
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-induced reversible hydrophobization of cationic gold nanoparticles via electrostatic adsorption of a photoacid†
Hang Zhang *,
Muhammad Junaid,
Kai Liu,
Robin H. A. Ras and
Olli Ikkala*
*,
Muhammad Junaid,
Kai Liu,
Robin H. A. Ras and
Olli Ikkala*
Department of Applied Physics, Aalto University, P.O. Box 15100, FI 02150 Espoo, Finland. E-mail: hang.zhang@aalto.fi; olli.ikkala@aalto.fi
First published on 10th July 2019
Abstract
The ability to switch the hydrophilicity/hydrophobicity of nanoparticles promises great potential for applications. Here we report a generic approach that allows hydrophobization of cationic surfaces by light-induced photoacid switching from the unbound zwitterionic form to the electrostatically bound anionic form. Importantly, this allows reversible assembly and disassembly of cationic AuNPs, with disassembly kinetics controlled by temperature. The AuNPs can be repeatedly transferred between aqueous and non-polar solvents using light, showing potential in purification processes. In the macroscopic scale, nontrivially, light triggers the in situ hydrophobization of a flat cationized gold surface. The current approach is generic and opens up a new way to control the surface properties and self-assembly of nanoparticles.
Introduction
Controlling the surface chemistry of nanoparticles has been a major focus due to its vital importance in, e.g., stabilizing the nanoparticles, guiding the self-assembly and plasmonic coupling, tuning the biocompatibility, and enabling sensitivity.1–4 The ability to reversibly switch the nanoparticles between the hydrophilic and hydrophobic states promises intriguing potential for applications such as purification of colloids, sensing of metals or biomolecules, and new routes for out-of-equilibrium self-assemblies.5–7 Most approaches so far are based on covalent grafting of photoresponsive ligands, use of light-controlled specific host–guest chemistry, or irreversible adsorption of charged surfactants.1,8–13 In particular, use of light to control the surface properties of nanoparticles is highly desirable due to the high temporal and spatial resolution of the light, and the potential to couple with other processes such as photomechanical actuation.14–19 Here we first report on the light-induced hydrophobization of cationic gold nanoparticles (AuNPs), based on the reversible electrostatic adsorption of a photoacid that can be switched between zwitterionic and anionic forms. In comparison with previous studies using covalently grafted spiropyrans,20–22 the present concept allows considerable design flexibility, since the components can be synthesized separately and different cationization methods can be used. The hydrophobization allows reversible assembly/disassembly of AuNPs, and the kinetics of the disassembly process can be easily controlled by temperature. Importantly, cationic AuNPs can be reversibly transferred between aqueous and non-polar solvents under the control of light, a challenging task for metal nanoparticles to date. The principle can further be extended to the macroscopic scale as shown by the in situ hydrophobization of a cationized flat gold surface.Results and discussion
Scheme 1 illustrates the light-induced hydrophobization process on cationic AuNPs. Upon irradiation (420 nm), the photoacid in Scheme 1 undergoes a transition from the protonated merocyanine form to the spiropyran form, meanwhile releasing a proton.23 Studies so far have been mainly based on the pH change, such as colloidal self-assembly14,15,24 or gelation of supramolecular hydrogels.25,26 In contrast, we utilize another notable feature of the photoacid: the transition from the zwitterionic merocyanine to anionic spiropyran upon irradiation. In the dark, the photoacid molecules are predominantly in their zwitterionic form and do not interact specifically with the cationic AuNPs. Upon irradiation, the transition leads to the adsorption of anionic spiropyran onto the nanoparticle surface, forming a hydrophobic shell. This process is reversible, as the photoacid spontaneously returns to its zwitterionic form and detaches from the surface of AuNPs in the dark. Consequently, the cationic AuNPs undergo reversible light-induced assembly/disassembly cycles, as shown experimentally in Fig. 1. | ||
| Scheme 1 Light-induced hydrophobization of cationic gold nanoparticles. (a) Reversible switching between the zwitterionic form and anionic form of the photoacid. (b) Cationic AuNPs. (c) Light-induced adsorption of the anionic photoacid on the cationic AuNPs. | ||
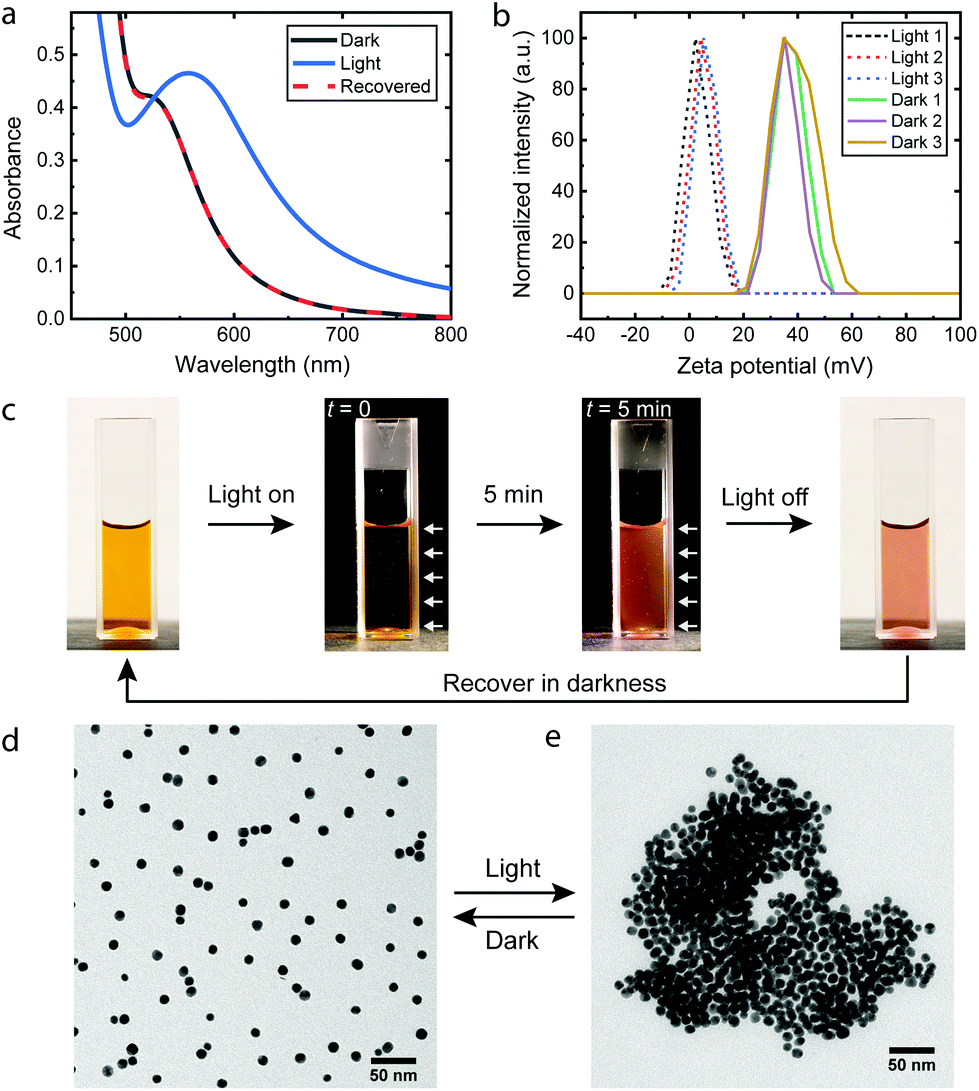 | ||
| Fig. 1 Light-induced reversible assembly of 10.9 nm cationic AuNPs. (a) UV-Vis spectra of AuNP/photoacid solution before and after irradiation. (b) Zeta potential change of AuNPs upon three cycles of irradiation. Irradiation: table lamp for 5 min. (c) Photographs of the AuNP/photoacid solution in a disposable cuvette. White arrows: direction of irradiation. (d), (e) Representative TEM images of cationic AuNPs in the dark and after irradiation. | ||
The AuNPs were synthesized by a seed-mediated method in the presence of tannic acid27 and subsequently modified by the cationic ligand (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide (MUTAB) by a two-step phase transfer method.28 The average diameter is 10.9 ± 1.2 nm as determined by transmission electron microscopy (TEM) (Fig. S1†). The AuNPs were dispersed in an aq. 0.2 mM photoacid solution, where the pH was adjusted to 2.70 with hydrochloric acid. The acidic conditions were chosen for two reasons. First, the stability of the photoacid in aqueous solutions is significantly enhanced by acidification,29 which allows storage of the solution in a fridge for several weeks. Second, the low pH suppresses the partial deprotonation of the merocyanine form23 and improves the reversibility of the assembly. In principle, reversible assemblies of AuNPs can be achieved in the pH range between 2.3 and 3.2 (Fig. S2†). At pH 2.70, irradiation only causes a minimal decrease of the pH to 2.66, close to the theoretical value (2.659) of full deprotonation of the photoacid. The ratio of photoacid molecules in the solution to cationic ligands on the AuNPs is estimated to be 10, assuming that each ligand occupies 0.214 nm2.15,30 Comparable results are achieved in the ratio range between 15 and 1.5 (Fig. S3†), demonstrating the robustness of the process. The cationic AuNPs are stable in photoacid solutions as proven by the plasmonic band around 520 nm and the zeta potentials of +37.2 ± 6.0 mV shown in Fig. 1a and b. After irradiation for 5 min, the UV-Vis spectrum shows a shifted plasmonic band around 557 nm and significantly increased absorbance above 520 nm, both indicating the formation of AuNP assemblies.3,14,31 Remarkably, the zeta potential of the AuNPs drops to +4.8 ± 4.3 mV, which confirms the adsorption of the anionic photoacid and neutralization of the surface charges. Once the irradiation is switched off, the photoacid molecules gradually desorb from the AuNPs and the electrostatic repulsion is restored. Consequently, the AuNP aggregates disassemble within one hour, as seen from the recovered UV-Vis spectrum and the zeta potential measurements shown in Fig. 1a and b. This assembly/disassembly process is highly reversible and also applicable for smaller AuNPs (5.1 nm), see Fig. S4 and S5.† Furthermore, the AuNPs/photoacid solution can be stored at room temperature for at least one week without significant changes of the response (Fig. S6†). In principle, other positively charged ligands, such as amine-terminated organothiols (Fig. S7†), can also be used for the hydrophobization process, showing the generic applicability of the approach.
Fig. 1c shows the photoacid solution containing AuNPs upon irradiation. A dark background was used to highlight the scattering of light during the irradiation. Due to the small size, the dispersed AuNPs show negligible scattering32 and therefore appears to be dark at the beginning of irradiation. After 5 minutes, aggregates of AuNPs are formed, which give rise to enhanced scattering of light and a bright appearance of the solution. The assemblies can be directly observed by TEM as shown in Fig. 1d, e and S8.† In the dark, the AuNPs are well dispersed, compared to the polydisperse aggregates after irradiation, which is commonly observed for destabilized nanoparticles.14,33–35
The ability to control the kinetics of transient assemblies has been a constant pursuit in out-of-equilibrium and dynamic systems.12,36,37 We next demonstrate that the disassembly kinetics of the metastable AuNP assemblies can be readily controlled by temperature. The kinetics of the AuNP disassembly depends on how fast the photoacid molecules desorb from the AuNP surface in the dark, as a result of the spontaneous recovery of the photoacid from the anionic spiropyran to zwitterionic merocyanine. This temperature-dependent process of the photoacid recovery can be followed by the change of absorbance of the aqueous photoacid solution at 420 nm as shown in Fig. 2a and S9.† Here a 0.1 mM solution was used due to the high absorbance of the photoacid. The recovery kinetics can be characterized by the half-life of the process (Fig. 2b), which increases from less than 10 s at 50 °C to almost 20 min at 15 °C. On the other hand, a strong dependence on temperature can be observed for the AuNP disassembly process, shown by the change of absorbance at 800 nm in Fig. 2c. For the range between 15 °C and 30 °C, the absorbance continues to increase after the irradiation has been removed. This could be the result of the slow photoacid recovery that leads to further assembly of the AuNPs. The time needed to reach the onset of re-dispersion is defined as the recovery time (t1) indicated in Fig. 2c. The recovery time demonstrates a linear relationship with the half-life of photoacid recovery (Fig. 2d), indicating the correlation between the two processes.
 | ||
| Fig. 2 Temperature-controlled transient stability of AuNP assemblies. (a) Recovery of the absorbance at 420 nm of a 0.1 mM photoacid solution after 5 min irradiation. (b) Half-life of the recovery process in (a). Dashed curve: Exponential fitting. (c) Change of absorbance at 800 nm of the AuNP/photoacid mixture after 5 min irradiation. The recovery time (t1) is exemplified for 40 °C. (d) The recovery time of the AuNPs vs. the half-life of the photoacid recovery. Dashed line: linear fitting. Irradiation for (a) and (c): 25 mW cm−2 at 455 nm. Irradiation stops at t = 1 min. | ||
Conventionally, the transfer of the nanoparticles between different solvent phases is achieved by surface modification such as ligand exchange, or adsorption of charged surfactants.9,10,38,39 However, the transfer is normally irreversible and sometimes accompanied by partial aggregation. Also light-controlled host–guest chemistry can be used, but sacrificing the generality and limiting its applicability only to small particles.13 We show that the solubility of the 10.9 nm AuNPs in water or a non-polar solvent (toluene) can be switched by light as shown in Fig. 3 (see Fig. S10† for 5.1 nm AuNPs). The AuNPs remain unchanged during repeated phase transfers, since the ligand (MUTAB) protection is kept intact throughout the process. In the dark, the AuNPs are positively charged and thus prefer to stay in the aqueous phase. Upon irradiation, the neutralized and hydrophobized AuNPs can be easily extracted into toluene by shaking. The transferred AuNPs in the toluene phase are stable as confirmed by the typical reddish color and TEM (Fig. S11†). In the dark, the AuNPs gradually regain positive charge and migrate back into the aqueous phase by gentle shaking. The time scale of the transfer is also temperature dependent. At 50 °C, less than 10 minutes are needed for the complete transfer of AuNPs back to the aqueous phase, compared to 1 hour at room temperature. The phase transfer process is remarkably quantitative: 97–98% of the AuNPs are transferred between the two phases as shown by the UV-Vis spectra in Fig. 3b and c. This ability to transfer the AuNPs between different phases can be potentially utilized for applications such as purification of colloids, as illustrated in Fig. S12.†
 | ||
| Fig. 3 Light-induced reversible transfer of cationic AuNPs between two solvents. (a) Photographs showing the transfer of AuNPs between aqueous and toluene phases. (b) UV-Vis spectra of the aqueous phase in the dark and after irradiation. (c) UV-Vis spectra of the toluene phase in the dark and after irradiation. Irradiation: 25 mW cm−2 at 455 nm, 5 min. | ||
The wettability of a surface is a highly relevant property for various applications. Although the possibility to reversibly switch the wettability has been shown,40in situ control of the contact angle is still challenging, requiring spontaneous flow of the residing droplet. Here we show that a macroscopic surface can also be hydrophobized (Fig. 4), as a further demonstration of the principle's versatility. A 150 nm gold film was evaporated on a silicon substrate (Fig. S13†), which was further modified by MUTAB. As shown in Fig. 4b and c, the contact angle of a photoacid solution on this surface is close to 0° before irradiation, as expected for a highly polar surface. Upon irradiation, the contact angle starts to increase within a few seconds and reaches 60.1 ± 0.7° in 50 s (Movie S1†), determined from 5 substrates. As the control, a bare gold surface shows no response upon irradiation, where a constant contact angle of 20° was observed. Intriguingly, the contact angle on the cationic surface did not show observable recovery in the dark, which may be attributed to the fact that the hydrophobized area (i.e. initial area covered by the droplet) is much larger than the area covered by the final droplet. The photoacid molecules on the dewetted surface could not detach from the surface due to the lack of contact with the liquid, and the surface surrounding the final droplet therefore remains hydrophobic.
 | ||
| Fig. 4 (a) Schematic illustration of the hydrophobization and change of the contact angle on a cationic gold surface. (b) Change of the contact angles of a photoacid solution (0.2 mM, pH 2.70) on a bare and a cationic gold surface upon irradiation. (c) Photographs of the photoacid solution on a cationic gold surface upon irradiation (455 nm, 8 mW cm−2) starting at 0 s. | ||
Conclusions
To sum up, we introduce the light-induced reversible hydrophobization of cationic AuNPs via electrostatic adsorption of an anionic photoacid, in contrast to previously reported methods using covalently grafted ligands or pH changes caused by the photoacid. The hydrophobization causes transient assembly of AuNPs in the aqueous phase, and the disassembly process can be kinetically controlled by temperature. This feature could be relevant in programming the lifetimes of assemblies in out-of-equilibrium and dissipative systems. The light-induced switching between cationic and hydrophobic states can be used to reversibly transfer the AuNPs between an aqueous and a non-polar organic phase with high efficiency, potentially useful for purification applications. This principle can also be extended to macroscopic surfaces, where the contact angle changes dramatically in situ from 0° to 60° upon irradiation. The proposed mechanism is generic and opens up a new way to control the surface properties and self-assembly of nanoparticles. Future work will focus on the extension to other colloidal systems and new application possibilities.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was carried out with the financial support from the Academy of Finland Centre of Excellence Programme (HYBER), European Research Council grant ERC-2016-CoG (725513-SuperRepel) and Advanced Grant DRIVEN (680083). We thank Dr Claudia Pigliacelli for preliminary review of the manuscript. We acknowledge the provision of facilities and technical support by Aalto University at OtaNano – Nanomicroscopy Center (Aalto-NMC).Notes and references
- M. C. M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- H. Guo, B. Xing, J. C. White, A. Mukherjee and L. He, Analyst, 2016, 141, 5261–5264 RSC.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef.
- S. M. Majedi, B. C. Kelly and H. K. Lee, Anal. Chim. Acta, 2013, 789, 47–57 CrossRef CAS.
- B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS PubMed.
- G. T. Wei, Z. Yang, C. Y. Lee, H. Y. Yang and C. R. C. Wang, J. Am. Chem. Soc., 2004, 126, 5036–5037 CrossRef CAS.
- J. Yang, J. Y. Lee and J. Y. Ying, Chem. Soc. Rev., 2011, 40, 1672–1696 RSC.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS.
- P. K. Kundu, S. Das, J. Ahrens and R. Klajn, Nanoscale, 2016, 8, 19280–19286 RSC.
- L. Peng, M. You, C. Wu, D. Han, I. Öçsoy, T. Chen, Z. Chen and W. Tan, ACS Nano, 2014, 8, 2555–2561 CrossRef CAS.
- D. Samanta and R. Klajn, Adv. Opt. Mater., 2016, 4, 1373–1377 CrossRef CAS.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- S. Palagi, A. G. Mark, S. Y. Reigh, K. Melde, T. Qiu, H. Zeng, C. Parmeggiani, D. Martella, A. Sanchez-Castillo, N. Kapernaum, F. Giesselmann, D. S. Wiersma, E. Lauga and P. Fischer, Nat. Mater., 2016, 15, 647–653 CrossRef CAS.
- H. Zhang, A. Mourran and M. Möller, Nano Lett., 2017, 17, 2010–2014 CrossRef CAS PubMed.
- H. Zeng, O. M. Wani, P. Wasylczyk and A. Priimagi, Macromol. Rapid Commun., 2018, 39, 1700224 CrossRef.
- M. P. M. Dicker, A. B. Baker, R. J. Iredale, S. Naficy, I. P. Bond, C. F. J. Faul, J. M. Rossiter, G. M. Spinks and P. M. Weaver, Sci. Rep., 2017, 7, 9197 CrossRef.
- B. I. Ipe, S. Mahima and K. G. Thomas, J. Am. Chem. Soc., 2003, 125, 7174–7175 CrossRef CAS.
- M. Ikbal, D. Balogh, E. Mervinetsky, R. Sfez and S. Yitzchaik, J. Phys. Chem. C, 2017, 121, 27176–27181 CrossRef CAS.
- Y. Shiraishi, E. Shirakawa, K. Tanaka, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Appl. Mater. Interfaces, 2014, 6, 7554–7562 CrossRef CAS.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS.
- D. Go, D. Rommel, Y. Liao, T. Haraszti, J. Sprakel and A. J. C. Kuehne, Soft Matter, 2018, 14, 910–915 RSC.
- C. Maity, W. E. Hendriksen, J. H. Van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998–1001 CrossRef CAS.
- P. Xue, J. Ding, M. Jin and R. Lu, J. Mater. Chem. C, 2017, 5, 5299–5303 RSC.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- J. Hassinen, V. Liljeström, M. A. Kostiainen and R. H. A. Ras, Angew. Chem., Int. Ed., 2015, 54, 7990–7993 CrossRef CAS.
- N. Abeyrathna and Y. Liao, J. Phys. Org. Chem., 2017, 30, e3664 CrossRef.
- C. M. Sorensen, in Nanoscale Materials in Chemistry, John Wiley & Sons, Inc, Hoboken, New Jersey, 2009 Search PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS.
- X. Liu, Y. Chen, H. Li, N. Huang, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS.
- S. Lotfizadeh, H. Aljama, D. Reilly and T. Matsoukas, Langmuir, 2016, 32, 4862–4867 CrossRef CAS.
- I. I. S. Lim, D. Mott, W. Ip, P. N. Njoki, Y. Pan, S. Zhou and C. J. Zhong, Langmuir, 2008, 24, 8857–8863 CrossRef CAS.
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS.
- K. S. Mayya and F. Caruso, Langmuir, 2003, 19, 6987–6993 CrossRef CAS.
- T. Zhang, J. Ge, Y. Hu and Y. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef CAS.
- X. Feng, L. Feng, M. Jin, J. Zhai, L. Jiang and D. Zhu, J. Am. Chem. Soc., 2004, 126, 62–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr05416b |
| This journal is © The Royal Society of Chemistry 2019 |