
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light induced emulsion photopolymerization with carbon nitride as a stabilizer and photoinitiator†
Qian
Cao
a,
Tobias
Heil
 a,
Baris
Kumru
a,
Markus
Antonietti
a and
Bernhard V. K. J.
Schmidt
*ab
a,
Baris
Kumru
a,
Markus
Antonietti
a and
Bernhard V. K. J.
Schmidt
*ab
aMax Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: bernhard.schmidt@mpikg.mpg.de; bernhard.schmidt@glasgow.ac.uk; Tel: (+44) 141 330 8469
bSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
First published on 4th September 2019
Abstract
Photopolymerization is a common method in the synthesis of polymers with various applications. Herein, a simple and effective route for surfactant-free emulsion photopolymerization (EPP) under visible light irradiation is described. Therein, graphitic carbon nitride (g-CN) was utilized as an stabilizer and a photoinitiator at the same time. As such, g-CN provides the starting point for polymer chain growth and particle formation. Notably, the as-prepared polymer latexes are directly crosslinked by g-CN, and the existence of g-CN is confirmed inside of the particle, as well as outside, where it forms relatively stable latexes. Moreover, surface functionalized g-CN was utilized to tailor the g-CN/monomer interactions for improved particle formation. g-CN quantum dots with enhanced photoluminescence properties were introduced in EPP as well, providing polymer latexes with enhanced photoluminescence. The obtained polymer nanoparticles might be promising candidates for bioimaging applications.
1. Introduction
Photopolymerization is an important area due to its wide application in industry, such as in coatings,1 inks,2 optical technologies3 and microelectronics.4 Very recently, with the rapid development of 3D printing, the photopolymerization process has also been applied for fabrication of 3D objects.5,6 Compared to thermally initiated polymerization, photopolymerization has several advantages, such as low polymerization temperature, low energy consumption, low environmental pollution, and spatial control.7–9 The utilized light sources can be classified according to the wavelength, commonly UV and visible light. However, issues such as ozone generation and safety concerns restrict broader utilization of UV light.10 Thus, various visible light initiating systems have been developed for free radical polymerization,11 cationic polymerization12 and controlled polymerization.13,14 UV light is frequently investigated for microemulsion, miniemulsion or Pickering emulsion polymerization,15–18 as in all three cases the light can reach the small organic droplets. Nevertheless for emulsion polymerization, environmentally harmless visible light shows better performance, due to less light scattering and absorption by large organic droplets,19 which has been investigated to a lesser extent in emulsion photopolymerization.19 For such an industrially relevant and environmentally friendly process, it is of high value to investigate the application of affordable visible light initiation systems instead of thermal initiation due to its low energy consumption and thermal load on molecules. In particular, Lacote and co-workers19 photopolymerized a polystyrene latex with a large particle size via visible light, using triazolylidene-borane and disulfide, producing a thiyl radical during the process. Moszner and co-workers9 studied bulk and emulsion polymerization of styrene using bis(acyl)germane or phosphine oxide as the photoinitiator (PI). Another area of growing interest in emulsion polymerization is surfactant-free emulsion polymerization as it helps to reduce the effect of potentially harmful surfactants;20,21 especially, reversible deactivation radical polymerization methods have been utilized in that regard.22,23Regarding visible light photopolymerization, the initiator plays a significant role.24–26 An efficient PI features a suitable absorption spectrum that matches well with the visible light emission spectrum to obtain higher photo-initiation efficiency. Thus, over the past few decades various researchers contributed to developing new initiators with lower energy requirements and longer wavelength sensitivity during processing.27–29 Most of the common PIs are organic molecules with a low molecular weight and are usually accompanied by relatively strong odor. Notably, new cleavable PIs have been reported, e.g., incorporating salt,30,31 amine11 or chloro triazine32 functions into the existing structures, in order to generate initiating radicals. For example, Pączkowski and co-workers investigated photoinitiation based on a three component PI system, consisting of a cyanine dye, borate and salt.31 Jean and co-workers added bis(trichloromethyl)-substituted-1,3,5-triazine to a dye/amine PI system and obtained an increased polymerization efficiency.33 However, problems such as complicated processing and material degradation might take place during processing.34
Recently, graphitic carbon nitride (g-CN) was introduced as a PI. Generally investigated as a metal-free photocatalyst,35,36 g-CN attracted significant interest due to its high physicochemical stability, appropriate electronic band gap structure and facile preparation.37 Since it was found that visible light irradiation can produce radicals via g-CN, researchers employed it as a radical initiator for free-radical or reversible deactivation radical polymerization. In particular, Yagci and co-workers applied mesoporous g-CN, combined with amine as a co-initiator, for free radical photopolymerization under visible light8 or together with copper(II) species and a ligand for reversible deactivation radical polymerization.38 Qiao and co-workers introduced g-CN as a photoinitiator for RAFT polymerization, producing linear polyacrylate and polyacrylamide without prior deoxygenation.39 The capability of photoinitiation was utilized in hydrogel formation as well.40–42 Grafting of polymer chains on the g-CN surface was realized during photografting and consumption of monomers in solution (HEMA), and g-CN entailing HEMA groups was employed for photoactive film formation.43 Later on, the very unique amphiphilic property of g-CN was found by Xu,44 indicating that g-CN presents remarkable stabilization of oil and water mixed interfaces. As a proof of concept, g-CN was previously applied as a stabilizer for thermally initiated emulsion polymerization to fabricate PS latexes with a tunable particle size.45
Inspired by these studies, we attempt here to utilize g-CN as an initiator and stabilizer for surfactant-free emulsion photopolymerization (EPP), as depicted in Scheme 1. Firstly, g-CN acts as a stabilizer for a monomer in a water emulsion system. The emulsion is subsequently illuminated with visible light, radicals are generated by g-CN under irradiation, and thus g-CN acts as a locus for initiation. Finally, polymer latexes are formed. Different species of g-CN are studied in this EPP process. The as-prepared polymer latex is crosslinked and characterized via scanning electron microscopy (SEM) and dynamic light scattering (DLS). The existence of g-CN in g-CN derived latex particles is further confirmed by scanning transmission electron microscopy (STEM), high-resolution transmission electron microscopy (HRTEM) tilt investigations and zeta potential measurements. Because of the inherent photoluminescence properties of g-CN, polymer latexes with fluorescence properties are obtained.
 | ||
| Scheme 1 The overall process of emulsion photopolymerization (EPP) with g-CN as a stabilizer and photoinitiator. | ||
2. Experimental section
2.1 Materials
Al2O3 (basic, Sigma Aldrich), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (AIBA, 97%, Sigma Aldrich), acetone (high-performance liquid chromatography (HPLC) grade, Sigma-Aldrich), 3-allyloxy-2-hydroxy-1-propanesulfonic acid sodium salt solution (40 wt%, AHPA, Sigma-Aldrich), benzyl methacrylate (96%, 50 ppm monomethyl ether hydroquinone as an inhibitor, Sigma Aldrich), cyanuric acid (98%, Sigma-Aldrich), 1-decene (94%, Sigma-Aldrich), 2,4-diamino-6-phenyl-1,3,5-triazine (Mp, 97%, Sigma-Aldrich), isopropyl alcohol (IPA, 99.7%, Sigma-Aldrich), 4-methyl-5-vinylthiazole (vTA, 97%, Sigma Aldrich), methyl methacrylate (MMA, 99%, ≤30 ppm MEHQ as an inhibitor, Sigma Aldrich), melamine (99%, Sigma Aldrich), and styrene (St, ≥99%, 4-tert-butylcatechol as stabilizer, Sigma Aldrich) were used. All the monomers were filtered through basic aluminium oxide to remove the inhibitor. Visible light irradiation was performed via two 50 W light-emitting diode (LED) chips (Foxpic High Power 50 W LED Chip Bulb Light DIY White 3800LM 6500 K) connected to a self-made circuit and a cooling system. Sonication was performed in a sonication bath from Elma (Transsonic T310). Various g-CN materials were utilized: CM (derived from the cyanuric acid/melamine complex),46 CMD (1-decene grafted CM),47 CMSO3 (AHPA modified CM),47 PhCMp (derived from a phenyl group modified cyanuric acid/melamine complex),48 and vTA-CMp (vTA grafted PhCMp)49 were synthesized according to the cited procedures.2.2 Preparation of g-CN/H2O suspensions
To obtain a well dispersed g-CN water suspension, ultrasonication was used to exfoliate the bulk carbon nitride. 40 mg g-CN of different types (CM, CMD PhCMp, vTA-CMp and CMSO3) and 20 mL of H2O were added to a 50 mL plastic centrifuge tube, the mixtures were treated with ultrasound with a power of 50% amplification and sonicated for 1 h, and 2 mg mL−1 of g-CN in H2O suspension (g-CN/H2O) were obtained for further utilization.2.3. EPP of polymers with g-CN
Firstly, 4 mL g-CN/H2O dispersion (2 mg mL−1) was mixed with 1 mL of monomers in a 15 mL round bottom flask. Then, under gentle stirring on a magnetic stirrer, a monomer emulsion formed quickly. Afterwards, the flask was sealed with a rubber septum, and the mixture was deoxygenated by purging with argon for 30 min. Then the emulsion was put between two 50 W LED daylight sources (20 cm apart from each other) and stirred continuously overnight. After reaction, a milky polymer latex including different g-CN species was obtained. Herein, g-CN/H2O dispersions of CM/H2O, CMD/H2O, PhCMp/H2O, vTA-CMp/H2O and CMSO3/H2O were utilized, and styrene, benzyl methacrylate (BMA) and methyl methacrylate (MMA) were chosen as monomers, respectively.2.4 Characterization
Zeta potential and particle size distribution of the latexes were determined using a Zeta Nanosizer instrument (Malvern Instruments, UK) at a fixed scattering angle of 90°. All measurements were repeated three times. Transmission electron microscopy (TEM) images were acquired using a Zeiss LEO 912 Omega TEM. Size exclusion chromatography (SEC) for the PS (CM)/THF solution was conducted in THF with toluene as an internal standard using a PSS 1260-Iso as a pump, a column system of a PSS SDV column (8 × 300 mm) with a PSS SDV precolumn (8 × 50 mm), a PSS-SECcurity-VWD and a PSS-SECcurity-RID as detectors and a calibration with PS standards from PSS. High resolution transmission electron microscopy (HR-TEM) and scanning transmission electron microscopy (STEM) images were acquired using a double Cs-corrected JEOL ARM200F, equipped with a cold field emission gun and an EDX detector. For the investigation, the acceleration voltage was set to 200 kV, the emission was set to 10 μA in order to reduce beam damage, and a condenser aperture with a diameter of 30 μm was used. With these settings, the microscope reaches a lattice resolution below 1 Å. The tilt images were acquired at 0°, 20°, 35°, −20°, and −35° tilt. Scanning electron microscopy (SEM) images for the determination of the morphology of the colloidal particles were obtained using a JEOL-7500F SEM equipped with an Oxford Instruments X-MAX 80 mm2 detector. Fluorescence images were obtained by confocal laser scanning microscopy (CLSM, TCS SP5, Leica, Germany). Surface tension of the emulsions was determined by ring RI 12 analysis using a LCD 4 × 40, 5 × 7 mold (KRÜSS, Germany). Solid state ultraviolet-visible (UV-Vis) spectroscopy for g-CN powders was performed via a Cary 500 Scan spectrophotometer equipped with an integrating sphere. g-CN suspensions were obtained via ultrasonication using a Sonifier 450 D (Branson). The surface tension of the final latex was determined with a Krüss tensiometer (Krüss, Hamburg) utilizing the du Noüy ring method. Elemental analysis was performed with a Vario ELIII device.3. Results and discussion
As a proof of concept, the very common and accessible CM (formed from cyanuric acid and melamine as precursors) was used as stabilizer and PI for EPP. CM was firstly dispersed in water and ultrasonicated, and a CM/H2O suspension with CM nanosheets of 160 nm (Fig. S1a, e and Table S1†) was obtained, absorbing all light between 250 and 440 nm (Fig. S1c†). Styrene and benzyl methacrylate (BMA) were chosen as monomers, and emulsions were formed by magnetically stirring monomers and g-CN/H2O to generate oil in water (oil/water) emulsions. EPP was carried out by using two 50 W LEDs as visible light sources. After overnight reaction, latexes were obtained, named PS (CM) and PBMA (CM). The morphology of PS (CM) and PBMA (CM) latexes was characterized via SEM (Fig. 1a and b); both of them consist of spherical particles with a relatively uniform size distribution. The specific size of PS (CM) and PBMA (CM) particles was further determined via DLS (Table S2†), with a hydrodynamic diameter of about 170 nm and 320 nm, respectively. To study the effect of g-CN surface chemistry, 1-decene modified CM (CMD) (Fig. S1b–d and Table S1†),47 which is organo dispersible, was employed for EPP as well. After the same EPP process as CM, the latexes PS (CMD) and PBMA (CMD) were successfully obtained (Fig. 1d and e). The particle sizes are about 150 nm and 740 nm (Table S2†), respectively. These results demonstrate the ability of g-CN to fulfill two different roles, a stabilizer and a PI during EPP, which effectively facilitates the process. From the uniformity of the obtained latex particles one can conclude very effective nucleation of the particle formation,50 which will be discussed in detail below.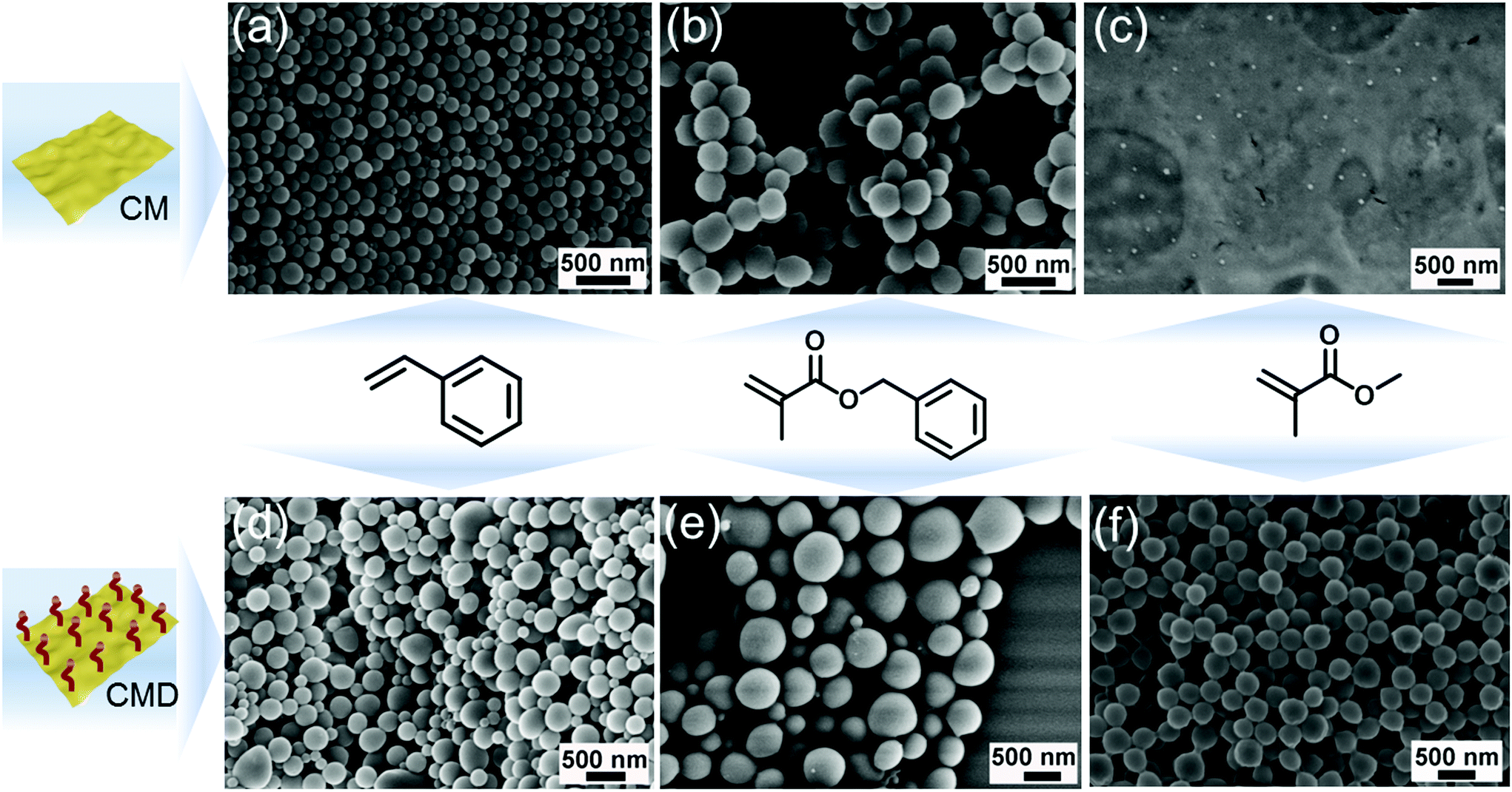 | ||
| Fig. 1 SEM images of PS (CM) (a) and PBMA (CM) (b) latex particles. (c) Ill-defined PMMA obtained from CM initiation. (d), (e) and (f) are SEM images of PS (CMD), PBMA (CMD) and PMMA (CMD) latex particles. | ||
Interestingly, in the case of MMA in contrast to aromatic monomers, no latex could be formed employing CM as a PI (Fig. 1c). We assume that the structure of the monomer significantly influences the g-CN initiated EPP process. In the case of phenyl containing monomers, there might be interactions between CN tri-s-triazine rings and the phenyl moiety, which contributes to a relatively stable emulsion and interaction of g-CN and monomer. As such, the phenyl containing monomers support the delamination of g-CN due to π–π interactions. Thus, polymerization can proceed via an adlayer process. In contrast, MMA does not contain functional groups capable of favored interactions with g-CN; thus, less stable emulsions were formed. This was further confirmed by simply adjusting the ratio of styrene and MMA as a co-monomer with CM as an emulsifier and PI. PS/PMMA latex particles with a well-defined shape were obtained when the ratio was 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (styrene:MMA) (Fig. S2a†). Only slight amounts of amorphous PMMA/PS were obtained when the ratio changed to 1:9; no particles were observed with TEM (Fig. S2b†). Besides, with organo dispersible CMD as an emulsifier and PI, the EPP of MMA could be successfully conducted (Fig. 1f), leading to uniform PMMA latexes with a particle size of about 320 nm (Table S2†). A possible explanation for this might be that CMD itself has an improved tendency to delaminate due to the grafted decyl groups,47 which is beneficial for the interaction between MMA and CMD. Moreover, it contributes to an improved and stable emulsion system for polymerization.
:1 (styrene:MMA) (Fig. S2a†). Only slight amounts of amorphous PMMA/PS were obtained when the ratio changed to 1:9; no particles were observed with TEM (Fig. S2b†). Besides, with organo dispersible CMD as an emulsifier and PI, the EPP of MMA could be successfully conducted (Fig. 1f), leading to uniform PMMA latexes with a particle size of about 320 nm (Table S2†). A possible explanation for this might be that CMD itself has an improved tendency to delaminate due to the grafted decyl groups,47 which is beneficial for the interaction between MMA and CMD. Moreover, it contributes to an improved and stable emulsion system for polymerization.
Usually, in order to achieve crosslinked products in emulsion polymerization, an additional heating process or addition of a cross-linker is required to crosslink the active species in the particles.51 As reported before, g-CN produces radicals under visible irradiation and can act as a cross-linker for hydrogel formation,42 even if the surface properties are tailored photochemically in a step ahead.52 Therefore, we investigated whether g-CN-based EPP leads to cross-linked latexes directly. After dispersing the dry PS (CM) latex in THF, a dispersion was formed instead of a solution (Fig. 2a and Fig. S3†) in contrast to the control sample (Fig. 2c) that formed a transparent solution. Thus, it can be confirmed that particles were crosslinked due to the existence of CM. Afterwards, the PS (CM)/THF dispersion was precipitated into H2O and stirred for 24 h to evaporate the THF completely. Subsequently, a reconstituted PS (CM) latex with a well-defined spherical particle shape was observed via SEM (Fig. 2b). In comparison, the control sample (Fig. S4†) showed an amorphous morphology and the particles dissolved in THF completely (Fig. 2d). Therefore, it was confirmed that CM also acts as a cross-linker during the polymerization, and a crosslinked PS (CM) latex was obtained without further cross-linker addition or heating processes. However, free polymers formed as well, probably due to radical transfer reactions with the monomer or solvent as well as initiation via hydroxyl radicals. The formation of free polymer chains was confirmed by measuring the SEC of the PS (CM)/THF solution after filtering the particles using a syringe filter (100 nm, PTFE) (Fig. S5†). The mass loss was 50% after filtering, which indicates that there was significant formation of free polymers.
 | ||
| Fig. 2 PS (CM) (a) and control sample PS (c) particles dispersed in THF. SEM images of PS (CM) (b) and control sample PS (d) after precipitation of the dispersion in water and evaporation of THF, respectively. | ||
As g-CN plays such a significant role in the EPP process, the whereabouts of g-CN in the final PS latex were further investigated. As shown with scanning transmission electron microscopy (STEM) (Fig. 3a), a region with increased electron contrast was found in the center of the PS (CM) latex. Corresponding elemental mapping of nitrogen (Fig. 3b) and carbon (Fig. 3c) showed a significant nitrogen signal coinciding with the higher density region in the particles. The related EDS analysis also showed a nitrogen signal (Fig. S6†), thus demonstrating the existence of CM in the final PS particles. Moreover, the amount of g-CN inside the particles was approximately 2.6% according to elemental analysis. Subsequently, the location of CM was confirmed via tilt investigation. With the sample holder tilted from 0° to −35° (Fig. 3d), −20° (Fig. 3e), 20° (Fig. 3f), and 35° (Fig. 3g), CM remains in the center of the PS latex, which indicates that the CM is located inside the PS latex. Further, from the STEM images the size of incorporated CM can be estimated as around 50–100 nm. The same phenomenon was also observed in the CMD stabilized and initiated PS latex (Fig. S7 and S8†). Thus, with g-CN as a stabilizer and PI, a PS latex with g-CN incorporation, defined as a PS–g-CN core–shell structure, was obtained. Moreover, the obtained PS (CM) latex emulsion showed significant stability. Over 90 days of observation (Fig. S9†), no coalescence and sedimentation was found. The extended stability of the latex is an indication for ongoing stabilization by g-CN. Therefore, in addition to g-CN introduction in the core of latex particles, a coverage of the surface with g-CN is indicated.
 | ||
| Fig. 3 (a) Dark-field STEM image of PS (CM) and (b) and (c) corresponding elemental mapping of nitrogen and carbon, respectively. Dark-field STEM images of PS (CM) with the sample holders tilted at the angles of −35° (d), −20° (e), 20° (f) and 35° (g). | ||
As for emulsion polymerization, it has been known for many years that adsorbed ions at the particle–water interface have significant effects on the latex stability, and emulsifiers and initiators usually attach to the surface of the latex to form a stable latex emulsion.53 These assumptions were then confirmed by zeta potential measurements. The surface zeta potential of final latex products usually depends on the zeta potential of the initiator or surfactant. The control sample which was formed with the azo-initiator AIBA for emulsion polymerization without a surfactant showed a zeta potential of 34 mV (Table S3†). CMSO3, (AHPA modified CM, −30 mV), namely a g-CN species with significant negative charge, was employed for EPP, which leads to a PS latex with a zeta potential of −30 mV. Moreover, with CM and CMD that have a zeta potential of −24 mV and −28 mV (Table S1†), the corresponding polymer latexes of PS (CM), PBMA (CM), PS (CMD), PBMA (CMD) and PMMA (CMD) emulsions have a zeta potential ranging from −20 to −30 mV. On top, the PS (CM) features the same surface tension (68.89 mV m−1, Table S4†) as the Pickering stabilizer CM; that is, it is proven that no low molecular weight amphiphilic species are created. Thus, we have to assume that a small amount of CM is also located on the surface of particles, and a stable particulate colloidal dispersion was formed using g-CN as a Pickering stabilizer at the particle–water interface. It should be noted that the measured surface tension is remarkably close to that of water itself, which is a strong indication for the surfactant-free polymerization process without in situ surfactant formation. Remarkably, although a surfactant-free process is performed rather small particles are formed.
The polymerization mechanism and particle formation during the EPP process in such Pickering systems free of low molecular weight surfactants are interesting to analyze as well.54–56 g-CN was generally employed as a photocatalyst, due to its suitable band gap that can generate electrons and holes under light excitation. The electrons and holes could be employed for formation of radicals (i.e., ˙OH, O2−, and HO2˙), which could initiate EPP. Hydrogen peroxide and triethanolamine were used as an electron scavenger (ES) and a hole scavenger (HS) to elucidate the polymerization process. With the addition of hydrogen peroxide to the system, PS particles can still be found (Fig. S10a†), while in the presence of triethanolamine, less polymer products were produced and no particles formed (Fig. S10b†). Thus, it is indicated that the radical polymerization process is mostly hole initiated; that is, the radical is created by oxidation. According to the mechanism of emulsion polymerization, the particle formation nucleates in CM stabilized micelles and in the course of polymerization the micelles swollen with the solubilized monomer are growing to form latex particles. To get further insight into the mechanism of the polymerization process, the EPP process was observed with SEM over different reaction times. In the case of styrene EPP with CM, after 30 min irradiation, no particles were formed (Fig. S11a†). After 1 h, random particles could be observed (Fig. S11b†). The amount of particles continuously increased with reaction times of 2.5 h (Fig. S11c†) and 4 h (Fig. S11d†), whereas the ill-defined particles could well be CM stabilizers. After an overnight reaction, uniform latexes were successfully formed (Fig. S11e†). With the kinetic observation, it seems like most of the particles grow around g-CN, which goes hand in hand with an adlayer polymerization mechanism. During the particle growth, the emulsifier g-CN stabilizes monomer droplets and provides a stabilizer for the micellar polymerization environment, which is indicated by the incorporation of CM in latex particles and cross-linking. As such, the common emulsion polymerization mechanism is followed, stating that the polymerization takes place inside the micelles.
Previously it was suggested that g-CN adhered to the monomer droplets, and formed a 3D network structure in the continuous water phase to form a stable emulsion.45 Thus, the adhered g-CN on monomer droplets might also act as loci for monomers to add on a free radical from g-CN and initiate chain growth, until it is terminated by interaction with another similarly produced radical chain. This process constitutes only a minor part of polymerization events, which is in accordance with the general emulsion polymerization mechanism. At the same time, as g-CN features an inhomogeneous size/diameter in the region from 50 nm to 500 nm (Fig. S1d and e†), larger size particles suspend in the continuous phase,45 smaller particles with diameters around 50 nm to 100 nm enter the particles and very small particles act as surface stabilizers, which was observed in STEM, cross-linking and zeta potential measurements. From a mechanistic point of view, the g-CN mediated EPP can be divided into various steps (Scheme 2a). First nucleation of latex particles proceeds at the surface of dispersed g-CN particles via hole driven adlayer polymerization. Polymer formation leads to a significant hydrophobization of the g-CN surface, which leads to a self-assembly process and formation of micellar structures. This process is very fast compared to the polymer propagation as indicated by the narrow particle size distributions.50 In the course of the reaction, monomers diffuse from the monomer droplets to swell the polymerization particles and facilitate continued particle growth. As such, g-CN forms the center of the latex particles. Moreover, dispersed g-CN originating either from the shrinking monomer droplets or from the continuous phase enables stable latex formation, as shown by zeta potential and surface tension measurements. From a mechanistic perspective (Scheme 2b), either free polymers or crosslinked polymers are formed during the polymerization process. On the one hand, radicals like ˙OH are produced with visible light excitation, which can directly initiate free radical polymerization and is in accordance with gravimetry results after THF treatment. On the other hand, reactions between the g-CN surface and monomers contribute to crosslinked particles. The crosslinking is due to the multifunctional nature of g-CN that contains various initiation sites due to its extended size.
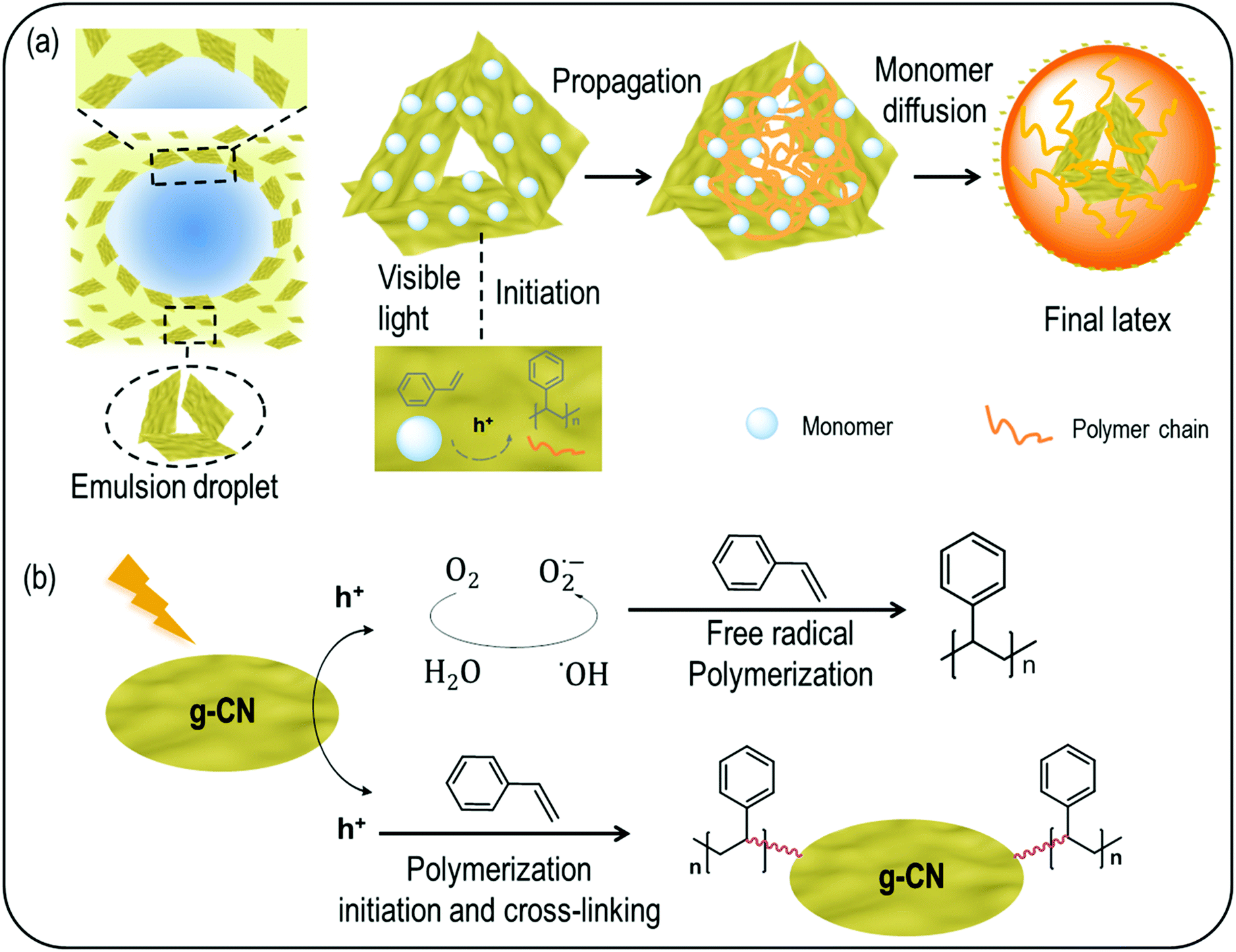 | ||
| Scheme 2 Mechanism illustration of particle formation (a) and the polymerization process (b) with g-CN as a polymerization locus. | ||
As mentioned before, no latex was observed for the polymerization of MMA with CM, whereas latex formation was observed for BMA. Therefore, it appeared that the monomer/g-CN interaction has a profound effect on the polymerization process. CMD polymerized MMA due to its improved tendency of delamination and monomer/g-CN interaction. To elucidate the process in more detail, the polymerization of styrene and MMA with CMD was followed kinetically as well. In the case of styrene, essentially the same growth phenomenon was observed as discussed above (Fig. S12†). Particles start to form after photoexcitation for 4 hours and a similar latex product is produced. However, with MMA (Fig. S13†), first signs of polymer formation were observed after 14 h reaction time and particles were finally formed after 17 h of polymerization. Apparently, the monomer structure has a significant impact on the photopolymerization process. As suggested before that phenyl containing molecules adhere to g-CN because of π–π interaction,44 there might be interactions between monomers and g-CN that profoundly affect the polymerization process, thus leading to a different reaction time. Herein, we assume that although CMD improved the balance of the monomer/water emulsion compared to CM, the slower conversion might be due to the still imperfect contact/interaction of MMA and CMD.
As g-CN has been widely recognized for its inherent optical properties, polymer latexes with in situ photoluminescence properties can be obtained without additional fluorescent labelling. Herein, two different g-CN materials with high quantum yields were employed for stabilizing and initiating the EPP process. One is phenyl group doped CMp (with cyanuric acid and 2,4-diamino-6-phenyl-1,3,5-triazine as precursors), named PhCMp (Fig. S14a, c and e†). The introduction of the phenyl group inhibits chain growth in the carbon nitride sheets and contributes to higher luminescence. The other one is 4-methyl-5-vinylthiazole (vTA) grafted CMp, named vTA-CMp (Fig. S14b–d†), with high green light luminescence around 540 nm. The EPP processes were similar to that observed before, and latexes of PS (PhCMp, Fig. 4a) and PS (vTA-CMp, Fig. 4b) were obtained, respectively. However, with vTA-CMp stabilized and initiated polymerization, a rather monodisperse latex with a dispersity of 0.038 was obtained. This might be due to the delocalized surface charge of vTA-CMp in the organic phase that caused an increased dispersibility of vTA-CMp in organic media. Thus, a more stable emulsion was formed before polymerization, which also benefits the swelling equilibrium, monomer diffusion and particle formation. Moreover, the inherent charge of vTA-Cmp most likely leads to an improved charge transfer interaction with styrene, which enhances the rate of the nucleation process and leads to more uniform latex formation. The photoluminescence of the latex was observed by confocal laser scanning microscopy (CLSM). Under the same measurement conditions, an increase in the fluorescence emission intensity was observed going from PS (CM) (Fig. 4c and d) to PS (PhCMp) (Fig. 4e and f) and PS (vTA-CMp) (Fig. 4g and h), which is due to the enhanced fluorescence efficiency of PhCMp and vTA-CMp compared to CM. Such a unique tunable fluorescence enables optical utilization of PS particles, as the PS latex has been widely regarded as a biocompatible material, utilized for food and medical products.57,58 The novel core–shell PS (g-CN) particles are promising potential candidates for bioimaging and biomedical applications.
 | ||
| Fig. 4 SEM images of PS (PhCMp) (a) and PS (vTA-CMp) (b) and CLSM and bright field images of PS (CM) (c and f), PS (PhCMp) (d and g) and PS (vTA-CMp) (e and h). | ||
4. Conclusions
In the present work, g-CN was applied as a Pickering stabilizer and photoinitiator for the emulsion polymerization process without extra modification and addition of further chemicals. A number of different delaminated carbon nitrides were utilized for radical initiation via visible light irradiation to obtain stable PS, PBMA and PMMA latex emulsions with relatively uniform particle sizes. TEM confirmed the presence of g-CN in the center of the particles, as well as small amounts on the particle surface that enable stable latex emulsions. Using two types of special g-CN quantum dots with unique photoluminescence properties, PhCMp and vTA-CMp, highly photoluminescent polymer particles could be produced. To the best of our knowledge, this research uses for the first time simple and affordable carbon nitride sheets as Pickering stabilizers for the combination of the two functions of a stabilizer and an initiator under visible light irradiation in one simple system. The whole EPP process is thereby highly simplified, as we only need water, monomers, and carbon nitride under light to generate controlled latex particles. The obtained latexes were endowed with tunable photoluminescence and have the potential to be useful for biomedicine and bioimaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from Max-Planck Society. The authors acknowledge Irina Shekova for surface tension measurements, Heike Runge for TEM measurements, Jianrui Zhang for assistance in CLSM and Yasemin Linde for elemental analysis Open Access funding provided by the Max Planck Society.Notes and references
- E. Amerio, M. Sangermano, G. Malucelli, A. Priola and B. Voit, Polymer, 2005, 46, 11241–11246 CrossRef CAS.
- J. Shi, M. B. Chan-Park, C. Gong, H. Yang, Y. Gan and C. M. Li, Chem. Mater., 2010, 22, 2341–2346 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- K. K. Baikerikar and A. B. Scranton, J. Appl. Polym. Sci., 2001, 81, 3449–3461 CrossRef CAS.
- M. Layani, X. Wang and S. Magdassi, Adv. Mater., 2018, 30, 1706344 CrossRef PubMed.
- C. Check, R. Chartoff and S. Chang, React. Funct. Polym., 2015, 97, 116–122 CrossRef CAS.
- Y. Y. Durmaz, M. Kukut, N. Moszner and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4793–4799 CrossRef CAS.
- B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546–549 CrossRef CAS.
- K. Krüger, K. Tauer, Y. Yagci and N. Moszner, Macromolecules, 2011, 44, 9539–9549 CrossRef.
- J. Shao, Y. Huang and Q. Fan, Polym. Chem., 2014, 5, 4195–4210 RSC.
- G. Temel, B. Enginol, M. Aydin, D. K. Balta and N. Arsu, J. Photochem. Photobiol., A, 2011, 219, 26–31 CrossRef CAS.
- J. Lalevée, M. El-Roz, X. Allonas and J. Pierre Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2008–2014 CrossRef.
- T. G. McKenzie, E. H. H. Wong, Q. Fu, A. Sulistio, D. E. Dunstan and G. G. Qiao, ACS Macro Lett., 2015, 4, 1012–1016 CrossRef CAS.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- K. Jain, J. Klier and A. B. Scranton, Polymer, 2005, 46, 11273–11278 CrossRef CAS.
- F. Jasinski, E. Lobry, A. Chemtob, C. Croutxé-Barghorn and A. Criqui, Macromol. Chem. Phys., 2013, 214, 1669–1676 CrossRef CAS.
- H. Lu, B. Fei, J. H. Xin, R. Wang and L. Li, J. Colloid Interface Sci., 2006, 300, 111–116 CrossRef CAS PubMed.
- X. Song, Y. Zhao, H. Wang and Q. Du, Langmuir, 2009, 25, 4443–4449 CrossRef CAS PubMed.
- F. Le Quemener, D. Subervie, F. Morlet-Savary, J. Lalevee, M. Lansalot, E. Bourgeat-Lami and E. Lacote, Angew. Chem., Int. Ed., 2018, 57, 957–961 CrossRef CAS PubMed.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 RSC.
- A. Schmid, P. Scherl, S. P. Armes, C. A. Leite and F. Galembeck, Macromolecules, 2009, 42, 3721–3728 CrossRef CAS.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- J. Rieger, W. Zhang, F. o. Stoffelbach and B. Charleux, Macromolecules, 2010, 43, 6302–6310 CrossRef CAS.
- A. Al Mousawi, C. Poriel, F. Dumur, J. Toufaily, T. Hamieh, J. P. Fouassier and J. Lalevée, Macromolecules, 2017, 50, 746–753 CrossRef CAS.
- S. Benedikt, J. Wang, M. Markovic, N. Moszner, K. Dietliker, A. Ovsianikov, H. Grützmacher and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 473–479 CrossRef CAS.
- J. Radebner, A. Eibel, M. Leypold, C. Gorsche, L. Schuh, R. Fischer, A. Torvisco, D. Neshchadin, R. Geier, N. Moszner, R. Liska, G. Gescheidt, M. Haas and H. Stueger, Angew. Chem., Int. Ed., 2017, 56, 3103–3107 CrossRef CAS PubMed.
- N. Moszner, U. K. Fischer, I. Lamparth, P. Fässler, J. Radebner, A. Eibel, M. Haas, G. Gescheidt and H. Stueger, J. Appl. Polym. Sci., 2018, 135, 46115 CrossRef.
- J. Wang, S. Stanic, A. A. Altun, M. Schwentenwein, K. Dietliker, L. Jin, J. Stampfl, S. Baudis, R. Liska and H. Grutzmacher, Chem. Commun., 2018, 54, 920–923 RSC.
- Y. Zhao, M. Yu, S. Zhang, Z. Wu, Y. Liu, C. H. Peng and X. Fu, Chem. Sci., 2015, 6, 2979–2988 RSC.
- X. Nan, Y. Huang, Q. Fan and J. Shao, Prog. Org. Coat., 2015, 81, 11–18 CrossRef CAS.
- J. Kabatc, M. Zasada and J. Pączkowski, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3626–3636 CrossRef CAS.
- J. Zhang, N. Zivic, F. Dumur, P. Xiao, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 445–451 CrossRef CAS.
- C. Grotzinger, D. Burget, P. J. Fouassier and P. Jean, Macromol. Chem. Phys., 2001, 202, 3513–3522 CrossRef CAS.
- A. Žukauskas, G. Batavičiūtė, M. Ščiuka, Z. Balevičius, A. Melninkaitis and M. Malinauskas, Opt. Mater., 2015, 39, 224–231 CrossRef.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. A. Tasdelen, B. Kiskan, X. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 675–681 CrossRef CAS.
- Q. Fu, Q. Ruan, T. G. McKenzie, A. Reyhani, J. Tang and G. G. Qiao, Macromolecules, 2017, 50, 7509–7516 CrossRef CAS.
- J. Sun, B. V. K. J. Schmidt, X. Wang and M. Shalom, ACS Appl. Mater. Interfaces, 2017, 9, 2029–2034 CrossRef CAS PubMed.
- B. Kumru, V. Molinari, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Soft Matter, 2018, 14, 2655–2664 RSC.
- J. Liu, T. An, Z. Chen, Z. Wang, H. Zhou, T. Fan, D. Zhang and M. Antonietti, J. Mater. Chem. A, 2017, 5, 8933–8938 RSC.
- B. Kumru, J. Barrio, J. Zhang, M. Antonietti, M. Shalom and B. V. K. J. Schmidt, ACS Appl. Mater. Interfaces, 2019, 11, 9462–9469 CrossRef CAS PubMed.
- J. Xu and M. Antonietti, J. Am. Chem. Soc., 2017, 139, 6026–6029 CrossRef CAS PubMed.
- Q. Cao, Q. Cui, Y. Yang, J. Xu, C. Han and L. Li, Chem. – Eur. J., 2018, 24, 2286–2291 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Langmuir, 2017, 33, 9897–9906 CrossRef CAS.
- Q. Cui, J. Xu, X. Wang, L. Li, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2016, 55, 3672–3676 CrossRef CAS PubMed.
- B. Kumru, D. Cruz, T. Heil, B. V. K. J. Schmidt and M. Antonietti, J. Am. Chem. Soc., 2018, 140, 17532–17537 CrossRef CAS PubMed.
- P. J. Colver, C. A. Colard and S. A. Bon, J. Am. Chem. Soc., 2008, 130, 16850–16851 CrossRef CAS PubMed.
- B. Kumru, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2017, 50, 1862–1869 CrossRef CAS.
- B. Kumru, V. Molinari, R. Dunnebacke, K. G. Blank and B. V. K. J. Schmidt, Macromol. Rapid Commun., 2019, 40, 1800712 Search PubMed.
- C. P. Roe, Ind. Eng. Chem., 1968, 60, 20–33 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 5775–5780 CrossRef CAS.
- S. Cauvin, P. J. Colver and S. A. F. Bon, Macromolecules, 2005, 38, 7887–7889 CrossRef CAS.
- A. Lotierzo and S. A. Bon, Polym. Chem., 2017, 8, 5100–5111 RSC.
- C. Loos, T. Syrovets, A. Musyanovych, V. Mailander, K. Landfester, G. U. Nienhaus and T. Simmet, Beilstein J. Nanotechnol., 2014, 5, 2403–2412 CrossRef.
- C. K. Poon, O. Tang, X. M. Chen, B. T. Pham, G. Gody, C. A. Pollock, B. S. Hawkett and S. Perrier, Biomacromolecules, 2016, 17, 965–973 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01157a |
| This journal is © The Royal Society of Chemistry 2019 |