
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical and diverse roles of phosphates in human bone formation
Erik A. B.
Hughes
 ab,
Thomas E.
Robinson
a,
David B.
Bassett
ac,
Sophie C.
Cox
a and
Liam M.
Grover
*a
ab,
Thomas E.
Robinson
a,
David B.
Bassett
ac,
Sophie C.
Cox
a and
Liam M.
Grover
*a
aSchool of Chemical Engineering, University of Birmingham, B15 2TT, UK. E-mail: l.m.grover@bham.ac.uk
bNIHR Surgical Rec and Microbiology Research Centre, Queen Elizabeth Hospital, Birmingham, UK
cDepartment of Physics, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
First published on 1st November 2019
Abstract
Humans utilise biomineralisation in the formation of bone and teeth. Human biomineralisation processes are defined by the transformation of an amorphous phosphate-based precursor to highly organised nanocrystals. Interestingly, ionic phosphate species not only provide a fundamental building block of biological mineral, but rather exhibit several diverse roles in mediating mineral formation in the physiological milieu. In this review, we focus on elucidating the complex roles of phosphate ions and molecules within human biomineralisation pathways, primarily referring to the nucleation and crystallisation of bone mineral.
1. Introduction
Phosphate is essential for terrestrial life. Along with water, carbon, nitrogen and oxygen, phosphates hold the same fundamental importance for the existence of life as we know it. For example, phosphate is a critical component of our genetic information. It comprises over 25% of our DNA by mass and bridges the deoxyribose molecules to maintain the complex double helix arrangement.1,2 However, the indispensable nature of phosphate is not limited to our genetic make-up. Physiological phosphate is present in soft tissues (14%) and extracellular fluid (1%).3 In soft tissue, intracellular phosphate can be found as part of not only nucleic acids, but also cell membrane phospholipids, phosphorylated amino acids (serine, threonine, and tyrosine in particular) and carbohydrates.4 Therefore, phosphate is a critically important chemical moiety in all four major classes of biomacromolecule. Within the extracellular fluid, 10% of phosphate is bound to protein, 33% is complexed with calcium or magnesium and the remainder is unbound.5Phosphates are also key components in biomineralisation. Biomineralisation allows for the sophisticated formation of highly organised, functional and often high-strength tissues by both vertebrates and invertebrates. In humans, the majority of phosphate (∼85%) is actually present in hard tissues that form through biomineralisation.5,6 Bones and teeth are prime examples of exceedingly durable and stiff tissues that exhibit hierarchical structuring from the nano- to the macro-scale. Bones provide a supportive framework, facilitate movement through interaction with muscles and protectively encase our organs. Less obvious, but equally important functions of bone, include acid–base and osmotic homeostasis, the storage of growth factors and the provision of space to generate blood cells.7 Although the specific mechanisms that dictate biomineralisation are still to be fully understood, it is largely agreed that such mechanisms are likely to have been conserved between many species over millennia.8
Our bones consist of a mineralised extracellular matrix, containing both organic and inorganic components, in addition to active populations of cells responsible for maintaining a functional and healthy tissue structure. Organic components contribute up to 30% of bone by mass. Approximately 90% of organic matter in hard tissue is accounted for by type-1 collagen, with non-collagenous proteins (NCPs), lipids and mass of hydration together making up the remaining 10%.9,10 Up to 70% of the remaining bone mass primarily consists of a nanocrystalline calcium phosphate-based mineral, generally described as hydroxyapatite ((Ca10(PO4)6(OH)2), HA). This mineral phase was first identified in bone during the 1920's by X-ray diffraction, which was later confirmed in the 1930's.11,12 HA is also the main mineral constituent of teeth in both enamel and dentine. The composition of dentine is similar to bone, whereas enamel is composed of a greater fraction of inorganic mineral (up to 96%). Enamel is therefore the most extreme example of human biomineralisation, as well as being the hardest material found within our bodies.13,14
Given that the formation of mineralised hard tissues in living systems is governed by a combination of cellularly driven processes and thermodynamics, biomineralisation should be considered both biological and chemical in nature. In the case of humans, phosphates are not only a key building block of biologically derived mineral, but free phosphates in the biological milieu also enact control over the formation of new mineral by influencing a wide variety of signalling molecules and enzymes. To this end, this paper reviews the chemistry and roles of different phosphates within biological microenvironments to better understand the critical and diverse roles of this versatile chemical moiety in the context of biomineralisation. The focus is primarily on inorganic phosphates species, the creation of phosphate-based bone mineral in addition to briefly touching upon mineral formation under pathological circumstances.
2. An overview of phosphate structures
2.1. Orthophosphates
Phosphates are not limited to one single form. Rather they exist in a range of forms that vary in size, chain length and structural arrangement. This gives rise to many types of phosphate molecules and ionic species. Orthophosphate refers to a phosphate molecule in possession of a single phosphorous atom. In its simplest form, a phosphorous atom is contained within a tetrahedron of electronegative oxygen atoms (M3PO4), where M is an electropositive monovalent ion such as hydrogen or sodium. Other elements may be present in place of oxygen, for example sulphur that yields thiophosphate species (PS4−xOx3−), with phosphorous remaining at the tetrahedral core. However, it is the orthophosphate molecule, and associated ions, composed of phosphorous, oxygen and hydrogen, that is of importance to biomineralisation.Tribasic orthophosphoric acid (H3PO4) serves as a progenitor to all forms of the orthophosphate anion that can exist in aqueous physiological environments. Subsequent deprotonation of the H3PO4 molecule can yield H2PO4−, HPO42− and PO43− anions (Fig. 1). This speciation is dependent on the pH and ionic strength, whereby H3PO4 predominates in strongly acidic conditions, H2PO4− predominates in weakly acidic and neutral conditions, HPO42− predominates in weakly basic conditions and PO43− predominates in strongly basic conditions.
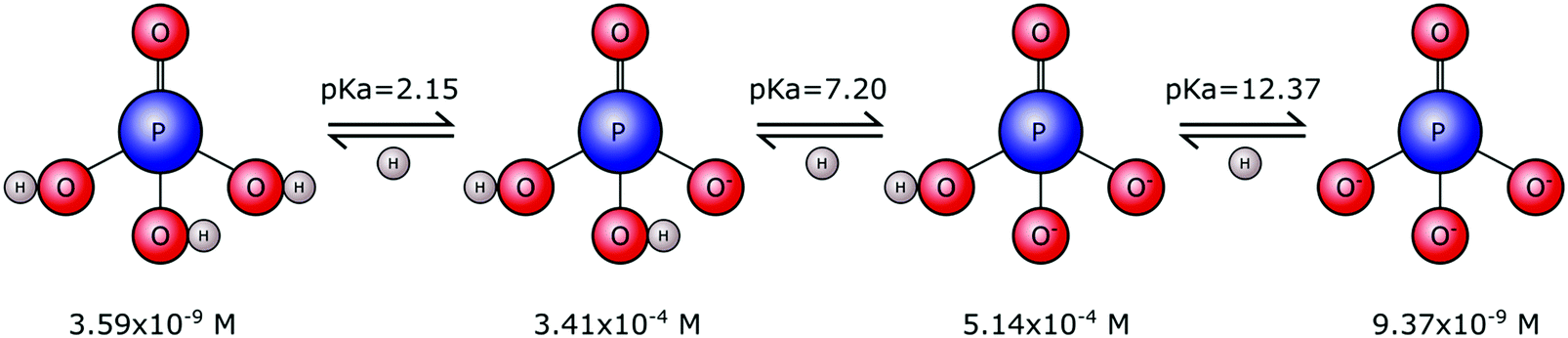 | ||
| Fig. 1 Orthophosphoric acid speciation. Physiological serum concentrations and pKa values of orthophosphate in vivo. The mono- and di-protonated forms are by far the most common at physiological pH, temperature and salt concentrations, consistent with a pKa value of 7.20. | ||
2.2. Condensed phosphates
Orthophosphate provides the monomer subunit for all phosphate-based polymers (Fig. 2A), which are also referred to as condensed phosphates. Condensed phosphates possess P–O–P bonds between orthophosphate molecules and molecular threads of P–O–P bonds are classed as polyphosphates (Mn+2PnO3n+1). Technically the shortest chain polyphosphate is a dimer consisting of two orthophosphate units, commonly referred to as pyrophosphate (M4P2O7) (Fig. 2B). Swedish chemist Jöns Jacob Berzelius was the first to synthesize pyrophosphate in 1816 by igniting orthophosphoric acid, resulting in its esterification.15 Biologically, inorganic pyrophosphate (P2O74−, PPi) plays an important role in the control of the human biomineralisation processes (Section 4.1).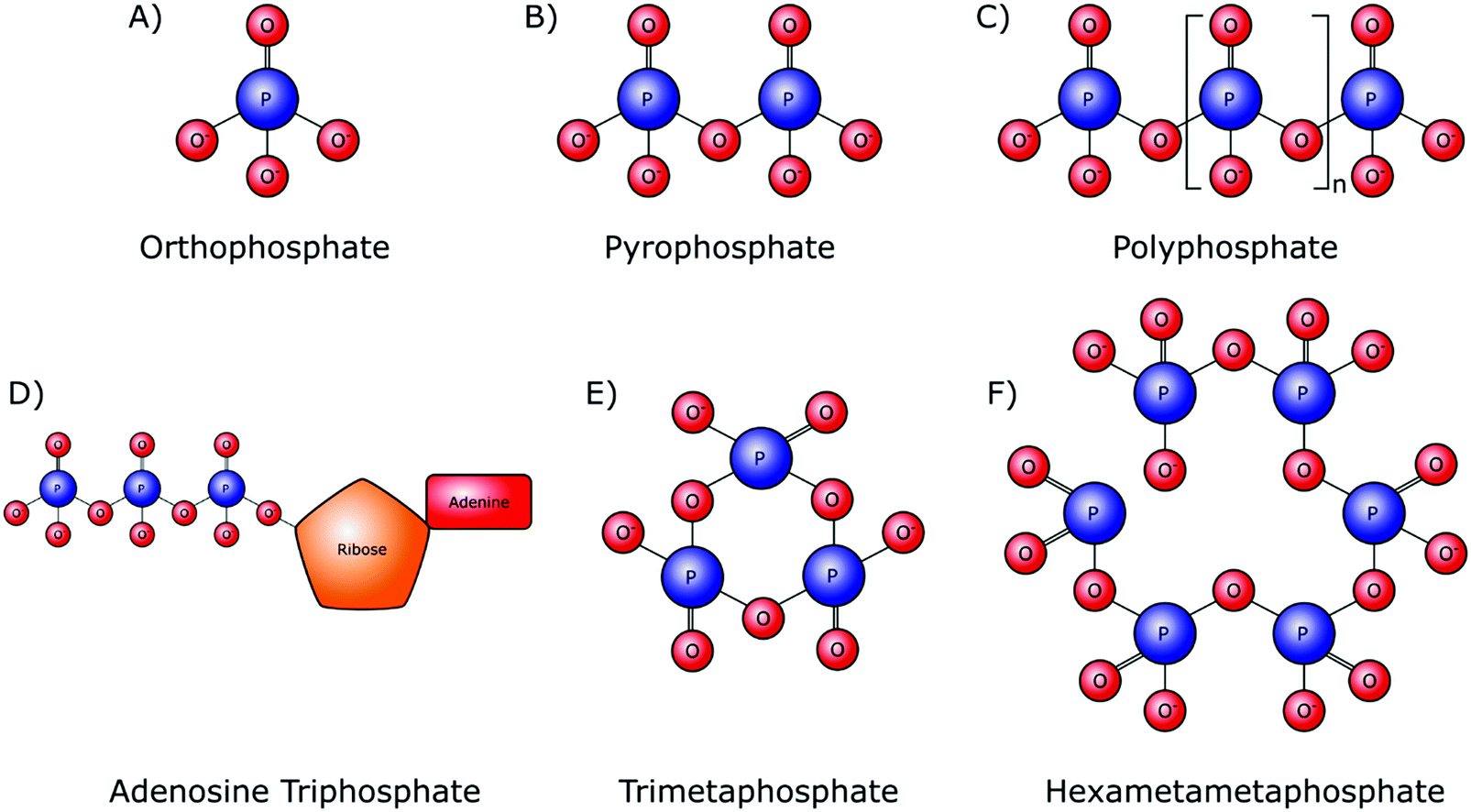 | ||
| Fig. 2 Chemical structures of linear and cyclic inorganic phosphate structures with varying chain lengths. (A) Orthophosphate (Pi). (B) Pyrophosphate (PPi). (C) Inorganic polyphosphate (polyP). (D) Adenosine triphosphate (ATP). (E) Trimetaphosphate (TMP). (F) Hexametaphosphate (HMP). | ||
Longer polyphosphate chains can consist of several residues, up to 1 × 106 orthophosphate monomers (Fig. 2C). Biological inorganic polyphosphate (polyP) was first discovered in cells in the late 19th century and is known to be present in both prokaryotes and eukaryotes. PolyP found in vivo is a linear polymer; however, large branched polymers can exist. These are termed ultraphosphates, though their structure may make them unstable due to rapid hydrolytic and enzymatic degradation, and as such they have not been found biologically. The possession of high energy phosphoanhydride bonds has led to suggestions that polyP may have a role in the origin of life as both a non-enzymatically produced energy carrier, as well as an orthophosphate donor.16,17 Several distinct biological roles of polyP have been identified in various cell types.18 Whilst this demonstrates the inter-domain ubiquity of polyP, it makes the exact role of these phosphate species in the origin of cellular life, and within specific cellularly influenced processes such as biomineralisation, difficult to pinpoint.
A polyphosphate chain consisting of three orthophosphate units is present in the structure of the organophosphate biomolecule adenosine triphosphate (ATP) (Fig. 2D), which is released by most cell types, including bone-forming osteoblasts. Hydrolytic cleavage of the high-energy phosphoanhydride bonds that link the individual orthophosphate residues together, as catalysed by phosphatase enzymes, releases the necessary chemical energy to fuel cellular activities and, by extension, all of our bodily functions. In addition to its capacity as the currency of energy in intracellular metabolism, ATP also has an important role as an extracellular signalling molecule in biomineralisation, being a source of both orthophosphate and PPi ions (Sections 4.1 and 4.2).
Although not involved in biological processes, polyphosphates can also form symmetrical open or closed ring structures. These cyclic polyphosphates are better known as metaphosphates (MnPnO3n) (Fig. 2E and F). Both enzymatic and H+ and OH− mediated hydrolysis mechanisms are capable of catalysing the degradation of metaphosphates to orthophosphates and shorter chains.
3. Orthophosphate and the formation of bone mineral
3.1. The phosphate required for bone formation
Orthophosphate is an essential building block of biological mineral. The orthophosphate required for biomineralisation is consumed through one's diet, with a typical Western diet correlating to the provision of between 1000–1600 mg per day, of which 3 mg per kg bodyweight per day enters the extracellular fluid, exchanging with bone as required.19 Serum phosphate concentration varies considerably with age, being greater in infants (1.5–2.65 mM) and declining toward adulthood (0.8–1.5 mM). This is because infants require additional levels of phosphate for bone growth and soft tissue.20,21In addition to biomineralisation, phosphates are involved in cell growth, migration, apoptosis, endocytosis and differentiation, and as such its presence is tightly regulated by the body.22 The homeostasis of both phosphate and calcium is governed by a network of complementary mechanisms interlinking kidney, intestinal and skeletal function (Fig. 3). The primary regulators of phosphate within this “parathyroid-kidney-intestine-bone” axis include the parathyroid hormone (PTH), calcitriol and fibroblast growth factor-23 (FGF-23), the roles of which are reviewed in detail elsewhere.5,22,23
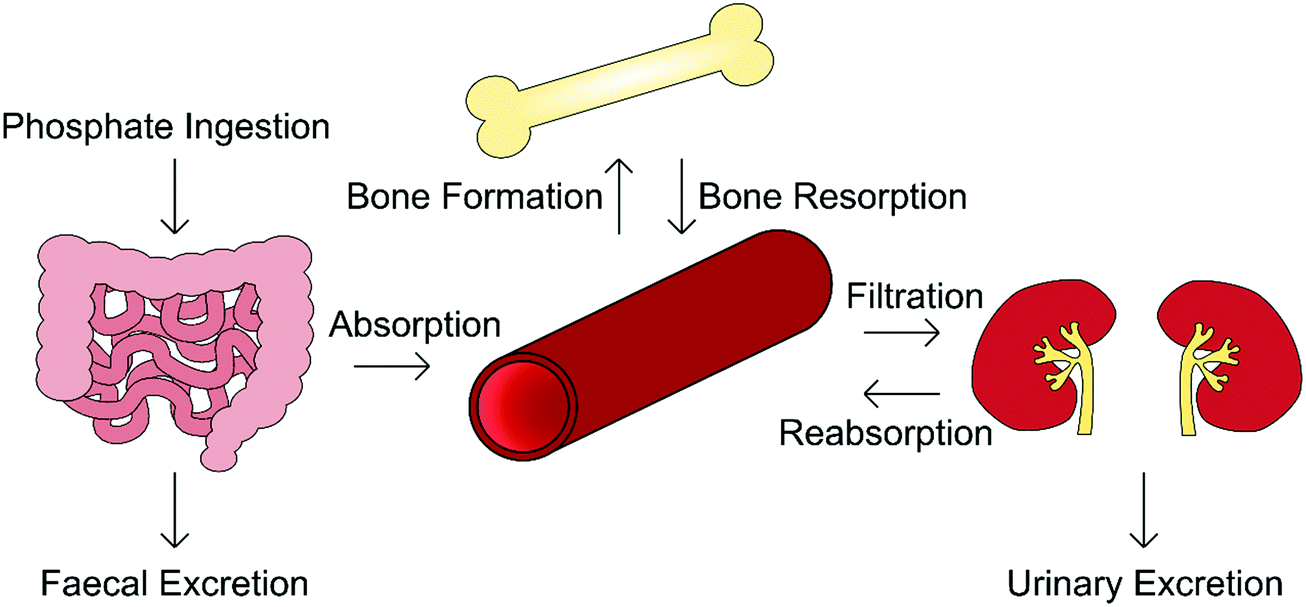 | ||
| Fig. 3 Overview of the “parathyroid-kidney-intestine-bone” axis that regulates physiological phosphate levels. Ingested in food, phosphate is absorbed through the intestines; that which is not absorbed is excreted in faeces. Phosphate is removed from the blood when bone is formed, and released when bone is resorbed. As blood is filtered by the kidneys, any phosphate that is not reabsorbed is excreted in the urine. These mechanisms serve to keep serum phosphate concentration constant. | ||
3.2. Nucleation of an amorphous mineral precursor
In humans, biologically derived mineral is formed by precipitation in the presence of proteins and polysaccharides within the mild physiological environment maintained at approximately pH 7.4 and near ambient temperature (37 °C).24 As H2PO4− and HPO42− ions are the most prominent orthophosphate species present in physiological solution (Fig. 1), they likely play an essential role in the early stages of biomineral formation. These orthophosphate ions are able to react avidly with divalent cations. Interaction with aqueous calcium ions for instance results in the nucleation and subsequent growth of biologically relevant mineral complexes, providing a chemical basis for biomineralisation (Fig. 4A).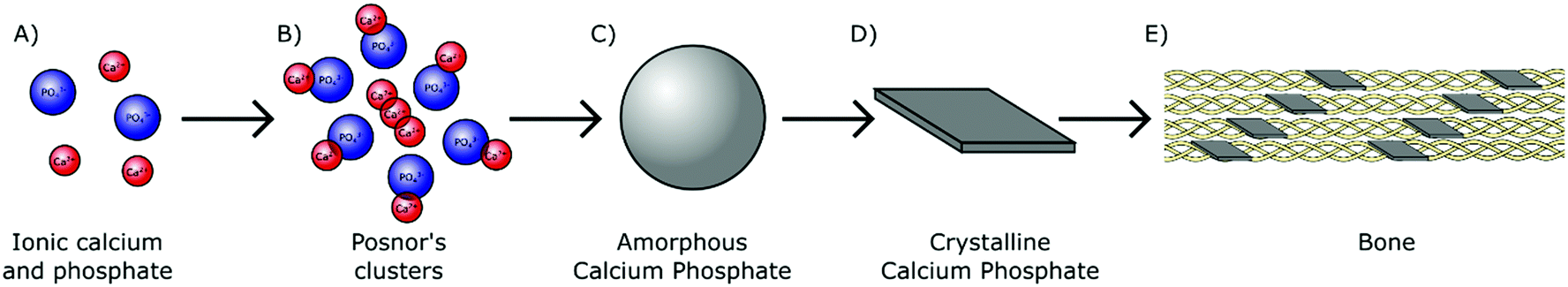 | ||
Fig. 4 From orthophosphate ions to bone mineral. (A) Calcium and phosphate ions are colocalised in the physiological milieu. (B) Ions associate to form Posner's clusters (0.7–1 nm), with a Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio of 1.5. (C) Posner's clusters agglomerate to form ACP particulates (30–100 nm). (D) ACP transforms into a crystalline HA phase with a Ca:P ratio of 1.67, which in bone possess a platelet morphology (50 nm × 25 nm × 3 nm). (E) In bone, the HA nanocrystals are incorporated into collagen fibrils, mineralising the organic scaffold. :P ratio of 1.5. (C) Posner's clusters agglomerate to form ACP particulates (30–100 nm). (D) ACP transforms into a crystalline HA phase with a Ca:P ratio of 1.67, which in bone possess a platelet morphology (50 nm × 25 nm × 3 nm). (E) In bone, the HA nanocrystals are incorporated into collagen fibrils, mineralising the organic scaffold. | ||
During the 1960s, an amorphous phase termed amorphous calcium phosphate (Ca3(PO4)2·nH2O, ACP) was first proposed as a precursor to crystalline calcium orthophosphate minerals in pseudo physiological systems.25 This includes HA, the major mineral component of both human bone and teeth.26 The presence of ACP in these tissues has since been confirmed in vivo.27,28 ACP has also been identified in the hard tissues of several other vertebrates29–31 and invertebrates,32,33 all but confirming the universal importance of this precursor within biomineralisation mechanisms across the animal kingdom.
ACP is comprised of spherical domains in the range of 0.7–1 nm in diameter that agglomerate together to form particulates.34 Betts and Posner first identified this nano-structuring during the 1970s and so these domains have become better known as Posner's clusters35 (Fig. 4B). The standard formula of Posner's clusters is given as Ca9(PO4)6, correlating to a calcium to phosphorous (Ca:P) ratio of 1.5, typical of ACP. The aggregation of Posner's clusters form spherical particles of ACP between 30–100 nm in diameter (Fig. 4C), which further aggregate into chains prior to transforming toward a more stable phase.36–39
In the late 1990s, it was proposed that crystalline HA forms via the step-wise assembly of Posner's clusters.34 However, speculation over the exact chemical composition of physiologically derived clusters and the applicability of this model for in vivo biomineral formation has only driven further efforts to understand the chemical structure of these early orthophosphate building blocks. Advances in analytical methods have led to the proposition that hydrated Posner's clusters with the formula Ca9(PO4)6(H2O)30 are the structural building blocks of ACP. Further, in the early stages of nucleation, these clusters may be calcium deficient, contain protonated phosphate ion ligands and are charge balanced by monovalent ions normally present in physiological solution such as Na+ and Cl−.40,41 Indeed, around 10–15% of the orthophosphate within ACP produced in vitro from neutral solution is protonated.42 Recently, computer simulations have shed-light on the formation of non-idealised clusters in the physiological environment rich in calcium and protonated phosphate ion species.43 Cluster development in the presence of HPO42− and H2PO4− was shown to be enhanced by Na+ ions, resulting in the generation of several varieties of calcium deficient cluster complexes (including, but not limited to, [Ca7(H2PO4)(HPO4)5]3+, [Ca3Na4(H2PO4)3(HPO4)3]+ and [CaNa6(HPO4)6]4−) that may aggregate through bridging phosphate ligands. Furthermore, it was hypothesised that these non-idealised clusters evolve through the ejection of protons, which in turn promotes association with Ca2+ ions to counter balance changes in overall charge, consequently increasing the Ca:P ratio toward 1.5. This is consistent with the fact that protonated orthophosphate species are present at the superficial surface of bone that is in contact with physiological solution, whereas unprotonated Pi species are prevalent in deep bone tissue.44,45 Taken together, the formation of precursors to crystalline HA comprising of protonated phosphate ions is seemingly consistent with the earliest nucleation events both in vitro and in vivo.
3.3. Biological mineralisation and the formation of bone crystals
The template for bone mineralisation is osteoid, which is deposited by osteoblasts. Osteoid is composed of bone's organic components, the major component being type-1 collagen. The exact role of this phase in the infiltration of mineral precursors and the subsequent evolution of highly oriented HA nanocrystals remains unknown, however there are several hypotheses. There is evidence that NCPs influence apatite nucleation, but also evidence that suggests that nucleation sites exist at the terminus of collagen molecules. A further body of evidence is growing to support the hypothesis that orthophosphate mineral precursors are formed separately before integrating with collagen.Several potential sites of mineral association with collagen fibrils have been identified.46 Mineralisation inhibitors, including osteopontin (OPN), polyaspartic acid and fetuin, may stabilise ACP as part of a net negatively charged complex, which can then interact with collagen at sites that possess a net positive charge.47–50 The mineral precursor may also exist in a fluidic state, referred to as polymer-induced liquid-precursor (PILP), which can infiltrate the supramolecular structure of collagen by capillary action before solidifying and undergoing crystallisation.51
As bone formation progresses, the mineral content undergoes a gradual transformation from amorphous to crystalline, resulting in HA nanocrystals. Any remaining amorphous phase may contribute toward maintaining ionic homeostasis. This transformation has previously been spatially resolved by mapping the transformation of ACP to mature mineral in zebrafish fin rays.52 While the transformation of ACP to crystalline HA has been proposed to proceed directly,53,54 there is also evidence to support the involvement of several other calcium phosphate phases. Acidic calcium orthophosphates, including dibasic calcium phosphate dihydrate (CaHPO4·2H2O, DCPD, also known as brushite) and octacalcium phosphate (Ca8(HPO4)2(PO4)4·5H2O, OCP), have long been implicated as transient phases that evolve from ACP prior to the formation of HA.55,56 The solubility isotherms of calcium phosphates predict that the most stable phase in physiological conditions is HA (−logKsp at 25 °C = 116.8). This suggests that the evolution of less stable and more soluble intermediate phases from ACP, such as highly soluble DCPD (−logKsp at 25 °C = 6.59) and metastable OCP (−logKsp at 25 °C = 96.6), prior to the most thermodynamically favoured composition of bone mineral, ensues through pathways that are more kinetically favourable.57 Indeed, several in vitro systems simulating pseudo physiological conditions have demonstrated the transformation of highly soluble DCPD to metastable OCP,57–60 with OCP further shown to undergo transformation to physiologically stable HA.61–67 Phase transformations can reportedly occur via various pathways, primarily hydrolysis and dissolution followed by reprecipitation reactions. Whilst intermediate phases remain difficult to detect during bone biomineralisation, several recent studies have discovered the presence of beta-tricalcium phosphate (β-Ca3(PO4)2, β-TCP), DCPD-like and OCP phases in the early stages of in vivo bone formation, providing some of the strongest evidence to date as to the crucial roles of transient orthophosphate phases that are part of the biologically mediated transformation of ACP to HA.68–71
Under correctly regulated conditions, biomineralisation nearly always results in crystals that exhibit specific size, shape and molecular organisation. Biological apatite crystals are typically formed on the nanoscale. HA nanocrystals formed in bone are described as platelets and are often assigned approximate dimensions of 50 nm × 25 nm × 3 nm that align on the c-axis parallel to the direction of collagen72 (Fig. 4D and E). It has been proposed that OCP, which has a triclinic crystal structure considered similar to that of bone mineral HA, may act as a template during crystal growth.73 The precise dimensions and organisation of bone HA crystals within a matrix of collagen, together with other NCPs, results in the mechanical properties of bone being greater than what would be expected given its constituent elements. Bone has both high strength and toughness, while its organic components are soft and ductile and inorganic mineral is considerably stiff and brittle.74–76
Biomacromolecules are believed to play crucial roles in enacting spatial and temporal control over the formation of mineral at the molecular scale. The precipitation of calcium and orthophosphate ions from supersaturated solution in the absence of biomacromolecules generally results in crystals of considerable size. In the physiological environment, however, nanocrystals may well be more kinetically stable whilst also being of ideal size to form interactions with protein structures and other physiological molecules. Bone possesses a sufficient concentration of NCPs, many of which are phosphoproteins, which may act to both control crystal growth and facilitate interfacial interaction between collagen and HA.77 NCPs of the small integrin-binding ligand N-glycosylation (SIBLING) family, which includes OPN, bone sialoprotein (BSP), dentin matrix protein 1 (DMP1), dentin sialophosphoprotein (DSPP) and matrix extracellular phosphoglycoprotein (MEPE), have the capacity to bind to collagen in a site-specific manner, as well as being able to form interactions with or accumulate Ca2+ ions.78 NCPs not of the SIBLING family include osteonectin (ON), osteocalcin (OC) and bone-GLA protein. Taken together, these different NCPs have been implicated in playing a highly orchestrated role in biomineralisation by means of promoting, inhibiting and regulating crystal growth.79,80 OPN and OC have been shown to influence developing HA crystals in vitro, however, the abundance of these proteins in vivo may not be sufficient to interact with all nanocrystal surfaces.81,82 Furthermore, OC has recently been found to complex with OCP crystals in bone tissue, demonstrating the role of NCPs in stabilising bone crystal nuclei during the embryonic stages of formation.71,83 Although a typically overlooked molecule for its role in bone mineral formation, it has been proposed that citrate (C6H5O73−) may play a major role in stabilising crystal thickening (up to 3 nm), whereas OC and OPN may instead be responsible for stabilising crystal lengthening (up to 50 nm).84
3.4. Biomineral composition
Pure stoichiometric HA (Ca10(PO4)6OH2) has a Ca:P ratio of 1.67. However, the composition of biologically derived HA crystals deviates widely from this ideal formula.85,86 In fact, pure stoichiometric HA is not known to form in biological systems.87 The crystal lattice of HA can withstand substantial distortion to accommodate naturally occurring ionic impurities, other than calcium and orthophosphate, whilst maintaining overall charge neutrality.88 For instance, approximately 20% of Ca2+ ions are substituted with other cations, such as Mg2+ and Na+, both of which are present in bone in minor concentrations.89 Other cations, including Sr2+, Pb2+ and Zn2+, are also capable of filling in lattice vacancies in place of Ca2+ but are only present within bone in trace amounts.90 In the absence of Ca2+, vacant spaces can remain unoccupied within the crystal unit cell, leading to calcium deficient HA (Ca9(PO4)5(HPO4)OH).91 However, carbonate (CO32−) is the most prevalent substitution in bone mineral. Carbonate makes up between 2–8% of bone mineral by mass and can take the place of either the hydroxyl groups (Ca10(PO4)6(OH)2−2x(CO3)x) or, more commonly, phosphate groups (Ca10−x(PO4)6−x(CO3)x(OH)2−x), referred to as A- and B-type substitution, respectively.92,93 Together, these substitutions heavily influence the resulting chemistry of bone mineral in terms crystallinity, thermodynamic stability, morphology, solubility and biological properties.94–97
Typically, HA exhibits two crystallographic forms; a hexagonal form that crystallises above 250 °C (P63/m space group with unit cell parameters a = b = 9.432 Å, c = 6.881 Å) and a monoclinic form that crystallises below 250 °C (P21/b space group with unit cell parameters a = 9.421 Å, b = 2 Å, c = 6.881 Å, β = 120°).98,99 Whilst impurities can cause the Ca:P ratio of biological mineral to fall as low as 1.3, the combination of foreign ion inclusions, ionic vacancies and Ca2+ deficiencies in fact contributes to stabilising biological HA in the hexagonal crystal form.87 As well as influencing crystal attributes on the atomic scale, impurities are capable of enacting differences in the physical and chemical attributes of biological mineral at greater length scales. This is exemplified by the substitution of fluoride (F−) into the HA lattice resulting in fluorapatite (Ca10(PO4)6F2), which beneficially contributes to making tooth mineral more resilient against acidic conditions and demineralisation.100 An overview of the elemental composition of mineralised tissues is provided in Table 1, highlighting the various proportions of trace elements found in bone, enamel and dentine. Expectedly, elemental phosphorus is a prominent component of each of these mineralised human tissues (15.2–17.7%), predominantly found in the form of orthophosphate. Whilst not a substituting component of the HA lattice, normal mineralised tissue also contains a small proportion of PPi (0.02–0.1%), which is important to normal bone formation and function (Section 4.1).
| Enamel | Dentine | Bone | HA | |
|---|---|---|---|---|
| a Ashed samples. b Non-ashed samples. | ||||
| Calcium (Ca)a | 36.5 | 35.1 | 34.5 | 39.6 |
| Phosphorous (P)a | 17.7 | 16.9 | 15.2 | 18.5 |
| Sodium (Na)a | 0.5 | 0.6 | 0.9 | — |
| Magnesium (Mg)a | 0.44 | 1.23 | 0.72 | — |
| Potassium (K)a | 0.08 | 0.05 | 0.03 | — |
| Carbonate (CO32−)b | 3.5 | 5.6 | 7.4 | — |
| Fluoride (F)a | 0.01 | 0.06 | 0.03 | — |
| Chloride (Cl)a | 0.30 | 0.01 | 0.13 | — |
| Pyrophosphate (P2O74−)b | 0.02 | 0.10 | 0.07 | — |
| Total water (H2O)b | 1.5 | 10 | 10 | — |
| Total organicb | 1.5 | 20 | 25 | — |
| Total inorganicb | 97 | 70 | 65 | 100 |
| Ca:P molar ratiob |
1.63 | 1.61 | 1.71 | 1.67 |
4. The multiple roles of condensed phosphates during biomineralisation
4.1. Pyrophosphate is both a regulator and promoter of biomineralisation
Pyrophosphate (P2O74−, PPi) has been identified as a potent regulator of the formation of a wide variety of biominerals in vivo. It has been most extensively studied in the context of mineralised tissue formation, with respect to bones and teeth, calcium oxalate deposition disorders such the formation of kidney stones,103 vascular calcification104–106 and pseudoxanthoma elasticum (PXE).107 To a lesser extent, PPi is implicated in joint chondrocalcinosis and pseudogout syndrome, a rare condition whereby crystals of calcium pyrophosphate dihydrate (Ca2P2O7·2H2O, CPPD) are errantly formed in the synovial fluid of affected joints.108In the context of calcium oxalate deposition, pyrophosphate is known to act as a nucleation and crystal growth inhibitor.109 However, the inhibitory action of PPi has been shown to be primarily due to Ca2+ complexation and surface interactions have only been confirmed for the monohydrate phase (CaC2O4·H2O).110,111 For the deposition of calcium phosphate mineral, primarily HA, the role of pyrophosphate is more complex as it acts as both a source of orthophosphate and has been postulated to act as a nucleation and growth inhibitor of HA through strong site specific interactions.112 In PXE, chondrocalcinosis and pseudogout, the PPi ion is directly involved in the precipitation of the mineral phase, having been allowed to locally reach conditions of supersaturation or through the lowering of barriers to nucleation.107,108 Whether all these effects are purely chemical processes or act upon biochemical pathways is a matter of debate. It is likely both pathways are important and pyrophosphate species certainly have a significant and critical role to play in the mineralisation of hard tissues nonetheless.
Alkaline phosphatase (ALP) is an enzyme responsible for dephosphorylating biomolecules. It is found in a wide range of organisms with the same general function but in different structural forms to suit specific environments.113,114 In humans, it is found in four forms, depending on its origin within the body; intestinal, placental, placental-like, and liver/bone/kidney (tissue-nonspecific alkaline phosphatase, TNAP).115 TNAP hydrolyses PPi and so is a key enzyme in the control of mineralisation. TNAP plays a crucial role promoting mineralisation of the extracellular matrix by restricting the concentration of PPi. Mutations in the gene encoding TNAP cause hypophosphatasia, an inheritable form of rickets and osteomalacia.116 PPi may be generated via the cell driven metabolism of ATP to adenosine monophosphate (AMP), for example via nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1), implicating ATP as a progenitor of bone mineralisation inhibition.107,117 However, cleavage of PPi by TNAP generates orthophosphate, whilst at the same time removing a proportion of inhibitory species. Thus, PPi can act as both a local inhibitor and source of orthophosphate to drive mineralisation.
As well as acting via inorganic pathways, PPi has been demonstrated to influence biomolecular mineralisation pathways. For example, PPi has been shown to upregulate the NCP osteopontin (OPN), found mainly in the bone matrix. OPN is also present in other cell types, such as hypertrophic chondrocytes, odontoblasts, cementoblasts, macrophages, as well as endothelial, smooth muscle and epithelial cells.118 As a potent inhibitor of apatite formation, it shows a dose dependent behaviour that is entirely depleted with removal of the phosphate group from OPN.81,119–121 The extended role of OPN with relation to PPi in the regulation of bone mineralisation is highlighted by two studies on mice unable to produce this protein.122,123 In the neonatal period, the mice showed no skeletal abnormalities; however, spleen cells were able to form osteoclasts far more easily than wild type spleen cells, which only became more pronounced with age. At the age of 4–6 months, OPN deficient mice had twice the volume of trabecular bone and three times the number of osteoclasts compared to wild type mice. Additionally, OPN deficient mice were more resistant to ovariectomy-induced bone resorption. Microcomputed tomography analysis indicated a 60% reduction in bone volume by ovariectomy in wild type mice, whereas the OPN deficient mice exhibited only a 10% reduction in trabecular bone volume after ovariectomy.
Studies have shown that TNAP activity increases in response to PPi.120,124 Meanwhile it has been concluded that TNAP's primary function in mineralising tissues is to act together with plasma cell membrane glycoprotein-1 to fine tune PPi concentrations to maintain steady-state levels and adequate control of mineralization.125 A further study has shown that OPN represents a natural substrate for TNAP and therefore it may regulate OPN function by dephosphorylation.126 As such it can be seen that PPi forms a complicated feedback loop that contributes to controlling biomineralisation in a number of ways.
4.2. The curious role of polyphosphates
Inorganic polyphosphates (polyP) are linear phosphate polymers. These molecules have been implicated in the origin of life as non-enzymatically produced energy carriers and phosphate donors, and several distinct biological roles have been identified since their discovery in cells in the late 19th century.17,18 PolyP is stored intracellularly in what are commonly referred to as ‘dense granules’, which are present in both prokaryotes and eukaryotes. In mammals, polyP is particularly concentrated in the granules of platelets; chains around 75 phosphate residues long are present in platelets at a concentration of 1.1 mM, 10–20 fold higher than in major organs.127 These granules also contain high concentrations of divalent cations; polyP allows storage of high concentrations of calcium and phosphate as an amorphous phase. The polyP in platelets has been implicated in the clotting process, which is particularly interesting for bone formation. Platelets are present in the haematoma formed around sites of bone healing following fracture, as well as the haematoma that develops prior to ectopic bone formation. Platelet-rich plasma is commonly studied pre-clinically, as well as currently used clinically, for repair of bone defects.128Dense polyP granules are found in many cell types, particularly within the mitochondria. This includes osteoblasts, where polyP is found in high concentrations (0.5 M in the dense granules in the mitochondria), and osteoclasts, which have a high number of mitochondria.129 These organelles have been implicated in bone formation for some time, as large amounts of energy are required both during bone formation and resorption.129
Biological reactions involving polyP can be grouped into two categories: chain-lengthening and chain-shortening. Chain-lengthening reactions are catalysed by the polyP kinase family of enzymes. These enzymes can polymerise orthophosphate to form polyP, or take phosphate residues from other molecules and add them to the polyP chain, for example from ATP.130 Chain shortening reactions are catalysed by several polyphosphatases, which can cleave the chain in the middle (endopolyphosphatases), or cleave the end phosphate (exophosphatases), which can then be attached to another molecule such as ADP, or utilised as free orthophosphate, such as in the formation of mineral.
The proposed role of polyP as a high-energy phosphate donor is somewhat analogous to ATP; the same high-energy (31 kJ mol−1) phosphoanhydride bonds are present in both molecules. Indeed, polyP has been shown to increase extracellular ATP and ADP generation in SaOS-2 (osteoblast-like) cells.131 In the same cell type, polyP was shown to promote proliferation and mineralisation under hypoxic conditions, those found in physiological and ectopic bone formation sites.132 This suggests that polyP can act as an alternative phosphate and metabolic energy source, particularly in extracellular or hypoxic conditions, such as those in bone formation where ATP is not readily available.133
In addition to its role as a substrate, polyP has also been shown to have a direct influence on biomineralisation pathways by inducing the expression of bone formation markers OPN, OC and ALP, leading to the mineralisation of pre-osteoblast MC3T3 cells.105,134 Increased ALP expression has also been shown in SaOS-2 cells, and an increase in intracellular calcium was seen when calcium–polyP was used as a substrate, but not with Ca, Pi, or polyP alone.135
As well as a role in bone formation, polyP has been shown to increase cartilage production, but for a short time before it is degraded by the polyphosphatases released by chondrocytes.136 This could enhance endochondral bone formation, by inducing chondrocytes to express more cartilage tissue to act as the scaffold for bone formation, then being degraded to halt cartilage production while leaving behind the building blocks for mineralisation.
An integral role of polyP in bone remodelling has been proposed.137 Firstly, osteoclasts use acids and enzymes to break down bone, releasing calcium and orthophosphate ions. Enzymes in the osteoclast then polymerise the orthophosphate into polyP. The calcium–polyP complex is left behind by the osteoclast in the bone resorption pit, or transferred through the extracellular space either freely or in vesicle compartments to a previous resorption pit. The calcium–polyP complex can then be used as a substrate by osteoblasts to produce bone mineral.
This general idea outlines the importance of polyP as both a substrate to be broken down, as well as a molecule capable of preventing mineral formation, but in doing so facilitates high concentrations of calcium and phosphate to coexist. However, this mechanism is somewhat speculative. The enzymes associated with osteoblasts, such as ALP, used to produce orthophosphate are well researched,124,138 as are the enzymes and processes used by osteoclasts to break down apatite and resorb bone.139 However, polyphosphate kinases, the enzymes used to synthesise polyP, have been mainly identified in bacteria. One of these enzymes has also been found in Dictyostelium discoideum, a unicellular eukaryote.140,141 Its conservation in eukaryotic cells suggests it may also be present in mammals. Nevertheless, the enzyme has not been found in mammalian cells to date, therefore the assumption that such an enzyme exists in mammalian osteoclasts remains unconfirmed.
This mechanism also fits with several current theories of bone formation. Recently, in addition to ACP, amorphous calcium carbonate (ACC) has been proposed as another pre-curser to bone mineral.142,143 Osteoblasts could lay down this ACC phase, which is then substituted for phosphate liberated from polyP. Furthermore, calcium–polyP may lead to the formation of ACP when enzymatically degraded, which over time may form HA. Another theory suggests that the calcium–polyP complex could directly infiltrate the collagen matrix by capillary action before being be broken down in situ by osteoblast-secreted enzymes to form HA. Alternatively, the polyP can be stored in and utilised by either osteoblasts or vesicles.144
Condensed linear phosphates are becoming increasingly recognised as multifunctional species with important physiological roles. Most notable are the ability of phosphate polymers to stimulate osteogenic markers, regulate the localised concentrations of other phosphate species in a given biological microenvironment, increase the levels of orthophosphate when broken down and antagonise biological crystallisation processes (Fig. 5).
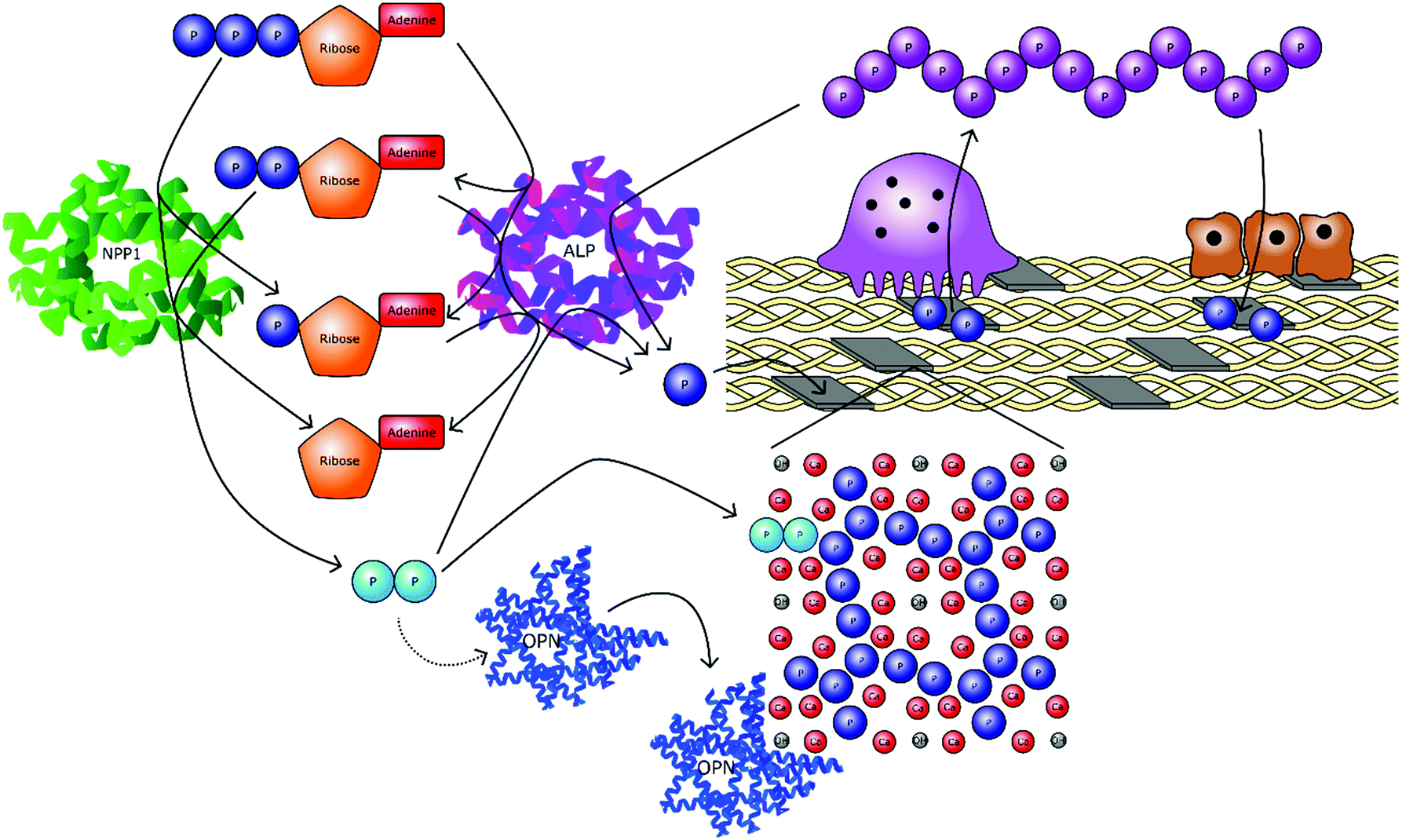 | ||
| Fig. 5 The interconnecting roles of phosphates in bone mineral formation. Orthophosphate ions (PO43−) are cleaved from organic molecules, primarily adenosine triphosphate (ATP), adenosine diphosphate (ADP) and adenosine monophosphate (AMP). In addition, inorganic phosphates are also broken down, including pyrophosphate (P2O74−) and polyphosphate (PnO3n+1(n+2)−). These processes occur enzymatically via alkaline phosphatase (ALP) and other exophosphatases. Orthophosphate is then utilised to form mineral phases, namely hydroxyapatite (HA), which are incorporated into a collagen scaffold to form bone. Cleavage by endophosphatases, primarily nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) results in the formation of pyrophosphate, which inhibits mineral growth by interrupting the crystal structure. Pyrophosphate also upregulates osteopontin (OPN), anther crystal growth inhibitor. When bone is resorbed by osteoclasts, the orthophosphate ions are released from the HA. This orthophosphate may then be recondensed by osteoclasts into polyP, before being transported extracellularly to osteoblasts for reutilisation in bone formation. | ||
5. Conclusions and future perspectives
Together, various phosphate-based structures and molecules play important roles in the formation and regulation of biological mineral. Orthophosphate ions provide a building block for the formation of bone mineral, whereas condensed phosphates have the ability to both regulate mineral formation and provide a source of additional orthophosphate as required. A comprehensive understanding of the complicated feedback loops involving phosphate species is necessary for progression of research, particularly in the area of regenerative medicine. Challenges remain in the preparation of synthetic forms of biological apatite mineral, thus investigating the use of several phosphate species in mineral formation as occurs in vivo may help to improve the clinical performance of bone-based grafting materials. As well as biomineralisation, the wider utilisation of phosphates throughout our bodies only adds to their complex involvement in biochemical and chemical mechanisms essential to the entirety of our bodily functions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is funded by the National Institute for Health Research (NIHR) Surgical Reconstruction and Microbiology Research Centre (SRMRC). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.References
- C. Maffeo, J. Yoo, J. Comer, D. B. Wells, B. Luan and A. Aksimentiev, J. Phys.: Condens. Matter, 2014, 26, 413101 CrossRef CAS PubMed.
- P. L. Privalov and C. Crane-Robinson, Prog. Biophys. Mol. Biol., 2018, 135, 30–48 CrossRef CAS PubMed.
- R. Villa-bellosta and J. Egido, Eur. Heart J., 2017, 38, 1801–1804 CAS.
- T. Hunter, Philos. Trans. R. Soc., B, 2012, 367, 2513–2516 CrossRef CAS PubMed.
- M. G. Penido and U. S. Alon, Pediatr. Nephrol., 2012, 27, 2039–2048 CrossRef PubMed.
- R. L. Wadsworth and S. Siddiqui, BJA Educ., 2016, 16, 305–309 CrossRef.
- G. A. Rodan, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13361 LP–13362 LP CrossRef PubMed.
- M. Cuéllar-Cruz, Prog. Cryst. Growth Charact. Mater., 2017, 63, 94–103 CrossRef.
- M. Tzaphlidou, J. Biol. Phys., 2008, 34, 39–49 CrossRef CAS PubMed.
- D. J. Hadjidakis and I. I. Androulakis, Ann. N. Y. Acad. Sci., 2006, 1092, 385–396 CrossRef CAS PubMed.
- W. F. de Jong, Recl. des Trav. Chim. des Pays-Bas, 1926, 45, 445–448 CrossRef CAS.
- H. H. Roseberry, A. B. Hastings and J. K. Morse, J. Biol. Chem., 1931, 90, 395–407 CAS.
- J. D. Currey and K. Brear, J. Mater. Sci.: Mater. Med., 1990, 1, 14–20 CrossRef.
- H. Chen and Y. Liu, in Advanced Ceramics for Dentistry, ed. J. Z. Shen and T. B. T.-A. C. D. Kosmač, Butterworth-Heinemann, Oxford, 2014, pp. 5–21 Search PubMed.
- J. J. Berzeilius, Ann. Phys., 1816, 53, 393 CrossRef.
- A. Kornberg, N. N. Rao and D. Ault-Riché, Annu. Rev. Biochem., 1999, 68, 89–125 CrossRef CAS PubMed.
- L. Achbergerová and J. Nahálka, Microb. Cell Fact., 2011, 10 Search PubMed.
- J. Jiménez, S. Bru, M. P. C. Ribeiro and J. Clotet, Curr. Genet., 2017, 63, 15–18 CrossRef.
- H. S. Tenenhouse, Annu. Rev. Nutr., 2005, 25, 197–214 CrossRef CAS.
- M. F. Burrit, J. M. Slockbower, R. W. Forsman, K. P. Offord, E. J. Bergstral and W. A. Smithson, Mayo Clin. Proc., 1990, 65, 329–336 CrossRef.
- J. B. Graham, R. W. Winters and B. G. Greenberg, J. Clin. Endocrinol. Metab., 1960, 20, 364–379 CrossRef PubMed.
- E. Takeda, Y. Taketani, N. Sawada, T. Sato and H. Yamamoto, The regulation and function of phosphate in the human body, 2004, vol. 21 Search PubMed.
- C. Bergwitz and H. Jüppner, Annu. Rev. Med., 2010, 61, 91–104 CrossRef CAS PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
- E. D. Eanes, I. H. Gillessen and A. S. Posnor, Nature, 1965, 208, 365–367 CrossRef CAS PubMed.
- E. D. Eanes and A. S. Posner, Trans. N. Y. Acad. Sci., 1965, 28, 233–241 CrossRef CAS.
- E. Beniash, R. A. Metzler, R. S. K. Lam and P. U. P. A. Gilbert, J. Struct. Biol., 2009, 166, 133–143 CrossRef CAS PubMed.
- O. A. Tertuliano and J. R. Greer, Nat. Mater., 2016, 15, 1195 CrossRef CAS.
- A. Akiva, G. Malkinson, A. Masic, M. Kerschnitzki, M. Bennet, P. Fratzl, L. Addadi, S. Weiner and K. Yaniv, Bone, 2015, 75, 192–200 CrossRef CAS.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748–12753 CrossRef CAS.
- E. S. Hara, M. Okada, N. Nagaoka, T. Hattori, T. Kuboki, T. Nakano and T. Matsumoto, ACS Biomater. Sci. Eng., 2018, 4, 617–625 CrossRef CAS.
- H. A. Lowenstam and S. Weiner, Science, 1985, 227, 51-LP–53-LP CrossRef PubMed.
- I. Lévêque, M. Cusack, S. A. Davis and S. Mann, Angew. Chem., Int. Ed., 2004, 43, 885–888 Search PubMed.
- K. Onuma and A. Ito, Chem. Mater., 1998, 3346–3351 CrossRef CAS.
- F. Betts and A. S. Posner, Mater. Res. Bull., 1974, 9, 353–360 CrossRef CAS.
- N. Kanzaki, G. Treboux, K. Onuma, S. Tsutsumi and A. Ito, Biomaterials, 2001, 22, 2921–2929 CrossRef CAS PubMed.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef.
- V. Čadež, I. Erceg, A. Selmani, D. D. Jurašin, S. Šegota, M. D. Lyons, D. Kralj and D. M. Sikirić, Crystals, 2018, 8, 254 CrossRef.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010 CrossRef CAS PubMed.
- Q. Zhang, Y. Jiang, B.-D. Gou, J. Huang, Y.-X. Gao, J.-T. Zhao, L. Zheng, Y.-D. Zhao, T.-L. Zhang and K. Wang, Cryst. Growth Des., 2015, 15, 2204–2210 CrossRef CAS.
- L.-W. Du, S. Bian, B.-D. Gou, Y. Jiang, J. Huang, Y.-X. Gao, Y.-D. Zhao, W. Wen, T.-L. Zhang and K. Wang, Cryst. Growth Des., 2013, 13, 3103–3109 CrossRef CAS.
- E. D. Eanes, in Calcium Phosphates in Biological and Industrial Systems, ed. Z. Amjad, Springer, US, Boston, MA, 1998, pp. 21–39 Search PubMed.
- G. Mancardi, C. E. Hernandez Tamargo, D. Di Tommaso and N. H. de Leeuw, J. Mater. Chem. B, 2017, 5, 7274–7284 RSC.
- S. Maltsev, M. J. Duer, R. C. Murray and C. Jaeger, J. Mater. Sci., 2007, 42, 8804–8810 CrossRef CAS.
- D. Laurencin, A. Wong, W. Chrzanowski, J. C. Knowles, D. Qiu, D. M. Pickup, R. J. Newport, Z. Gan, M. J. Duer and M. E. Smith, Phys. Chem. Chem. Phys., 2010, 12, 1081–1091 RSC.
- W. J. Landis and F. H. Silver, Cells Tissues Organs, 2009, 189, 20–24 CrossRef CAS PubMed.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- A. S. Deshpande and E. Beniash, Cryst. Growth Des., 2008, 8, 3084–3090 CrossRef CAS PubMed.
- P. A. Price, D. Toroian and J. E. Lim, J. Biol. Chem., 2009, 284, 17092–17101 CrossRef CAS PubMed.
- H. C. Margolis, S.-Y. Kwak and H. Yamazaki, Front. Physiol., 2014, 5, 339 Search PubMed.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- J. Mahamid, B. Aichmayer, E. Shimoni, R. Ziblat, C. Li, S. Siegel, O. Paris, P. Fratzl, S. Weiner and L. Addadi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6316 LP–6321 LP CrossRef PubMed.
- J. A. Stammeier, B. Purgstaller, D. Hippler, V. Mavromatis and M. Dietzel, Methods, 2018, 5, 1241–1250 Search PubMed.
- A. Lotsari, A. K. Rajasekharan, M. Halvarsson and M. Andersson, Nat. Commun., 2018, 9, 4170 CrossRef PubMed.
- W. E. Brown, J. P. Smith, J. R. Lehr and A. W. Frazier, Nature, 1962, 196, 1050–1055 CrossRef CAS.
- M. S. Johnsson and G. H. Nancollas, Crit. Rev. Oral Biol. Med., 1992, 3, 61–82 CrossRef CAS PubMed.
- A. Bannerman, R. L. Williams, S. C. Cox and L. M. Grover, Sci. Rep., 2016, 6, 32671 CrossRef CAS PubMed.
- M. S. Tung, L. C. Chow and W. E. Brown, J. Dent. Res., 1985, 64, 2–5 CrossRef CAS PubMed.
- L. Perez, L. J. Shyu and G. H. Nancollas, Colloids Surf., 1989, 38, 295–304 CrossRef CAS.
- N. Temizel, G. Girisken and A. C. Tas, Mater. Sci. Eng., C, 2011, 31, 1136–1143 CrossRef CAS.
- I. Y. Pieters, E. A. P. De Maeyer and R. M. H. Verbeeck, Inorg. Chem., 1996, 35, 5791–5797 CrossRef CAS.
- N. Ito, M. Kamitakahara, S. Murakami, N. Watanabe and K. Ioku, J. Ceram. Soc. Jpn., 2010, 118, 762–766 CrossRef CAS.
- D. G. A. Nelson and J. D. McLean, Calcif. Tissue Int., 1984, 36, 219–232 CrossRef CAS PubMed.
- M. Iijima, H. Kamemizu, N. Wakamatsu, T. Goto, Y. Doi and Y. Moriwaki, J. Cryst. Growth, 1997, 181, 70–78 CrossRef CAS.
- A. Bigi, E. Boanini, G. Falini, S. Panzavolta and N. Roveri, J. Inorg. Biochem., 2000, 78, 227–233 CrossRef CAS PubMed.
- H. Shi, J. Zhang, X. Ye, T. Wu, T. Yu and J. Ye, CrystEngComm, 2019, 21, 5174–5184 RSC.
- R. Horváthová, L. Müller, A. Helebrant, P. Greil and F. A. Müller, Mater. Sci. Eng., C, 2008, 28, 1414–1419 CrossRef.
- N. J. Crane, V. Popescu, M. D. Morris, P. Steenhuis and M. A. Ignelzi, Bone, 2006, 39, 434–442 CrossRef CAS.
- A. Vyalikh, C. Elschner, C. M. Schulz, R. Mai and U. Scheler, Magnetochemistry, 2017, 3, 39 CrossRef.
- R. Xin, Y. Leng and N. Wang, Adv. Eng. Mater., 2010, 12, B552–B557 CrossRef.
- P. Simon, D. Grüner, H. Worch, W. Pompe, H. Lichte, T. El Khassawna, C. Heiss, S. Wenisch and R. Kniep, Sci. Rep., 2018, 8, 13696 CrossRef.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2014, 14, 23 CrossRef PubMed.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- B. Ji and H. Gao, J. Mech. Phys. Solids, 2004, 52, 1963–1990 CrossRef.
- F. Bouville, E. Maire, S. Meille, B. Van de Moortèle, A. J. Stevenson and S. Deville, Nat. Mater., 2014, 13, 508 CrossRef CAS PubMed.
- E. A. Zimmermann, B. Busse and R. O. Ritchie, BoneKEy Rep., 2015, 4, 743 Search PubMed.
- S. R. Stock, Calcif. Tissue Int., 2015, 97, 262–280 CrossRef CAS PubMed.
- C. Jiang, K. Zurick, C. Qin and M. T. Bernards, Connect. Tissue Res., 2018, 59, 274–286 CAS.
- K. A. Staines, V. E. Macrae and C. Farquharson, J. Endocrinol., 2012, 214, 241–255 CAS.
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed.
- G. K. Hunter, C. L. Kyle and H. A. Goldberg, Biochem. J., 1994, 300, 723–728 CrossRef CAS PubMed.
- E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009, 21, 425–430 CrossRef CAS PubMed.
- W. N. Addison, D. L. Masica, J. J. Gray and M. D. McKee, J. Bone Miner. Res., 2010, 25, 695–705 CrossRef CAS PubMed.
- Y.-Y. Hu, A. Rawal and K. Schmidt-Rohr, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22425–22429 CrossRef CAS PubMed.
- J.-P. Bonjour, J. Am. Coll. Nutr., 2011, 30, 438S–48S CrossRef CAS.
- L. T. Kuhn, M. D. Grynpas, C. C. Rey, Y. Wu, J. L. Ackerman and M. J. Glimcher, Calcif. Tissue Int., 2008, 83, 146–154 CrossRef CAS.
- V. S. Dorozhkin, Materials, 2009, 2, 399–498 CrossRef.
- V. Uskoković, RSC Adv., 2015, 5, 36614–36633 RSC.
- A. Farzadi, F. Bakhshi, M. Solati-Hashjin, M. Asadi-Eydivand and N. A. A. Osman, Ceram. Int., 2014, 40, 6021–6029 CrossRef CAS.
- J. A. Spadaro, R. O. Becker and C. H. Bachman, Calcif. Tissue Res., 1970, 6, 49–54 CrossRef CAS PubMed.
- M. Vallet-Regi and D. A. A. Navarrete, Biomimetic Nanoceramics in Clinical Use: From Materials to Applications, The Royal Society of Chemistry, 2008, pp. 61–121 Search PubMed.
- A. Bigi, G. Cojazzi, S. Panzavolta, A. Ripamonti, N. Roveri, M. Romanello, K. Noris Suarez and L. Moro, J. Inorg. Biochem., 1997, 68, 45–51 CrossRef CAS.
- F. C. M. Driessens, Bull. Soc. Chim. Belg., 1980, 89, 663–689 CrossRef CAS.
- J. Kolmas, A. Jaklewicz, A. Zima, M. Bućko, Z. Paszkiewicz, J. Lis, A. Ślósarczyk and W. Kolodziejski, J. Mol. Struct., 2011, 987, 40–50 CrossRef CAS.
- S. M. Barinov, I. V. Fadeeva, D. Ferro, J. V. Rau, S. N. Cesaro, V. S. Komlev and A. S. Fomin, Russ. J. Inorg. Chem., 2008, 53, 164–168 CrossRef.
- S. Kannan and J. M. F. Ferreira, Chem. Mater., 2006, 18, 198–203 CrossRef CAS.
- B. B. Hole, D. S. Keller, W. M. Burry and J. A. Schwarz, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1847–1850 CrossRef CAS PubMed.
- G. Ma and X. Y. Liu, Cryst. Growth Des., 2009, 9, 2991–2994 CrossRef CAS.
- R. Pérez-Solis, J. J. Gervacio-Arciniega, B. Joseph, E. M. Mendoza and A. Moreno, Crystals, 2018, 8, 458 CrossRef.
- Restorative Dentistry, ed. A. D. Walmsley, T. F. Walsh, P. J. Lumley, F. J. T. Burke, A. C. C. Shortall, R. Hayes-Hall and I. Pretty, Churchill Livingstone, Edinburgh, 2007, pp. 73–87 Search PubMed.
- S. V. Dorozhkin, Prog. Biomater., 2015, 5, 9–70 CrossRef.
- M. Chieruzzi, S. Pagano, S. Moretti, R. Pinna, E. Milia, L. Torre and S. Eramo, Nanomaterials, 2016, 6, 134 CrossRef PubMed.
- G. W. Drach, A. D. Randolph and M. D. John, J. Urol., 1978, 119, 99–103 CrossRef CAS.
- A. P. Sage, Y. Tintut and L. L. Demer, Nat. Rev. Cardiol., 2010, 7, 528 CrossRef CAS.
- Y. Kawazoe, T. Shiba, R. Nakamura, A. Mizuno, K. Tsutsumi, T. Uematsu, M. Yamaoka, M. Shindoh and T. Kohgo, J. Dent. Res., 2004, 83, 613–618 CrossRef CAS.
- L. L. Demer and T. Y. Yin, Arterioscler., Thromb., Vasc. Biol., 2014, 34, 715–723 CrossRef CAS.
- R. S. Jansen, S. Duijst, S. Mahakena, D. Sommer, F. Szeri, A. Váradi, A. Plomp, A. A. Bergen, R. P. Oude Elferink, P. Borst and K. van de Wetering, Arterioscler., Thromb., Vasc. Biol., 2014, 34, 1985–1989 CrossRef CAS.
- K. P. H. Pritzker, Hum. Pathol., 1986, 17, 543–545 CrossRef CAS.
- H. Fleisch, Kidney Int., 1978, 13, 361–371 CrossRef CAS.
- G. W. Drach, W. G. Robertson, D. S. Scurr and A. D. Randolph, World J. Urol., 1983, 1, 146–149 CrossRef.
- Y. Shirane and S. Kagawa, J. Urol., 1993, 150, 1980–1983 CrossRef CAS.
- C. J. Ibsen Steenberg and H. Birkedal, Minerals, 2018, 8(2), 65 CrossRef.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed.
- J. L. Millán, Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology, Wiley-VCH, 2006 Search PubMed.
- J. L. Millán and M. P. Whyte, Calcif. Tissue Int., 2016, 98, 398–416 CrossRef PubMed.
- M. C. Yadav, A. Maria, S. Simão, S. Narisawa, C. Huesa, M. D. Mckee, C. Farquharson and J. L. Millán, J. Bone Miner. Res., 2011, 26, 286–297 CrossRef CAS PubMed.
- I. R. Orriss, M. L. Key, M. O. R. Hajjawi and T. R. Arnett, PLoS One, 2013, 8, e69057–e69057 CrossRef CAS PubMed.
- M. D. McKee and W. G. Cole, Pediatric Bone, Academic Press, 2nd edn, 2012, pp. 9–37 Search PubMed.
- A. L. Boskey, M. Maresca, W. Ullrich, S. B. Doty, W. T. Butler and C. W. Prince, Bone Miner., 1993, 22, 147–159 CrossRef CAS.
- W. N. Addison, F. Azari, E. S. Sørensen, M. T. Kaartinen and M. D. McKee, J. Biol. Chem., 2007, 282, 15872–15883 CrossRef CAS PubMed.
- S. Jono, C. Peinado and C. M. Giachelli, J. Biol. Chem., 2000, 275, 20197–20203 CrossRef CAS PubMed.
- S. R. Rittling, H. N. Matsumoto, M. D. Mckee, A. Nanci, X.-R. An, K. E. Novick, A. J. Kowalski, M. Noda and D. T. Denhardt, J. Bone Miner. Res., 1998, 13, 1001–1111 CrossRef PubMed.
- H. Yoshitake, S. R. Rittling, D. T. Denhard and A. Noda, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8156–8160 CrossRef CAS PubMed.
- M. Pujari-Palmer, S. Pujari-Palmer, X. Lu, T. Lind, H. Melhus, T. Engstrand, M. Karlsson-Ott and H. Engqvist, PLoS One, 2016, 11, e0163530 CrossRef PubMed.
- L. Hessle, K. A. Johnson, H. C. Anderson, S. Narisawa, A. Sali, J. W. Goding, R. Terkeltaub and J. L. Millá, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9445–9449 CrossRef CAS PubMed.
- S. Narisawa, M. C. Yadav and J. L. Millán, J. Bone Miner. Res., 2013, 28, 1587–1598 CrossRef CAS PubMed.
- F. A. Ruiz, C. R. Lea, E. Oldfield and R. Docampo, J. Biol. Chem., 2004, 279, 44250–44257 CrossRef CAS PubMed.
- A. Roffi, B. Di Matteo, G. S. Krishnakumar, E. Kon and G. Filardo, Int. Orthop., 2017, 41, 221–237 CrossRef PubMed.
- K. Väänänen, Adv. Drug Delivery Rev., 2005, 57, 959–971 CrossRef PubMed.
- S. J. Omelon and M. D. Grynpas, Chem. Rev., 2008, 108, 4694–4715 CrossRef CAS PubMed.
- W. E. G. Müller, S. Wang, M. Neufurth, M. Kokkinopoulou, Q. Feng, H. C. Schröder and X. Wang, J. Cell Sci., 2017, 130, 2747–2756 CrossRef PubMed.
- W. E. G. Müller, H. C. Schröder, E. Tolba, B. Diehl-Seifert and X. Wang, FEBS J., 2016, 283, 74–87 CrossRef PubMed.
- W. E. G. Müller, E. Tolba, H. C. Schröder and X. Wang, Macromol. Biosci., 2015, 15, 1182–1197 CrossRef PubMed.
- K. Kato, K. Morita, I. Hirata, K. Doi, T. Kubo, K. Kato and K. Tsuga, In Vitro Cell. Dev. Biol.: Anim., 2018, 54, 449–457 CrossRef CAS PubMed.
- W. E. G. Müller, X. Wang, B. Diehl-Seifert, K. Kropf, U. Schloßmacher, I. Lieberwirth, G. Glasser, M. Wiens and H. C. Schröder, Acta Biomater., 2011, 7, 2661–2671 CrossRef PubMed.
- J.-P. St-Pierre, Q. Wang, S. Q. Li, R. M. Pilliar and R. A. Kandel, Tissue Eng., Part A, 2012, 18, 1282–1292 CrossRef CAS PubMed.
- S. Omelon, J. Georgiou, Z. J. Henneman, L. M. Wise, B. Sukhu, T. Hunt, C. Wynnyckyj, D. Holmyard, R. Bielecki and M. D. Grynpas, PLoS One, 2009, 4, e5634 CrossRef PubMed.
- Y. H. Kim, D. S. Yoon, H. O. Kim and J. W. Lee, Stem Cells Dev., 2012, 21, 2958–2968 CrossRef CAS PubMed.
- A. Cappariello, A. Maurizi, V. Veeriah and A. Teti, Arch. Biochem. Biophys., 2014, 558, 70–78 CrossRef CAS PubMed.
- L. Eichinger, J. A. Pachebat, G. Glöckner, M.-A. Rajandream, R. Sucgang, M. Berriman, J. Song, R. Olsen, K. Szafranski, Q. Xu, B. Tunggal, S. Kummerfeld, M. Madera, B. A. Konfortov, F. Rivero, A. T. Bankier, R. Lehmann, N. Hamlin, R. Davies, P. Gaudet, P. Fey, K. Pilcher, G. Chen, D. Saunders, E. Sodergren, P. Davis, A. Kerhornou, X. Nie, N. Hall, C. Anjard, L. Hemphill, N. Bason, P. Farbrother, B. Desany, E. Just, T. Morio, R. Rost, C. Churcher, J. Cooper, S. Haydock, N. van Driessche, A. Cronin, I. Goodhead, D. Muzny, T. Mourier, A. Pain, M. Lu, D. Harper, R. Lindsay, H. Hauser, K. James, M. Quiles, M. Madan Babu, T. Saito, C. Buchrieser, A. Wardroper, M. Felder, M. Thangavelu, D. Johnson, A. Knights, H. Loulseged, K. Mungall, K. Oliver, C. Price, M. A. Quail, H. Urushihara, J. Hernandez, E. Rabbinowitsch, D. Steffen, M. Sanders, J. Ma, Y. Kohara, S. Sharp, M. Simmonds, S. Spiegler, A. Tivey, S. Sugano, B. White, D. Walker, J. Woodward, T. Winckler, Y. Tanaka, G. Shaulsky, M. Schleicher, G. Weinstock, A. Rosenthal, E. C. Cox, R. L. Chisholm, R. Gibbs, W. F. Loomis, M. Platzer, R. R. Kay, J. Williams, P. H. Dear, A. A. Noegel, B. Barrell and A. Kuspa, Nature, 2005, 435, 43–57 CrossRef CAS PubMed.
- H. Zhang, M. R. Gomez-Garcia, X. Shi, N. N. Rao and A. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16486–16491 CrossRef CAS PubMed.
- X. Wang, H. C. Schröder, U. Schlossmacher, M. Neufurth, Q. Feng, B. Diehl-Seifert and W. E. G. Müller, Calcif. Tissue Int., 2014, 94, 495–509 CrossRef CAS PubMed.
- E. Tolba, W. E. G. Müller, B. M. Abd El-Hady, M. Neufurth, F. Wurm, S. Wang, H. C. Schröder and X. Wang, J. Mater. Chem. B, 2016, 4, 376–386 RSC.
- Y. C. Chai, A. Carlier, J. Bolander, S. J. Roberts, L. Geris, J. Schrooten, H. Van Oosterwyck and F. P. Luyten, Acta Biomater., 2012, 8, 3876–3887 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |