
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical constituents from the stems of Machilus philippinensis Merr. and the neuroprotective activity of cinnamophilin†
Shih-Huang Tai‡
a,
Ping-Chung Kuo‡ b,
Sio Hong Lamb,
Shiow-Chyn Huangc,
Yi-Zhuan Kuod,
Hsin-Yi Hungb,
Meei-Jen Lioue,
Po-Chuen Shiehf,
E.-Jian Lee*a and
Tian-Shung Wu*bf
b,
Sio Hong Lamb,
Shiow-Chyn Huangc,
Yi-Zhuan Kuod,
Hsin-Yi Hungb,
Meei-Jen Lioue,
Po-Chuen Shiehf,
E.-Jian Lee*a and
Tian-Shung Wu*bf
aDepartment of Surgery, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan 701, Taiwan
bSchool of Pharmacy, College of Medicine, National Cheng Kung University, Tainan 701, Taiwan. E-mail: tswu@mail.ncku.edu.tw
cDepartment of Pharmacy, Chia-Nan University of Pharmacy and Science, Tainan 717, Taiwan
dDepartment of Chemistry, National Cheng Kung University, Tainan 701, Taiwan
eDepartment of Applied Chemistry, Providence University, Taichung 433, Taiwan
fDepartment of Pharmacy, College of Pharmacy and Health Care, Tajen University, Pingtung 907, Taiwan
First published on 11th July 2019
Abstract
The Machilus genus (Lauraceae) had been extensively utilized in folk medicine due to its broad range of bioactivities. In the present study, a series of chromatographic separations of the methanol extract of stems of M. philippinensis led to the identification of thirty eight compounds totally. Among these, biscinnamophilin (1), machilupins A–C (2–4), machilutone A (5), and machilusoxide A (6) were new compounds reported for the first time. In addition, 5 was characterized with a unprecedented carbon skeleton. Other known compounds, including the major compounds cinnamophilin (7) and meso-dihydroguaiaretic acid (8), are identified by comparison of their physical and spectroscopic data with reported values. One of the reported compounds, cinnamophilin A (10), should be revised as dehydroguaiaretic acid (9) after careful comparison of all the 1H and 13C NMR data. Moreover, the neuroprotective activity of cinnamophilin (7) was examined in a primary cortical neuron culture and the results indicated that 7 was effective against glutamate induced excitotoxicity.
Introduction
Stroke, a major public health problem in the world, usually generates miscellaneous neurological issues including focal motor weakness, sensory loss, visual damage, impaired speech comprehension and cognitive and/or memory disturbances. Current treatment modality for acute ischemic stroke, however, is confined to thrombolysis and supportive treatment, and can benefit only a small proportion of stroke patients.1 Therefore, development of a more applicable and efficient therapy is highly demanded.2,3 Cinnamophilin (CINN), (8R,8′S)-4,4′-dihydroxy-3,3′-dimethoxy-7-oxo-8,8′-neolignan from M. philippinensis, is a novel antioxidant and anti-inflammatory, and readily passes through the blood brain barrier with a slow decay in the brain.4–7 CINN not only reduces brain infarction and oxidative stress, but also improves behavioral outcome, as assessed at 24 h post-stroke.7 CINN protects against ischemic brain damage with a therapeutic window up to 6 h in vivo and in vitro, which may be attributed to its direct antioxidant and anti-inflammatory effects.8,9 M. philippinensis had been utilized in folk medicine due to its broad range of bioactivities, including cytotoxic,10–13 antioxidant,14,15 antiinflammatory,8,16 antiplatelet aggregatory,17,18 and neuro-protective effects.7–9,19,20 In our continuing program aimed to screen for natural neuroprotective lead compounds from Formosan Lauraceous plants, exploring the complete chemical composition of M. philippinensis and searching for possible candidates for the treatment of stroke should be performed according to our previous experimental results.7–9 In the present study, six new compounds 1–6 were characterized through spectroscopic and spectrometric analyses and the structural elucidations are discussed below.Results and discussion
The powdered stems of M. philippinensis were reflux with methanol, and combined extracts were concentrated to produce the methanol extract. Processed with liquid–liquid partition and continuous conventional chromatographic techniques combination, totally thirty-eight compounds were characterized and among them compounds 1–6 were new (Fig. 1). Other known compounds were identified by comparison of their experimental data with those in the literature, including seven neolignans, cinnamophilin (7),17 meso-dihydroguaiaretic acid (8),21,22 dehydroguaiaretic acid (9),23 oleiferin D,24 lyoniresol,25 lyoniside,26 and 9,9′-O-diferuloyl-seco-isolarici-resinol;27 three catechins, (+)-catechin,28 (−)-epicatechin,28 and (+)-epicatechin;29 three coumarins, scopoletin,30 isofraxidin,31 and scopolin;32 ten benzenoids, methyl syringate,33 vanillic acid,34 syringic acid,34 3,4,5-trimethoxyphenyl-β-D-glucopyranoside,35 5,6-dimeth-oxybenzene-1,3-diol,36 4′-hydroxy-2,3-dihydrocinnamic acid penta-cosyl ester,37 (E)-ferulic acid octacosyl ester,38 methyl-4-hydroxybenzoate,39 homovanillyl alcohol,40 and 2,4,6-trimethoxy-phenol;41 two amides, N-trans-feruloyltyramine42 and N-cis-feruloyltyramine;43 three isoquinolines: tetrahydroisoquinolinone,44 6,7-dimethoxyisoquinoline,45 and thalifoline;46 two sesquiterpenes, grasshopper ketone47 and sesquichamaenol;48 and two steroids, β-sitosterol and β-sitosteryl glucoside,49 respectively. | ||
| Fig. 1 Chemical structures of new compounds 1–6 and 7. | ||
The pseudomolecular formula of compound 1 was assigned as C40H46O10Na by HR-ESI-MS analysis which showed an ion adduct peak at m/z 709.2992. Its UV absorption maxima (λmax) were at 280 and 227 nm and IR absorption bands at 3393 and 1665 cm−1 were consistent with the presence of 7-oxo-8,8′-neolignan basic skeleton.50 The 1H NMR spectrum (Table 1) revealed one set of ABX-type tri-substituted aromatic ring at δ 7.42 (br s), 7.28 (d, J = 8.4 Hz), and 6.83 (d, J = 8.4 Hz); one set of 1,3,4,5-tetrasubstituted aromatic ring at δ 6.73 (br s) and 6.70 (br s); two methoxy groups at δ 3.92 and 3.86; and one aliphatic C6 fragment evidenced by COSY spectral analysis at δ 3.40 (m), 2.57 (dd, J = 13.5, 7.4 Hz), 2.51 (dd, J = 13.5, 7.4 Hz), 2.27 (m), 1.14 (d, J = 7.8 Hz), 0.88 (d, J = 6.6 Hz), respectively. The 2J, 3J-HMBC correlations from H-2 (δ 7.42) and H-6 (δ 7.28) to carbonyl C-7 (δ 202.9) suggested the tri-substituted aromatic ring was attached to C-7. The NOESY spectral analysis also exhibited NOE correlations between H-2 (δ 7.42) and OCH3-3 (δ 3.92), and between H-2′ (δ 6.73) and OCH3-3′ (δ 3.86), confirmed the locations of methoxy groups. The ECD spectrum which displayed a positive Cotton effect at 277 nm and a negative Cotton effect at 235 nm indicated the 8R and 8′S configurations.17 On the basis of above elucidations, the structure of compound 1 was very similar with cinnamophilin (7).17 Since the molecular formula C40H46O10 revealed the occurrence of two units of 7 (C20H24O5) and the minor spectral difference observed in 1 was disappearance of H-5′, the chemical structure of 1 was established as shown (Fig. 1) and the full assignments of 1H and 13C NMR signals (Tables 1 and 2) were determined from the NOESY and HMBC spectral analyses (Fig. 2). Conclusively, compound 1 was named trivially as 5,5′-biscinnamophilin.
| Position | 1a | 2b | 3c | 4d | 5a |
|---|---|---|---|---|---|
| a 1H NMR data (δ) were measured in chloroform-d at 300 MHz.b 1H NMR data (δ) were measured in chloroform-d at 400 MHz.c 1H NMR data (δ) were measured in acetone-d6 at 400 MHz.d 1H NMR data (δ) were measured in acetone-d6 at 300 MHz. | |||||
| 1 | — | — | — | 6.82 (br d, 7.5) | — |
| 2 | 7.42 (s) | 6.74 (s) | 7.21 (s) | 6.69 (d, 7.5) | 6.66 (s) |
| 3 | — | — | — | — | — |
| 4 | — | — | — | — | — |
| 5 | 6.83 (d, 8.4) | 6.90 (s) | 7.31 (s) | 6.99 (d, 1.8) | 7.47 (s) |
| 6 | 7.28 (d, 8.4) | — | — | — | — |
| 7 | — | 3.02 (dd, 13.6, 7.2) | 10.39 (s) | 3.38 (m) | 6.45 (s) |
| 2.60 (dd, 13.6, 7.2) | |||||
| 8 | 3.40 (m) | 2.90 (tq, 7.2, 6.8) | — | 1.76 (m) | — |
| 9 | 1.14 (d, 7.8) | 1.03 (d, 6.8) | 2.03 (s) | 0.72 (d, 6.9) | 1.77 (s) |
| 1′ | — | — | — | — | — |
| 2′ | 6.73 (s) | 7.52 (d, 2.0) | 7.10 (d, 2.0) | 7.03 (d, 1.8) | 6.75 (d, 1.2) |
| 3′ | — | — | — | — | — |
| 4′ | — | — | — | — | — |
| 5′ | — | 6.89 (d, 8.0) | 6.73 (d, 8.4) | 6.69 (d, 7.5) | 6.73 (d, 7.6) |
| 6′ | 6.70 (s) | 7.21 (dd, 8.0, 2.0) | 6.92 (dd, 8.4, 2.0) | 6.82 (br d, 7.5) | 6.77 (dd, 7.6, 1.2) |
| 7′ | 2.57 (dd, 13.5, 7.4) | — | 5.18 (d, 7.2) | 3.57 (d, 12.0) | — |
| 2.51 (dd, 13.5, 7.4) | |||||
| 8′ | 2.27 (m) | — | 3.74 (m) | 2.69 (m) | — |
| 9′ | 0.88 (d, 6.6) | 2.03 (s) | 1.03 (d, 6.8) | 0.65 (d, 6.7) | 1.67 (s) |
| OCH3-3 | 3.92 (s) | 3.96 (s) | 3.98 (s) | 3.83 (s) | 3.96 (s) |
| OCH3-3′ | 3.86 (s) | 3.93 (s) | 3.81 (s) | 3.83 (s) | 3.75 (s) |
| OH | 6.06, 6.11 | 5.54, 6.10 | 7.45, 8.05 | 7.24, 7.26 | — |
| 1a | 2b | 3c | 4d | 5b | |
|---|---|---|---|---|---|
| a 13C NMR data (δ) were measured in chloroform-d at 75 MHz.b 13C NMR data (δ) were measured in chloroform-d at 100 MHz.c 13C NMR data (δ) were measured in acetone-d6 at 100 MHz.d 13C NMR data (δ) were measured in acetone-d6 at 75 MHz. | |||||
| 1 | 129.5 | 131.3 | 128.0 | 121.2 | 133.3 |
| 2 | 110.3 | 113.4 | 116.9 | 115.1 | 107.7 |
| 3 | 146.7 | 146.6 | 153.2 | 148.1 | 151.9 |
| 4 | 150.1 | 142.9 | 145.8 | 145.4 | 144.9 |
| 5 | 113.7 | 115.8 | 111.0 | 112.3 | 112.8 |
| 6 | 123.3 | 132.1 | 141.1 | 138.3 | 121.2 |
| 7 | 202.9 | 36.2 | 190.9 | 66.9 | 121.4 |
| 8 | 42.8 | 48.9 | 211.6 | 36.6 | 145.4 |
| 9 | 11.6 | 16.5 | 28.8 | 9.9 | 19.8 |
| 1′ | 132.5 | 130.8 | 134.9 | 137.6 | 132.6 |
| 2′ | 111.3 | 111.2 | 113.0 | 112.2 | 109.3 |
| 3′ | 147.1 | 147.6 | 148.3 | 148.1 | 146.5 |
| 4′ | 141.0 | 150.4 | 146.1 | 145.4 | 144.7 |
| 5′ | 124.3 | 113.6 | 115.8 | 115.1 | 114.2 |
| 6′ | 123.8 | 126.5 | 121.0 | 121.4 | 119.5 |
| 7′ | 41.5 | 196.3 | 46.6 | 56.7 | 200.6 |
| 8′ | 37.7 | 212.6 | 52.0 | 36.6 | 55.0 |
| 9′ | 15.5 | 29.1 | 16.8 | 12.1 | 22.0 |
| OCH3-3 | 56.0 | 56.1 | 56.5 | 56.2 | 56.0 |
| OCH3-3′ | 56.1 | 56.2 | 56.3 | 56.2 | 55.8 |
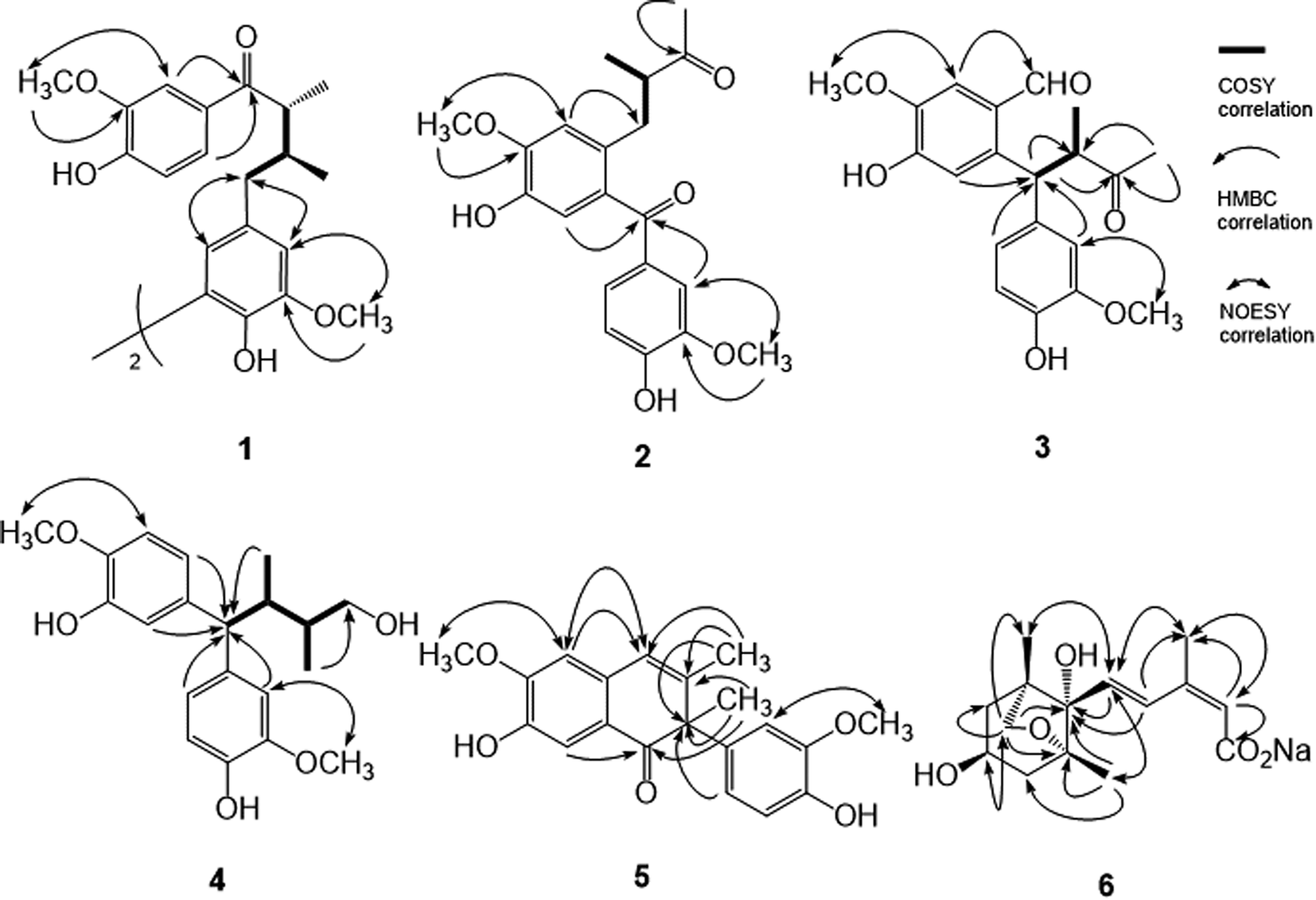 | ||
| Fig. 2 Significant COSY, HMBC and NOESY correlations of 1–6. | ||
The UV, IR, 1H, and 13C NMR spectral characteristics of 2–5 were similar to those of 1. It indicated those compounds also to be lignan derivatives. Both machilupins A (2) and B (3) were obtained as colorless syrup and their molecular formula were determined as C20H22O6 by HREIMS analysis. The 1H NMR spectrum of 2 (Table 1) exhibited one set of ABX-type mutually coupled protons at δ 7.52 (1H, d, J = 2.0 Hz), 7.21 (1H, dd, J = 8.0, 2.0 Hz), and 6.89 (1H, d, J = 8.0 Hz); two aromatic singlets at δ 6.90 and 6.74; and two methoxy singlets at δ 3.96 and 3.93, respectively. In addition, one aliphatic 2-methylbutan-3-one fragment [CHCH(CH3)COCH3] displayed proton signals at δ 3.02 (1H, dd, J = 13.6, 7.2 Hz), 2.90 (1H, tq, J = 7.2, 6.8 Hz), 2.60 (1H, dd, J = 13.6, 7.2 Hz), 2.03 (3H, s), and 1.03 (3H, d, J = 6.8 Hz), and its arrangement was evidenced by COSY and HMBC analysis. In the HMBC analysis, there were correlations from H-2 to C-7, from H-5 to C-7′, from H-2′ to C-7′, and from CH3-9′ to C-8′. The NOESY spectrum also showed NOE correlations of H-2/OCH3-3 and H-2′/OCH3-3′. Therefore, the chemical structure of 2 was constructed as shown (Fig. 1). The 13C spectrum of 3 (Table 2) combined with HSQC NMR analysis revealed 20 signals, of which were two carbonyls at δ 211.6 and 190.9, twelve aromatic carbons, two methyls (δ 28.8 and 16.8), and two methoxy groups (δ 56.5 and 56.3), respectively. Not only the same molecular formula, but the 1H NMR spectral signals of 3 were also similar to those of 2, including three ABX-type mutually coupled protons, two aromatic singlets, two methoxy singlets, and one 2-methylbutan-3-one aliphatic fragment. The observed difference was one more formyl group δ 10.39 (1H, s) in 3. The HMBC spectrum of 3 displayed correlations from H-2 to C-7, from H-5 to C-7′, from H-2′ to C-7′, from H-6′ to C-7′, from H-7′ to C-8 and C-8′, and from CH3-9′ to C-8 and C-8′, respectively. NOESY analysis of 3 determined the methoxy function substitutions and finally furnished the planar structure assignment of 3 (Fig. 1).
Machilupin C (4) was afforded as optically active colorless powder with a molecular formula of C20H26O5 determined by HREIMS analysis. Proton signals including two sets of ABX-type mutually coupled protons at δ 7.03 (1H, d, J = 1.8 Hz), 6.99 (1H, d, J = 1.8 Hz), 6.82 (2H, br d, J = 7.5 Hz), and 6.69 (2H, d, J = 7.5 Hz); two methoxy singlets at δ 3.83; and one 2,3-dimethylbutan-4-ol aliphatic fragment [CHCH(CH3)CH(CH3)CH2OH] evidenced by COSY analysis at δ 3.57 (1H, d, J = 12.0 Hz), 3.38 (2H, m), 2.69 (1H, m), 1.76 (1H, m), 0.72 (3H, d, J = 6.9 Hz), and 0.65 (3H, d, J = 6.7 Hz), were appeared in the 1H NMR spectrum of 4. Analysis of its HMBC spectrum (Fig. 2) revealed correlations from H-1 to C-7′, from H-5 to C-7′, from H-2′ to C-7′, and from H-6′ to C-7′ determined that the aliphatic moiety was connected through C-7′ with C-6 and C-1′ of the aromatic rings. The NOESY spectrum exhibited NOE correlations of H-2/OCH3-3 and H-2′/OCH3-3′ determined the locations of methoxy groups. The ECD spectrum of 4 exhibited a positive Cotton effect at 276 nm and a negative Cotton effect at 235 nm also indicated the 8R and 8′S configurations. Consequently, the chemical structure of 4 was established as shown (Fig. 1).
Machilutone A (5) was obtained as colorless syrup with the pseudomolecular formula of C20H20O5Na determined by HRESIMS ion peak at m/z 363.1206, representing eleven unsaturation equivalents which was one more than that of 2. Although the 1H NMR spectrum of 5 also showed one set of ABX-type mutually coupled protons at δ 6.77 (1H, dd, J = 7.6, 1.2 Hz), 6.75 (1H, d, J = 1.2 Hz), and 6.73 (1H, d, J = 7.6 Hz); two aromatic singlets at δ 7.47 and 6.66; and two methoxy singlets at δ 3.96 and 3.75, respectively, some more spectral differences would be observed. One more aromatic singlet at δ 6.45 and two methyl singlets at δ 1.77 and 1.67 appeared in its 1H NMR spectrum. However, there were not any signals representing the aliphatic moieties commonly found in 2–4. These NMR spectral and mass spectrometric information suggested the change of the side chain in 5. Analysis of its HMBC spectrum (Fig. 2) displayed the correlations from H-2 to C-7, from H-5 to C-7′, from CH3-9 to C-7, C-8, and C-8′, from H-2′ to C-8′, from H-6′ to C-8′, and from CH3-9′ to C-8, C-7′, and C-8′, respectively. NOESY analysis also located the methoxy groups at C-3 and C-3′. Therefore, all the spectral evidences supported the structure of 5 as shown (Fig. 1) and the full assignments of 1H and 13C NMR signals were completed by the assistance of 2D NMR experiments. The chemical structure of 5 was characterized as unprecedented carbon skeletons from the natural sources.
Machilusoxide A (6) was isolated as optically active colorless syrup. The HRESIMS analytical result of 6 exhibited a pseudo-molecular ion peak at m/z 281.1382. The UV spectrum showed the absorption maxima at 273 and 249 nm. The IR spectrum indicated the presence of hydroxyl (3507 cm−1) and carbonyl groups (1713 cm−1). The 1H-NMR spectrum exhibited signals for one vinyl proton at δ 5.85 (1H, s), two trans olefinic protons at δ 7.70 (1H, d, J = 16.2 Hz) and 6.23 (1H, d, J = 16.2 Hz), three methyl singlets at δ 1.98, 1.15, and 0.92, one oxymethine at δ 4.09 (1H, tdd, J = 10.8, 6.6, 3.3 Hz), two sets of methylene protons at δ 2.04 (2H, m) and 1.83 (2H, m), respectively. The 13C-NMR spectrum exhibited a carboxylic acid group at δ 175.4; four olefinic carbons at δ 139.7, 132.6, 129.5, and 128.1; and four oxygenated carbons at δ 87.2, 82.7, 76.8, and 63.7. The above spectral data evidenced the structure of 6 was similar to dihydrophaseic acid,51 an abscisic acid (ABA) derivative commonly occurred in the phytohormones, which played the key role in biotic and abiotic stress responses.52 The substitution pattern was further confirmed through the HMBC experiment, in which displayed the 2J, 3J-HMBC correlations from H-2 to C-1 and C-6; from H-4 to C-1′ and CH3-6; from H-5 to C-1′; from CH3-6 to C-2 and C-4; from CH3-7′ to C-1′, C-2′, and C-3′; and from CH2-8′ to C-1′, C-2′, C-5′, C-6′, and CH3-9′, respectively. The relative stereochemistry was determined by the NOESY analysis which revealed NOE correlations of H-2/CH3-6, H-5/CH3-6, H-5/CH3-7′, H-5/CH3-9′, and H-4′/CH2-8′ (Fig. 2). Therefore, the side chain at C-1′, the hydroxyl groups at C-4′, and methyl substituent at C-2′ and C-6′ all were in cis configuration. The C-2/C-3 carbon–carbon double bond was in Z and the C-2-C-5 fragment was in S-trans configurations, respectively. The presence of 6 as a sodium salt was evidenced by the ICPMS analytical data, in which [Na+] was equal to 1 ppm in the 10 ppm sample. Comparison of the spectral data of 6 with that of dihydrophaseic acid,51 upfield shift of H-4 (δ 7.96 to 7.70) further supported the occurrence of sodium carboxylate. After acidification of 6, the resulted sample produced totally the same 1H-NMR spectrum as that of dihydrophaseic acid.51 Conclusively, the structure of 6 was assigned as sodium dihydrophaseate based on the above-mentioned elucidations.
Dehydroguaiaretic acid (9) was obtained as colorless needles. The 1H-NMR spectrum exhibited signals for two aromatic singlets at δ 7.50 and 7.06; one set of ABX system coupled protons at δ 7.03 (1H, d, J = 8.4 Hz), 6.74 (1H, dd, J = 8.4, 2.0 Hz), and 6.72 (1H, d, J = 2.0 Hz); two methoxy groups at δ 4.00 (3H, s) and 3.86 (3H, s); and two methyl singlets at δ 2.45 (3H) and 2.13 (3H). These spectral data were coincided well with those reported for cinnamophilin A (10).53 However, with careful evaluation of all the 1H and 13C NMR data (Table 3), it should be revised as dehydroguaiaretic acid (9) which had already been published in the literature.23,54
|
|||
|---|---|---|---|
| Position | Dehydroguaiaretic acid (9) | Cinnamophilin A (10) | |
| δC | δH (multi., J in Hz) | δH (multi., J in Hz) | |
| 1 | 138.0 | — | — |
| 2 | 133.4 | — | — |
| 3 | 132.6 | — | — |
| 4 | 126.7 | 7.50 (s) | 7.49 (s) |
| 4a | 129.0 | — | — |
| 5 | 115.2 | 7.06 (s) | 7.05 (s) |
| 6 | 147.5 | — | — |
| 7 | 145.6 | — | — |
| 8 | 109.5 | 6.82 (s) | 6.81 (s) |
| 8a | 128.3 | — | — |
| 9 | 18.3 | 2.13 (s) | 2.11 (s) |
| 10 | 21.9 | 2.45 (s) | 2.45 (s) |
| 1′ | 134.1 | — | — |
| 2′ | 123.9 | 6.72 (d, 2.0) | 6.73 (d, 1.6) |
| 3′ | 147.3 | — | — |
| 4′ | 145.3 | — | — |
| 5′ | 105.9 | 7.03 (d, 8.4) | 7.02 (d, 8.0) |
| 6′ | 113.5 | 6.74 (dd, 8.4, 2.0) | 6.72 (dd, 8.0, 1.6) |
| OCH3-6 | 56.7 | 4.00 (s) | 4.00 (s) |
| OCH3-3′ | 56.8 | 3.86 (s) | 3.85 (s) |
The plausible biosynthetic mechanism of above-mentioned new lignans was proposed as shown (Fig. 3). Cyclization of the dibenzylbutane type lignan meso-dihydroguaiaretic acid (8) resulted in a aryltetralin intermediate 11. Successive 7′,8′-cleavage of 11 and further oxidation of C-7′ and C-8′ in 12 yielded machilupin A (2). In comparison, 7,8-cleavage of the cyclic intermediate 11 and further oxidation of C-7 and C-8 in 13 produced machilupin B (3). 1,7-Cleavage of aryltetralin intermediate 11 and hydroxylation of the terminal alkene in 14 afforded machilupin C (4). Machilutone A (5) was resulted from the phenyl migration from C-7′ to C-8′ of cyclic intermediate 11 and further oxidation on C-7′ and C-7/C-8 of 15.
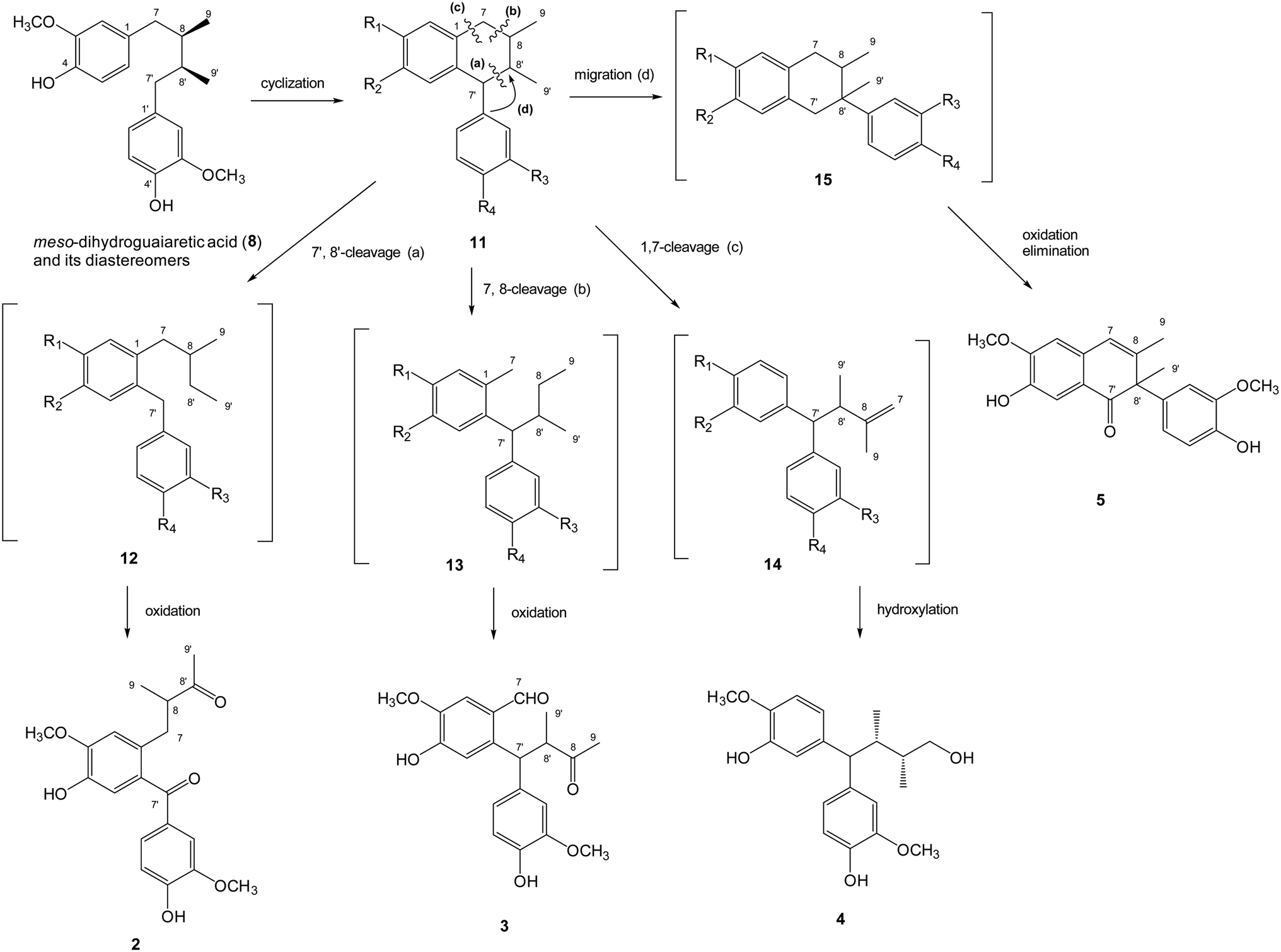 | ||
| Fig. 3 The plausible biosynthetic mechanism of new lignans 2–5. | ||
Among these isolated compounds, several lignans exhibited the significant contents in the stem extracts of M. philippinensis, such as cinnamophilin (7) and meso-dihydroguaiaretic acid (8). In the previous reports, cinnamophilin (7) exhibited the ability to block Na+ and Ca2+ inward currents in rat cardiac cells, and also been demonstrated to protect against ischemic-reperfusion injury of muscle in vivo.7 In addition, it also offered prolonged neuro-protection against gray and white matter damage and improved functional and electrophysiological outcomes after transient focal cerebral ischemia.9 To further evaluate the neuroprotective potential of cinnamophilin (7), its bioactivity was examined in the primary cortical neurons culture by glutamate-induced oxidative stress assay.55 Both the trypan blue stain and lactate dehydrogenase release data displayed that 7 was effective against the glutamate induced excitotoxicity, with the IC50 values of 4.3 ± 2.0 and 3.5 ± 1.7 μM, respectively (Fig. 4–7). These experimental results supported the further development of the stem extracts of M. philippinensis and related principles as the natural neuroprotective lead compounds.
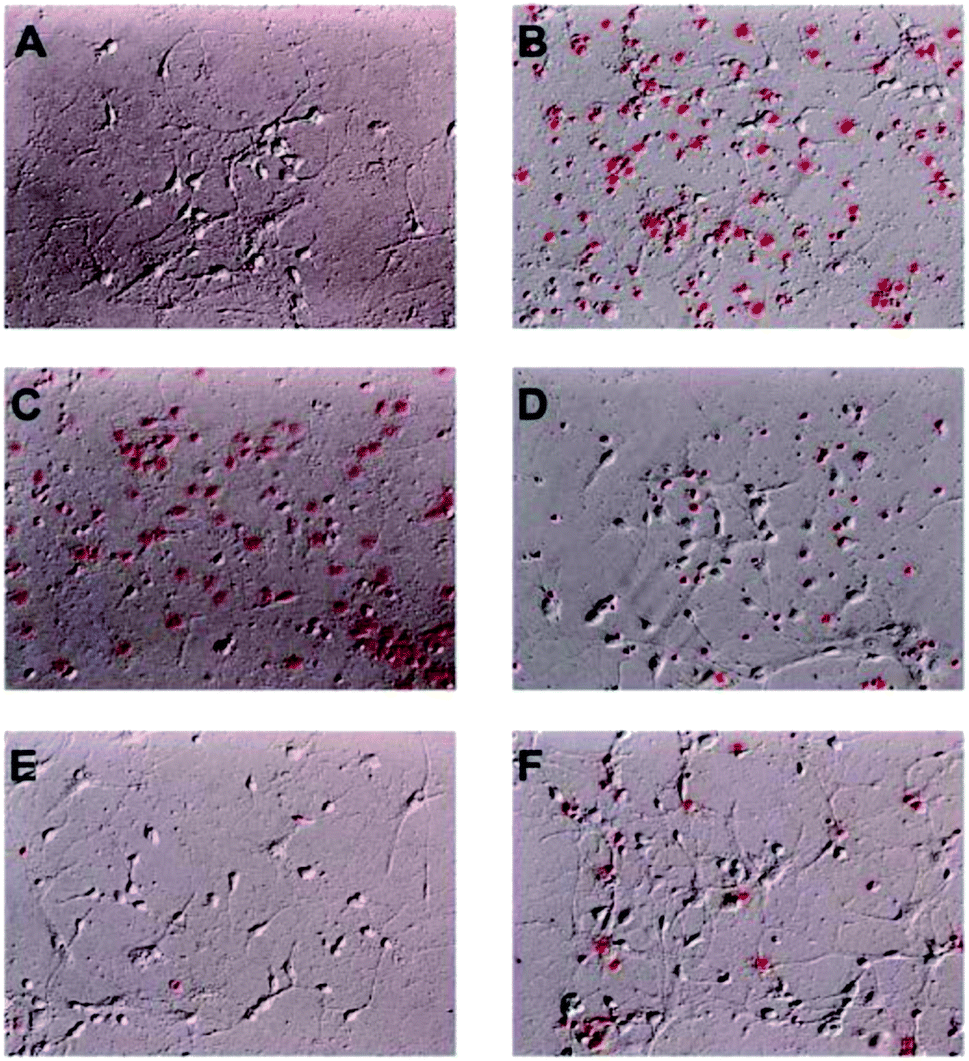 | ||
| Fig. 4 Cinnamophilin (7) reduced the glutamate-stimulated neuronal death in the concentration-dependent manner. PI uptake in cell culture was shown. (A) Primary cortical neurons treated DMSO 20 min then replaced with fresh medium as control. (B) Primary cortical neurons treated DMSO, (C) 1, (D) 3, (E) 10 μM 7 and (F) 20 μM MK-801, after 20 min added 300 μM glutamate for 6 h, n = 6. | ||
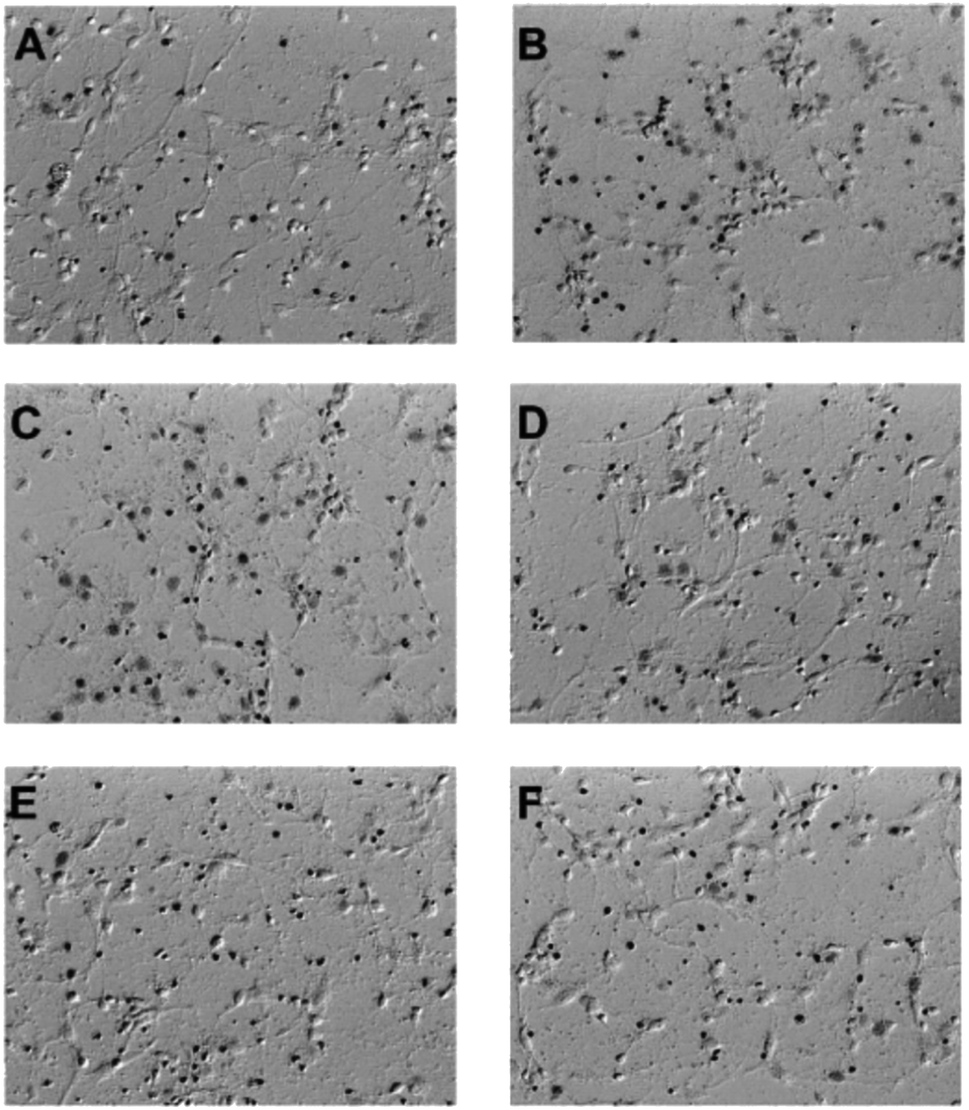 | ||
| Fig. 5 Cinnamophilin (7) reduced the glutamate-stimulated neuronal death in the concentration-dependent manner. Trypan blue uptake in cell culture was shown. (A) Primary cortical neurons treated DMSO 20 min then replaced with fresh medium as control. (B) Primary cortical neurons treated DMSO, (C) 1, (D) 3, (E) 10 μM 7 and (F) 20 μM MK-801, after 20 min added 300 μM glutamate for 6 h, n = 4. | ||
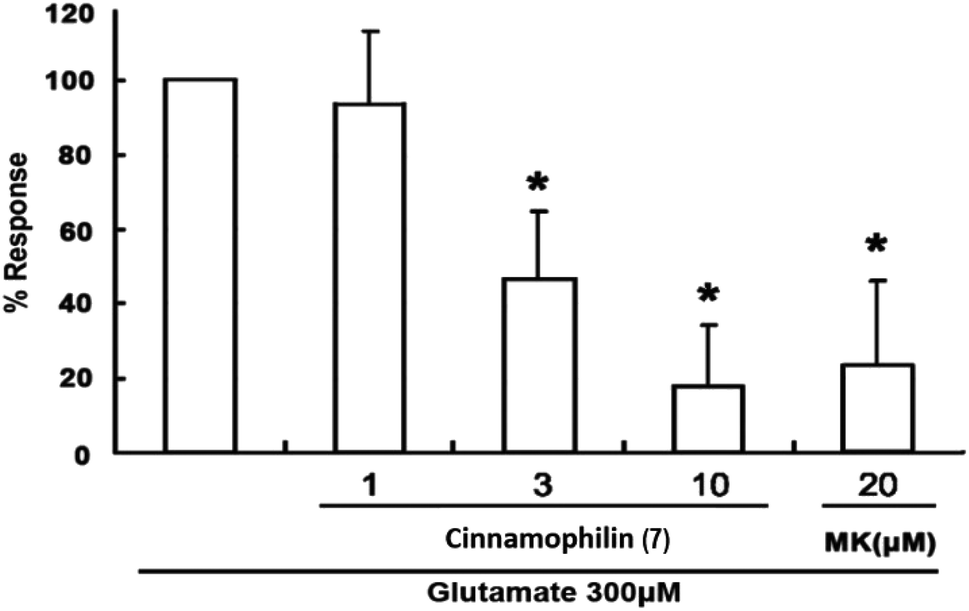 | ||
| Fig. 6 Cinnamophilin (7) reduced the glutamate-stimulated neuronal death in the concentration-dependent manner. Trypan blue in cell released out by treating 1% Triton X-100 and measured the absorbance at 600 nm. Data are expressed as percentage of control optical density (OD) values. Bars represent the mean ± SD, n = 4. *Significantly different from activated control cultures (one-way ANOVA followed by LSD test, P < 0.05). | ||
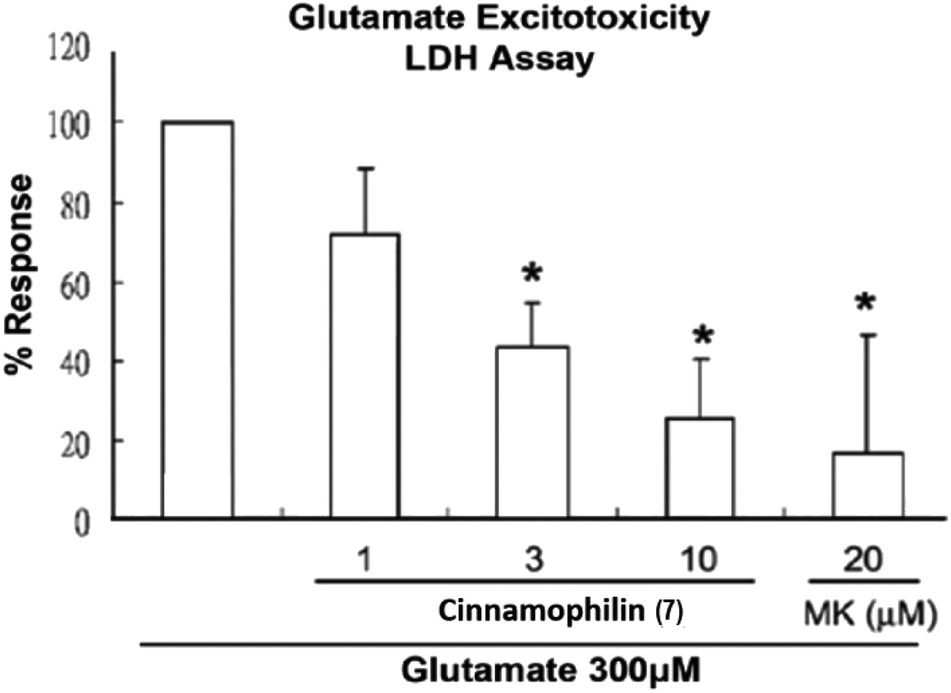 | ||
| Fig. 7 Cinnamophilin (7) reduced the glutamate-stimulated neuronal death in the concentration-dependent manner. After primary cortical neurons in culture were treated 7 (1, 3 and 10 μM) or MK-801 (20 μM) and all were exposed to glutamate (300 μM), culture medium was removed and then sampled for LDH by measuring the absorbance at 490 nm. Data are expressed as percentage of control optical density (OD) values. Bars represent the mean ± SD, n = 6. *Significantly different from activated control cultures (one-way ANOVA followed by LSD test, P < 0.05). | ||
Experimental
General experimental procedures
Melting points were determined using Yanagimoto MP-S3 micro melting point apparatus without correction. Optical rotations were measured using a JASCO DIP-370 digital polarimeter. UV spectra were examined at room temperature on a Hitachi UV-3210 spectrophotometer. IR spectra were obtained with a Shimadzu FTIR Prestige-21 spectrophotometer. 1H and 13C NMR spectra were recorded on the Bruker Advance 300 and AMX-400 NMR spectrometers. Chemical shifts are shown in δ values (ppm) with tetramethylsilane as an internal standard. The EIMS and HREIMS were measured on a VG 70-250S mass spectrometer, and the ESIMS and HRESIMS were taken on a Bruker APEX II FT-MS spectrometer (positive-ion mode). Inductively coupled plasma (ICP) MS was recorded on a Thermo-Element XR ICPMS spectrometer, and three elements (Na, K, Ca) were examined for their concentration by comparison with the standard curves. ECD spectra were obtained on a JASCO J-720 spectrometer. X-ray single crystal diffraction was measured in National Chung-Hsing University with a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector system equipped with a Cu Incoatec IμS microfocus source (λ = 1.54178 Å). Column chromatography (CC) was performed on silica (70–230 mesh and 230–400 mesh, Merck) and Diaion HP-20 (Mitsubishi) gels. Preparative thin-layer chromatography (pTLC) was conducted on Merck precoated silica gel 60 F254 plates, using UV light to visualize the spots. High-performance liquid chromatography (HPLC) was performed on a Shimadzu LC-20AT series pumping system equipped with a Shimadzu SPD-20A UV-Vis detector, and a SIL-10AF autosampling system at ambient temperature and a RP-18 column (Ascentis C18, 5 μm, 10 mm × 25 cm).Plant material
The stems of M. philippinensis Merr. was collected in Pingtung County, Taiwan, in July 2005 and identified by Prof. Chang-Sheng Kuoh, Department of Life Science, National Cheng Kung University (NCKU), Tainan, Taiwan. A voucher specimen (TSWu 200507001) has been deposited in the Herbarium of School of Pharmacy, NCKU.Extraction and isolation
The dried stems of M. philippinensis (34 kg) were powdered and consecutively extracted with methanol. The methanol was removed and evaporated in vacuo to yield dried extract (1.9 kg). The syrup was suspended in water and then extracted successively with chloroform and n-butanol to produce the chloroform (600 g), n-butanol (B, 500 g), and water (800 g) layers, respectively. The chloroform layer was further acidified by 2% HCl(aq) and the combined organic solvent was concentrated to afford non-alkaloid chloroform fraction (NC, 570 g). The acidic hydrophilic layer was neutralized by ammonia water and the pH value was adjusted to about 10. Again this aqueous solution was extracted by chloroform to yield alkaloid chloroform fraction (AC, 10 g) and residue (10 g) which was combined with water fraction to form water solubles (W, 810 g). The resulted NC and AC layers were subjected to silica gel column chromatography (SiO2 CC) eluted with step-gradient n-hexane/ethyl acetate (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) and chloroform/methanol (9:1 to 1:1) to afford eleven (NC-1 to -11) and eight (AC-1 to -8) fractions. The n-butanol layer and water solubles were purified on Diaion HP-20 CC with step-gradient water/methanol (9:1 to 1:9) to give six (B-1 to -6) and six (W-1 to -6) fractions, respectively. Further purifications were performed on the fractions displayed significant spots based on the TLC profiles.
:1 to 1:1) and chloroform/methanol (9:1 to 1:1) to afford eleven (NC-1 to -11) and eight (AC-1 to -8) fractions. The n-butanol layer and water solubles were purified on Diaion HP-20 CC with step-gradient water/methanol (9:1 to 1:9) to give six (B-1 to -6) and six (W-1 to -6) fractions, respectively. Further purifications were performed on the fractions displayed significant spots based on the TLC profiles.
Fractions NC-4, -5, and -6 were isolated by SiO2 CC eluted with a solvent mixture of n-hexane/ethyl acetate (15:1 to 1:1) and further recrystallization of the resulting subfractions of NC-4 and -5 afforded 4′-hydroxy-2,3-dihydrocinnamic acid pentacosyl ester (4.2 g). Subfraction NC-6-1 was recrystallized with chloroform/methanol to produce β-sitosterol (3.68 g). NC-6-2 was resolved on SiO2 CC eluted with a step gradient mixture of n-hexane/acetone (100:1 to 1:1) and further purification of the resulting minor fractions with pTLC to yield oleiferin D (0.6 mg), methyl-4-hydroxybenzoate (0.4 mg), and sesquichamaenol (6.2 mg). Fraction NC-7 was purified by SiO2 CC eluted with a solvent mixture of n-hexane/acetone (5:1 to 1:1) to afford five subfractions. NC-7-2 was recrystallized with chloroform/acetone to produce dehydroguaiaretic acid (9) (1.24 g). NC-7-4 was isolated on SiO2 CC eluted with a step gradient mixture of n-hexane/acetone (10:1 to 1:1) and the resulting minor fractions were resolved by pTLC eluted with solvent mixtures of n-hexane/ethyl acetate (3:1) and n-hexane/acetone (3:1) to afford methyl syringate (0.7 mg), machilupin A (2) (2.6 mg), machilupin B (3) (3.8 mg), respectively. Fractions NC-8 and -9 were isolated by SiO2 CC eluted with a solvent mixture of n-hexane/ethyl acetate (3:1 to 1:1) and further resolved by recrystallization, repeated SiO2 CC, and pTLC for the resulting subfractions produced cinnamophilin (7) (70.9 g), (E)-ferulic acid octacosyl ester (7.6 mg), 5,5′-biscinnamophilin (1) (2.2 mg), scopoletin (4.3 mg), and isofraxidin (2.4 mg), respectively. Fraction NC-10 was carried out SiO2 CC purification eluted with a solvent mixture of chloroform/methanol (19:1 to 1:1) to afford five subfractions. NC-10-2 was recrystallized with chloroform/acetone to yield 9,9′-O-diferuloyl-seco-isolariciresinol (275.5 mg). NC-10-3 was isolated by SiO2 CC eluted with a solvent mixture of chloroform/methanol (9:1 to 1:1) and further resolved by pTLC eluted with chloroform/methanol (6:1) to result in machilupin C (4) (4.5 mg), N-trans-feruloyltyramine (0.8 mg), and N-cis-feruloyltyramine (0.6 mg). Recrystallization of the last fraction NC-11 afforded β-sitosteryl-3-O-β-glucopyranoside (2.7 g).
Most of the alkaloid chloroform fractions (AC-2 to -7) were further separated by SiO2 CC. Fractions AC-2 and -3 were eluted by n-hexane/acetone (6:1) and further resolved by recrystallization of the resulting subfractions yielded meso-dihydroguaiaretic acid (8) (6.2 mg). Fraction AC-4 was eluted by n-hexane/acetone (5:1) to afford five subfractions. AC-4-2 was further resolved by repeated SiO2 CC and pTLC to produce machilutone A (5) (3.1 mg). AC-4-4 was purified by pTLC to afford tetrahydroisoquinolinone (2.4 mg), thalifoline (0.2 mg), and 6,7-dimethoxyisoquinoline (1.2 mg). Fractions AC-5, -6, and -7 were combined and separated by SiO2 CC eluted with a solvent mixture of chloroform/methanol (19:1). Recrystallization of the resulting subfractions afforded homovanillyl alcohol (3.5 mg).
The polar fractions B-1 and -2 were combined and applied to a reverse-phase Sephadex LH-20 column eluted with a step gradient of water and methanol (10:0, 7:3, 5:5, 3:7, 0:10) to result in several subfractions. Further resolved by repeated Sephadex LH-20 CC and recrystallization of the minor fractions produced (+)-catechin (4.0 mg), (−)-epicatechin (1.3 mg), and (+)-epicatechin (0.7 mg). Fraction B-3 was separated by SiO2 CC eluted with a solvent mixture of chloroform/methanol (5:1 to 1:1) and yielded five subfractions. Subfraction B-3-2 was further resolved by SiO2 CC eluted with chloroform/methanol (5:1) solvent mixture and recrystallization of the minor fractions yielded (−)-epicatechin (6.4 mg), scopolin (2.1 mg), and 5,6-dimethoxybenzene-1,3-diol (2.4 mg). Subfractions B-3-3 and B-3-4 were purified by pTLC eluted with chloroform/methanol (5:1) to afford vanillic acid (3.6 mg), syringic acid (5.5 mg); and grasshopper ketone (5.7 mg), respectively. B-3-4 was further isolated by SiO2 CC eluted with chloroform/methanol (5:1) and pTLC separation of the resulting minor fractions yielded machilusoxide A (6) (2.3 mg). Fractions B-4, B-5, and B-6 was respectively separated by SiO2 CC eluted with a solvent mixture of chloroform/methanol (9:1 to 1:1) and further CC and recrystallization of the subfractions produced totally lyoniside (8.6 mg), 2,4,6-trimethoxyphenol (17.2 mg), lyoniresol (9.2 mg), 3,4,5-trimethoxyphenyl-β-D-glucopyranoside (7.2 mg).
The water soluble fractions W-1 and -2 were combined and applied to a reverse-phase Sephadex LH-20 column eluted with a step gradient of water and methanol (10:0, 7:3, 5:5, 3:7, 0:10) to result in several subfractions. Recrystallization of the subfractions yielded vanillic acid (0.8 mg) and syringic acid (1.2 mg). Fractions W-3 and -4 were purified by SiO2 CC eluted with a solvent mixture of chloroform/methanol (6:1 to 1:1), and further CC and recrystallization of the minor fractions produced (−)-epicatechin (2.3 mg) and lyoniresol (1.8 mg). Fractions W-5 and -6 were subjected to SiO2 CC eluted with a solvent mixture of chloroform/methanol (9:1 to 1:1), and further recrystallization of the subfractions of W-5 generated grasshopper ketone (2.2 mg). Subfraction W-6-2 was isolated by SiO2 CC eluted with a solvent mixture of chloroform/methanol (9:1 to 1:1) and further pTLC purification of the minor fractions eluted with chloroform/methanol (5:1) afforded scopoletin (0.5 mg), lyoniresol (1.2 mg), and isofraxidin (1.1 mg).
ε) 280 (2.9), 227 (3.5) nm; CD (MeOH, c 8.33 × 10−4 M) (mol. CD) 277 (+20.3), 235 (−8.9), 217 (+15.7) nm; IR (KBr) νmax 3393, 2960, 2931, 1708, 1665, 1592 cm−1; 1H and 13C NMR, see Tables 1 and 2; ESIMS (rel. int.) m/z 709 ([M + Na]+, 100); HRESIMS m/z 709.2992 ([M + Na]+ calcd for C40H46O10Na, 709.2989).ε) 316 (3.2), 285 (3.2), 235 (3.5) nm; IR (KBr) νmax 3402, 2924, 2858, 1703, 1643, 1588, 1546 cm−1; 1H and 13C NMR, see Tables 1 and 2; EIMS (rel. int.) m/z 358 ([M]+, 17), 315 (36), 286 (100); HREIMS m/z 358.1414 ([M]+, calcd for C20H22O6, 358.1416).ε) 317 (2.8), 282 (3.2), 237 (3.5) nm; IR (KBr) νmax 3610, 1711, 1682, 1514, 1488 cm−1; 1H and 13C NMR, see Tables 1 and 2; EIMS (rel. int.) m/z 358 ([M]+, 34), 340 (20), 287 (100), 177 (12), 43 (21); HREIMS m/z 358.1416 ([M]+, calcd for C20H22O6, 358.1416).ε) 281 (2.6) nm; CD (MeOH, c 5.00 × 10−4 M) (mol. CD) 276 (+20.5), 235 (−10.5), 217 (+22.0) nm; IR (KBr) νmax 3447, 2961, 2928, 1607, 1513 cm−1; 1H and 13C NMR, see Tables 1 and 2; EIMS (rel. int.) m/z 346 ([M]+, 5), 259 (100); HREIMS m/z 346.1783 ([M]+, calcd for C20H26O5, 346.1780).ε) 281 (3.8), 259 (3.6) nm; IR (KBr) νmax 3396, 1660, 1598, 1511 cm−1; 1H and 13C NMR, see Tables 1 and 2; EIMS (rel. int.) m/z 340 ([M]+, 100), 325 (32), 309 (27), 293 (19), 120 (26); HRESIMS m/z 363.1206 ([M + Na]+, calcd for C20H20O5Na, 363.1208).ε) 273 (2.9), 249 (2.8) nm; IR (KBr) νmax 3509, 3003, 2938, 1713, 1422, 1362, 1224 cm−1; 1H-NMR (CD3OD, 300 MHz) δ 7.70 (1H, d, J = 16.2 Hz, H-4), 6.23 (1H, d, J = 16.2 Hz, H-5), 5.85 (1H, s, H-2), 4.09 (1H, tdd, J = 10.8, 6.6, 3.3 Hz, H-4′), 3.79 (1H, d, J = 6.4 Hz, H-8′), 3.68 (1H, d, J = 6.4 Hz, H-8′), 2.04 (1H, m, H-3′), 1.98 (3H, s, CH3-6), 1.83 (1H, m, H-5′), 1.15 (3H, s, CH3-9′), 0.92 (3H, s, CH3-7′); 13C-NMR (CD3OD, 75 MHz) δ 175.0 (C-1), 139.7 (C-3), 132.6 (C-4), 129.5 (C-5), 128.1 (C-2), 87.2 (C-2′), 82.7 (C-1′), 76.8 (C-8′), 65.6 (C-6′), 63.7 (C-4′), 45.4 (C-3′), 44.0 (C-5′), 19.9 (C-6), 19.1 (C-7′), 15.8 (C-9′); HRESIMS m/z 281.1382 ([M − Na]− calcd for C15H21O5, 281.1389).Primary cortical neuronal culture
Cultured cortical cells were collected from the cerebral cortices of Sprague-Dawley rats (1 day-old) under pentobarbital anesthesia according to the previous report.55 The cortices were kept in ice-cold Dulbecco's modified Eagle's medium (DMEM; GIBCO) and minced. Tissue chunks were then incubated in a papain solution (0.6 mg mL−1 papain and DNase I in HBSS) at 37 °C for 30 min to dissociate the cells, and the reaction was stopped by addition of heat-inactivated horse serum. After the cell suspensions were centrifuged at 800 g, pellets were plated onto poly-D-lysine-coated Petri dishes. Dissociated cells were suspended in DMEM with 10% horse serum and incubated at 37 °C in a humidified incubator with 5% CO2. Three hours after plating, the culture medium was replaced by a serum-free neurobasal medium containing 25 μM glutamate, 0.5 mM L-glutamine, and 2% B27 supplement (17504-044; Invitrogen Corp., Carlsbad, CA, USA). Cultured cells were allowed to grow for approximately 7–14 days. These cultures contained 80% neurons and 20% astrocytes, as determined by immunohistochemistry using monoclonal antibodies against microtubule-associated protein-2 (MAP-2; Boehringer Mannheim, Mannheim, Germany) and glial fibrillary acidic protein (GFAP, Sigma-Aldrich).Glutamate-induced oxidative stress assay
For cytotoxicity studies, cells were plated onto poly-D-lysine-coated 24 well dishes at a density of 2 × 106 cells per well. All studies were initiated within 7–14 day after plating. Cinnamophilin (7) was dissolved in dimethylsulfoxide (DMSO) (final concentration < 0.1%) or MK-801 (20 μM) which is a glutamate receptor inhibitor56 was co-applied with L-glutamate to the cultures. Neuronal cultures were exposed to 300 μM glutamate (Sigma) in the absence or presence of compounds in a CO2-buffered incubator at 37 °C for 24 hours. Cell damage was assessed by fluorescent image analysis of propidium iodide (PI) uptake and by differential interference contrast (DIC) image analysis of trypan blue uptake (Sigma). After co-applied incubation for 24 hours, cells were washed with Earle's balanced salt solution three times, 1% 10 μg mL−1 PI and 4% trypan blue was added to the cultures and incubated for 20 min and 5 min. Trypan blue in cell released out by treating 1% Triton X-100 and measured the absorbance at 600 nm.Lactate dehydrogenase (LDH) release assay
Cell death was quantified by LDH levels in the extracellular medium and measured by using a LDH assay kit (Promega, Madison, WI, USA). Following the procedures provided by the vendor, the values of LDH were measured by the optical density (OD) taken at 490 nm in a plate reader (Stat Fax 2100; Awareness Technology, Inc.). Data are expressed as a value of the response%, i.e. a percentage relative to the values obtained from the medium of the glutamate-treated control neuronal cultures.Statistical analysis
The data were expressed as the mean ± standard error of the mean (S.E.M.). Paired Students' t test was used to evaluate the response to a change in conditions, and one-way analysis of variance (one-way ANOVA) with Fisher's protected least significant difference post hoc comparison was used to evaluate differences between groups. P < 0.05 was selected for statistical significance.Conclusions
In summary, one dimeric lignin biscinnamophilin (1), four lignans machilupins A-C (2–4) and machilutone A (5), along with one ionone machilusoxide A (6) are characterized with spectroscopic and spectrometric analysis. Although there were a few papers reported the chemical constituents from this species,57 the present report was the first study about its comprehensive chemical composition. The most abundant compound, cinnamophilin (7), was examined for its bioactivity in the primary cortical neurons culture by glutamate-induced oxidative stress assay for the first time. The experimental data evidenced the stem extracts of M. philippinensis and related principles as the potential neuroprotective drug candidates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is sponsored by the Ministry of Science and Technology (MOST), Taiwan.Notes and references
- B. M. Demaerschalk and T. R. Yip, Stroke, 2005, 36, 2500–2503 CrossRef.
- D. J. Gladstone, S. E. Black and A. M. Hakim, Stroke, 2002, 33, 2123–2136 CrossRef.
- B. Schaller, Free Radical Biol. Med., 2005, 38, 411–425 CrossRef CAS.
- S. M. Yu, F. N. Ko, T. S. Wu, J. Y. Lee and C. M. Teng, Eur. J. Pharmacol., 1994, 256, 85–91 CrossRef CAS.
- S. M. Yu, T. S. Wu and C. M. Teng, Br. J. Pharmacol., 1994, 111, 906–912 CrossRef CAS PubMed.
- G. Hsiao, C. M. Teng, J. R. Sheu, Y. W. Cheng, K. K. Lam, Y. M. Lee, T. S. Wu and M. H. Yen, Biochim. Biophys. Acta, 2001, 1525, 77–88 CrossRef CAS.
- E. J. Lee, H. Y. Chen, M. Y. Lee, T. Y. Chen, Y. S. Hsu, Y. L. Hu, G. L. Chang and T. S. Wu, Free Radical Biol. Med., 2005, 39, 495–510 CrossRef CAS PubMed.
- E. J. Lee, H. Y. Chen, Y. C. Hung, T. Y. Chen, M. Y. Lee, S. C. Yu, Y. H. Chen, I. C. Chuang and T. S. Wu, Exp. Neurol., 2009, 217, 74–83 CrossRef CAS.
- T. Y. Chen, S. H. Tai, E. J. Lee, C. C. Huang, A. C. Lee, S. Y. Huang and T. S. Wu, Crit. Care Med., 2011, 39, 1130–1137 CrossRef CAS.
- I. L. Tsai, J. H. Chen, C. Y. Duh and I. S. Chen, Planta Med., 2000, 66, 403–407 CrossRef CAS.
- I. L. Tsai, J. H. Chen, C. Y. Duh and I. S. Chen, Planta Med., 2001, 67, 559–561 CrossRef CAS.
- M. J. Cheng, B. Jayaprakasam, T. Ishikawa, H. Seki, I. L. Tsai, J. J. Wang and I. S. Chen, Helv. Chim. Acta, 2002, 85, 1909–1914 CrossRef CAS.
- G. Li, C. S. Lee, M. H. Woo, S. H. Lee, H. W. Chang and J. K. Son, Biol. Pharm. Bull., 2004, 27, 1147–1150 CrossRef CAS.
- Y. U. Yu, S. Y. Kang, H. Y. Park, S. H. Sung, E. J. Lee, S. Y. Kim and Y. C. Kim, J. Pharm. Pharmacol., 2000, 52, 1163–1169 CrossRef CAS.
- W. C. Hou, R. D. Lin, K. T. Cheng, Y. T. Hung, C. H. Cho, C. H. Chen, S. Y. Hwang and M. H. Lee, Phytomedicine, 2003, 10, 170–175 CrossRef CAS.
- N. Y. Kim and J. H. Ryu, Phytother. Res., 2003, 17, 372–375 CrossRef CAS PubMed.
- T. S. Wu, Y. L. Leu, Y. Y. Chan, S. M. Yu, C. M. Teng and J. D. Su, Phytochemistry, 1994, 36, 785–788 CrossRef CAS.
- T. S. Wu, C. M. Teng and S. M. Yu, US Pat. 5656274 A12, 1997.
- C. J. Ma, S. H. Sung and Y. C. Kim, Planta Med., 2004, 70, 79–80 CrossRef CAS.
- C. J. Ma, S. R. Kim, J. Kim and Y. C. Kim, Br. J. Pharmacol., 2005, 146, 752–759 CrossRef CAS.
- K. Reyes-Melo, A. García, A. Romo-Mancillas, E. Garza-González, V. M. Rivas-Galindo, L. D. Miranda, J. Vargas-Villarreal, J. M. J. Favela-Hernández and M. D. R. Camacho-Corona, Bioorg. Med. Chem., 2017, 25, 5247–5259 CrossRef CAS.
- J. M. J. Favela-Hernández, A. García, E. Garza-González, V. M. Rivas-Galindo and M. D. R. Camacho-Corona, Phytother. Res., 2012, 26, 1957–1960 CrossRef.
- M. M. M. Pinto, A. Kujoa, I. O. Mondranondra and A. B. G. Werner, Phytochemistry, 1988, 29, 1985–1990 CrossRef.
- N. Nakatani, K. Ikeda, H. Kikuzaki, M. Kido and Y. Yamaguchi, Phytochemistry, 1988, 27, 3127–3129 CrossRef CAS.
- H. Shibuya, Y. Takeda, R. S. Zhang, A. Tanitame, Y. L. Tsai and I. Kitagawa, Chem. Pharm. Bull., 1992, 40, 2639–2646 CrossRef CAS.
- S. Inoshiri, M. Sasaki, H. Otsuka and K. Yamasaki, Phytochemistry, 1987, 26, 2811–2814 CrossRef CAS.
- H. Karikome, Y. Mimaki and Y. Sashida, Phytochemistry, 1991, 30, 315–319 CrossRef CAS.
- R. W. Hemingway, F. L. Tobiason, G. W. McGraw and J. P. Steynberg, Magn. Reson. Chem., 1996, 34, 424–433 CrossRef CAS.
- T. Masuda, S. Mizuguguchi, T. Tanaka, K. Iritani and Y. Takeda, J. Agric. Food Chem., 2000, 48, 1479–1484 CrossRef CAS.
- H. Ishii, T. Ishikawa, H. Wada, H. Miyazaki, Y. Kaneko and T. Harayama, Chem. Pharm. Bull., 1992, 40, 2614–2619 CrossRef CAS.
- D. V. Banthorpe and G. D. Brown, Phytochemistry, 1989, 28, 3003–3007 CrossRef CAS.
- W. W. Li and W. Barz, Planta Med., 2006, 72, 248–254 CrossRef CAS.
- P. A. Spencer, A. Tanaka and G. H. N. Towers, Phytochemistry, 1990, 29, 3785–3788 CrossRef CAS.
- J. Tan, P. Bednarek, J. Liu, B. Schneider, A. Svatos and K. Hahlbrock, Phytochemistry, 2004, 65, 691–699 CrossRef CAS.
- H. Shimomura, Y. Sashida, M. Oohara and H. Tenma, Phytochemistry, 1988, 27, 644–646 CrossRef CAS.
- Y. Li, M. R. Meselhy, L. Q. Wang, C. M. Ma, N. Nakamura and M. Hattroi, Chem. Pharm. Bull., 2000, 48, 1239–1241 CrossRef CAS.
- Y. Hashidoko, S. Tahara and J. Mizutani, Phytochemistry, 1992, 31, 3282–3283 CrossRef CAS.
- Y. R. Govindachari, P. C. Parthasrathy, H. K. Desai and P. A. Mohamed, Indian J. Chem., 1971, 63, 1027 Search PubMed.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464–1470 CrossRef CAS.
- S. Christophoridu, P. Dais, L. H. Tseng and M. Spraul, J. Agric. Food Chem., 2005, 53, 4667–4679 CrossRef PubMed.
- M. Lucarini, P. Pedrielli, G. F. Pedulli, S. Cabiddu and C. Fattuoni, J. Org. Chem., 1996, 61, 9259–9263 CrossRef CAS.
- F. R. Chang, C. Y. Chen, T. J. Hsieh, C. P. Cho and Y. C. Wu, J. Chin. Chem. Soc., 2000, 47, 913–920 CrossRef CAS.
- B. N. Su, E. J. Park, D. Nikolic, J. S. Vigo, J. G. Graham, F. Cabieses, R. B. Breemen, H. H. S. Fong, N. R. Farnsworth, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 2003, 66, 1089–1093 CrossRef CAS.
- B. C. Michele, M. D. Paolis and J. Zhu, Tetrahedron Lett., 2001, 42, 3427–3430 CrossRef.
- S. Hibino, E. Sugino, Y. Adachi, K. Nomi and K. Sato, Heterocycles, 1989, 28, 275–282 CrossRef CAS.
- C. T. Chen, F. R. Chang, C. M. Teng and Y. C. Wu, J. Chin. Chem. Soc., 1999, 46, 77–86 CrossRef CAS.
- B. Y. Hwang, B. N. Su, H. Chai, Q. Mi, L. B. S. Kardono, J. J. Afriastini, S. Riswan, B. D. Santarsiero, A. D. Mesecar, R. Wild, C. R. Fairchild, G. D. Vite, W. C. Rose, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto, S. M. Swanson and A. D. Kinghorn, J. Org. Chem., 2004, 69, 3350–3358 CrossRef CAS.
- V. Singh, A. Khurana, I. Kaur, V. Sapehiyla, G. L. Kad and J. Singh, J. Chem. Soc., Perkin Trans. 1, 2002, 1766–1768 RSC.
- B. Talapatra, S. Goswami, A. Ghash and S. K. Talapetra, J. Indian Chem. Soc., 1982, 59, 1364–1368 CAS.
- V. J. C. Martinez, S. L. E. Cuca, M. A. J. Santana, E. Pombo-Villar and B. I. Golding, Phytochemistry, 1985, 24, 1612–1614 CrossRef.
- D. Wang, Y. Mu, H. Dong, H. Yan, C. Hao, X. Wang and L. Zhang, Molecules, 2018, 23, 72 CrossRef.
- W. J. Davies and H. G. Jones, in Abscisic acid: Physiology and Biochemistry, BIOS Scientific Publishers, Oxford, 1991 Search PubMed.
- C. Y. Chen, Y. T. Yeh and Y. R. Hsui, Chem. Nat. Compd., 2011, 47, 519–520 CrossRef CAS.
- J. C. T. Reddel, W. Wang, K. Koukounas and R. J. Thomson, Chem. Sci., 2017, 8, 2156–2160 RSC.
- S. H. Tai, Y. C. Hung, E. J. Lee, A. C. Lee, T. Y. Chen, C. C. Shen, H. Y. Chen, M. Y. Lee, S. Y. Huang and T. S. Wu, J. Pineal Res., 2011, 50, 292–303 CrossRef CAS.
- R. L. Kraus, R. Pasieczny, K. Lariosa-Willingham, M. S. Turner, A. Jiang and J. W. Trauger, J. Neurochem., 2005, 94, 819–827 CrossRef CAS.
- Y. R. Hsui, H. L. Chen, C. T. Chen and C. Y. Chen, Chem. Nat. Compd., 2013, 49, 79–80 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data of compounds 1–6. See DOI: 10.1039/c9ra03514a |
| ‡ S. H. Tai and P. C. Kuo contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |