1, 1121 cm−1 and 1376 cm−1 shown from the intensity of the corresponding loadings plot in Fig. 5c where 1121 cm−1 and 1376 cm−1 are more intense in the spiked sample. The peak at 1046 cm−1, which is increased in the glycated and control, is assigned to C–O vibration of lysozyme in the Raman spectrum summarised in Table S8.† It can also be seen in FTIR-ATR (Fig. 3). Therefore, PC 1 separates spectra based on the free glucose in solution from the glucose spiked in sample. PC 2, on the y-axis, accounts for only 4% of the variance for lysozyme, but still accurately separates the samples. The PC 2 loadings in Fig. 5c highlight peaks at 1040 cm−1 and 1121 cm−1 as contributing to the most variance. Both these bands are increased in the glycated sample, but not in control samples. Thus 1040 cm−1 and 1121 cm−1 appear to distinguish between glycated and control samples with or without buffer subtraction.
O of the aldehyde group in the open form of glucose. Similar spectral differences can be picked out in Fig. 5d for albumin, where 1121 cm−1 is the main peak shown in the glucose spike and subtle spectral shifts again are seen for CH2/CH3 and the amide I region. We again used PCA to pull out smaller differences in the spectra for both lysozyme and albumin. Fig. 5b and e show the three spectral repeats of each sample. In both PCA plots, PC1 accounts for by far the largest total explained variance. In general the PCA plots are very similar, showing a mirror image of one another along the y-axis suggesting the spectral differences in the samples follow a similar pattern of separation. For lysozyme, the most significant peaks that led to the separation of the data points in lysozyme across PC 1 are 1046 cm−1, 1121 cm−1 and 1376 cm−1 shown from the intensity of the corresponding loadings plot in Fig. 5c where 1121 cm−1 and 1376 cm−1 are more intense in the spiked sample. The peak at 1046 cm−1, which is increased in the glycated and control, is assigned to C–O vibration of lysozyme in the Raman spectrum summarised in Table S8.† It can also be seen in FTIR-ATR (Fig. 3). Therefore, PC 1 separates spectra based on the free glucose in solution from the glucose spiked in sample. PC 2, on the y-axis, accounts for only 4% of the variance for lysozyme, but still accurately separates the samples. The PC 2 loadings in Fig. 5c highlight peaks at 1040 cm−1 and 1121 cm−1 as contributing to the most variance. Both these bands are increased in the glycated sample, but not in control samples. Thus 1040 cm−1 and 1121 cm−1 appear to distinguish between glycated and control samples with or without buffer subtraction.
In albumin, PC1 is dominated by an increase in peaks at 1624 cm−1 and 1365 cm−1 in the glycated samples (Fig. 5f). The peak at 1624 cm−1 is usually assigned to changes of protein structure and can indicate increased β-sheet formation. Alternatively it may report on Tyr/Trp/Phe, where an increase in intensity would suggest unfolding. Ultimately these results suggest that incubation of lysozyme at high concentration may be destabilising the protein; this seems to be reversible as Fig. 5d shows no secondary structure change after free glucose removal from glycated albumin.38 The peak at 1365 cm−1 is assigned to the CH2 groups of free glucose. PC 1 also shows that there is loss of a peak at 937 cm−1 in the glucose spiked samples assigned to N–C–C which is usually vibrations from the protein backbone. However, a peak at 940 cm−1 is also dominant in PC 2 in the glycated samples and therefore it could be N–C–C vibrations from the formation of Amadori products. PC 2 also highlights peaks at 1040 cm−1 and 1083 cm−1 in the sugar region. The peak at 1040 cm−1, similarly to lysozyme, shows an increase in intensity, whereas the peak at 1086 cm−1 is only present in the glycated samples in PC 2. These peaks are not assigned to free glucose in the same buffer (Fig. S8†). Overall the peak at 1027/1030 cm−1 in FTIR-ATR for glycated lysozyme and the peaks at 1040/1038 cm−1 and 1121 cm−1 in Raman for both lysozyme and albumin seem to be increasing in intensity in glycated only samples. Albumin shows a further peak at 1086 cm−1 that is only present in glycated samples. These peaks all originate from the C–O vibrations and are within the sugar region of a Raman spectrum.
Quantification of glycation
In order to determine the ability of Raman to quantify the extent of glycation in lysozyme, we used PLSR to generate a linear predictive model. To produce samples for liquid Raman analysis, a larger incubation volume was needed in which lysozyme was glycated using the same conditions as before, but in two large vials of 100 mL each for control (without glucose) and glycated (with 0.5 M glucose). The samples were again filter buffer exchanged into 25 mM MOPS, 115 mM NaCl at pH 7.4 and were concentrated to 12 mg mL−1. From these samples, a concentration gradient was created (Table S7†), where we had concentrations of glycated mixed with control samples to create a total volume of 360 μL and a concentration gradient of 0% to 100% glycated in increments of ∼6%. As the lysozyme could have anywhere between 0–3 lysine residues glycated, the known % glycation was an approximation. This generated a total of 19 samples, three of which were removed due to being outliers, leaving a total of 16 samples, 8 of which were used for a training set and 8 for the test set.The results for PLSR model are shown in Fig. 6 and the statistics summarised in Table 3 based on the 3 PLS factors using leave-one-out cross validation. The model chosen uses the minimum number of PLS factors needed in the leave-one-out cross validation to obtain a low root mean square error of cross validation (RMSEcv), which in this case was 3. Here the R2 value provides a measure of how well a linear model fits the training data and the Q2 is the same linear correlation coefficient for the two types of model. Q2cv is the performance on prediction based on leave-one-out cross validation where as Q2p is the performance prediction of the model on the test data set; these are the 8 samples left out of the model and not used during cross validation. A robust model seeks to have Q2 values close to the R2, with a perfect linear relationship being 1. Overall, the training set fits the model with an R2 value of 0.99 and a root mean square error of covariance (RMSEc) of 2.80%. The model, with 3 PLS factors, had a Q2cv of 0.96 and Q2p of 0.97. The root mean squared error for cross validation for predictive ability of the model (RMSEp) is 5.53%, showing that we had a very robust model for the prediction of glycation % from spectroscopic data.
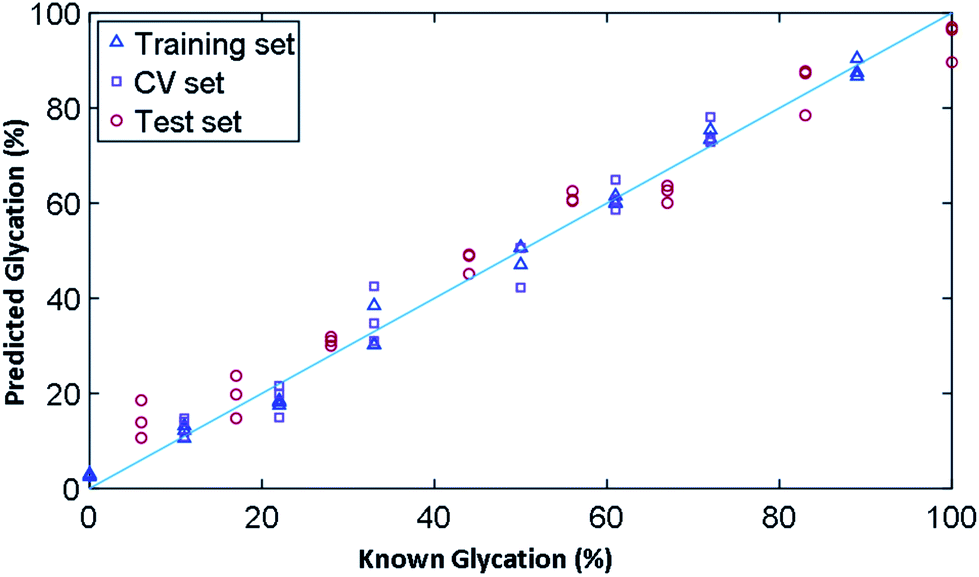 | ||
| Fig. 6 PLSR linear predictive model of % glycation of lysozyme from 0 to 100% in ∼6% increments totaling 16 concentrations. The model was built on 8 data sets each with three spectral repeats. The model was tested on the remaining 8 data sets. The model test set had a Q2p of 0.97 and an RMSEp of 5.53%. | ||
| PLSR statistics | Value | |
|---|---|---|
| Training set | R 2 | 0.99 |
| RMSEc | 2.80 | |
| Cross-validation set | Q 2cv | 0.96 |
| RMSEcv | 5.31 | |
| Test set | Q 2p | 0.97 |
| RMSEp | 5.53 |
Conclusion
Vibrational spectroscopy has successfully been used to distinguish between glycated and non-glycated proteins. For the first time, we have demonstrated the sensitivity of Raman spectroscopy to quantify glycation of proteins in solution. Furthermore, we used a 96 well plate set-up suggesting the potential for high throughput, automated analysis of protein samples. Raman spectroscopy analysis of glycated proteins in less than two minutes allows rapid screening thus providing proof-of-concept for the development of Raman spectroscopy for in-line protein characterisation.Lysozyme was incubated with glucose over a 30 day period and samples were removed at regular intervals for a time course analysis. Mass spectrometry was used to determine the number of bound glucose molecules to the protein. Lysozyme was glycated to some extent within 24 hours. By day 30, the most abundant species was lysozyme with one glucose molecule bound though up to 4 was possible. Lyophilised samples from each time course interval were anlaysed using FTIR-ATR. PCA identified a peak at 1027 cm−1 which correlated with glycation levels. This peak was not present in the control lysozyme or the lysozyme sample spiked with glucose and therefore we assign 1027 cm−1 to bound glucose.
Raman spectroscopy analysis required a larger incubation set-up using larger sample volumes required for solution analysis. These were generated using lysozyme incubated with glucose and a non-glycated control incubated under the same environmental conditions with no glucose. Both were incubated for 30 days. MS analysis revealed that after 30 days the main species present was again lysozyme with one glucose bound (47%) up to a maximum of three (6%). It can be noted here that the populations of glycated species in large set-ups differ to those in the small scale set-up for FTIR (36% one glucose bound, 14% for three glucose bound) and therefore incubation factors such as volume may affect the glycation rate. These samples were buffer exchanged and concentrated to 12 mg mL−1 and then used to create 16 samples from 0 to 100% glycated at ∼6% intervals. Using a 96 well plate set-up, the samples were analysed with Raman spectroscopy via automation on a Raman microscope. PCA revealed a more complex variance, with more contributing peaks, across PC 1 and PC 2 compared to FTIR-ATR samples, due to the addition of vibrations of buffer and water from using samples in solution. Both the Raman spectra in Fig. 5a and loadings plots in Fig. 5c highlighted increased intensity of peaks at 1038 cm−1, 1060 cm−1 and 1121 cm−1 compared to that of the peaks in the control. Furthermore, the peaks highlighted at 1038 cm−1 and 1060 cm−1 were not found in free glucose alone (Fig. S8†). To confirm the relevance of these peaks to other glycated proteins, albumin was glycated using the same conditions. In Fig. 5 we compared both glycated albumin and lysozyme with controls and glucose spiked in to the control protein. The spectra and PCA loadings again highlighted peak intensities at 1040 cm−1 and 1121 cm−1 as important to distinguish between glycated and non-glycated control protein. Albumin, which was found to be most abundant with 17 glucose bound, had a peak at 1086 cm−1 which was only present in the glycated samples and therefore could indicate bound glucose only. The sensitivity of Raman spectroscopy to quantify and predict glycation was investigated using multivariate linear regression model. The concentration gradient was used to build a PLSR model which produced an accurate predictive model with an RMSEp of 5.53%. Raman spectroscopy, when used with chemometrics, therefore has the capability to rapidly quantify protein glycation.
Overall, these studies show the scope of Raman spectroscopy in the identification and quantification of glycation in proteins central to disease diagnostics and quality control of protein therapeutics. The development of Raman spectroscopy in these fields would allow for rapid sample analysis in solution and therefore provide the foundation for evolving on-line and in-line analytical and diagnostic tools.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a BBSRC CASE award with UCB Pharma BB/L014734/1. RG thanks the UK BBSRC (BB/L014823/1) for support for Raman spectroscopy.References
- L. C. Maillard, C. R. Hebd. Seances Acad. Sci., 1912, 154, 66–68 CAS.
- J. S. Ramalho, C. Marques, P. C. Pereira and M. C. Mota, Eur. J. Ophthalmol., 1996, 6, 155–161 CrossRef CAS PubMed.
- C. Iannuzzi, G. Irace and I. Sirangelo, Front. Mol. Biosci., 2014, 1, 9 CrossRef PubMed.
- J. M. Ashraf, S. Ahmad, G. Rabbani, A. Tasleem Jan, E. Ju Lee, R. Hasan Khan and I. Choi, IUBMB Life, 2014, 66, 686–693 CrossRef CAS.
- M. Luthra and D. Balasubramanians, J. Biol. Chem., 1993, 268, 18119–18127 CAS.
- S. Ghosh, N. K. Pandey, A. Singha Roy, D. R. Tripathy, A. K. Dinda and S. Dasgupta, PLoS One, 2013, 8, e74336 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko, L. Stryer and L. Stryer, Biochemistry, W.H. Freeman, New York, 5th edn, 2002 Search PubMed.
- Q. Zhang, J. M. Ames, R. D. Smith, J. W. Baynes and T. O. Metz, J. Proteome Res., 2009, 8, 754–769 CrossRef CAS PubMed.
- A. Goldin, J. A. Beckman, A. M. Schmidt and M. A. Creager, Circulation, 2006, 114, 597–605 CrossRef CAS PubMed.
- V. P. Singh, A. Bali, N. Singh and A. S. Jaggi, Korean J. Physiol. Pharmacol., 2014, 18, 1–14 CrossRef CAS PubMed.
- A. Rahmadi, N. Steiner and G. Mü, Clin. Chem. Lab. Med., 2011, 49, 385–391 CAS.
- C. Quan, E. Alcala, I. Petkovska, D. Matthews, E. Canova-Davis, R. Taticek and D. Ma, Anal. Biochem., 2008, 373, 179–191 CrossRef CAS PubMed.
- M. K. Joubert, M. Deshpande, J. Yang, H. Reynolds, C. Bryson, M. Fogg, M. P. Baker, J. Herskovitz, T. J. Goletz, L. Zhou, M. Moxness, G. C. Flynn, L. O. Narhi and V. Jawa, PLoS One, 2016, 11, e0159328 CrossRef PubMed.
- D. M. Kennedy, A. W. Skillen and C. H. Self, Clin. Exp. Immunol., 1994, 98, 245–251 CrossRef CAS PubMed.
- R. Dolhofer, E. A. Siess and O. H. Wieland, Biol. Chem. Hoppe-Seyler, 1985, 366, 361–366 CrossRef CAS PubMed.
- ICH, ICH Topic Q 6 B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, 1999 Search PubMed.
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) Guideline on similar biological medicinal products containing monoclonal antibodies-non-clinical and clinical issues, http://www.ema.europa.eu, (accessed 17 June 2019).
- FDA, Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry DRAFT GUIDANCE, https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-regulatory-information-biologics/biologics-guidances, (accessed 17 June 2019).
- N. Alt, T. Y. Zhang, P. Motchnik, R. Taticek, V. Quarmby, T. Schlothauer, H. Beck, T. Emrich and R. J. Harris, Biologicals, 2016, 44, 291–305 CrossRef CAS PubMed.
- B. Wei, K. Berning, C. Quan and Y. T. Zhang, mAbs, 2017, 9, 586–594 CrossRef CAS PubMed.
- C. Quan, E. Alcala, I. Petkovska, D. Matthews, E. Canova-Davis, R. Taticek and S. Ma, Anal. Biochem., 2008, 373, 179–191 CrossRef CAS PubMed.
- L. J. Brady, A. Theresa Martinez and A. Balland, Anal. Chem., 2007, 79, 9403–9413 CrossRef CAS PubMed.
- N. C. Dingari, G. L. Horowitz, J. W. Kang, R. R. Dasari and I. Barman, PLoS One, 2012, 7, e32406 CrossRef CAS PubMed.
- Y. Han, J. Chen, T. Pan and G. Liu, Chemom. Intell. Lab. Syst., 2015, 145, 84–92 CrossRef CAS.
- J. V. Glenn, J. R. Beattie, L. Barrett, N. Frizzell, S. R. Thorpe, M. E. Boulton, J. J. McGarvey and A. W. Stitt, FASEB J., 2007, 21, 3542–3552 CrossRef CAS.
- T. Pan, M. Li, J. Chen and H. Xue, J. Innovative Opt. Health Sci., 2014, 07, 1350060 CrossRef.
- K. A. Esmonde-White, M. Cuellar, C. Uerpmann, B. Lenain and I. R. Lewis, Anal. Bioanal. Chem., 2017, 409, 637–649 CrossRef CAS PubMed.
- T. De Beer, A. Burggraeve, M. Fonteyne, L. Saerens, J. P. Remon and C. Vervaet, Int. J. Pharm., 2011, 417, 32–47 CrossRef CAS.
- J. Müller, K. Knop, M. Wirges and P. Kleinebudde, J. Pharm. Biomed. Anal., 2010, 53, 884–894 CrossRef.
- H. F. Bunn, R. Shapiro, M. Mcmanus, L. Garrick, M. J. Mcdonald, P. M. Gallop, K. H. Gabbay, P. Bent and B. Hospital, J. Biol. Chem., 1979, 254, 3892–3898 CAS.
- B. H. Shilton, R. L. Campbell and D. J. Walton, Eur. J. Biochem., 1993, 215, 567–572 CrossRef CAS PubMed.
- O. Anjos, M. G. Campos, P. C. Ruiz and P. Antunes, Food Chem., 2015, 169, 218–223 CrossRef CAS PubMed.
- K. J. Welsh, M. S. Kirkman and D. B. Sacks, Diabetes Care, 2016, 39, 1299–1306 CrossRef CAS PubMed.
- N. A. Ansari, A. S. Moinuddin and R. Ali, Dis. Markers, 2011, 30, 317–324 CrossRef CAS.
- M. E. Rubio-Ruiz, E. Diaz-Diaz, M. Cardenas-Leon, R. Arguelles-Medina, P. Sanchez-Canales, F. Larrea-Gallo, E. Soria-Castro and V. Guarner-Lans, Glycobiology, 2008, 18, 517–525 CrossRef CAS.
- B.-S. Lee, S. Krishnanchettiar, S. S. Lateef and S. Gupta, Int. J. Mass Spectrom., 2007, 260, 67–74 CrossRef CAS.
- A. Frolov and R. Hoffmann, Anal. Bioanal. Chem., 2010, 397, 2349–2356 CrossRef CAS PubMed.
- J. Juárez, P. Taboada and V. Mosquera, Biophys. J., 2009, 96, 2353–2370 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an02318f |
| This journal is © The Royal Society of Chemistry 2020 |