
Cu and Zn interactions with Aβ peptides: consequence of coordination on aggregation and formation of neurotoxic soluble Aβ oligomers
Monika
Rana
and
Anuj Kumar
Sharma
 *
*
Department of Chemistry, Central University of Rajasthan, NH-8, Bandarsindri, Ajmer 305817, India. E-mail: anuj.sharma@curaj.ac.in
First published on 14th September 2018
Abstract
The coordination chemistry of transition metal ions (Fe, Cu, Zn) with the amyloid-β (Aβ) peptides has attracted a lot of attention in recent years due to its repercussions in Alzheimer's disease (AD). Aβ peptide undergoes self-aggregation to form amyloid plaques and soluble oligomers, which are believed to play a central role in AD pathology. Metal ions and Aβ-metal adducts can generate toxic radical species capable of modifying biomolecules, ultimately causing death of the neuronal cells. The impact of these metal ions on Aβ aggregation and neurotoxic species formation is still not well understood. A large number of reports indicate that transition metal ions such as Cu and Zn have significant effects on Aβ peptide aggregation and stabilization of neurotoxic soluble Aβ aggregates; some of these reports are contradictory too. A review on the effects of Cu and Zn metal ions on Aβ peptide aggregation due to their coordination with these peptides is presented herein. The review includes brief discussions with regards to the levels of Cu and Zn in an AD affected brain, structures of Aβ peptides, and the coordination of Aβ peptides with Cu and Zn. In the current scenario, a consensus appears to be emerging regarding the coordination mode and the effect of metal ions (especially Cu and Zn) on the aggregation of Aβ peptides. Towards the end, a short discussion on current research trends in the field of bifunctional metal chelators for tackling the metal effect on Aβ aggregation and toxicity has also been presented. It is expected that this review would aid in gaining a better understanding of the current status of various aspects of AD from an inorganic chemistry perspective.
 Monika Rana | Monika Rana received her BSc (Honours) degree in Chemistry from Miranda House, University of Delhi in 2013 and her MSc degree in Chemistry from the Central University of Rajasthan in 2015 which included a dissertation entitled “Ascertaining the fluoride ions by selective and sensitive Chemosensors”. Currently, she is pursuing her PhD from the same university with Dr Anuj K. Sharma as her supervisor. Her research interest mainly includes synthesis of multifunctional chelators and studying their coordination chemistry and biological properties with relevance to metal-induced neurodegenerative disorders, specifically Alzheimer's disease. |
 Anuj Kumar Sharma | Dr Anuj K. Sharma obtained his MSc in Chemistry from the Indian Institute of Technology, Roorkee, in 2004 and PhD from the Indian Institute of Technology, Kanpur with Prof. Rabindranath Mukherjee in September 2009. Then he joined as postdoctoral fellow at Washington University in St. Louis, USA, with Prof. Liviu M. Mirica. During his post doc he worked on metal ion–amyloid interactions and to develop multifunctional compounds for controlling metal mediated problems (aggregation, neurotoxicity etc.) in Alzheimer's disease. He also worked on development of 64Cu based PET imaging agents for early detection of Alzheimer's disease. Currently, he is an Assistant Professor of Chemistry at the Central University of Rajasthan, India. |
1. Introduction
Alzheimer's disease (AD), first described in 1906, is a degenerative brain disorder and the most common cause of dementia in elderly people.1 AD has become a major socio-economic problem in many countries as the patients need constant attention from their caregivers. It is currently affecting more than 30 million people worldwide and more than 100 million people are estimated to be affected by 2050, mainly due to the rapidly escalating elderly population in developing countries (http://www.alz.co.uk). While many countries already have a significant level of ageing population, some of the developing countries like India have ∼70% of their population in the 30–40 years age bracket, and may be expected to face serious AD related problems in a few decades' time. The scientific community appears to have taken note of this enormous challenge as evidenced by the sharp increase in the number of publications in AD research in the recent past.The causes of AD as well as other neurodegenerative disorders are not well understood. The disease is generally characterized by the presence of extracellular senile plaques, intracellular tangles and altered levels of neurotransmitters in the brain of those affected with it.2,3 Senile plaques, commonly known as amyloid plaques, are composed of insoluble aggregated peptides called amyloid-β (Aβ), which are constituted of 39–43 amino acid residues.4 The Aβ peptides are formed after sequential cleavage of the APP by the proteolytic enzymes α-, β- and γ-secretase.5,6 Formation of aggregates of these peptides into amyloid plaques (amyloid cascade hypothesis2) or smaller soluble oligomeric species (the oligomer hypothesis7) is believed to play a key role in AD pathology. It remains a topic of debate as to which of the two forms – the final form of aggregates, the amyloid plaques, or the initial smaller soluble oligomers – is more toxic. However, according to recent research, diffused smaller oligomeric aggregates are suggested to have higher neurotoxicity than the fully grown amyloid fibrils.7–12
The inorganic chemistry aspects of Alzheimer's disease (AD) have certainly caught the attention of chemists in recent years.4,13–15 Post-mortem examination of the brain of AD affected patients suggested that metal ions such as Cu(II), Fe(III), and Zn(II) were found in high concentrations (∼400, ∼950, and ∼1100 μM, respectively) within the core and periphery of senile plaque deposits.16,17 Many of these transition metal ions can be found in healthy individuals too, as the human body has an elaborate system for managing and regulating the amount of these key metals circulating in the blood and stored inside the cells. Metal ions are believed to play an important role in the peptide aggregation and misfolding mechanisms.18–20 However, these interactions and mechanisms are not completely understood even today.8,9,21 Many amyloid forming proteins can interact with the transition metal ions such as zinc, copper and iron, and this binding can bring about changes to the structures and functions of these amyloidogenic proteins.4,22,23 In an improperly functioning system, the ratios of these metal ions reach abnormal levels, a condition that is generally known as metal ion dyshomeostasis.4 Under these conditions, the levels of copper and iron, both redox metals, increase in the brain of the affected people, normally aged ones. Some reports have shown these metal ions to promote Aβ aggregation as well as take part in the formation of reactive oxygen species (ROS), leading to oxidative stress.24 The redox nature of these metal ions and the presence of oxygen in our body lead to ROS formation in Fenton type reactions.22,25,26 These ROS can cause oxidative modifications of biomolecules, especially in the brain, in the case of AD conditions.27 The formation of misfolded protein aggregates and the role of metal ions in this misfolding process have led to the hypothesis in AD that interactions between metal ions and amyloidogenic protein are connected to metal dyshomeostasis.28,29
Metal ions are believed to coordinate with the hydrophilic part of amyloid forming peptides. The coordination chemistry of these peptides with transition metal ions, especially Fe, Cu and Zn, is well documented. There are several well written research articles in the literature lucidly describing the Aβ–M (M = Fe, Cu, Zn) coordination modes studied using NMR and other spectroscopic techniques.19,20,30–34 Another aspect of metal–Aβ interaction is the effect that it has on aggregation processes and formation of metal-associated species. There have been concerted efforts by some research groups to understand the nature of these metal affected aggregates of Aβ peptides. In this regard, a careful study reveals that the experimental conditions and methods used to characterize the aggregates formed vary from one research group's report to another.9 Further, summarizing the studies on metal ions affecting the aggregation of Aβ peptides has not been as extensive in comparison to the reviews on other aspects related to AD research. We have therefore chosen to focus on this aspect and review the excellent efforts that have been carried out by researchers in this regard. The primary focus of this report shall thus revolve around metal ions affecting the aggregation process of Aβ peptides, especially the longer and more neurotoxic Aβ1–42 peptide. Aβ1–42 is established as more neurotoxic than Aβ1–40 and is also more likely to generate hydrogen peroxide, which has been proposed to contribute to cytotoxicity.35,36 In addition to this aspect, a brief discussion has also been presented on metal–Aβ coordination to set the stage for various explanations. Towards the end, the possible treatment/strategies suggested for combating the problems related to metal ions in Alzheimer's disease are also presented.
2. Formation, structure and functions of Aβ peptides.
The Aβ peptides consist of a hydrophilic N-terminus and a hydrophobic C-terminus2,13 and form after sequential cleavage of the Amyloid Precursor Protein (APP), the post-translationally modified protein containing 770 residues.37–39 Three proteolytic enzymes α-, β- and γ-secretase cleave APP at different sites to produce Aβ peptides comprising 39–43 amino acid residues. The β-secretase cleaves APP to form the N-termini, whereas the γ-secretase does so to generate the C termini of Aβ leading to 39–43 variants (Fig. 1). These Aβ peptides undergo self-aggregation as well as metal induced aggregation, forming soluble Aβ oligomers and insoluble amyloid plaques. The normal function of Aβ is not well understood. Although some animal studies have shown that the absence of Aβ does not lead to any obvious loss of physiological functions,40,41 several potential activities have been discovered for Aβ, including activation of kinase enzymes,42 protection against oxidative stress,43,44 and anti-microbial activity that may potentially be associated with Aβ's pro-inflammatory activity.45 Aβ is the main component of amyloid plaques, the extracellular deposits found in the brains of patients with Alzheimer's disease.41,46 Aβ1–40 is reported to be more abundant as compared to the longer version i.e., Aβ1–42. The Aβ1–40/Aβ1–42 ratio in the soluble fractions was suggested to be ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the soluble fractions across the cortex in AD brains.47 However, some recent reports describing the biochemical distribution and accumulation of Aβ are contradictory and indicate that Aβ1–42 is more abundant than Aβ1–40.48 The ratio of Aβ1–40/Aβ1–42 in plaques was suggested to be ∼1:2; Aβ1–42 is considered more toxic as compared to Aβ1–40, displaying a higher propensity to aggregate, and initiate Aβ aggregation.20,49–51 The increased proportion of Aβ1–42, which is more prone than Aβ1–40 to aggregate is thought to initiate the disease process.52,53 Aβ1–42 is known to form soluble neurotoxic oligomers, whereas it has been suggested that Aβ1–40 does not form such oligomers.9,54 The sites of action of α-, β, and γ-secretases on APP and the amino acid sequence for Aβ1–42 peptide fragments are shown in Fig. 1. Amino acids which participate in metal binding are shown in blue and bold in the sequence of the full length Aβ1–42.
:1 in the soluble fractions across the cortex in AD brains.47 However, some recent reports describing the biochemical distribution and accumulation of Aβ are contradictory and indicate that Aβ1–42 is more abundant than Aβ1–40.48 The ratio of Aβ1–40/Aβ1–42 in plaques was suggested to be ∼1:2; Aβ1–42 is considered more toxic as compared to Aβ1–40, displaying a higher propensity to aggregate, and initiate Aβ aggregation.20,49–51 The increased proportion of Aβ1–42, which is more prone than Aβ1–40 to aggregate is thought to initiate the disease process.52,53 Aβ1–42 is known to form soluble neurotoxic oligomers, whereas it has been suggested that Aβ1–40 does not form such oligomers.9,54 The sites of action of α-, β, and γ-secretases on APP and the amino acid sequence for Aβ1–42 peptide fragments are shown in Fig. 1. Amino acids which participate in metal binding are shown in blue and bold in the sequence of the full length Aβ1–42.
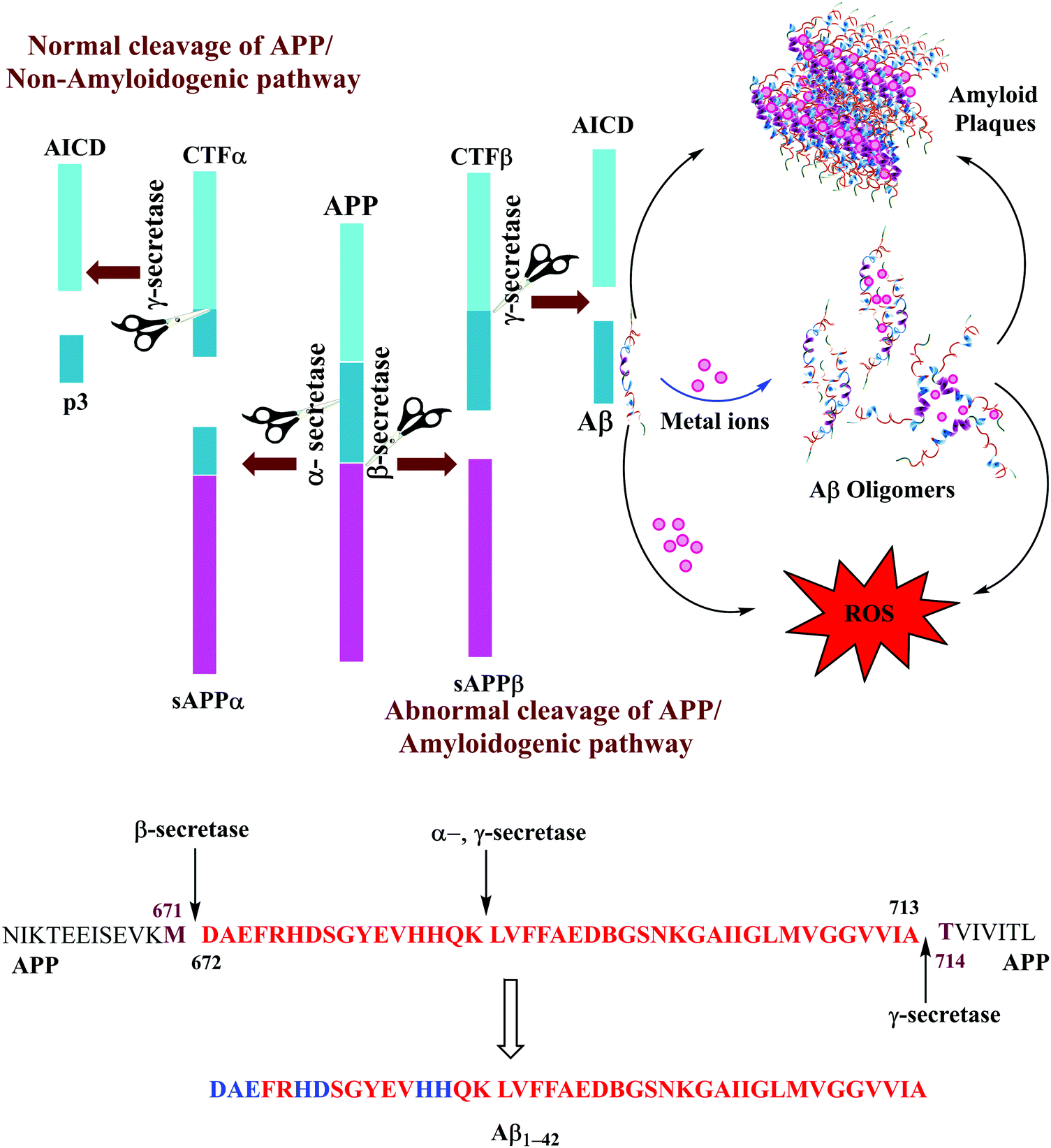 | ||
| Fig. 1 (top) Normal (α-, γ-secretases) and abnormal (β-, γ-secretases) cleavage pathways of APP. Aβ are formed by an abnormal mode of APP cleavage. (bottom) Showing the sites of action of α-, β, and γ-secretases on APP and amino acid sequence for Aβ1–42 peptide fragments (in red); amino acids that participate in metal binding are shown in blue and bold. | ||
3. Cu and Zn levels in Alzheimer's disease
Initial reports suggested that zinc (Zn), and copper (Cu) concentrations in cerebrospinal fluid (CSF) and serum were found increased in patients affected with neurodegenerative diseases. These concentrations were determined with atomic absorption spectrophotometry in 74 patients suffering from various neurological diseases, and in 28 healthy controls.55 Another report by Basun et al. indicated no observable changes in CSF Zn concentrations; however a 2-fold increased level of Cu in CSF was reported.56 Molina et al. reported that the CSF Zn level is significantly decreased in AD when compared to age-matched controls.57 Deibel et al. studied the levels of Cu, Fe, and Zn in five different brain regions (amygdala, hippocampus, inferior parietal lobule, superior and middle temporal gyri, and cerebellum) by instrumental neutron activation analysis (INAA) in samples from Alzheimer's disease (AD) patients and age-matched control subjects. They found a significant decrease in Cu, and significant increases in Zn and Fe in AD hippocampus and amygdala, areas showing severe histopathologic alterations in AD.58 A few other studies observed a significant low level of brain Cu in AD, including some recent reports.58–60 X-ray fluorescence microprobe studies have indicated that deposits of copper and zinc co-localize with amyloid plaques in human AD brain tissue and amyloid plaque rim.17,61 Other techniques like ICP-MS in combination of flow injection on amyloid plaques of Alzheimer's disease-affected brains suggested that the concentrations of Cr, Mn, Ni, Cu, and Pb were in the 0.2–0.8 mg L−1 range, whereas that of Al, Fe, and Zn were 2–20 mg L−1 in the plaque sample.62 Later in 2009, Rajendran et al. suggested that it is more accurate to combine the three ion beam techniques of scanning transmission ion microscopy, Rutherford back scattering spectrometry and particle induced X-ray emission in conjunction with a high energy (MeV) proton microprobe to determine the concentration of metal ions in amyloid plaques. Their data also suggested increased metal concentrations within the amyloid plaques compared with the surrounding tissues.63 Roberts et al. described that Cu levels in AD-affected cortical tissues were decreased compared to normal age neuropil.64 They suggested that due to Cu association in plaques, the remaining brain tissues have a lower metal concentration. Further, they also reported variations in Zn concentration in different tissues of the brain. Miller et al. used synchrotron Fourier transform infrared micro-spectroscopy (FTIRM) to image the in situ secondary structure of the amyloid plaques in brain tissue of AD patients. They showed a strong spatial correlation between elevated β-sheet content in Aβ plaques and accumulated Cu and Zn ions, emphasizing an association of metal ions with amyloid formation in AD.61 The same group later compared the metal content in the plaques and the iron speciation in the cortex of three mouse models, two of which had significant neurodegeneration and one with very little neurodegeneration.65 The content and speciation of Fe, Cu and Zn were determined by synchrotron X-ray fluorescence microscopy (XFM) and X-ray absorption spectroscopy (XAS), respectively. The plaques from the mouse models with significant neurodegeneration contained ∼25% more Cu as compared to the one with little neurodegeneration, indicating that redox-active metal ions may be inducing oxidative damage and influencing neurodegeneration.61 In all these studies, the most common observation was the presence of a higher concentration of metal ions in amyloid plaques. The minor differences in results may be speculated as due to the source of samples, human AD brain tissues or tissues from mouse models, different techniques used or even due to the number of samples. Becker et al. has elegantly summarized the pros and cons of various techniques used for detection of metals in biological tissues.66 The authors mentioned that “most analytical techniques for the visualization of inhomogeneous distributions of elements in tissues (e.g., histological or histochemical staining techniques) possess poor detection limits, that means, no element can be detected at trace and ultra-trace concentrations.” They suggested that other analytical techniques such as scanning electron microscopy with energy disperse X-ray analysis, microproton-induced X-ray emission and autoradiography also cannot be used for trace element detection due to their low detection power. Secondary ion mass spectrometry (SIMS) or laser ablation inductively coupled plasma mass spectrometry (LA-ICPMS) have been described as sensitive surface analytical techniques for the determination of element distribution on the surfaces of biological samples.67,68 However, it is out of the scope of this review to compare each technique. In fact, we do not feel competent enough to comment on the pros and cons of some of the techniques. It is worth mentioning here that the content of metal ions in the brain is rigorously regulated, as is their movement across the BBB. The increased implications of metal ions in interactions with the major protein components of neurodegenerative diseases is not merely due to increased exposure to metals, but rather due to a breakdown in the homeostatic mechanisms that compartmentalize and regulate metals.694. ROS production by redox metal ions in AD
Elevated levels of redox active metal ions (Cu/Fe) may result in the generation of reactive oxygen species (ROS), DNA damage, and mitochondrial dysfunction.70 These metal ions are known to cause neuronal cell damage by promoting the production of reactive oxygen species (ROS)/nitrogenated species (RNS) species bearing unpaired electrons, mainly represented by superoxide anion radical, hydroxyl, alkoxyl and lipid peroxyl radicals, nitric oxide and peroxynitrite.17,49,69 Oxidative stress is an imbalance in the generation and clearance of ROS, which are otherwise an inherent part of healthy metabolism, eventually ending up in the unusual degradation of biomolecules. Fenton type chemistry has been proposed to play roles in production of ROS.71–74 Redox active metal ions, in both the free and loosely bound forms, are well known to be involved in the formation of ROS species via the Fenton reaction (Scheme 1: reactions (1) and (2)) and the Haber–Weiss reaction (Scheme 1: reaction (3)) as shown below.75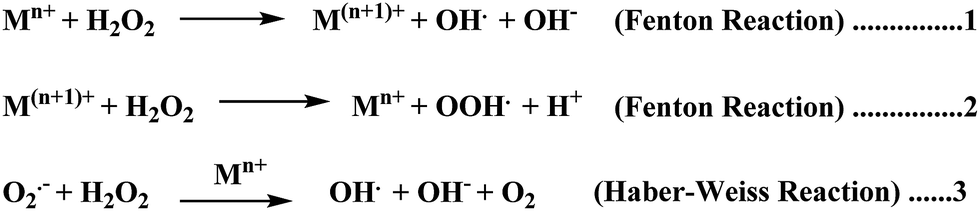 | ||
| Scheme 1 | ||
Fe and Cu are both known to coordinate with Aβ peptide, and such Fe–Aβ and Cu–Aβ species have been implicated in the production of ROS. However, ROS production has been more commonly studied with Cu–Aβ, since Fe–Aβ has a lower redox activity.71,76 Indeed, in vitro studies suggested that Cu–Aβ is able to catalyze the production of H2O2 and HO˙ in the presence of a biologically relevant reducing agent such as ascorbate and dioxygen.8,77 There have also been discussions on whether Aβ is an anti-oxidant or pro-oxidant. It is considered as an antioxidant on the basis of the fact that free Cu catalyzes ROS production more efficiently than Cu–Aβ. However, “free” Cu concentrations in biology are extremely low; Nature has evolved a complex system of metal transporters and chaperones that tightly regulate metal homeostasis, and pools of ‘free’ metal ions, which in a biological context refers to metal ions bound by labile, low affinity, and therefore, readily exchangeable, ligands, are rare. Derreumaux et al. has suggested that an antioxidant activity of Aβ compared to “free” copper might be relevant only under particular conditions where free Cu reaches much higher concentrations.78 However, chances for such a situation to occur are rare when taking into account the presence of high concentrations of potential ligands (such as glutathione, histidine, cysteine, etc.). When there is an imbalance of Cu, it would be more relevant to compare the efficiency of Cu–Aβ with that of the Cu pool from which Aβ obtains Cu in AD instead of free Cu.78 A catalytic role of the Cu–Aβ complex in various redox reactions is depicted in Fig. 2. Naturally occurring reducing agents, i.e. ascorbate, glutathione etc., reduce these metal ions to their lower oxidation states e.g., Cu(I) and Fe(II). These reduced form of metal ions in amyloid plaques then trigger the initiation of ROS production by reacting with molecular oxygen.75
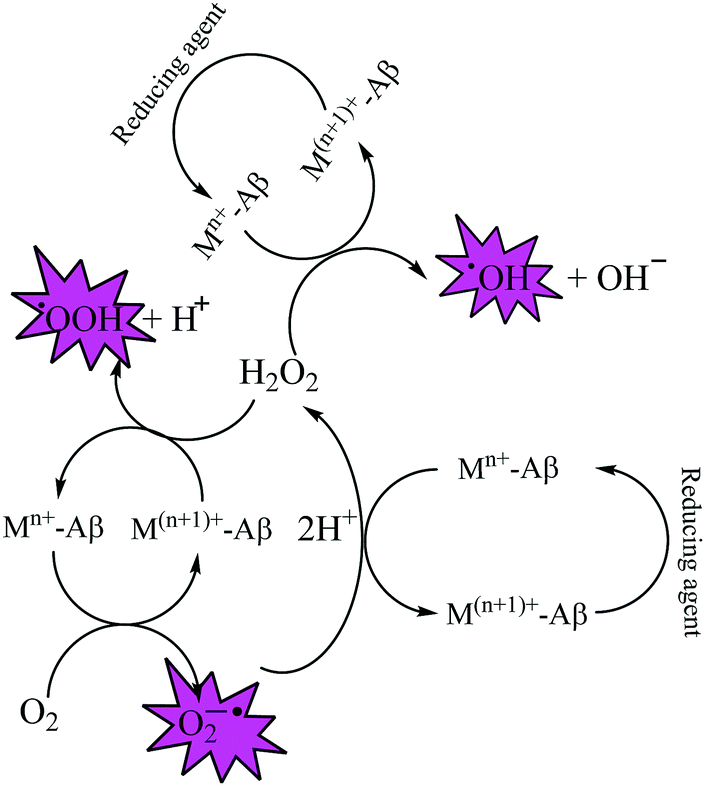 | ||
| Fig. 2 Depiction of the catalytic role of Cu–Aβ in ROS production by Fenton-type reactions. | ||
In addition, it is proposed that production of ROS by Cu–Aβ adducts leads to the oxidative damage of Aβ peptide itself and also the surrounding biomolecules.79 Sites for oxidative damage have been studied widely; Asp1, His13, His14, Try10 and phenylalanine have been identified as main targets which undergo oxidation leading to formation of various oxidative products.71 Dityrosine cross linking between two tyrosine residues is widely indicated as a marker for oxidative stress in different neurodegenerative disorders.80 Furthermore, in the redox atmosphere in amyloid plaques, binding of heme to Aβ peptides has also been reported to play a key role in AD.39,81–84
5. Coordination of Aβ with metal ions
Coordination chemistry of transition metal ions like Fe, Cu and Zn with Aβ peptide and its aggregates has been widely explored in recent years.85 There are several divergent reports on the coordinating amino acid residues of Aβ peptides that form the coordination sphere of metal ions. The binding of different amino acids to metal ions depends on various experimental conditions such as pH, buffer medium and temperature etc.86,87 Numerous studies have investigated the metal coordination environment of Aβ–Mn+ complexes; very few reports have studied the full-length Aβ40/Aβ42 peptides,88,89 shorter non-aggregating forms like Aβ1–16 or Aβ1–28 being used more commonly.20,32,34,89,90 The EPR spectra of the Cu(II) complexes with Aβ1–16/1–28 and Aβ1–40 are similar, suggesting that the high affinity metal binding site lies on the N-terminus, and more specifically, in the first 16 amino acids.91–94 Due to this, Aβ1–16 has been considered as a decent model for studying coordination properties with metal ions.5.1. Copper(II) interactions with Aβ peptides
Cu(II) has a d9 electronic configuration, which is Jahn–Teller distorted to produce a tetragonal geometry dominated by square planar coordination modes.95,96 Cu(II) is known to bind Aβ using various coordination modes and the coordination sphere for these Aβ–M complexes is very dynamic.20 Different models of coordination have been proposed and recent review articles have summarized such models.4,23,71,72,85,97–99 Typical models involve three N donor atoms and at least one O donor, but, precisely which of the N/O donors are involved in the coordination is still debatable. Among the very early reports, Huang et al. in 1999 proposed a 3N1O model for copper coordination to Aβ peptide using the computer simulations of the experimental EPR spectrum and correlations with other Cu protein spectra.36 Curtain et al. in 2001 by means of NMR, EPR and CD spectroscopy supported the proposed 3N1O model for copper coordination to Aβ peptide with the involvement of three histidine units (His6, His13, His14). The fourth coordination by an oxygen could be a carboxyl or a hydroxyl side chain, water/hydroxo, or phosphate from the buffer, as suggested by EPR spectra of Aβ–Cu species.88 Karr and co-workers also concurred with this model, as EPR parameters obtained were consistent with a characteristic Type2 Cu(II) centre with three nitrogen and one oxygen donor ligands.59,89,91,100,101 Another study shed light into the importance of the N-terminus in metal binding, where it was observed that even small changes in the N-terminus affected the metal coordination and also indicated that the O donor atom does not involve tyrosine.89,91 Syme et al., using various spectroscopic techniques, studied the effect of pH on coordination of Cu(II) to Aβ and assigned a 4N coordination involving three histidine units and an N-terminus at physiological pH in a square planar geometry; by presumably deprotonating the amide nitrogen at a higher pH, they suggested that Tyr10 is not involved in coordination.86 Later, XAS and EXAFS studies assigned a model in which Cu has a five coordinate geometry to Aβ1–40, involving three nitrogen atoms from three histidine (His-6, His-13 and His-14) residues, one oxygen coordination from Tyr 10 and a second oxygen coordination possibly from water (or OH−) or from other amino acids.90,102 In 2008, Streltsov et al. also ruled out Tyr as the oxygen donor ligand and reported that the Cu(II) binds to Aβ in a distorted six-coordinated (3N3O) geometry. The coordination sphere comprises three histidine residues (His-6, His-13 and His-14), an oxygen (from glutamic or aspartic acid) and axial ligands that come from a water molecule and a carboxylate oxygen from either Glu-11 or Asp-1, in an almost equatorial planar arrangement.103 Drew and co-workers, who studied the site-specific labelling of amino acids together with continuous wave electron paramagnetic resonance (CW-EPR) and hyperfine sublevel correlation (HYSCORE), provided support for a 3N1O coordination environment. Their CW-EPR studies for Cu(II)/Aβ species at a comparatively lower pH (between 6 and 7), suggest two equally contributing forms in 3N1O coordinate modes intact upon the amino terminus, CO from Asp1–Ala2 and the imidazole side chain of His6, with the third nitrogen swapping between imidazole side chains of His13 and His14. The authors referred to these two forms as component Ia and component Ib. In component Ia, His13 provides the coordinating site, whereas His14 is the coordinating ligand for component Ib (Fig. 3, component I). At a higher pH of ∼8.0, a different coordination geometry was observed, which was referred to as component II. In this component, the coordination sphere was described comprising all three histidine residues, along with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group from the Ala2–Glu3 peptide bond, while the amino terminus no longer coordinates. Tyr10 does not provide a phenolate oxygen ligand in any of the modes.104–107 The proportions of the components I and II were described to be pH-sensitive;86,108 as the pH increases, the intensity of component II also increases, while that of component I decreases. Ghosh et al. described that Cu–Aβ shows an equilibrium between these two species in the range of pH 6.5–9.5 with a pKa of ∼8.1.109 Another report by Furlan et al. suggested the N terminus, N− (Asp1–Ala2), CO (Ala2–Glu3), His6 and a water molecule to be the Cu(II) coordinating ligands at higher pH (Fig. 3, component II).110 Further reports were divergent in the involvement of different amino acids in the coordination sphere of Cu(II) in component II.15,32,85,105–107 Another study suggested that Cu(II) is bound to the –NH2 group of Asp1 (amino terminus), the amidyl N− from the Asp1–Ala2 peptide bond, carbonyl from Ala2–Glu3 and with only one of the imidazole units from the three His residues.106 Silva et al. reported the presence of a single histidine by ESEEM analysis and determined that in component II, His13 and His14 (equal contribution) are more favoured as equatorial ligands compared to His6; they also quantified the contribution from each histidine, with His13 and His14 (each contributing 40% and His6 20% (Fig. 3)).111 The above contradiction in the coordination sphere can be attributed to the varying experimental conditions, which comprise the use of various buffers, pH, temperature and concentration of peptides etc. However, in spite of the contradiction, the coordinating amino acids are now understood to a major extent for such complex systems.
O group from the Ala2–Glu3 peptide bond, while the amino terminus no longer coordinates. Tyr10 does not provide a phenolate oxygen ligand in any of the modes.104–107 The proportions of the components I and II were described to be pH-sensitive;86,108 as the pH increases, the intensity of component II also increases, while that of component I decreases. Ghosh et al. described that Cu–Aβ shows an equilibrium between these two species in the range of pH 6.5–9.5 with a pKa of ∼8.1.109 Another report by Furlan et al. suggested the N terminus, N− (Asp1–Ala2), CO (Ala2–Glu3), His6 and a water molecule to be the Cu(II) coordinating ligands at higher pH (Fig. 3, component II).110 Further reports were divergent in the involvement of different amino acids in the coordination sphere of Cu(II) in component II.15,32,85,105–107 Another study suggested that Cu(II) is bound to the –NH2 group of Asp1 (amino terminus), the amidyl N− from the Asp1–Ala2 peptide bond, carbonyl from Ala2–Glu3 and with only one of the imidazole units from the three His residues.106 Silva et al. reported the presence of a single histidine by ESEEM analysis and determined that in component II, His13 and His14 (equal contribution) are more favoured as equatorial ligands compared to His6; they also quantified the contribution from each histidine, with His13 and His14 (each contributing 40% and His6 20% (Fig. 3)).111 The above contradiction in the coordination sphere can be attributed to the varying experimental conditions, which comprise the use of various buffers, pH, temperature and concentration of peptides etc. However, in spite of the contradiction, the coordinating amino acids are now understood to a major extent for such complex systems.
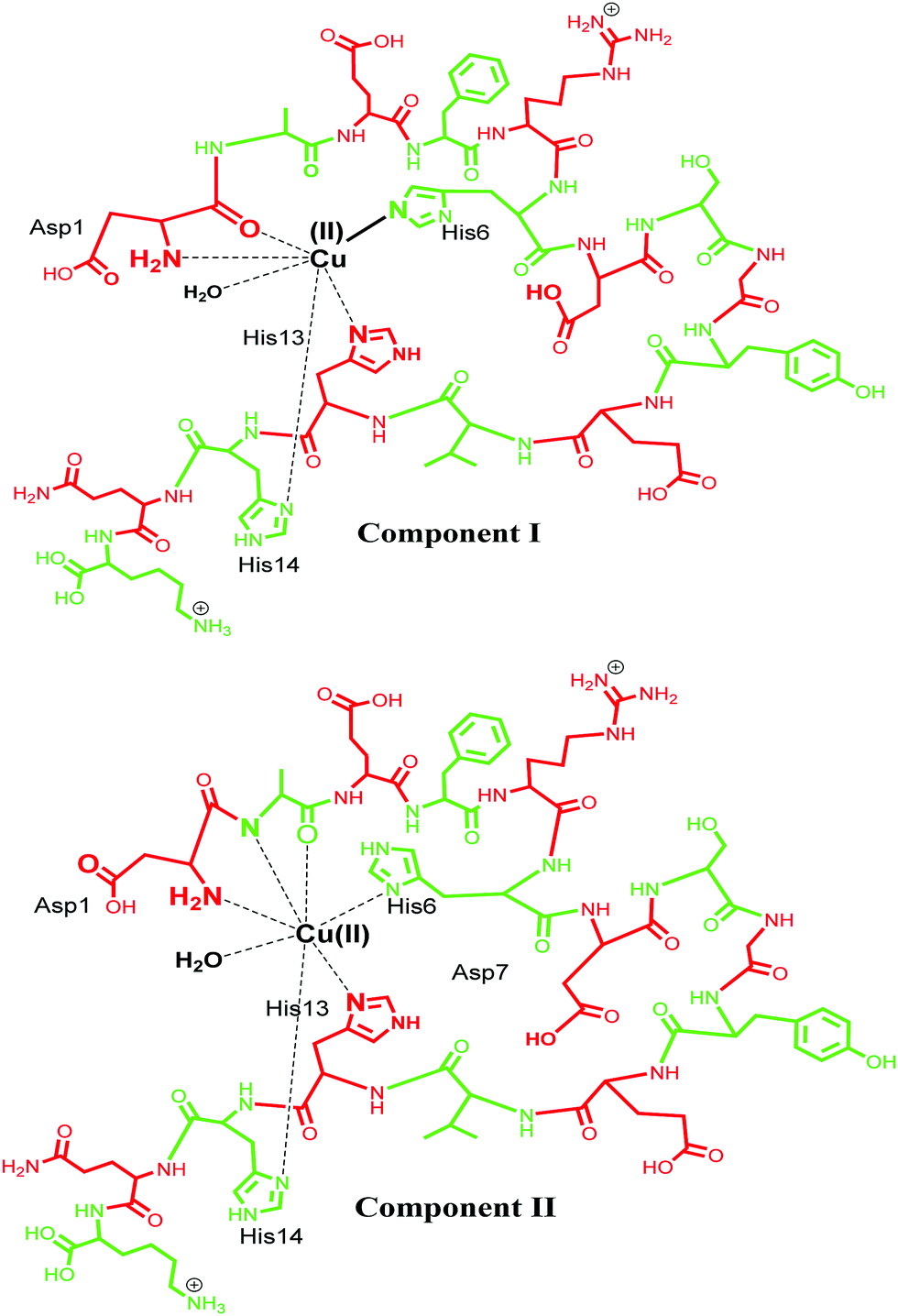 | ||
| Fig. 3 Representation of Cu(II) coordination sphere at low pH (component I) with Aβ1–16 fragment (top) and at high pH (component II) (bottom). | ||
5.2. Copper(I) interaction with Aβ peptides
As compared to Cu(II), only a few reports are available on the coordination of Cu(I) with different fragments of Aβ. Karlin and co-workers, using their model His–His dipeptide, hypothesised a His–His ligation of a Cu(I) ion in a linear two-coordinate geometry (NHis–CuI–NHis). The copper(I) complex of these ligands was observed as a linear, two-coordinate complex by X-ray absorption spectroscopy and found to be quite robust toward oxidation by O2.112 They further demonstrated the two-coordinate geometry of Cu(I) ions with N-terminal region fragments of Aβ even in the presence of potentially coordinating residues of histidine (His-6, His-13 and His-14) and other amino acids (Tyr10, Asp7, Glu11, Ser8, backbone carbonyl O and amide N).113 Simultaneously, Shearer et al. studied the X-ray absorption near-edge spectrum region of AβCu(I), which displayed an intense pre-edge feature at 8984.1(2) eV; carrying out a data-fitting operation yielded a two-coordinate geometry, with two imidazole ligands coordinated to Cu(I) at 1.877(2) Å in a linear geometry.114 In agreement with these results, tandem mass spectrometry studies also identified His13 and His14 as binding sites for Cu(I) with Aβ1–16.115 However, an early computational study suggested contradictory results where Cu(I) forms a tricoordinated complex with two imidazole units and one carbonyl group.116 However, experimental evidence supports the linear two coordinate geometry around Cu(I). The above discussion indicates that Aβ binds to Cu(I) with two His residues, with a majority of the binding coming from His13 and His14. The commonly accepted binding model is shown in Fig. 4.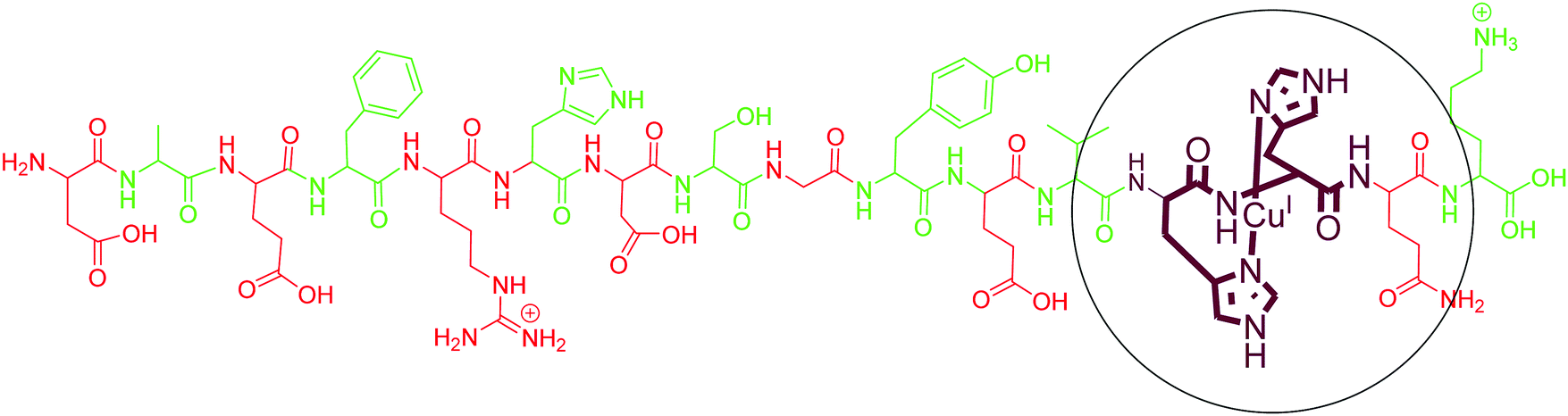 | ||
| Fig. 4 Depiction of the chemical structure of an Aβ1–16–Cu(I) complex. His13 and His14 are shown as coordinating residues. | ||
5.3. Zinc(II) coordination with Aβ peptides
A zinc binding site for Aβ peptide is located in the N-terminus hydrophilic region (Asp1–Lys16), similar to Cu(II). It is relatively more challenging to define the exact coordination for Zn(II), considering its closed shell d10 electronic structure and its silent behaviour towards most of the usual spectroscopic techniques. Insights into the coordination mode have been mainly achieved by NMR and EXAFS spectroscopy. Early reports established that Zn(II) forms a 1:1 complex with Aβ with a mononuclear binding site.117,118 Zn coordination was proposed to involve four to six ligands with Zn(II) at the centre.28,97 By means of Raman spectroscopy, in 2000, Miura et al. demonstrated that Zn(II) binds to Aβ peptide with His–Zn–His bridges through an inter-peptide aggregation mode.101 In agreement with this, a four-coordinated Zn(II) model was proposed, with four histidine units coordinating simultaneously but from adjacent peptides. It favours an inter-peptide linkage due to lack of a fourth coordination site as histidine along the Aβ sequence.90,119 Another coordination sphere was determined by the use of NMR spectroscopy, where Zn(II) was found to have a tetrahedral coordination environment with the involvement of three histidine residues (His-6, His-13 and His-14)88 and an additional ligand. The identity of the fourth ligand is still under scrutiny, with Asp1, Arg5, Glu11, Try10 and an exogenous water molecule as probable candidates. Glu 11 as an oxygen donor with Aβ1–16 at pH 6.5, as well as Arg 5 and Asp1 have been suggested, whereas Tyr10 has been ruled out.118,120,121 Gaggelli et al. carried out further investigations on the structure and binding mode of Zn(II) metal with rat and human Aβ1–28 using NMR spectroscopy. A tetrahedral coordinated complex was suggested for rat Aβ with Asp1-amine, His6, Glu11 and His13 being the coordinating amino acid residues. Whereas for human Aβ, a penta-coordination was suggested with amino acid residues His6, His13, His14, Asp1-amine and Glu11 as carboxylate in the Zn(II) coordination sphere.122 Recently, Alies et al. has reported a tetrahedral Zn(II) coordination site with the involvement of two histidine (His6, His13 or His14) residues along with two carboxylate side chains (Glu11 and Asp1 or Glu3 or Asp7); an equilibrium between different forms was observed with a marginal preference for Asp1.30 In conclusion, different models of Zn(Aβ) coordination have been observed, where Zn is suggested to have a coordination number between four and six. In the first coordination mode, the involvement of three histidines (His6, His13 and His14) has been suggested, along with extra donor ligands (Asp1/Glu3/Asp7) (in Fig. 5, the coordination mode of Zn(II) with Aβ1–16 is represented in line with this model). In the other coordination mode, it has been proposed that Zn coordinates to four histidines with inter-peptide linkage along with adjacent peptides. Also, in recent reports, a tetrahedral geometry has been proposed, involving two histidine residues and two carboxylate chains Asp1.30,123
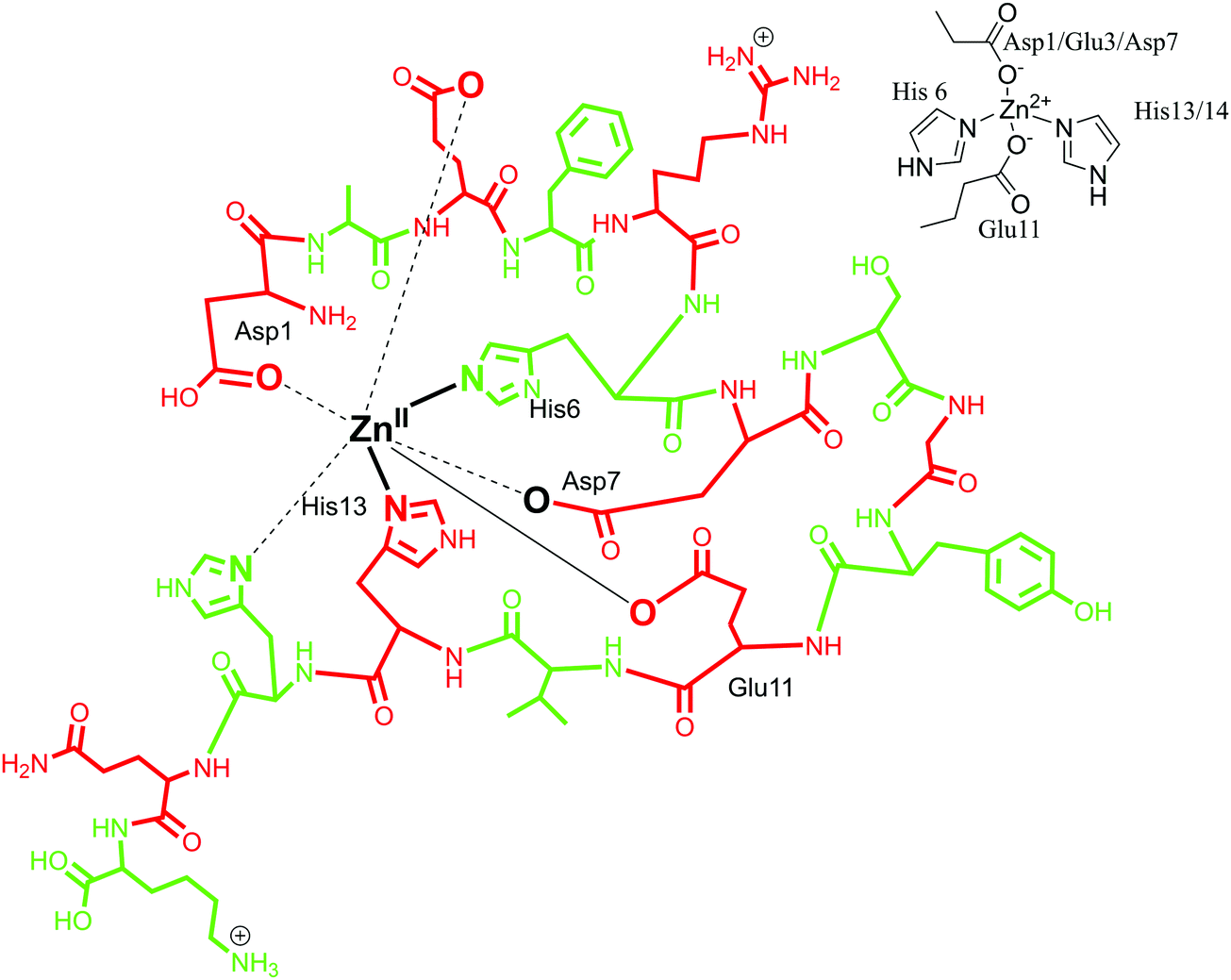 | ||
| Fig. 5 Depiction of Zn(II) coordination with Aβ1–16 peptide; only the coordination sphere is shown as inset for clarity. | ||
6. Role of metal ions in Aβ aggregation
Altered concentrations of transition metal ions, mainly Cu, Zn and Fe, are found to be associated with amyloid plaques as discussed earlier. Particularly, Cu and Zn are well known to bind to the Aβ peptides and affect their self-aggregation.17,61,124 These metal ions can coordinate with the Aβ peptides and bring structural and conformational changes as discussed and represented above in Fig. 3–5. The Aβ–metal supra–metal complexes are expected to alter the normal Aβ aggregation processes.23,125 Various opinions have been offered regarding the exact role of metals in aggregation of these peptides. Metal induced effects are diverse and sometimes contradictory to each other.9,97 In this section, selected studies on metal induced aggregation of Aβ peptides are discussed. Since the Aβ1–40 does not form neurotoxic soluble species, the discussion focuses primarily on the aggregation of Aβ1–42 peptide.9,126 It is important to discuss that Aβ peptides are known to form polymorphic aggregates which differ significantly from each other: three types of aggregates are commonly observed, the most structured fibrillar aggregates, soluble oligomers, and large amorphous aggregates.127 In some studies, metal ions are observed to increase the aggregation of Aβ peptides, expediting the formation of amyloid fibrils or amorphous aggregates.128,129 In contrast, in other studies, the metal ions were observed to reduce the Aβ aggregation and stabilize the smaller aggregates, including soluble neurotoxic Aβ oligomers, especially with the Cu(II).130,131 The molecular mechanisms of metal–Aβ species interactions are not completely understood, especially for the more neurotoxic Aβ1–42 peptide, and the role of metal ions in the aggregation of Aβ is still under debate. In the next subsections, a detailed discussion is presented regarding the various studies that have been carried out on Cu and Zn induced Aβ aggregation.6.1. Aβ aggregation in the presence of copper(II); stabilization of neurotoxic soluble Aβ42 oligomers
In this section, we shall be focusing majorly on the influence of Cu(II) on the aggregation of Aβ peptides. According to some studies, Cu(II) has been suggested to be involved in disrupting the larger aggregates or reducing Aβ1–42 aggregation,131–135 whereas, in stark contrast, other studies report a dramatic enhancement of aggregation in the presence of Cu(II).129,136,137 The cell toxicity studies in the presence of transition metal ions also showed both increased35,138–140 as well as decreased43,133 toxicity of Aβ peptides. It was believed from very early on that Cu(II) ions stabilize the toxic early-stage aggregates of Aβ.101 In 2001, Zou et al. studied the effect of Cu(II) on Aβ1–42 aggregation by means of ThT fluorescence and AFM imaging and observed an inhibitory effect of Cu(II) on Aβ1–42 aggregation.132 Exley et al. reported that Al(III), Fe(III), Zn(II), and Cu(II) can influence the in vitro formation of amyloid fibrils of Aβ1–42.130,141 In this study, Cu(II) was shown to inhibit the formation of amyloid fibrils in both freshly prepared and aged solutions. They monitored the fibrillization of Aβ1–42 in the presence of Al(III), Fe(III), Zn(II), and Cu(II) over a period of 32 weeks. The research group also investigated the dissolution of these aged peptide aggregates in the presence of both desferrioxamine (DFO) and ethylenediaminetetraacetic acid (EDTA), the two well-known metal chelators. Aβ1–42 aggregation was followed by the methods of ThT fluorescence and TEM images. In 2007, Park et al. studied the morphological behaviour of Aβ1–42 aggregates formed on a solid template incubated at pH 7.4, 6.0, or 4.6 in the presence of different metal ions. The authors wrote about a synthetic template by immobilizing Aβ oligomers onto an N-hydroxysuccinimide ester-activated solid surface. The methods of ex situ atomic force microscopy (AFM), Fourier transform infrared spectroscopy (FT-IR), and ThT fluorescence spectroscopy were used in this study. The authors observed that Cu(II) and Zn(II) ions accelerated both Aβ1–40 and Aβ1–42 deposition but resulted only in the formation of “amorphous” aggregates. In contrast, Fe(III) induced the deposition of “fibrillar” amyloid plaques at neutral pH. Under mildly acidic environments, the formation of fibrillar amyloid plaques was not induced by any metal ion tested in this work.142 In 2008, Barnham et al. reported that the presence of Cu(II) significantly promotes and stabilizes the formation of the soluble oligomeric β-sheet structures. However, in the absence of added Cu(II), more amyloid plaques were formed.54In 2009, Exley et al. wrote that Cu(II) is bound by monomeric Aβ1–42 and, upon precipitation of the copper–peptide complex, prevents Aβ1–42 from adopting a β-sheet secondary structure. In this report, they investigated the influence of this interaction on the conformation of the precipitated peptide and suggested that copper significantly reduced the thioflavin T fluorescence of aged Aβ1–42 samples. Transmission electron microscopy also showed that copper significantly reduced the quantities of amyloid fibrils. Congo red staining and polarized light demonstrated a copper-induced abolition of apple-green birefringence. The authors wrote that “while we can agree that fibrillar Aβ1–42 binds Cu(II) with great avidity, we can now add that binding actually results in the abolition of its β-sheet structure.”131 The same group in 2013 reported that methods of fluorimetry and transmission electron microscopy provide unequivocal evidence that, under near-physiological conditions, both sub- and super-stoichiometric concentrations of Cu(II) prevented the assembly of Aβ1–42 into ThT-positive β-sheet rich amyloid fibrils.141 Another study by Garai et al. showed that low concentrations of Cu(II) (1 μM) and Zn(II) (4 μM) selectively eliminate the oligomeric population (within ∼2 h) of Aβ, by using fluorescence correlation spectroscopy to resolve the various soluble species.143 The authors suggested that physiological concentrations of Cu(II) and Zn(II) ions can critically alter the stability of the toxic Aβ oligomers and can potentially control the course of neurodegeneration. This was in stark contrast to the reports related to the stabilization of Aβ1–42 oligomers by Cu(II).143 Zatta et al. studied the aggregation of Aβ1–42 in the presence of Al, Fe, Cu and Zn; they found that all the four metals differentially altered the aggregation properties of Aβ1–42 peptide. Their studies suggested that Aβ1–42–Cu and Aβ1–42–Zn show a similar profile and form amorphous aggregates, whereas Aβ1–42–Al and Aβ1–42–Fe promoted the formation of oligomers.144 Pederson et al. reported that the Cu(II):Aβ ratio in solution has a major influence on the aggregation kinetics/mechanism of Aβ and the morphology of the aggregates, which shifted from fibrillar to non-fibrillar with increasing Cu(II):Aβ ratios. The authors suggested that the presence of a slow-aggregating soluble Aβ–Cu(II) oligomer could act as a rate-limiting step for the overall aggregation process and may help in understanding the Aβ–Cu(II)-mediated neurotoxicity in AD.135
In 2013, Mirica et al. employed a wide range of techniques to study the effect of metal ions on the oligomerization and aggregation of the more neurotoxic Aβ1–42 peptide.9,21 ThT fluorescence, native gel electrophoresis and Western blotting, transmission electron microscopy (TEM), and cellular toxicity studies were used to determine the formation of both soluble and insoluble Aβ1–42 aggregates in the presence as well as absence of Cu(II) and Zn(II). Most importantly, they correlated these in vitro results with neurotoxicity studies, in order to address the potential in vivo role of metal–Aβ1–42 interactions in AD. A significantly low ThT fluorescence was observed for Cu(II) and Zn(II) containing Aβ aggregates. TEM and native gel/Western blotting studies suggested that Cu(II) and Zn(II) both inhibit fibrillization. Interestingly in this report, Zn(II) was shown to form amorphous aggregates, whereas Cu(II) stabilized the soluble Aβ1–42 species. The cellular toxicity studies suggested that while Zn(II) reduces the Aβ1–42 toxicity by formation of non-toxic insoluble amorphous aggregates, Cu(II) significantly increases Aβ1–42 neurotoxicity due to the formation of neurotoxic soluble Aβ1–42 oligomers. These studies ruled out a preventive role of Cu(II), and suggested that Cu(II) may lead to an increased Aβ1–42 neurotoxicity in vivo due to the formation of Aβ1–42 oligomers. The authors proposed that this Cu(II) promoted increased neurotoxicity of soluble Aβ1–42 species may play an important role in the aetiology of AD (see the schematic representation in Fig. 6).
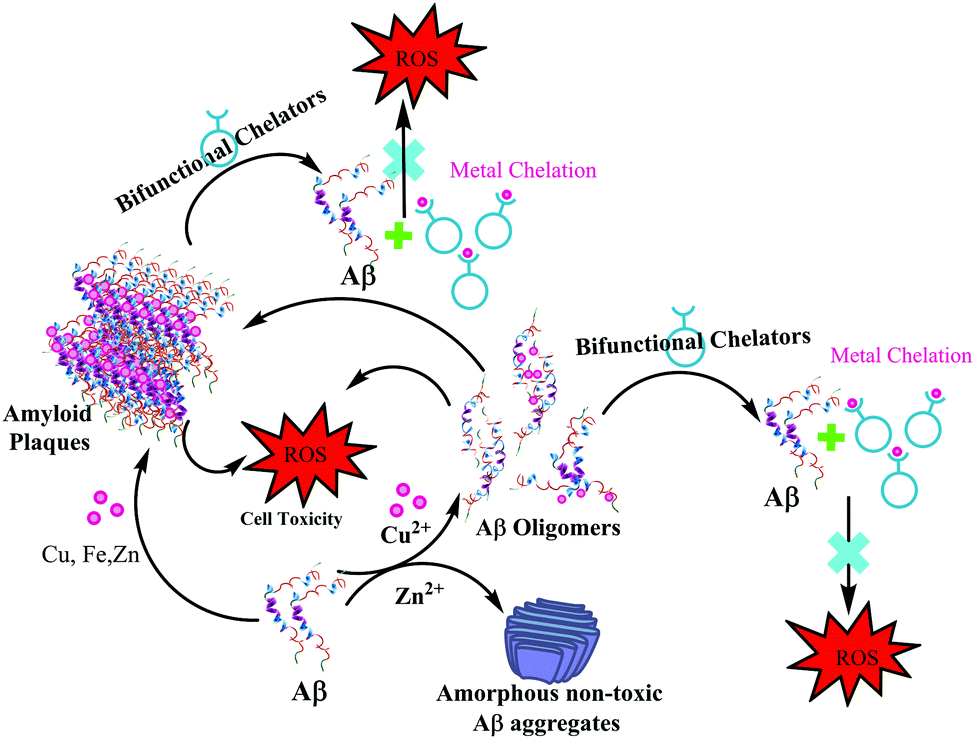 | ||
| Fig. 6 Depiction of metal effects on Aβ1–42 aggregation and proposed role of bifunctional chelators in metal chelation therapy. | ||
In another study by Viles et al. in 2015, it was reported that substoichiometric Cu(II) was observed to affect the misfolding pathway of Aβ1–40, and Aβ1–42, by distinctly different mechanisms. Cu(II) was observed to accelerate Aβ1–40 fibrils formation; however, with Aβ1–42, substoichiometric levels of Cu(II) exclusively promote the formation of oligomeric and protofibrillar assemblies. In addition, the mature Aβ1–42 fibrils were disassembled into oligomers when Cu(II) was added. The authors also reported that these Cu(II) stabilized oligomers of Aβ1–42 interact with the lipid bilayer, disrupting the membrane and increasing permeability. In addition, investigation of Aβ1–40/Aβ1–42 mixtures with Cu(II) revealed that Aβ1–40 neither contributed to nor perturbed the formation of Aβ1–42 oligomers, although Cu(II)–Aβ1–42 does frustrate Cu(II)–Aβ1–40 fibre growth. The authors also mentioned that small amounts of Cu(II) accentuated the differences in the propensity of Aβ1–40 and Aβ1–42 to form toxic oligomers, providing an explanation for the connection between disrupted Cu(II) homoeostasis and elevated Aβ1–42 neurotoxicity in Alzheimer's disease.145
6.2. Aβ aggregation in the presence of Zn(II) and neuroprotective role of Zn
Although there are several studies that report the effect of both Cu and Zn on Aβ aggregation, there are few reports exclusively on the effect of Zn(II) ions on aggregation of Aβ1–40 and/or Aβ1–42. Zinc was shown to specifically bind Aβ1–40 with a 1:1 stoichiometry, displaying a high affinity binding with Kd = 107 nM and another low affinity binding with Kd = 5.2 μM.128,146 Early reports from Bush et al. showed that human Aβ1–40 peptide rapidly forms amyloid in the presence of more than 300 nM concentrations of Zn(II).128,147 Some reports suggested the involvement of specific amino acids that interact with Zn(II) in the rapid precipitation of Aβ peptides.148 Zn(II) at a concentration of a few micromolar, which is too dilute to affect the precipitation equilibrium of Aβ, can destabilize these aggregates.143 Nevertheless, a growing number of reports indicate that zinc inhibits Aβ-mediated cytotoxicity. Garai et al. observed reduced Aβ toxicity in the presence of Zn(II) by selectively precipitating aggregation intermediates.149 In the same report, it was shown that the larger soluble aggregates (size > 10 nm) form only in supersaturated Aβ solutions, implying that they are intermediates in the pathway toward fibril formation. Zn(II) was observed to destabilize these intermediates by accelerating their aggregation kinetics. The resulting change in the size distribution of the Aβ solution is sufficient to eliminate its toxicity to cultured mammalian neurons, lending support to the observations that at a concentration of a few micromolar, Zn(II), significantly reduces Aβ toxicity.149
Zinc and copper may also exert a combined effect. A report by Lee et al. systematically characterized Zn–Aβ aggregates by incubating equimolar Aβ with Zn. The authors reported that Zn–Aβ1–40 and Zn–Aβ1–42 both form spherical oligomers with a diameter of ∼12–14nm composed of reduced β-sheet content. In addition, Zn–Aβ and Aβ derived diffusible ligands (ADDLs) were found to be distinct as examined by analytical ultracentrifugation, hydrophobic exposure by BisANS spectra, and their immunoreactivity. Moreover, Zn–Aβ oligomers were shown to exhibit stronger toxicity than ADDLs by cytotoxicity assays.150 Very recently, Zhang et al. made use of a combination of analytical ultracentrifugation (AUC), circular dichroism (CD) spectroscopy, thioflavin T (ThT) assay and atomic force microscopy (AFM) imaging to analyze the impact of stoichiometric Zn(II) on the aggregation process of Aβ1–42. It was observed that the Aβ1–42 aggregates found in the presence of Zn(II) were smaller in size, non-fibrillary and showed less β-sheet structures than aggregates formed in the absence of Zn(II). Their studies showed that Zn(II) was capable of retaining monomeric Aβ1–42 in solution. The authors wrote that “our results provide further evidence that Zn(II) shifts the self-association of Aβ1–42 toward a non-fibrillary pathway by interfering with the aggregation process at multiple levels”.151 Exley et al. in 2004 showed that fibrilization observed in the presence of Zn(II) was very slow and suggested that the fibrillar structures formed slowly in the presence of Zn(II) were less “ThT-reactive”. The authors suggested that this lack of reactivity may be due to either a conformational state which was specific to Zn(II) binding or a competition between Zn(II) and ThT for a specific binding region.141 In contrast, studies by Mirica et al., as discussed above, suggested that Zn induces precipitation of amorphous aggregates of Aβ1–42 as well as of Aβ1–40. It was also found that the Zn induced aggregates were not toxic to the Neuro2A cells.9
7. Discussion on sample preparation and techniques used to study Aβ aggregation
Overall, the above discussion gives an overview of the current status of metal effects on Aβ aggregation. In this regard, sample preparation and techniques used for carrying out the studies are important factors to be taken into consideration. Metal-induced aggregation has been widely studied under different pH conditions, temperature, concentration/metal to peptide ratio, etc. A few important factors which can potentially affect the Aβ aggregation results may also be brought into the discussion. Aβ peptide available from commercial sources is normally not 100% pure. Aβ peptide is either chemically synthesized or a recombinant peptide. Chemically synthesized peptides contain counter ions, typically trifluoroacetate (TFA) from HPLC purification. Purification at lower pH (∼2) results in protonation of histidine and carboxylates. If such a peptide is dissolved in water at higher concentrations (mM), the presence of counter anions will be resulting in quite low pH.152 There can be a significant drop in pH even when the peptide is dissolved in a high concentration of buffer; hence, it is important that the buffer used and the pH be chosen with care. Aggregation is generally studied under the so-called fibrillation conditions.9,77 The most common methods use Aβ monomeric films, which are prepared by dissolving the peptides in HFIP to break the preformed aggregates and obtain the monomeric form.153 The aliquots of this solution after evaporation and vacuum centrifugation form monomeric films, which are generally stored at −80 °C and used for months. The aggregation of these Aβ films is initiated by first dissolving them in DMSO or dilute aq. NH4OH, followed by diluting into the appropriate buffer medium and incubating for a sufficiently long time at 25–37 °C with or without continuous agitation.8,77 The concentration of the peptide is usually evaluated using UV spectral data, as Tyr absorbs at 275 nm with an extinction coefficient of 1400 M−1 cm−1. UV-vis is not always a valuable method to determine the peptide concentration, since metals bound to the peptide could also absorb in the same region as that of the aromatic amino acids. For metal-containing fibrils, the corresponding metal ions are normally mixed before the initiation of the fibrillation conditions. The methods employed for fibrillization may vary marginally in the reports from different research groups. For example, another method by Dahlgren et al. that has been used for fibrillar conditions is using 10 mM HCl to bring the peptide to a final concentration of 100 μM and incubated for 24 h at 37 °C.10 The same reports also described the method for the oligomer preparation as follows: monomeric film of Aβ1–42 was dissolved in anhydrous DMSO and diluted in DMEM:F12 media (1:1 v:v, without phenol red, Invitrogen). After that the Aβ1–42 solution (50–100 μM) is incubated at 4 °C for 24 h and then centrifuged at 10000g for 10 min. The supernatant can be used as a solution of soluble Aβ1–42 oligomers.10,153
Techniques used for monitoring the metal–Aβ interaction and aggregation are wide-ranging and have been documented in detail elsewhere.152 A brief summary of selected methods is presented here for the benefit of the reader. Fluorescence spectroscopy has emerged as one of the most important diagnostic tools for analysis and imaging of amyloids. The most widely used method to monitor the extent of fibrillation is thioflavin-T (ThT) fluorescence assay. ThT was first introduced as a fluorescent marker for amyloid fibrils in 1965 by Levine and has been in use ever since.154 However, the exact mechanism, information regarding specific interactions, and stoichiometry have not yet been completely understood.155 The dye is weakly fluorescent in an aqueous environment but increases by several folds upon binding to amyloid fibrils. This protocol has been largely unchanged and is perhaps the most widely employed method for monitoring Aβ aggregation. ThT is the most commonly used dye but lacks the specificity for various amyloids and detects only the final aggregates, the amyloid fibrils. Early stage aggregates, which are suggested to be much more neurotoxic, cannot be determined by ThT assay. The presence of paramagnetic metal ions like Cu(II) may affect the emission.9 Hence, ThT fluorescence assay should not be used in isolation to determine the nature and extent of aggregation.
Another routinely employed technique to quantify fibril content and inhibitory ability of small molecules toward amyloids is Congo Red (CR) spectral shift assays.156 It is known that CR has two binding sites in amyloid, parallel and antiparallel to the β-sheet and the binding ratio may depend on the type of amyloid under investigation. Binding of CR to the extensive β-sheet structures results in an enhanced absorption as well as a bathochromic shift in its absorption spectrum from ∼480 nm to 540 nm. When crossed polarizers are used (polarized light), green birefringence is observed upon binding to the fibrils and has been typically used as a qualitative measure of fibril formation.157 There are several reports indicating CR as an inhibitor of amyloid aggregation.158
A more quantitative analysis of the Aβ aggregation studies is provided by native gel electrophoresis/Western blot analysis and transmission electron microscopy (TEM) or atomic force microscopic (AFM) techniques. While the former type of analysis reveals the presence of smaller, soluble Aβ aggregates and their molecular weight distribution, the latter method allows the characterization of the larger, insoluble Aβ aggregates that cannot be analyzed by gel electrophoresis. In general, TEM images should be reported with great care as the image taken from just one region of the surface of TEM grid may not represent the entire sample. Therefore, images which are representative of the entire surface of TEM grids that show a homogeneous distribution of the Aβ species should be presented. The use of all these methods together provides a more comprehensive picture of the extent and pathways of Aβ aggregation under various conditions.131,132,134,136,141,159 However, only a few reports have employed all these techniques together under similar experimental conditions to provide a detailed picture of the neurotoxicity implications of the Cu(II) and Zn(II) mediated formation of soluble and/or insoluble Aβ1–42 aggregates. More comprehensive studies on the effect of metal ions on Aβ1–42 aggregation and neurotoxicity are required for a better understanding of their role in AD progression.
8. Rational design of therapeutics to tackle metal ions in AD
Research into the field of neurodegenerative diseases has rapidly advanced in the last two decades as is evident from an exponential growth in research articles published in this domain. Despite the continuous efforts of a large body of researchers in the field of neurodegenerative diseases and more specifically Alzheimer's disease, the exact cause for the pathology of AD is still not known, and as such, there is no treatment available for AD to date. The five FDA approved medications for Alzheimer's disease help only in alleviating the symptoms or slow them down from worsening. Out of the five available medications, donepezil, galantamine and rivastigmine belong to the “cholinesterase inhibitors” class of drugs.160–162 These drugs help by increasing the levels of acetylcholine, an important neurotransmitter for learning and memory. Considering the involvement of multiple factors in the pathogenesis of AD, therapies or drugs targeting a single cause are inadequate to address disease progression. It is becoming more evident from the trends in recent research that the next generation of therapies must have multiple functions to fight the various mechanisms of disease succession. Individuals with neurodegenerative diseases can be benefited by these new drug molecules being developed, which can target multiple problems.163,164 To function as an effective drug for such diseases, these molecules should possess more than one of the following properties: control over ROS formation, metal chelating ability, affinity for their uptake to cross the blood–brain barrier, antioxidant capabilities, lower Aβ peptide aggregation, or inhibit enzymes associated with disease pathways such as acetylcholinesterase and monoamine oxidase, etc.165,166 Current AD therapies primarily focus on various underlying factors like modulating neurotransmission, tau based therapies, amyloid based strategies, modulating intracellular signalling cascades, oxidative stress reduction and metal chelation therapy. Considering the crucial role played by metal ions, metal chelation therapy is expected to play a pivotal role in AD therapeutic research. Keeping the essential role of metal ions in biology in the background, designing selective and specific metal chelators is highly challenging. 2,3-Dimercaptopropanol (British anti-Lewisite, BAL) was the first metal chelator which was developed in the 1940's against the toxicity associated with arsenic.14,167 It was followed by the use of ethylenediaminetetraacetic acid (EDTA), introduced in the 1950s for the treatment of lead toxicity.14 Much later, the sequence of diethylenetriaminepentaacetic acid (DTPA), triethylenetetraamine (TETA) and standard iron chelator desferrioxamine (DFO) emerged in the late 1980s and 1990s.14 It was followed by the discovery of the first generation of metal chelators towards the end of 1990s and the introduction of a small lipophilic molecule clioquinol (5-chloro-7-iodo-8-hydroxyquinoline, CQ).168,169 It significantly reduced Aβ plaque deposits, leading to improved cognitive behavior, in early phase II clinical trials.16,22,26,137 Unfortunately, the long-term use of CQ is limited due to its adverse side effects, especially the sub-acute myelo-optic neuropathy.169,170 However, it has led to the development of several CQ based metal chelators extensively studied in the context of metal chelation therapy for AD.13Bifunctional metal chelators (BFC) having functionalities that have not just metal chelating ability but are also capable of binding with amyloids were proposed to tackle the multifactorial nature of AD (Fig. 6). Considerable progress has been made in this field in the last decade.8,13,171,172 Thioflavin-T (ThT) is well known to bind most amyloids and, as discussed earlier, has been used as the most important fluorescent dye indicator to detect the presence of amyloids.154 The ThT molecular framework has been extensively explored for multifunctional molecule design.8,163,173 XH1 was the first bifunctional molecule based on this design principle to link different molecular fragments with different functions to form a hybrid molecule (Fig. 7).174 Its structure comprised two terminal neutral ThT-derived moieties linked via a diethylenetriaminepentaacetic acid (DTPA) binding unit.
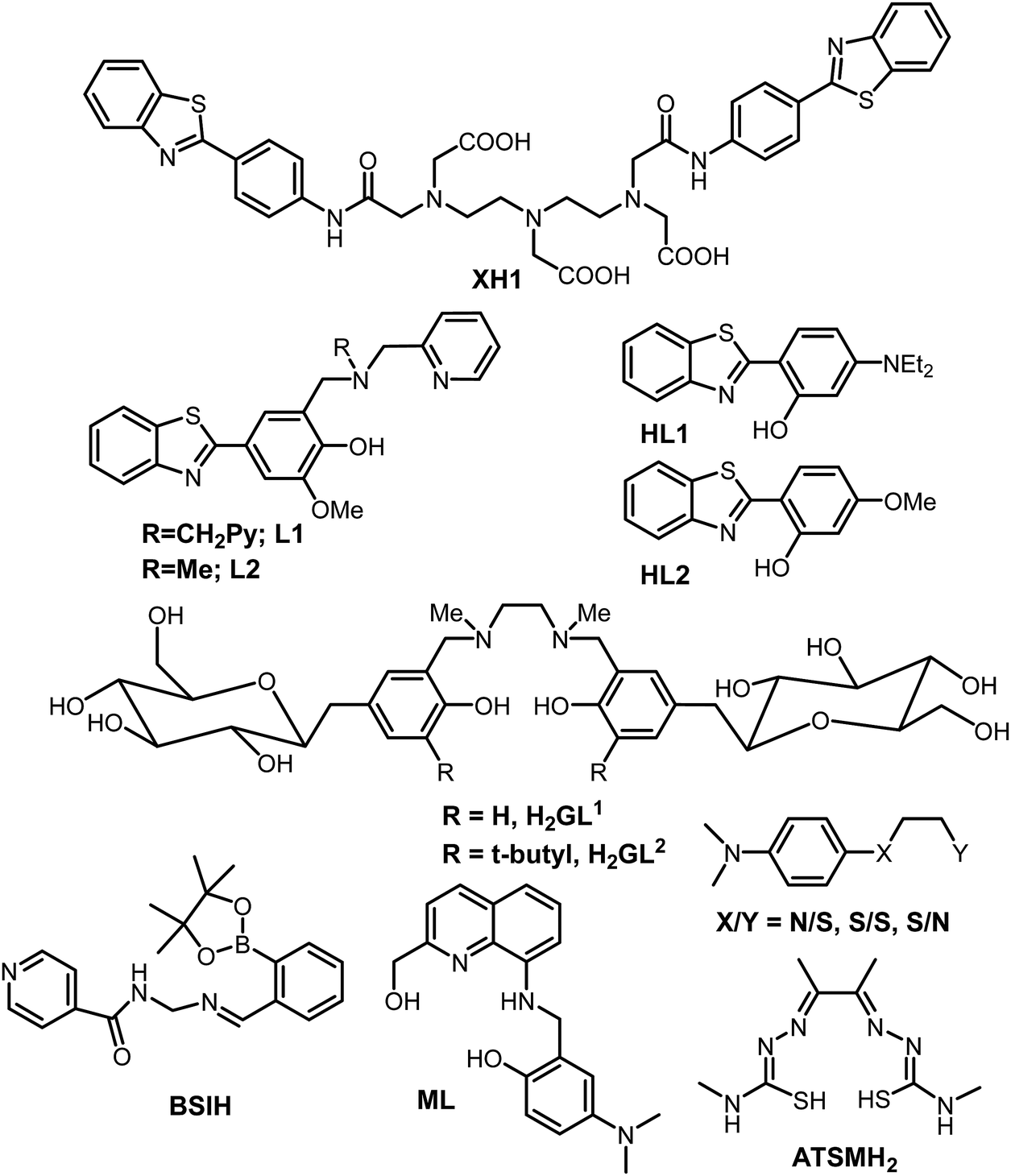 | ||
| Fig. 7 Structures of selected bifunctional chelators/prochelators reported from different research groups as discussed in the text. | ||
Mirica et al. reported the role of metal ions in AD and new bifunctional compounds based on phenylbenzothiazole (inspired by ThT), linking them with small metal chelators such as dipicolylamine or pyrinophane type metal chelators (Fig. 7).8,163,175,176 In addition to metal binding and Aβ fibril binding properties, the effects on Aβ1–42 aggregation in the presence and absence of metal ions were also investigated.8 Bifunctional compounds having ThT and CQ type chelation properties, which have the capability of converting neurotoxic soluble oligomers into less toxic fibrils, were reported by the same group. A novel and effective strategy was introduced by Franz and co-workers in 2006 to design prochelators. The rationale in the strategy was that the chelators get activated only in the presence of oxidative stress and that they do not chelate metal ions until a protective mask is selectively removed by oxidative stress. This approach proved to be excellent in addressing the selective removal of metal ions and preventing loss of essential metal ions from metalloproteins. Boronic ester (BSIH), a first generation prochelator containing a boronic ester as a mask in place of phenolic oxygen, an excellent metal affinity group, was designed following this approach. It gets activated to salicylaldehyde isonicotinoyl hydrazone (SIH), a known iron chelator in the presence of H2O2.177 Different analogues for BSIH with various substitutions as boronic esters as well as acids were explored with respect to their ability as metal chelators.178 Later, the same group also utilized this strategy for various promising molecular scaffolds and investigated them for their effect on multiple factors responsible for AD.179–182
A series of rationally designed bifunctional compounds from several families with various frameworks have been reported by Lim et al. IMPY and stilbene derived compounds were studied for their metal binding properties, and effect on Aβ40 aggregation in the presence and absence of metal ions.77,183 Very recently, novel small molecules (2,2′-bipyridine (bpy) derivatives and other N,N-dimethylanalines (DMA) with bidentate ligands) were reported as chemical tools and were shown to be effective in targeting multiple pathological factors associated with Alzheimer's disease.184,185 The effects of various flavonoids including myricitin and epigallocatechin gallate (EGCG) molecules have also been reported.186–189 SALEN type Schiff-bases linked with carbohydrate moieties were reported for the first time by Orvig et al. in addition to other metal chelators by the same group.13,164,190–194 The most advanced chelator in terms of reaching the patients has been PBT2, a second generation 8-hydroxyquinoline based scaffold which is inspired from CQ but lacking iodine. It is a metal binding agent which has a binding affinity for copper and zinc, while also exhibiting good antioxidant properties.13,69,195 PBT2 also has a good blood brain barrier permeability, is more effective than CQ in cognition effects and is a superior ionophore. In vitro, PBT2 significantly reduces H2O2 production and prevents Cu- and Zn-induced formation of Aβ aggregates. Treatment with PBT2 displayed inhibition to metal-induced damage, and importantly, did not lead to any changes in the concentration of metal ions in other parts such as the brain, kidney, liver or plasma.196 PBT2 reverses the Alzheimer phenotype within days by significantly decreasing insoluble Aβ levels by ∼30% and affects both Tau phosphorylation and synaptophysin protein levels. Upon PBT2 treatment, total insoluble tau levels were decreased while the soluble tau level was increased.13 Another important ligand to be mentioned here is diacetylbis(N(4)-methylthiosemicarbazonato), commonly known as ATSM (see the structure in Fig. 7). The Cu(ATSM) compound was found to play a protective role against peroxynitrite-induced nitrosative damage and prolongs survival in the Amyotrophic Lateral Sclerosis (ALS) mouse model.197 However, ATSM has not been much evaluated for metal chelation or protection from ROS in the context of AD. It is important to mention that numerous multifunctional molecules capable of simultaneously targeting and combating multiple pathological factors of neurodegenerative disorders have been rationally designed and evaluated for their efficacies employing different relevant studies; such molecular frameworks have been well summarized elsewhere.13,171,198 Finally, it is important to mention that, despite the enormous progress that has been made in the development of multifunctional molecules as therapeutics, very few have made it to human clinical trials. Although the above strategy of using multifunctional-molecules to tackle the multifactorial nature of these disorders seems quite promising, the potential issues causing clinical failure have to be understood; a summary of probable causative factors are reviewed elsewhere.199,200 Overall, it is an attractive strategy that is making progress and hopefully delivers better therapeutics for AD in the near future.
9. Conclusions
In summary, most aspects of Cu and Zn interactions with Aβ peptides have been reviewed. A brief discussion on the formation, structure and functions of Aβ peptide as well as the levels of metal ions in AD affected brains and related studies has been presented. A discussion on the redox nature of metal ions and role of the Cu–Aβ complex in ROS formation has also been presented to highlight this chemistry. Whereas excellent articles in great detail are available in the literature, a concise summary of Cu(II), Cu(I) and Zn(II) coordination chemistry has been presented here. In addition, the rarely reviewed topic of aggregation of Aβ peptides in the presence of metal ions has been a major focus of this review. In general, it has been observed that while most of the initial studies were carried out with Aβ1–40 peptide, it was understood that it has a limited role in the toxicity involved in AD. It is the longer version, the Aβ1–42 that forms more soluble neurotoxic oligomeric aggregates, and studies with Aβ1–42 are therefore becoming more common. In this review, the effect of Cu(II) on Aβ aggregation and especially on the stabilization of neurotoxic soluble oligomerization has been presented. Similarly, the effect of Zn(II) on Aβ aggregation and its proposed neuroprotective roles has also been reviewed. It is comprehensible from the above discussion that, although there were conflicting results obtained by various groups regarding the coordination of metal ions to Aβ, a consensus appears to be emerging as of today. Similarly, the aggregation of Aβ peptide and especially Aβ1–42 aggregation in the presence of metal ions has led to several contradictory studies. However again, in the present day scenario, researchers evidently concur that Cu(II) stabilizes the smaller soluble oligomers, while, on the other hand, Zn(II) has been suggested to form larger, amorphous less toxic aggregates. The reason for these conflicting studies may be attributed to the inherent nature of Aβ peptides to form different conformations even upon minimal changes in the experimental conditions, leading to different results; the environmental factors in the studies comprise the pH, metal ions, ionic strength and buffer used, presence of metal ions, choice of peptide (Aβ1–40vs. Aβ1–42), concentration of peptide or concentration of metal ions etc. In addition, the methods of study and the techniques used to monitor the aggregation also differ from one study to another. Research groups have to come on the same platform with regards to the experimental conditions and techniques to be employed, so as to avoid conflicting results for such important issues.Abbreviations
| Aβ | Amyloid-β |
| AD | Alzheimer's disease |
| ADDLs | Aβ derived diffusible ligands |
| AFM | Atomic force microscopy |
| ALS | Amyotrophic lateral sclerosis |
| ANS | 1-Anilinonaphthalene-8-sulphonate |
| APP | Amyloid precursor protein |
| AUC | Analytical ultracentrifugation |
| BAL | British anti-Lewisite |
| BBB | Blood brain barrier |
| BFC | Bifunctional chelators |
| CD | Circular dichroism |
| CQ | 5-Chloro-7-iodo-8-hydroxyquinoline |
| CR | Congo red |
| CSF | Cerebrospinal fluid |
| CW-EPR | Continuous wave electron paramagnetic resonance |
| DFO | Desferrioxamine |
| DMEM | Dulbecco's modified eagle's medium |
| DTPA | Diethylenetriaminepentaacetic acid |
| EGCG | Epigallocatechin gallate |
| EPR | Electron paramagnetic resonance |
| ESEEM | Electron spin-echo envelope modulation |
| EXAFS | Extended absorption fine structure |
| HFIP | Hexafluoroisopropanol |
| HYSCORE | Hyperfine sublevel correlation |
| ICP-MS | Inductively coupled plasma – mass spectrometry |
| FTIRM | Fourier transform infrared micro-spectroscopy |
| INAA | Instrumental neutron activation analysis |
| LA-ICPMS | Laser ablation inductively coupled plasma mass spectrometry |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogenated species |
| SIH | Salicylaldehyde isonicotinoyl hydrazone |
| TEM | Transmission electron microscopy |
| TFA | Trifluoroacetate |
| TETA | Triethylenetetraamine |
| ThT | Thioflavin T |
| NMR | Nuclear magnetic resonance |
| SIMS | Secondary ion mass spectrometry |
| XFM | X-ray fluorescence microscopy |
| XAS | X-ray absorption spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. S. acknowledges the Department of Science and Technology (DST, Govt. of India) and Science and Engineering Research Board (SERB) for financial support (grant reference numbers EMR/2016/001452). The authors acknowledge the help and suggestions from Prof. Liviu M. Mirica and Dr Easwar Srinivasan in improving the manuscript. We thank the reviewers of this manuscript for their valuable comments and advice. Authors appreciate the help of Dr Anurag Prakash Sunda in preparation of figures of the manuscript.References
- 2018, Alzheimer's Disease Facts and Figures: Annual Report from http://www.alz.org.
- J. A. Hardy and G. A. Higgins, Alzheimer's disease: the amyloid cascade hypothesis, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- K. P. Kepp, Bioinorganic Chemistry of Alzheimer's Disease, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- J. T. Jarrett, E. P. Berger and P. T. Lansbury, The C-Terminus of the β Protein is Critical in Amyloidogenesisa, Ann. N. Y. Acad. Sci., 1993, 695, 144–148 CrossRef CAS PubMed.
- T. Hartmann, S. C. Bieger, B. Brühl, P. J. Tienari, N. Ida, D. Allsop, G. W. Roberts, C. L. Masters, C. G. Dotti, K. Unsicker and K. Beyreuther, Distinct sites of intracellular production for Alzheimer's disease Aβ40/42 amyloid peptides, Nat. Med., 1997, 3, 1016–1020 CrossRef CAS.
- S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, A specific amyloid-beta protein assembly in the brain impairs memory, Nature, 2006, 440, 352–357 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, The effect of Cu2+ and Zn2+ on the Aβ42 peptide aggregation and cellular toxicity, Metallomics, 2013, 5, 1529–1536 RSC.
- K. N. Dahlgren, A. M. Manelli, W. B. Stine, L. K. Baker, G. A. Krafft and M. J. LaDu, Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability, J. Biol. Chem., 2002, 277, 32046–32053 CrossRef CAS PubMed.
- Y. S. Gong, L. Chang, K. L. Viola, P. N. Lacor, M. P. Lambert, C. E. Finch, G. A. Krafft and W. L. Klein, Alzheimer's disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10417–10422 CrossRef CAS.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis, Science, 2003, 300, 486–489 CrossRef CAS.
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases, Coord. Chem. Rev., 2012, 256, 2308–2332 CrossRef.
- L. E. Scott and C. Orvig, Medicinal Inorganic Chemistry Approaches to Passivation and Removal of Aberrant Metal Ions in Disease, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS.
- I. W. Hamley, The Amyloid Beta Peptide: A Chemist's Perspective. Role in Alzheimer's and Fibrillization, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- A. I. Bush, The metallobiology of Alzheimer's disease, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- P. Faller and C. Hureau, Metal ions in neurodegenerative diseases, Coord. Chem. Rev., 2012, 256, 2127–2128 CrossRef CAS.
- P. Faller and C. Hureau, Bioinorganic chemistry of copper and zinc ions coordinated to Aβ peptide, Dalton Trans., 2009, 1080–1094 RSC.
- P. Faller, Copper and Zinc Binding to Amyloid-β: Coordination, Dynamics, Aggregation, Reactivity and Metal-Ion Transfer, ChemBioChem, 2009, 10, 2837–2845 CrossRef CAS.
- Y. Zhang, D. L. Rempel, J. Zhang, A. K. Sharma, L. M. Mirica and M. L. Gross, Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14604–14609 CrossRef CAS.
- K. J. Barnham and A. I. Bush, Metals in Alzheimer's and Parkinson's Diseases, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS.
- J. H. Viles, Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion diseases, Coord. Chem. Rev., 2012, 256, 2271–2284 CrossRef CAS.
- X. Zhu, B. Su, X. Wang, M. Smith and G. Perry, Causes of oxidative stress in Alzheimer disease, Cell. Mol. Life Sci., 2007, 64, 2202–2210 CrossRef CAS PubMed.
- T. Lynch, R. A. Cherny and A. I. Bush, Oxidative processes in Alzheimer's disease: the role of Aβ-metal interactions, Exp. Gerontol., 2000, 35, 445–451 CrossRef CAS.
- K. J. Barnham, C. L. Masters and A. I. Bush, Neurodegenerative diseases and oxidative stress, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS.
- R. E. Pacifici and K. J. A. Davies, Protein-Degradation as an Index of Oxidative Stress, Methods Enzymol., 1990, 186, 485–502 CAS.
- V. Tõugu and P. Palumaa, Coordination of zinc ions to the key proteins of neurodegenerative diseases: Aβ, APP, α-synuclein and PrP, Coord. Chem. Rev., 2012, 256, 2219–2224 CrossRef.
- V. Tõugu, A. Karafin, K. Zovo, R. S. Chung, C. Howells, A. K. West and P. Palumaa, Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1–42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators, J. Neurochem., 2009, 110, 1784–1795 CrossRef.
- B. Alies, A. Conte-Daban, S. Sayen, F. Collin, I. Kieffer, E. Guillon, P. Faller and C. Hureau, Zinc(II) Binding Site to the Amyloid-β Peptide: Insights from Spectroscopic Studies with a Wide Series of Modified Peptides, Inorg. Chem., 2016, 55, 10499–10509 CrossRef CAS PubMed.
- B. Alies, C. Bijani, S. Sayen, E. Guillon, P. Faller and C. Hureau, Copper Coordination to Native N-Terminally Modified versus Full-Length Amyloid-β: Second-Sphere Effects Determine the Species Present at Physiological pH, Inorg. Chem., 2012, 51, 12988–13000 CrossRef CAS.
- B. Alies, H. Eury, C. Bijani, L. Rechignat, P. Faller and C. Hureau, pH-Dependent Cu(II) Coordination to Amyloid-β Peptide: Impact of Sequence Alterations, Including the H6R and D7N Familial Mutations, Inorg. Chem., 2011, 50, 11192–11201 CrossRef CAS.
- F. Bousejra-ElGarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Iron(II) Binding to Amyloid-β, the Alzheimer's Peptide, Inorg. Chem., 2011, 50, 9024–9030 CrossRef CAS PubMed.
- H. Eury, C. Bijani, P. Faller and C. Hureau, Copper(II) Coordination to Amyloid β: Murine versus Human Peptide, Angew. Chem., Int. Ed., 2011, 50, 901–905 CrossRef CAS.
- X. D. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS.
- X. D. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. A. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, Cu(II) potentiation of Alzheimer A beta neurotoxicity – Correlation with cell-free hydrogen peroxide production and metal reduction, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Copper homeostasis and neurodegenerative disorders (Alzheimer's, prion, and Parkinson's diseases and amyotrophic lateral sclerosis), Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- A. Rauk, The chemistry of Alzheimer's disease, Chem. Soc. Rev., 2009, 38, 2698–2715 RSC.
- D. Pramanik, C. Ghosh, S. Mukherjee and S. G. Dey, Interaction of amyloid β peptides with redox active heme cofactor: Relevance to Alzheimer's disease, Coord. Chem. Rev., 2013, 257, 81–92 CrossRef CAS.
- Y. Luo, B. Bolon, M. A. Damore, D. Fitzpatrick, H. Liu, J. Zhang, Q. Yan, R. Vassar and M. Citron, BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time, Neurobiol. Dis., 2003, 14, 81–88 CrossRef CAS.
- S. Sadigh-Eteghad, M. Talebi, M. Farhoudi, S. E. J. Golzari, B. Sabermarouf and J. Mahmoudi, Beta-amyloid exhibits antagonistic effects on alpha 7 nicotinic acetylcholine receptors in orchestrated manner, J. Med. Hypotheses Ideas, 2014, 8, 49–52 CrossRef CAS.
- M. Tabaton, X. Zhu, G. Perry, M. A. Smith and L. Giliberto, Signaling effect of amyloid-β42 on the processing of AβPP, Exp. Neurol., 2010, 221, 18–25 CrossRef CAS.
- K. Zou, J.-S. Gong, K. Yanagisawa and M. Michikawa, A Novel Function of Monomeric Amyloid β-Protein Serving as an Antioxidant Molecule against Metal-Induced Oxidative Damage, J. Neurosci., 2002, 22, 4833–4841 CrossRef CAS.
- R. Baruch-Suchodolsky and B. Fischer, Aβ40, either Soluble or Aggregated, Is a Remarkably Potent Antioxidant in Cell-Free Oxidative Systems, Biochemistry, 2009, 48, 4354–4370 CrossRef CAS.
- S. J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hyman, M. A. Burton, L. E. Goldstein, S. Duong, R. E. Tanzi and R. D. Moir, The Alzheimer's Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide, PLoS One, 2010, 5, e9505 CrossRef.
- S. Sadigh-Eteghad, B. Sabermarouf, A. Majdi, M. Talebi, M. Farhoudi and J. Mahmoudi, Amyloid-Beta: A Crucial Factor in Alzheimer's Disease, Med. Princ. Pract., 2015, 24, 1–10 CrossRef.
- E. Hellström-Lindahl, M. Viitanen and A. Marutle, Comparison of Aβ levels in the brain of familial and sporadic Alzheimer's disease, Neurochem. Int., 2009, 55, 243–252 CrossRef.
- B. R. Roberts, M. Lind, A. Z. Wagen, A. Rembach, T. Frugier, Q.-X. Li, T. M. Ryan, C. A. McLean, J. D. Doecke, C. C. Rowe, V. L. Villemagne and C. L. Masters, Biochemically-defined pools of amyloid-β in sporadic Alzheimer's disease: correlation with amyloid PET, Brain, 2017, 140, 1486–1498 CrossRef.
- P. Faller, C. Hureau and O. Berthoumieu, Role of Metal Ions in the Self-assembly of the Alzheimer's Amyloid-β Peptide, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van Der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, Neurotoxicity of Alzheimer's disease A[beta] peptides is induced by small changes in the Aβ42 to Aβ40 ratio, EMBO J., 2010, 29, 3408–3420 CrossRef CAS.
- K. Pauwels, T. L. Williams, K. L. Morris, W. Jonckheere, A. Vandersteen, G. Kelly, J. Schymkowitz, F. Rousseau, A. Pastore, L. C. Serpell and K. Broersen, Structural Basis for Increased Toxicity of Pathological Aβ42:Aβ40 Ratios in Alzheimer Disease, J. Biol. Chem., 2012, 287, 5650–5660 CrossRef CAS.
- J. T. Jarrett, E. P. Berger and P. T. Lansbury, The carboxy terminus of the.beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer's disease, Biochemistry, 1993, 32, 4693–4697 CrossRef CAS.
- M. Citron, Alzheimer's disease: strategies for disease modification, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS.
- D. J. Tew, S. P. Bottomley, D. P. Smith, G. D. Ciccotosto, J. Babon, M. G. Hinds, C. L. Masters, R. Cappai and K. J. Barnham, Stabilization of Neurotoxic Soluble β-Sheet-Rich Conformations of the Alzheimer's Disease Amyloid-β Peptide, Biophys. J., 2008, 94, 2752–2766 CrossRef CAS.
- E. Kapaki, J. Segditsa and C. Papageorgiou, Zinc, copper and magnesium concentration in serum and CSF of patients with neurological disorders, Acta Neurol. Scand., 1989, 79, 373–378 CrossRef CAS.
- H. Basun, L. G. Forssell, L. Wetterberg and B. Winblad, Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer's disease, J. Neural Transm.: Parkinson's Dis. Dementia Sect., 1991, 3, 231–258 CAS.
- J. A. Molina, F. J. Jiménez-Jiménez, M. V. Aguilar, I. Meseguer, C. J. Mateos-Vega, M. J. González-Muñoz, F. de Bustos, J. Porta, M. Ortí-Pareja, M. Zurdo, E. Barrios and M. C. Martínez-Para, Cerebrospinal fluid levels of transition metals in patients with Alzheimer's disease, J. Neural Transm., 1998, 105, 479–488 CrossRef CAS.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: possible relation to oxidative stress, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS.
- L. O. Plantin, U. Lying-Tunell and K. Kristensson, Trace elements in the human central nervous system studied with neutron activation analysis, Biol. Trace Elem. Res., 1987, 13, 69–75 CrossRef CAS.
- A. C. Leskovjan, A. Lanzirotti and L. M. Miller, Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer's disease, NeuroImage, 2009, 47, 1215–1220 CrossRef.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer's disease, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS.
- D. Beauchemin and R. Kisilevsky, A Method Based on ICP-MS for the Analysis of Alzheimer's Amyloid Plaques, Anal. Chem., 1998, 70, 1026–1029 CrossRef CAS.
- R. Rajendran, R. Minqin, M. D. Ynsa, G. Casadesus, M. A. Smith, G. Perry, B. Halliwell and F. Watt, A novel approach to the identification and quantitative elemental analysis of amyloid deposits—Insights into the pathology of Alzheimer's disease, Biochem. Biophys. Res. Commun., 2009, 382, 91–95 CrossRef CAS.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, The role of metallobiology and amyloid-β peptides in Alzheimer's disease, J. Neurochem., 2012, 120, 149–166 CrossRef CAS.
- M. W. Bourassa, A. C. Leskovjan, R. V. Tappero, E. R. Farquhar, C. A. Colton, W. E. Van Nostrand and L. M. Miller, Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer's disease that exhibit neurodegeneration, Biomed. Spectrosc. Imaging, 2013, 2, 129–139 CAS.
- J. S. Becker, M. V. Zoriy, C. Pickhardt, N. Palomero-Gallagher and K. Zilles, Imaging of Copper, Zinc, and Other Elements in Thin Section of Human Brain Samples (Hippocampus) by Laser Ablation Inductively Coupled Plasma Mass Spectrometry, Anal. Chem., 2005, 77, 3208–3216 CrossRef CAS.
- J. Feldmann, A. Kindness and P. Ek, Laser ablation of soft tissue using a cryogenically cooled ablation cell, J. Anal. At. Spectrom., 2002, 17, 813–818 RSC.
- A. M. Ghazi, J. C. Wataha, N. L. O’Dell, B. B. Singh, R. Simmons and S. Shuttleworth, Quantitative concentration profiling of nickel in tissues around metal implants: a new biomedical application of laser ablation sector field ICP-MS, J. Anal. At. Spectrom., 2002, 17, 1295–1299 RSC.
- A. I. Bush, Drug Development Based on the Metals Hypothesis of Alzheimer's Disease, J. Alzheimer's Dis., 2008, 15, 223–240 CAS.
- V. Desai and S. G. Kaler, Role of copper in human neurological disorders, Am. J. Clin. Nutr., 2008, 88, 855S–858S CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Oxidative stress and the amyloid beta peptide in Alzheimer's disease, Redox Biol., 2018, 14, 450–464 CrossRef CAS.
- E. Atrián-Blasco, A. Conte-Daban and C. Hureau, Mutual interference of Cu and Zn ions in Alzheimer's disease: perspectives at the molecular level, Dalton Trans., 2017, 46, 12750–12759 RSC.
- A. Georgi, M. Velasco Polo, K. Crincoli, K. Mackenzie and F.-D. Kopinke, Accelerated Catalytic Fenton Reaction with Traces of Iron: An Fe–Pd-Multicatalysis Approach, Environ. Sci. Technol., 2016, 50, 5882–5891 CrossRef CAS.
- M. A. Greenough, J. Camakaris and A. I. Bush, Metal dyshomeostasis and oxidative stress in Alzheimer's disease, Neurochem. Int., 2013, 62, 540–555 CrossRef CAS.
- B. Halliwell, Oxidative stress and neurodegeneration: where are we now?, J. Neurochem., 2006, 97, 1634–1658 CrossRef CAS.
- M. Nakamura, N. Shishido, A. Nunomura, M. A. Smith, G. Perry, Y. Hayashi, K. Nakayama and T. Hayashi, Three Histidine Residues of Amyloid-β Peptide Control the Redox Activity of Copper and Iron, Biochemistry, 2007, 46, 12737–12743 CrossRef CAS PubMed.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. H. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, Small Molecule Modulators of Copper-Induced A beta Aggregation, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N.-V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Amyloid β Protein and Alzheimer's Disease: When Computer Simulations Complement Experimental Studies, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS.
- H. Bayir, Reactive oxygen species, Crit. Care Med., 2005, 33, S498–S501 CrossRef.
- S. Mukherjee, E. A. Kapp, A. Lothian, A. M. Roberts, Y. V. Vasil’ev, B. A. Boughton, K. J. Barnham, W. M. Kok, C. A. Hutton, C. L. Masters, A. I. Bush, J. S. Beckman, S. G. Dey and B. R. Roberts, Characterization and Identification of Dityrosine Cross-Linked Peptides Using Tandem Mass Spectrometry, Anal. Chem., 2017, 89, 6136–6145 CrossRef CAS.
- C. Ghosh, S. Mukherjee, M. Seal and S. G. Dey, Peroxidase to Cytochrome b Type Transition in the Active Site of Heme-Bound Amyloid β Peptides Relevant to Alzheimer's Disease, Inorg. Chem., 2016, 55, 1748–1757 CrossRef CAS.
- D. Pramanik, C. Ghosh and S. G. Dey, Heme–Cu Bound Aβ Peptides: Spectroscopic Characterization, Reactivity, and Relevance to Alzheimer's Disease, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS.
- S. Mukherjee, C. Ghosh, M. Seal and S. G. Dey, Copper induced spin state change of heme–Aβ associated with Alzheimer's disease, Dalton Trans., 2017, 46, 13171–13175 RSC.
- C. Ghosh, M. Seal, S. Mukherjee and S. Ghosh Dey, Alzheimer's Disease: A Heme–Aβ Perspective, Acc. Chem. Res., 2015, 48, 2556–2564 CrossRef CAS.
- K. P. Kepp, Alzheimer's disease: How metal ions define β-amyloid function, Coord. Chem. Rev., 2017, 351, 127–159 CrossRef CAS.
- C. D. Syme, R. C. Nadal, S. E. J. Rigby and J. H. Viles, Copper binding to the amyloid-beta (A beta) peptide associated with Alzheimer's disease - Folding, coordination geometry, pH dependence, stoichiometry, and affinity of A beta-(1-28): Insights from a range of complementary spectroscopic techniques, J. Biol. Chem., 2004, 279, 18169–18177 CrossRef CAS.
- G. Z. Eskici and P. H. Axelsen, Copper and Oxidative Stress in the Pathogenesis of Alzheimer's Disease, Biochemistry, 2012, 51, 6289–6311 CrossRef CAS.
- C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, K. Beyreuther, C. J. Barrow, C. L. Masters, A. I. Bush and K. J. Barnham, Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits, J. Biol. Chem., 2001, 276, 20466–20473 CrossRef CAS.
- J. W. Karr, L. J. Kaupp and V. A. Szalai, Amyloid-beta binds Cu2+ in a mononuclear metal ion binding site, J. Am. Chem. Soc., 2004, 126, 13534–13538 CrossRef CAS.
- V. Minicozzi, F. Stellato, M. Comai, M. D. Serra, C. Potrich, W. Meyer-Klaucke and S. Morante, Identifying the Minimal Copper- and Zinc-binding Site Sequence in Amyloid-β Peptides, J. Biol. Chem., 2008, 283, 10784–10792 CrossRef CAS.
- J. W. Karr, H. Akintoye, L. J. Kaupp and V. A. Szalai, N-terminal deletions modify the Cu2+ binding site in amyloid-beta, Biochemistry, 2005, 44, 5478–5487 CrossRef CAS.
- L. Guilloreau, L. Damian, Y. Coppel, H. Mazarguil, M. Winterhalter and P. Faller, Structural and thermodynamical properties of CuII amyloid-β16/28 complexes associated with Alzheimer's disease, JBIC, J. Biol. Inorg. Chem., 2006, 11, 1024–1038 CrossRef CAS.
- C. C. Curtain, F. E. Ali, D. G. Smith, A. I. Bush, C. L. Masters and K. J. Barnham, Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-beta peptide with membrane lipid, J. Biol. Chem., 2003, 278, 2977–2982 CrossRef CAS.
- T. Kowalik-Jankowska, M. Ruta, K. Wiśniewska and L. Łankiewicz, Coordination abilities of the 1–16 and 1–28 fragments of β-amyloid peptide towards copper(II) ions: a combined potentiometric and spectroscopic study, J. Inorg. Biochem., 2003, 95, 270–282 CrossRef CAS.
- B. J. Hathaway and D. E. Billing, The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- B. J. Hathaway, A New Look at the Stereochemistry and Electronic Properties of Complexes of the Copper(II) Ion, Struct. Bonding, 1984, 57, 55–118 CrossRef CAS.
- Y. Miller, B. Ma and R. Nussinov, Metal binding sites in amyloid oligomers: Complexes and mechanisms, Coord. Chem. Rev., 2012, 256, 2245–2252 CrossRef CAS.
- C. Hureau, Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 1: An overview, Coord. Chem. Rev., 2012, 256, 2164–2174 CrossRef CAS.
- C. Hureau and P. Dorlet, Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 2: Dependence of Cu(II) binding sites with Aβ sequences, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS.
- J. W. Karr and V. A. Szalai, Role of Aspartate-1 in Cu(II) Binding to the Amyloid-β Peptide of Alzheimer's Disease, J. Am. Chem. Soc., 2007, 129, 3796–3797 CrossRef CAS.
- T. Miura, K. Suzuki, N. Kohata and H. Takeuchi, Metal binding modes of Alzheimer's amyloid beta-peptide in insoluble aggregates and soluble complexes, Biochemistry, 2000, 39, 7024–7031 CrossRef CAS.
- F. Stellato, G. Menestrina, M. D. Serra, C. Potrich, R. Tomazzolli, W. Meyer-Klaucke and S. Morante, Metal binding in amyloid β-peptides shows intra- and inter-peptide coordination modes, Eur. Biophys. J., 2006, 35, 340–351 CrossRef CAS.
- V. A. Streltsov, S. J. Titmuss, V. C. Epa, K. J. Barnham, C. L. Masters and J. N. Varghese, The Structure of the Amyloid-β Peptide High-Affinity Copper II Binding Site in Alzheimer Disease, Biophys. J., 2008, 95, 3447–3456 CrossRef CAS.
- S. C. Drew, C. J. Noble, C. L. Masters, G. R. Hanson and K. J. Barnham, Pleomorphic Copper Coordination by Alzheimer's Disease Amyloid-β Peptide, J. Am. Chem. Soc., 2009, 131, 1195–1207 CrossRef CAS.
- S. C. Drew, C. L. Masters and K. J. Barnham, Alanine-2 Carbonyl is an Oxygen Ligand in Cu2+ Coordination of Alzheimer's Disease Amyloid-β Peptide − Relevance to N-Terminally Truncated Forms, J. Am. Chem. Soc., 2009, 131, 8760–8761 CrossRef CAS.
- D. Pierre, G. Serge, F. Peter and H. Christelle, Pulse EPR Spectroscopy Reveals the Coordination Sphere of Copper(II) Ions in the 1–16 Amyloid-β Peptide: A Key Role of the First Two N-Terminus Residues, Angew. Chem., Int. Ed., 2009, 48, 9273–9276 CrossRef PubMed.
- H. Christelle, C. Yannick, D. Pierre, S. P. Lorenzo, S. Stéphanie, G. Emmanuel, S. Laurent and F. Peter, Deprotonation of the Asp1-Ala2 Peptide Bond Induces Modification of the Dynamic Copper(II) Environment in the Amyloid-β Peptide near Physiological pH, Angew. Chem., Int. Ed., 2009, 48, 9522–9525 CrossRef.
- C. J. Sarell, C. D. Syme, S. E. J. Rigby and J. H. Viles, Copper(II) Binding to Amyloid-β Fibrils of Alzheimer's Disease Reveals a Picomolar Affinity: Stoichiometry and Coordination Geometry Are Independent of Aβ Oligomeric Form, Biochemistry, 2009, 48, 4388–4402 CrossRef CAS.
- C. Ghosh and S. G. Dey, Ligand-Field and Ligand-Binding Analysis of the Active Site of Copper-Bound Aβ Associated with Alzheimer's Disease, Inorg. Chem., 2013, 52, 1318–1327 CrossRef CAS.
- S. Furlan, C. Hureau, P. Faller and G. La Penna, Modeling Copper Binding to the Amyloid-β Peptide at Different pH: Toward a Molecular Mechanism for Cu Reduction, J. Phys. Chem. B, 2012, 116, 11899–11910 CrossRef CAS.
- K. I. Silva, B. C. Michael, S. J. Geib and S. Saxena, ESEEM Analysis of Multi-Histidine Cu(II)-Coordination in Model Complexes, Peptides, and Amyloid-β, J. Phys. Chem. B, 2014, 118, 8935–8944 CrossRef CAS.
- R. A. Himes, G. Y. Park, A. N. Barry, N. J. Blackburn and K. D. Karlin, Synthesis and X-ray Absorption Spectroscopy Structural Studies of Cu(I) Complexes of HistidylHistidine Peptides: The Predominance of Linear 2-Coordinate Geometry, J. Am. Chem. Soc., 2007, 129, 5352–5353 CrossRef CAS.
- R. A. Himes, G. Y. Park, G. S. Siluvai, N. J. Blackburn and K. D. Karlin, Structural Studies of Copper(I) Complexes of Amyloid-β Peptide Fragments: Formation of Two-Coordinate Bis(histidine) Complexes, Angew. Chem., Int. Ed., 2008, 47, 9084–9087 CrossRef CAS.
- J. Shearer and V. A. Szalai, The Amyloid-β Peptide of Alzheimer's Disease Binds CuI in a Linear Bis-His Coordination Environment: Insight into a Possible Neuroprotective Mechanism for the Amyloid-β Peptide, J. Am. Chem. Soc., 2008, 130, 17826–17835 CrossRef CAS.
- Y. Lu, M. Prudent, L. Qiao, M. A. Mendez and H. H. Girault, Copper(I) and copper(II) binding to β-amyloid 16 (Aβ16) studied by electrospray ionization mass spectrometry, Metallomics, 2010, 2, 474–479 RSC.
- D. F. Raffa, G. A. Rickard and A. Rauk, Ab initio modelling of the structure and redox behaviour of copper(I) bound to a His–His model peptide: relevance to the β-amyloid peptide of Alzheimer's disease, JBIC, J. Biol. Inorg. Chem., 2007, 12, 147–164 CrossRef CAS.
- S. A. Kozin, S. Zirah, S. Rebuffat, G. H. Hoa and P. Debey, Zinc binding to Alzheimer's Abeta(1-16) peptide results in stable soluble complex, Biochem. Biophys. Res. Commun., 2001, 285, 959–964 CrossRef CAS.
- S. Zirah, S. Rebuffat, S. A. Kozin, P. Debey, F. Fournier, D. Lesage and J.-C. Tabet, Zinc binding properties of the amyloid fragment Aβ(1–16) studied by electrospray-ionization mass spectrometry, Int. J. Mass Spectrom., 2003, 228, 999–1016 CrossRef CAS.
- C. D. Syme and J. H. Viles, Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer's disease, Biochim. Biophys. Acta, 2006, 1764, 246–256 CrossRef CAS.
- Y. Mekmouche, Y. Coppel, K. Hochgräfe, L. Guilloreau, C. Talmard, H. Mazarguil and P. Faller, Characterization of the ZnII binding to the peptide amyloid-beta1-16 linked to Alzheimer's disease, ChemBioChem, 2005, 6, 1663–1671 CrossRef CAS.
- D. Jens, P. Roberta, B. Lucia and G. Astrid, High-resolution NMR studies of the zinc-binding site of the Alzheimer's amyloid β-peptide, FEBS J., 2007, 274, 46–59 CrossRef.
- E. Gaggelli, A. Janicka-Klos, E. Jankowska, H. Kozlowski, C. Migliorini, E. Molteni, D. Valensin, G. Valensin and E. Wieczerzak, NMR Studies of the Zn2+ Interactions with Rat and Human β-Amyloid (1–28) Peptides in Water-Micelle Environment, J. Phys. Chem. B, 2008, 112, 100–109 CrossRef CAS.
- S. Noël, S. Bustos Rodriguez, S. Sayen, E. Guillon, P. Faller and C. Hureau, Use of a new water-soluble Zn sensor to determine Zn affinity for the amyloid-β peptide and relevant mutants, Metallomics, 2014, 6, 1220–1222 RSC.
- C. Opazo, X. D. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, Metalloenzyme-like activity of Alzheimer's disease beta-amyloid - Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS.
- R. Roychaudhuri, M. Yang, M. M. Hoshi and D. B. Teplow, Amyloid β-Protein Assembly and Alzheimer Disease, J. Biol. Chem., 2009, 284, 4749–4753 CrossRef CAS.
- V. Borutaite, R. Morkuniene and G. Valincius, Beta-amyloid oligomers: recent developments, Biomol. Concepts, 2011, 2, 211–222 CAS.
- Y. Miller, B. Ma and R. Nussinov, Polymorphism in Alzheimer Aβ Amyloid Organization Reflects Conformational Selection in a Rugged Energy Landscape, Chem. Rev., 2010, 110, 4820–4838 CrossRef CAS.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. D. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Rapid Induction of Alzheimer a-Beta Amyloid Formation by Zinc, Science, 1994, 265, 1464–1467 CrossRef CAS.
- C. S. Atwood, R. D. Moir, X. D. Huang, R. C. Scarpa, N. M. E. Bacarra, D. M. Romano, M. K. Hartshorn, R. E. Tanzi and A. I. Bush, Dramatic aggregation of Alzheimer A beta by Cu(II) is induced by conditions representing physiological acidosis, J. Biol. Chem., 1998, 273, 12817–12826 CrossRef CAS.
- M. Mold, L. Ouro-Gnao, B. M. Wieckowski and C. Exley, Copper prevents amyloid-β1–42 from forming amyloid fibrils under near-physiological conditions in vitro, Sci. Rep., 2013, 3, 1256 CrossRef.
- E. House, M. Mold, J. Collingwood, A. Baldwin, S. Goodwin and C. Exley, Copper Abolishes the β-Sheet Secondary Structure of Preformed Amyloid Fibrils of Amyloid-β_{42}, J. Alzheimer's Dis., 2009, 18, 811–817 CAS.
- J. Zou, K. Kajita and N. Sugimoto, Cu2+ inhibits the aggregation of amyloid beta-peptide(1-42) in vitro, Angew. Chem., Int. Ed., 2001, 40, 2274–2277 CrossRef CAS.
- Y. Yoshiike, K. Tanemura, O. Murayama, T. Akagi, M. Murayama, S. Sato, X. Y. Sun, N. Tanaka and A. Takashima, New insights on how metals disrupt amyloid beta-aggregation and their effects on amyloid-beta cytotoxicity, J. Biol. Chem., 2001, 276, 32293–32299 CrossRef CAS.
- M. Innocenti, E. Salvietti, M. Guidotti, A. Casini, S. Bellandi, M. L. Foresti, C. Gabbiani, A. Pozzi, P. Zatta and L. Messori, Trace Copper(II) or Zinc(II) Ions Drastically Modify the Aggregation Behavior of Amyloid-β1-42: An AFM Study, J. Alzheimer's Dis., 2010, 19, 1323–1329 CAS.
- J. T. Pedersen, J. Ostergaard, N. Rozlosnik, B. Gammelgaard and N. H. Heegaard, Cu(II) mediates kinetically distinct, non-amyloidogenic aggregation of amyloid-beta peptides, J. Biol. Chem., 2011, 286, 26952–26963 CrossRef CAS.
- C. J. Sarell, S. R. Wilkinson and J. H. Viles, Substoichiometric Levels of Cu2+ Ions Accelerate the Kinetics of Fiber Formation and Promote Cell Toxicity of Amyloid-β from Alzheimer Disease, J. Biol. Chem., 2010, 285, 41533–41540 CrossRef CAS.
- A. I. Bush, Metal complexing agents as therapies for Alzheimer's disease, Neurobiol. Aging, 2002, 23, 1031–1038 CrossRef CAS.
- D. P. Smith, G. D. Ciccotosto, D. J. Tew, M. T. Fodero-Tavoletti, T. Johanssen, C. L. Masters, K. J. Barnham and R. Cappai, Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer's disease amyloid-beta peptide, Biochemistry, 2007, 46, 2881–2891 CrossRef CAS.
- F. Kuperstein and E. Yavin, Pro-apoptotic signaling in neuronal cells following iron and amyloid beta peptide neurotoxicity, J. Neurochem., 2003, 86, 114–125 CrossRef CAS.
- C. A. Rottkamp, A. K. Raina, X. W. Zhu, E. Gaier, A. I. Bush, C. S. Atwood, M. Chevion, G. Perry and M. A. Smith, Redox-active iron mediates amyloid-beta toxicity, Free Radicals Biol. Med., 2001, 30, 447–450 CrossRef CAS.
- E. House, J. Collingwood, A. Khan, O. Korchazkina, G. Berthon and C. Exley, Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Aβ42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease, J. Alzheimer's Dis., 2004, 6, 291–301 CAS.
- C. Ha, J. Ryu and C. B. Park, Metal Ions Differentially Influence the Aggregation and Deposition of Alzheimer's β-Amyloid on a Solid Template†, Biochemistry, 2007, 46, 6118–6125 CrossRef CAS.
- K. Garai, P. Sengupta, B. Sahoo and S. Maiti, Selective destabilization of soluble amyloid β oligomers by divalent metal ions, Biochem. Biophys. Res. Commun., 2006, 345, 210–215 CrossRef CAS.
- S. Bolognin, L. Messori, D. Drago, C. Gabbiani, L. Cendron and P. Zatta, Aluminum, copper, iron and zinc differentially alter amyloid-Aβ1-42 aggregation and toxicity, Int. J. Biochem. Cell Biol., 2011, 43, 877–885 CrossRef CAS.
- C. J. Matheou, N. D. Younan and J. H. Viles, Cu2+ accentuates distinct misfolding of Aβ(1–40) and Aβ(1–42) peptides, and potentiates membrane disruption, Biochem. J., 2015, 466, 233–242 CrossRef CAS.
- A. I. Bush, G. Multhaup, R. D. Moir, T. G. Williamson, D. H. Small, B. Rumble, P. Pollwein, K. Beyreuther and C. L. Masters, A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer's disease, J. Biol. Chem., 1993, 268, 16109–16112 CAS.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J.-P. Vonsattel, R. E. Tanzi and A. I. Bush, Zinc-induced Alzheimer's Aβ1–40 Aggregation Is Mediated by Conformational Factors, J. Biol. Chem., 1997, 272, 26464–26470 CrossRef CAS.
- S. T. Liu, G. Howlett and C. J. Barrow, Histidine-13 is a crucial residue in the zinc ion-induced aggregation of the A beta peptide of Alzheimer's disease, Biochemistry, 1999, 38, 9373–9378 CrossRef CAS.
- K. Garai, B. Sahoo, S. K. Kaushalya, R. Desai and S. Maiti, Zinc Lowers Amyloid-β Toxicity by Selectively Precipitating Aggregation Intermediates†, Biochemistry, 2007, 46, 10655–10663 CrossRef CAS.
- M.-C. Lee, W.-C. Yu, Y.-H. Shih, C.-Y. Chen, Z.-H. Guo, S.-J. Huang, J. C. C. Chan and Y.-R. Chen, Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer's disease, Sci. Rep., 2018, 8, 4772 CrossRef.
- T. Zhang, T. Pauly and L. Nagel-Steger, Stoichiometric Zn2+ interferes with the self-association of Aβ42: Insights from size distribution analysis, Int. J. Biol. Macromol., 2018, 113, 631–639 CrossRef CAS.
- P. Faller, C. Hureau, P. Dorlet, P. Hellwig, Y. Coppel, F. Collin and B. Alies, Methods and techniques to study the bioinorganic chemistry of metal–peptide complexes linked to neurodegenerative diseases, Coord. Chem. Rev., 2012, 256, 2381–2396 CrossRef CAS.
- W. L. Klein, A beta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets, Neurochem. Int., 2002, 41, 345–352 CrossRef CAS.
- H. LeVine 3rd, Quantification of beta-sheet amyloid fibril structures with thioflavin T, Methods Enzymol., 1999, 309, 274–284 CAS.
- N. Amdursky, Y. Erez and D. Huppert, Molecular Rotors: What Lies Behind the High Sensitivity of the Thioflavin-T Fluorescent Marker, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS.
- I. Maezawa, H.-S. Hong, R. Liu, C.-Y. Wu, R. H. Cheng, M.-P. Kung, H. F. Kung, K. S. Lam, S. Oddo, F. M. LaFerla and L.-W. Jin, Congo red and thioflavin-T analogs detect Aβ oligomers, J. Neurochem., 2008, 104, 457–468 CAS.
- L. P. Jameson, N. W. Smith and S. V. Dzyuba, Dye-Binding Assays for Evaluation of the Effects of Small Molecule Inhibitors on Amyloid (Aβ) Self-Assembly, ACS Chem. Neurosci., 2012, 3, 807–819 CrossRef CAS.
- H.-S. Hong, S. Rana, L. Barrigan, A. Shi, Y. Zhang, F. Zhou, L.-W. Jin and D. H. Hua, Inhibition of Alzheimer's Amyloid Toxicity with a Tricyclic Pyrone Molecule In Vitro and In Vivo, J. Neurochem., 2009, 108, 1097–1108 CrossRef CAS.
- B. Alies, P.-L. Solari, C. Hureau and P. Faller, Dynamics of ZnII Binding as a Key Feature in the Formation of Amyloid Fibrils by Aβ11-28, Inorg. Chem., 2011, 51, 701–708 CrossRef PubMed.
- A. Kochi, T. J. Eckroat, K. D. Green, A. S. Mayhoub, M. H. Lim and S. Garneau-Tsodikova, A novel hybrid of 6-chlorotacrine and metal–amyloid-β modulator for inhibition of acetylcholinesterase and metal-induced amyloid-β aggregation, Chem. Sci., 2013, 4, 4137–4145 RSC.
- R. Bartus, R. Dean, B. Beer and A. Lippa, The cholinergic hypothesis of geriatric memory dysfunction, Science, 1982, 217, 408–414 CrossRef CAS.
- R. Schliebs and T. Arendt, The significance of the cholinergic system in the brain during aging and in Alzheimer's disease, J. Neural Transm., 2006, 113, 1625–1644 CrossRef CAS.
- A. K. Sharma, J. Kim, J. T. Prior, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Small Bifunctional Chelators That Do Not Disaggregate Amyloid β Fibrils Exhibit Reduced Cellular Toxicity, Inorg. Chem., 2014, 53, 11367–11376 CrossRef CAS.
- H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, K. H. Thompson and C. Orvig, Combating Alzheimer's disease with multifunctional molecules designed for metal passivation, Angew. Chem., Int. Ed., 2007, 46, 1716–1718 CrossRef CAS.
- K. Rajasekhar, C. Madhu and T. Govindaraju, Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity, ACS Chem. Neurosci., 2016, 7, 1300–1310 CrossRef CAS PubMed.
- M. D. Bartholomä, Recent developments in the design of bifunctional chelators for metal-based radiopharmaceuticals used in Positron Emission Tomography, Inorg. Chim. Acta, 2012, 389, 36–51 CrossRef.
- R. A. Peters, L. A. Stocken and R. H. S. Thompson, British Anti-Lewisite (BAL), Nature, 1945, 156, 616 CrossRef CAS.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. S. Kim, X. D. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice, Neuron, 2001, 30, 665–676 CrossRef CAS.
- L. Cahoon, The curious case of clioquinol, Nat. Med., 2009, 15, 356–359 CrossRef CAS.
- A. M. Mancino, S. S. Hindo, A. Kochi and M. H. Lim, Effects of Clioquinol on Metal-Triggered Amyloid-beta Aggregation Revisited, Inorg. Chem., 2009, 48, 9596–9598 CrossRef CAS.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, The Ongoing Search for Small Molecules to Study Metal-Associated Amyloid-β Species in Alzheimer's Disease, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS.
- M. Rana, H.-J. Cho, T. K. Roy, L. M. Mirica and A. K. Sharma, Azo-dyes based small bifunctional molecules for metal chelation and controlling amyloid formation, Inorg. Chim. Acta, 2018, 471, 419–429 CrossRef CAS.
- W. E. Klunk, H. Engler, A. Nordberg, Y. M. Wang, G. Blomqvist, D. P. Holt, M. Bergstrom, I. Savitcheva, G. F. Huang, S. Estrada, B. Ausen, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Langstrom, Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B, Ann. Neurol., 2004, 55, 306–319 CrossRef CAS.
- A. Dedeoglu, K. Cormier, S. Payton, K. A. Tseitlin, J. N. Kremsky, L. Lai, X. H. Li, R. D. Moir, R. E. Tanzi, A. I. Bush, N. W. Kowall, J. T. Rogers and X. D. Huang, Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer's amyloidogenesis, Exp. Gerontol., 2004, 39, 1641–1649 CrossRef CAS.
- A. K. Sharma, J. W. Schultz, J. T. Prior, N. P. Rath and L. M. Mirica, Coordination Chemistry of Bifunctional Chemical Agents Designed for Applications in 64Cu PET Imaging for Alzheimer's Disease, Inorg. Chem., 2017, 56, 13801–13814 CrossRef CAS PubMed.
- N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers and L. M. Mirica, Evaluation of 64Cu-Based Radiopharmaceuticals that Target Aβ Peptide Aggregates as Diagnostic Tools for Alzheimer's Disease, J. Am. Chem. Soc., 2017, 139, 12550–12558 CrossRef CAS.
- L. K. Charkoudian, D. M. Pham and K. J. Franz, A Pro-Chelator Triggered by Hydrogen Peroxide Inhibits Iron-Promoted Hydroxyl Radical Formation, J. Am. Chem. Soc., 2006, 128, 12424–12425 CrossRef CAS.
- L. K. Charkoudian, D. M. Pham, A. M. Kwon, A. D. Vangeloff and K. J. Franz, Modifications of boronic ester pro-chelators triggered by hydrogen peroxide tune reactivity to inhibit metal-promoted oxidative stress, Dalton Trans., 2007, 5031–5042 RSC.
- M. G. Dickens and K. J. Franz, A Prochelator Activated by Hydrogen Peroxide Prevents Metal-Induced Amyloid β Aggregation, ChemBioChem, 2010, 11, 59–62 CrossRef CAS.
- D. S. Folk and K. J. Franz, A Prochelator Activated by β-Secretase Inhibits Aβ Aggregation and Suppresses Copper-Induced Reactive Oxygen Species Formation, J. Am. Chem. Soc., 2010, 132, 4994–4995 CrossRef CAS.
- L. M. Hyman and K. J. Franz, A cell-permeable fluorescent prochelator responds to hydrogen peroxide and metal ions by decreasing fluorescence, Inorg. Chim. Acta, 2012, 380, 125–134 CrossRef CAS.
- L. R. Perez and K. J. Franz, Minding metals: Tailoring multifunctional chelating agents for neurodegenerative disease, Dalton Trans., 2010, 39, 2177–2187 RSC.
- J.-S. Choi, J. J. Braymer, S. K. Park, S. Mustafa, J. Chae and M. H. Lim, Synthesis and characterization of IMPY derivatives that regulate metal-induced Aβ aggregation, Metallomics, 2011, 3, 284–291 RSC.
- Y. Ji, H. J. Lee, M. Kim, G. Nam, S. J. C. Lee, J. Cho, C.-M. Park and M. H. Lim, Strategic Design of 2,2′-Bipyridine Derivatives to Modulate Metal–Amyloid-β Aggregation, Inorg. Chem., 2017, 56, 6695–6705 CrossRef CAS.
- J. Han, H. J. Lee, K. Y. Kim, S. J. C. Lee, J.-M. Suh, J. Cho, J. Chae and M. H. Lim, Tuning Structures and Properties for Developing Novel Chemical Tools toward Distinct Pathogenic Elements in Alzheimer's Disease, ACS Chem. Neurosci., 2018, 9, 800–808 CrossRef CAS.
- A. S. DeToma, J.-S. Choi, J. J. Braymer and M. H. Lim, Myricetin: A Naturally Occurring Regulator of Metal-Induced Amyloid-β Aggregation and Neurotoxicity, ChemBioChem, 2011, 12, 1198–1201 CrossRef CAS.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, EGCG remodels mature β ± -synuclein and amyloid-β fibrils and reduces cellular toxicity, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS.
- X. He, H. M. Park, S.-J. Hyung, A. S. DeToma, C. Kim, B. T. Ruotolo and M. H. Lim, Exploring the reactivity of flavonoid compounds with metal-associated amyloid-β species, Dalton Trans., 2012, 41, 6558–6566 RSC.
- S.-J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J.-S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-β species, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743–3748 CrossRef CAS PubMed.
- L. E. Scott, M. Telpoukhovskaia, C. Rodriguez-Rodriguez, M. Merkel, M. L. Bowen, B. D. G. Page, D. E. Green, T. Storr, F. Thomas, D. D. Allen, P. R. Lockman, B. O. Patrick, M. J. Adam and C. Orvig, N-Aryl-substituted 3-([small beta]-D-glucopyranosyloxy)-2-methyl-4(1H)-pyridinones as agents for Alzheimer's therapy, Chem. Sci., 2011, 2, 642–648 RSC.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, Synthesis, characterization, and metal coordinating ability of multifunctional carbohydrate-containing compounds for Alzheimer's therapy, J. Am. Chem. Soc., 2007, 129, 7453–7463 CrossRef CAS.
- T. Storr, K. H. Thompson and C. Orvig, Design of targeting ligands in medicinal inorganic chemistry, Chem. Soc. Rev., 2006, 35, 534–544 RSC.
- T. Storr, L. E. Scott, M. L. Bowen, D. E. Green, K. H. Thompson, H. J. Schugar and C. Orvig, Glycosylated tetrahydrosalens as multifunctional molecules for Alzheimer's therapy, Dalton Trans., 2009, 3034–3043 RSC.
- L. E. Scott, B. D. G. Page, B. O. Patrick and C. Orvig, Altering pyridinone N-substituents to optimise activity as potential prodrugs for Alzheimer's disease, Dalton Trans., 2008, 6364–6367 RSC.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q.-X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Rapid Restoration of Cognition in Alzheimer's Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ, Neuron, 2008, 59, 43–55 CrossRef CAS.
- P. J. Crouch, M. S. Savva, L. W. Hung, P. S. Donnelly, A. I. Mot, S. J. Parker, M. A. Greenough, I. Volitakis, P. A. Adlard, R. A. Cherny, C. L. Masters, A. I. Bush, K. J. Barnham and A. R. White, The Alzheimer's therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity, J. Neurochem., 2011, 119, 220–230 CrossRef CAS.
- C. P. W. Soon, P. S. Donnelly, B. J. Turner, L. W. Hung, P. J. Crouch, N. A. Sherratt, J.-L. Tan, N. K.-H. Lim, L. Lam, L. Bica, S. Lim, J. L. Hickey, J. Morizzi, A. Powell, D. I. Finkelstein, J. G. Culvenor, C. L. Masters, J. Duce, A. R. White, K. J. Barnham and Q.-X. Li, Diacetylbis(N(4)-methylthiosemicarbazonato) Copper(II) (CuII(atsm)) Protects against Peroxynitrite-induced Nitrosative Damage and Prolongs Survival in Amyotrophic Lateral Sclerosis Mouse Model, J. Biol. Chem., 2011, 286, 44035–44044 CrossRef CAS.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis in the Last Decade, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.8b00138.
- A. J. Doig, M. P. del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer's Disease?, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef CAS PubMed.
- S. C. Drew, The Case for Abandoning Therapeutic Chelation of Copper Ions in Alzheimer's Disease, Front. Neurosci., 2017, 11, 317 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |