
A nanolayer coating on polydimethylsiloxane surfaces enables a mechanistic study of bacterial adhesion influenced by material surface physicochemistry†
Fei
Pan
 ab,
Stefanie
Altenried
a,
Mengdi
Liu
ab,
Dirk
Hegemann
c,
Ezgi
Bülbül
c,
Jens
Moeller
d,
Wolfgang W.
Schmahl
b,
Katharina
Maniura-Weber
a and
Qun
Ren
*a
ab,
Stefanie
Altenried
a,
Mengdi
Liu
ab,
Dirk
Hegemann
c,
Ezgi
Bülbül
c,
Jens
Moeller
d,
Wolfgang W.
Schmahl
b,
Katharina
Maniura-Weber
a and
Qun
Ren
*a
aLaboratory for Biointerfaces, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: Qun.Ren@empa.ch
bDepartment of Earth- and Environmental Sciences, Ludwig Maximilian University of Munich, Theresienstrasse 41, 80333 Munich, Germany
cLaboratory of Advanced Fibers, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland
dLaboratory of Applied Mechanobiology, Department of Health Sciences and Technology, ETH Zurich, 8093 Zurich, Switzerland
First published on 2nd September 2019
Abstract
To control material-associated bacterial infections, understanding the underlying mechanisms of bacteria and surface interactions is essential. Here we focused on studying how material mechanical and chemical properties can impact bacterial adhesion, using polydimethylsiloxane (PDMS) as a model material. To this end, PDMS surfaces of different stiffness were coated with a 2 nm highly cross-linked PDMS-like polymer film to confer comparable surface chemistry, while retaining similar mechanical properties for coated and uncoated samples. The uncoated samples showed increased interfacial adhesion force with the decrease of Young's modulus, whereas the nanolayer deposition yielded a comparable adhesion force for all surfaces. The Gram negative strains Escherichia coli, its fimbriae mutants and Pseudomonas aeruginosa as well as the Gram positive strain Staphylococcus epidermidis were analysed for their adhesion on these surfaces. For each bacterial strain similar numbers were found on the coated surfaces of different PDMS species, whereas the numbers on the uncoated surfaces increased several fold with the decrease of material modulus. Similar adhesion behaviour was also observed for the negatively charged abiotic polystyrene beads of similar size to bacteria. These results strongly suggest that the interfacial chemistry of the PDMS rather than the material mechanical properties plays a critical role in bacterial adhesion.
New conceptsA nanolayer coating through plasma polymerization was utilized in this work to study the mechanism of bacterial adhesion on soft matter. The so-far reported work ascribed the important roles of material stiffness and bacterial mechanosensing in bacterial initial colonization. Our work presented here challenges the previous conclusion and demonstrates that interfacial chemistry (molecular bridging) rather than material stiffness or bacterial mechanosensing plays a decisive role in regulating bacterial adhesion upon contacting viscoelastic surfaces. This demonstration was achieved by coating material (PDMS) surfaces of different stiffness with a 2 nm PDMS-like polymer film. Similar chemistry could be obtained on all coated surfaces even though the Young's moduli of the coated sample interfaces remained different. Similar numbers of the adhered bacteria (abiotic polystyrene beads as well) were found on all coated surfaces, but significantly different numbers were found on the uncoated surfaces similar to the previous reports. Thus, interfacial chemistry was concluded as a critical factor to influence bacterial adhesion at the initial colonization stage. These findings provide new fundamental insights into the influence of material properties on bacterial adhesion. The knowledge gained here will facilitate the development of antimicrobial materials with tailored physicochemical properties. |
Biofilms, aggregates of bacteria embedded in a self-produced matrix,1,2 are considered as the leading factor to cause persistent and chronic bacterial infections due to their enhanced resistance towards host defence mechanisms and antibiotics.3,4 Biofilms are established through stages that are characterised by reversible bacteria adhesion, irreversible adhesion, biofilm development, biofilm maturation and biofilm dispersal.5,6 In the process of biofilm formation, irreversible bacteria adhesion plays a crucial role and is affected by multiple factors, including surface chemistry,7–10 surface roughness,11–13 topography,14–16 and material stiffness.17–20 It has been reported that bacteria, i.e. Escherichia coli, Staphylococcus aureus, Pseudoalteromonas sp., and Bacillus sp. adhere in higher numbers on stiff than on soft agarose, agar and poly(ethylene glycol) dimethacrylate (PEGDMA) hydrogels.20–22 However, it was also shown that on polyacrylamide hydrogels, S. aureus cells adhered in higher numbers on soft than on stiff substrates.23 Song and coworkers also reported that on polydimethylsiloxane (PDMS), a widely utilised biomaterial with low toxicity, good biocompatibility and durability,24E. coli and P. aeruginosa adhered in higher numbers on soft PDMS.25,26 These contradicting results are very likely caused by different physicochemical and mechanical properties of the materials, different bacterial strains and different experimental conditions used in those studies.23
Previously, it has been shown that fimbriae play a critical role in bacterial adhesion.27,28 Recently we found that the intrinsic material properties associated with PDMS substrates of different stiffness strongly influence bacterial adhesion, complementing the previously reported theory on active bacterial mechanosensing.29 We further discovered that the material viscosity, not only stiffness (elasticity), greatly impacts the initial adhesion of E. coli.30 The high viscosity of soft PDMS confers a high degree of stickiness to the material in addition to the high degree of deformatibility.30 Thus, one of the remaining questions is, whether the observed higher number of adhered bacteria on soft PDMS is due to larger interaction area caused by potential material deformation, or due to interfacial chemical properties such as more available and/or longer polymer chains in soft materials thus supporting molecular bridging with the bacteria. To address this question, we designed our work as illustrated in Fig. 1, aiming to decouple the above mentioned two factors by coating PDMS substrates of different stiffness with a chemically PDMS-like nanolayer. While the interfacial and bulk mechanical properties of each PDMS species did not vary much before and after the nanolayer coating, the adhesion profiles of different bacterial strains (E. coli, its fimbriae mutants, P. aeruginosa and S. epidermidis) were compared on PDMS having five different Young's moduli in the range of 64.2 to 2326.8 kPa with and without the nanolayer. Similar to what has been reported previously25,29 a higher number of adhered bacteria was found on soft (64.2 kPa) than on stiff (2326.8 kPa) PDMS for the uncoated samples. By contrast, on the five nanolayer-coated PDMS samples the tested individual bacterial strain adhered in comparable numbers with varying numbers for different strains. The negatively charged abiotic polystyrene (PS-COOH) beads also exhibited an adhesion profile similar to that of bacteria. These results strongly suggested that the interfacial chemistry rather than the material stiffness plays a predominant role in the initial stage of bacteria colonization of a PDMS surface. This work provides novel insights for an improved understanding of how bacteria interact with engineered material surfaces. Those findings can be potentially used to tune the material surface properties in such a way that we can tailor bacterial adhesion to the specific application.
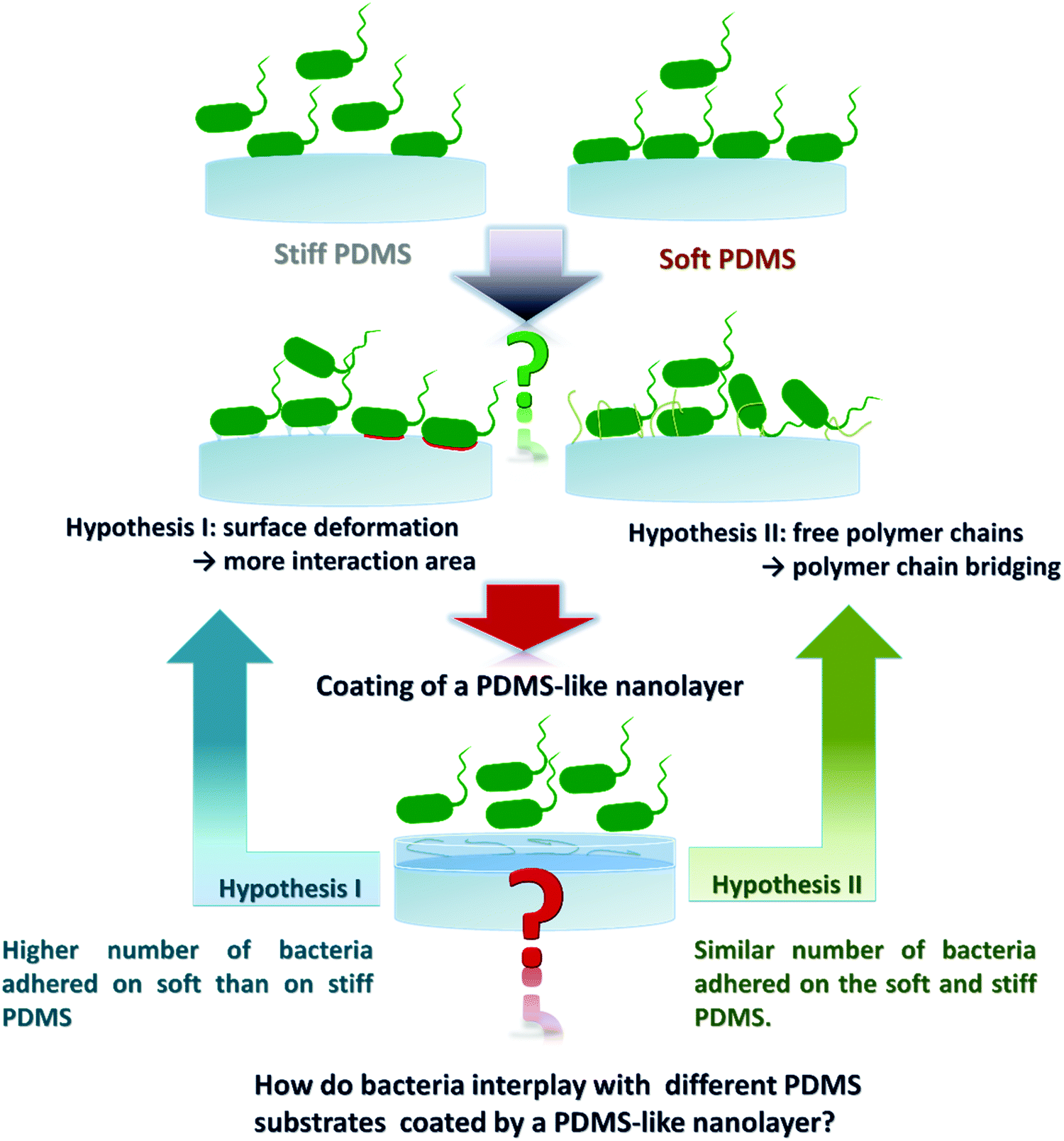 | ||
| Fig. 1 Approach to decouple two possible mechanisms involved in bacterial adhesion on PDMS substrates of varying stiffness. Hypothesis I considers the deformation of the interface of PDMS, where soft PDMS would deform more than the stiff substrates. Hypothesis II proposes that the surface chemistry impacts bacterial adhesion by the availability of free PDMS polymer chains and PDMS polymer chain ends. To evaluate these two mechanisms, a 2 nm layer of crosslinked PDMS-like plasma polymer (HMDSO) was applied on the top of the different PDMS substrates to confer similar surface chemical properties, and subsequently bacteria adhesion assays were performed. | ||
In this work, plasma polymerization was used to generate a hexamethyldisiloxane-derived (HMDSO, (CH3)3Si–O–Si(CH3)3) nanolayer coating on PDMS surfaces of different stiffness. HMDSO is one of the most commonly used compounds applied in plasma polymerization.31 In the process of plasma polymerization, the covalent bonds of HMDSO can be broken and film-forming radicals ˙(CH3)2Si–O˙ are generated.32 After adjusting the plasma polymerization parameters such as gas flow rate (HMDSO/Ar 4/20 sccm), power input (50 W) and pressure (7 Pa), a highly crosslinked PDMS-like and nanometer-thin layer could be achieved on the PDMS surfaces.33 Plasma polymer films were deposited at constant plasma conditions with varying deposition time on PDMS and silicon substrates to derive the substrate-specific deposition rate (Fig. S1, ESI†). The deposition time was adjusted to obtain a 2 nm thin nanolayer on all PDMS species of different stiffness, which was confirmed by ellipsometry analysis (Table 1) based on the different refractive indices of plasma-coated HMDSO layers and PDMS substrates.34,35 The chemical composition of the coated PDMS samples was analysed by XPS. As expected, similar chemical compositions of uncoated and coated PDMS surfaces were found (Table S1, ESI†). Plasma treatment of PDMS with HMDSO was previously shown to potentially induce not only surface oxidation and polymer chain scission but also crosslinking and formation of a silica-like layer during surface activation.36,37 Therefore, careful selection of plasma activation and deposition conditions was required to optimise the deposition thickness of PDMS-like nanolayers.38 Moreover, the plasma-coated layer can act as a barrier to inhibit PDMS chain migration to the surface.39,40 In this work analysis and characterisation of the coated PDMS samples were performed immediately after plasma coating.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 being stiffest and uncoated and coated PDMS 40:1 softest. The loss factor, the ratio of loss modulus over storage modulus, revealed that uncoated and coated PDMS species with lower crosslinker content exhibited a higher viscosity and lower elasticity. Three independent measurements were conducted with three replicates of one sample in one measurement. Standard deviation was calculated based on a total of 9 measurements carried out for one sample
:1 being stiffest and uncoated and coated PDMS 40:1 softest. The loss factor, the ratio of loss modulus over storage modulus, revealed that uncoated and coated PDMS species with lower crosslinker content exhibited a higher viscosity and lower elasticity. Three independent measurements were conducted with three replicates of one sample in one measurement. Standard deviation was calculated based on a total of 9 measurements carried out for one sample
| PDMS species (elastomer: curing agent, wt/wt) | Coating layer thickness [nm] | Shear complex modulus (G*) [kPa] | Storage modulus (G′) [kPa] | Loss modulus (G′′) [kPa] |
|---|---|---|---|---|
| 5:1 |
0 | 67.7 ± 0.38 | 67.6 ± 0.38 | 3.3 ± 0.02 |
| 2.0 ± 0.12 | 65.2 ± 0.43 | 65.1 ± 0.43 | 3.0 ± 0.03 | |
| 10:1 |
0 | 53.9 ± 0.50 | 53.8 ± 0.50 | 2.9 ± 0.06 |
| 2.0 ± 0.13 | 50.2 ± 0.47 | 50.1 ± 0.47 | 2.9 ± 0.03 | |
| 20:1 |
0 | 40.5 ± 0.06 | 40.5 ± 0.06 | 1.9 ± 0.01 |
| 2.0 ± 0.11 | 41.6 ± 0.18 | 41.5 ± 0.18 | 2.0 ± 0.02 | |
| 30:1 |
0 | 25.1 ± 0.02 | 25.0 ± 0.02 | 1.7 ± 0.01 |
| 2.0 ± 0.20 | 24.0 ± 0.05 | 24.0 ± 0.05 | 1.5 ± 0.02 | |
| 40:1 |
0 | 4.3 ± 0.02 | 4.2 ± 0.02 | 1.0 ± 0.04 |
| 2.0 ± 0.10 | 3.9 ± 0.08 | 3.8 ± 0.08 | 0.9 ± 0.03 |
The thin HMDSO plasma polymer coating was further investigated for its impact on the interfacial hydrophobicity. Unlike the uncoated PDMS which showed a slight increase in water contact angle (in a range of 109–121°) with the decrease of crosslinker content (Fig. 2A), the coated PDMS species exhibited similar water contact angle values in a narrow range of 108–113° indicative of successful HMDSO coating and thus the similar interfacial chemistry. The results of the water contact angle measurements indicated that coating of PDMS surfaces with a 2 nm thin film could block the free polymer chains and polymer chain ends on PDMS surfaces, reflected by the reduced water contact angle of the coated surfaces compared to the uncoated counter samples and the similar contact angle values for all coated samples (Fig. 2A). Similar observations have been reported previously.36,37,40 We further analysed the surface roughness of all substrates by AFM (Fig. 2B). The HMDSO coating resulted in an increase of surface roughness, from an average roughness (Ra) range of 1.5–5.8 nm before to that of 11.8–12.1 nm after coating. The increase in surface roughness after plasma coating is consistent with previous reports.41,42 The slight difference of about 4 nm measured for the uncoated samples is unlikely to affect bacterial adhesion.43–45 The surfaces of all coated PDMS samples were flat without characteristic morphologies (Fig. S2, ESI†).
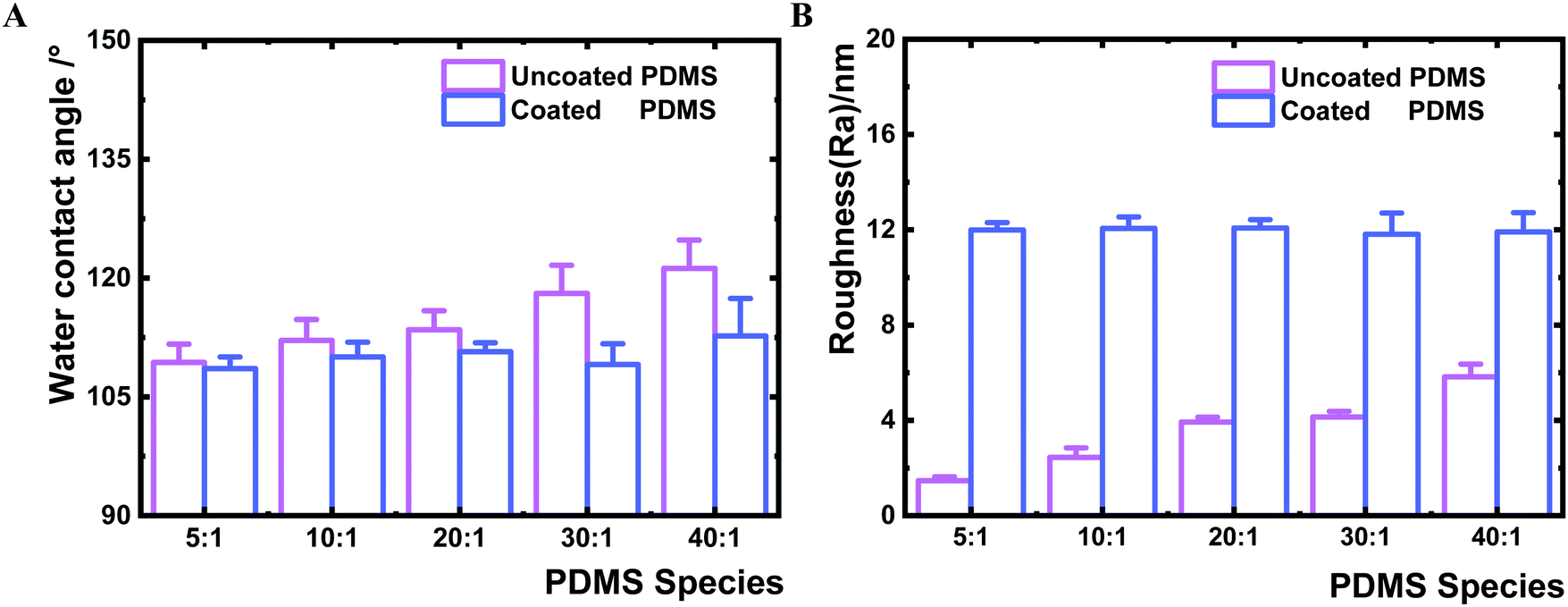 | ||
| Fig. 2 Surface characterisation of uncoated and coated PDMS samples. A. Water contact angle measurement. Four samples of each PDMS species were measured and each sample was measured six times. Error bars represent the standard deviations of 24 measurements. B. Surface average roughness (Ra) measured by AFM. Each sample was measured 5 times and independent measurements were conducted three times. Error bars represent the standard deviations of 15 measurements. Student's t-test (p < 0.01) revealed a significant difference among all uncoated samples, no significant difference was found among all coated samples. | ||
To ensure that the nanolayer coating on the PDMS surfaces does not change the bulk material properties, the bulk mechanical properties of coated and uncoated PDMS samples were measured by rheometry. The shear complex modulus, a measure of the materials viscoelasticity, was similar for a given PDMS species before and after coating (Table 1 and Fig. S3, ESI†). Furthermore, the deposition of the HMDSO nanolayer had no impact on the bulk deformability of the PDMS material (Fig. S4, ESI†). The maximum bulk adhesion forces of uncoated and coated PDMS samples were compared as well. The adhesion force of the uncoated samples increased from 0.6 to 30.2 N with the decrease of the crosslinking agent. Likewise, the maximum bulk adhesion forces of the coated PDMS showed a very comparable range of 0.6 to 31.0 N (Fig. 3A). Hence, the maximum adhesion forces measured for the same kind of PDMS remained constant before and after HMDSO coating. Moreover, the adhesion energies of coated and uncoated bulk materials were found to be similar for a given PDMS species before and after plasma coating (Fig. 3B). Yet, there was a clear trend that the adhesion energy of bulk materials increased for both coated and uncoated PDMS species with the decrease of crosslinker content (Fig. 3B). The bulk adhesion energies for uncoated and coated PDMS 40:1 were larger than 15 J m−2, while those for uncoated and coated PDMS species 5:1, 10:1, 20:1 and 30:1 were about 0.1, 0.2, 3.0 and 9.0 J m−2, respectively.
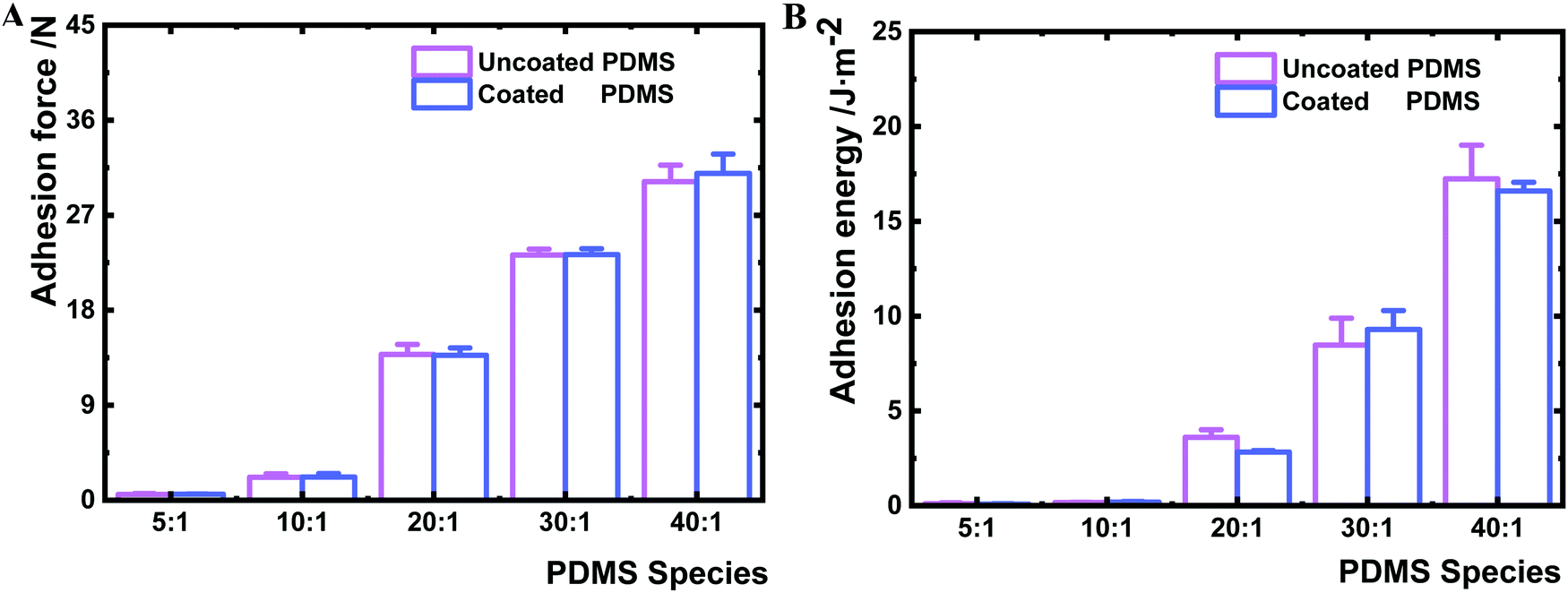 | ||
| Fig. 3 Comparison of the bulk adhesion force (A) and adhesion energy (B) of PDMS before and after HMDSO plasma coating. Three repeated measurements were performed for one sample and each sample type was analysed in triplicate. | ||
In the next step, we investigated the interfacial Young's modulus and adhesion force of the PDMS samples before and after coating by AFM (Fig. 4). The interfacial Young's modulus of the coated PDMS increased from 163.8 to 3630.8 kPa with increasing crosslinker content. This trend was similar for the uncoated PDMS substrates (increase from 64.2 to 2326.8 kPa) (Fig. 4A). By contrast, the interfacial maximum adhesion force, measured by AFM-force spectroscopy assessing near-surface (nm scale) properties during cantilever retraction from the sample surface,46 was substantially different before and after coating (Fig. 4B). While the maximum adhesion force of uncoated PDMS surfaces increased from 1 to 283 nN between the 5:1 and 40:1 samples, the adhesion forces for the coated samples varied only between 12 and 38 nN for 5:1 and 40:1, respectively (Fig. 4B). The narrow range of adhesion forces for modified PDMS demonstrated that all PDMS species exhibit similar interfacial adhesion properties after plasma coating. Higher adhesion forces present on uncoated PDMS species with lower crosslinker content indicated that the surface physicochemical properties were altered by decreasing the crosslinking degree.
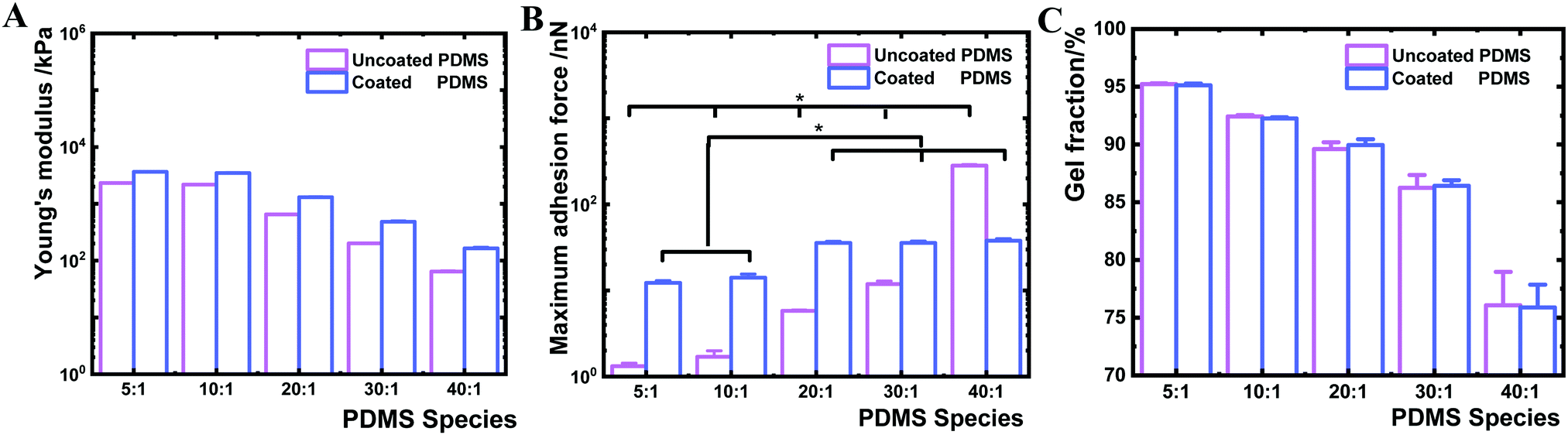 | ||
| Fig. 4 Interfacial characterisation of uncoated and coated PDMS samples. Young's modulus (A) was measured by AFM-force spectroscopy. Maximum surface adhesion force (B) was determined by AFM-force spectroscopy during cantilever retracting. Three replicates of uncoated and coated PDMS of every species were characterised and 576 indentations of every replicate were performed to derive interface Young's modulus and adhesion force, and corresponding standard deviation. Student's t-test (p < 0.01) in (A) revealed a significant difference among all the uncoated samples, all coated samples and before and after coating of the same PDMS species. * in (B) denotes p < 0.01 according to Student's t-test. (C) Quantification of uncrosslinked PDMS polymer chains inside bulk materials by gel fraction analysis. Three repeated measurements were performed for one sample and each sample type was analysed in triplicate. | ||
We further sought to derive the amount of free polymer chains in PDMS and the length of the polymer chain ends on PDMS surfaces. The gel fraction was determined based on sample mass change before and after ethanol extraction for one week. Coated and uncoated PDMS 40:1 showed a gel fraction of about 76 wt%, indicating that 24 wt% of uncrosslinked PDMS polymers were extracted (Fig. 4C). By contrast, higher levels in gel fraction were observed in stiffer uncoated and coated PDMS substrates (PDMS 5:1 of 95 wt% and PDMS 10:1 of 92 wt%). These results suggested that uncoated and coated soft 40:1 PDMS contain a high amount of free or uncrosslinked PDMS polymer chains. The length of the chain ends was theoretically estimated based on a previously reported method and Monte Carlo simulation,47 which revealed that the length of the polymer chain ends decreased with increasing crosslinking degree. Thereby, the PDMS chain ends would increase with decreasing crosslinker content on uncoated PDMS samples.47 Uncoated PDMS 40:1 would therefore have the longest and uncoated PDMS 5:1 the shortest chain ends.
In summary, the material mechanical properties of PDMS bulk materials of varying degree of crosslinking appeared very similar before and after coating of the 2 nm thin layer with respect to shear complex modulus, loss factor, maximum adhesion force and adhesion energy (Table 1 and Fig. 3). By contrast, the interfacial properties of the samples with and without the HMDSO layer exhibited a pronounced difference in water contact angle and interfacial adhesion force (Fig. 2A and 4B). The similar interfacial adhesion forces, hydrophobicity (water contact angle) and surface roughness (Ra) of all coated PDMS substrates strongly suggest that the amount of free polymer chains and polymer chain ends, derived from uncrosslinked polymer content, were blocked after HMDSO coating. This expectation is also supported by previous reports that plasma treatment can lead to surface polymer chain scission and chain crosslinking.36,37 After deposition, the crosslinked HMDSO layer can act as a physical barrier to limit the PDMS polymer chains migrating from the bulk material to the surface.40
To test if the nanolayer barrier of the surfaces can impact bacterial adhesion, the PDMS samples were exposed to different bacterial strains immediately after plasma coating. Similar to what has been reported previously,25E. coli BW25113 adhered to uncoated PDMS more with decreased crosslinker content, from 910 cells mm−2 on PDMS 5:1 to 6070 cells mm−2 on PDMS 40:1 under the examined conditions (Fig. 5A). Interestingly, on all nanolayer-coated PDMS samples, bacteria adhered in similar numbers in a range of 2480–2820 cells mm−2. The uncoated PDMS 40:1 allowed almost 6.7 times higher number of adherent bacteria compared to PDMS 5:1, whereas the coated 40:1 samples attracted only 1.1 times of the adhered cells observed for the coated PDMS 5:1, thus showing the same range of bacteria adhesion independent of crosslinking degree. To investigate whether components of entire type1 fimbriae, which are used by bacteria to adhere to biotic and abiotic surfaces,28,48 play a role in the observed adhesion profile, the isogenic FimH knockout mutant JW4283-3 of E. coli BW25113 was analysed (Fig. 5B). The adhesion of the FimH mutant displayed a similar profile, with 6.5 fold more cells on uncoated PDMS 40:1 than on the uncoated PDMS 5:1, and only 1.2 times difference in cell number between the coated PDMS 5:1 and 40:1. The type 1 fimbriae null E. coli mutant AAEC191A and the recombinant AAEC191A-pSH2 strain that constitutively express type 1 fimbriae were also tested for their adhesion. Similar results were obtained, showing that the number of adherent bacteria varied with the change of PDMS cross-linking degree independent of the presence of fimbriae, whereas no difference could be seen on all examined coated PDMS species (Fig. 5C and D). The obtained difference in the absolute number of the adhered BW25113 and AAEC191A bacteria is likely due to different origins of these two E. coli strains. Other Gram negative strain P. aeruginosa and Gram positive strain S. epidermidis were analysed as well and similar adhesion profiles were observed to E. coli strains (Fig. 5E, F and 6A, B), suggesting that adhesion on nanolayer coated PDMS surfaces independent of crosslinking degree is a general phenomenon for various bacteria species.
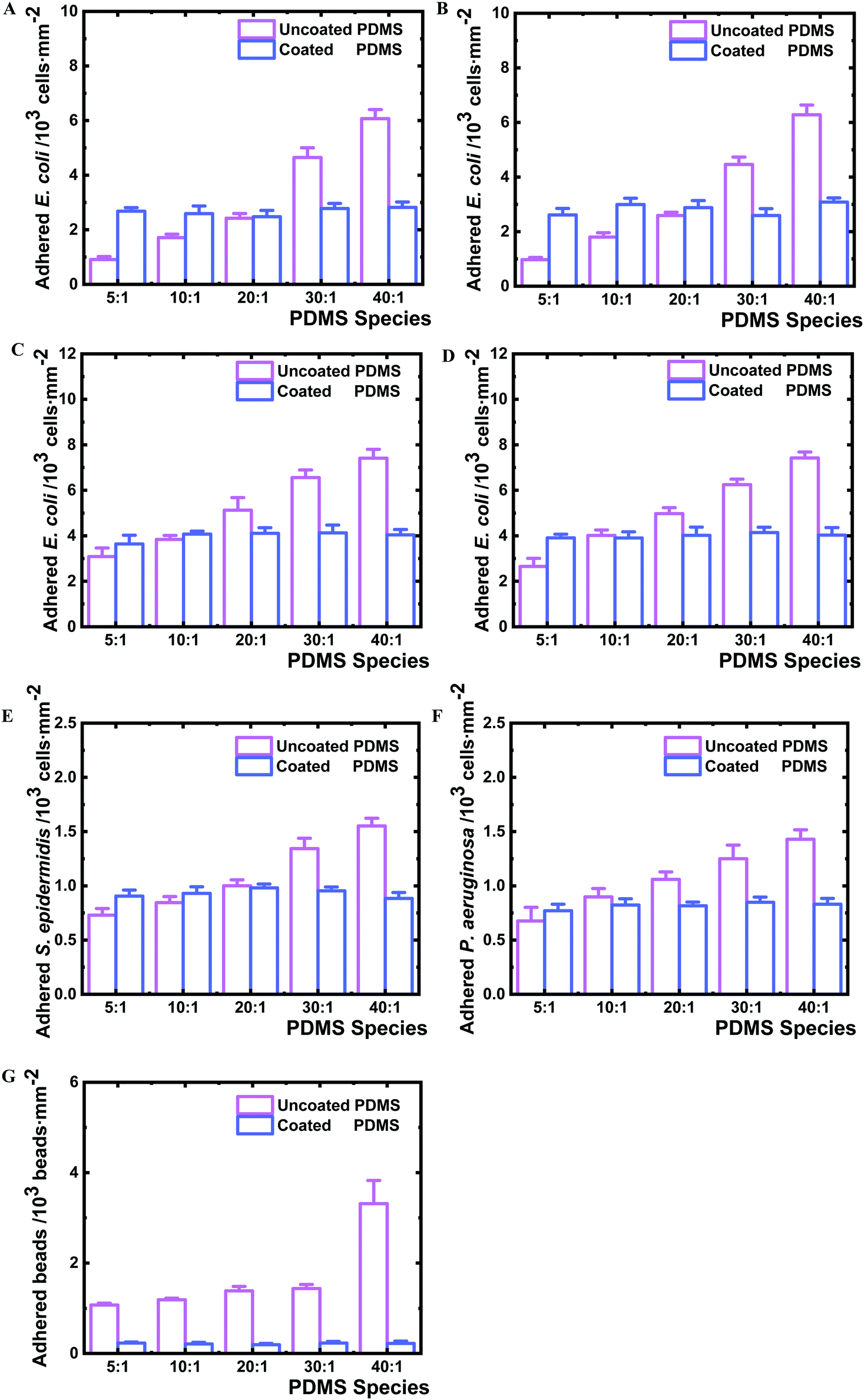 | ||
| Fig. 5 Adhesion of different bacteria species and mutants as well as polystyrene beads on coated and uncoated PDMS samples. A. E. coli BW25113, B. isogenic FimH knockout mutant JW4283-3 of BW25113, C. fimbriae null E. coli mutant AAEC191A, D. fimbriae complemented recombinant AAEC191A (pSH2), E. S. epidermidis, F. P. aeruginosa and G. carboxylate-modified polystyrene beads on coated and uncoated PDMS samples. The bacteria and beads were incubated with PDMS samples for 2 hours in PBS under static conditions at 37 °C, followed by careful removal of the suspensions. Subsequently all samples were rinsed with fresh PBS twice to remove unattached bacteria or beads. Adhered bacteria and beads were imaged by optical microscopy and fluorescent microscopy respectively and then counted. Three samples were analysed for each PDMS of different species and five images at random locations were taken for each sample. Independent experiments were performed three times and one set of results was displayed. Error bars represent the standard deviations of the total 15 measurements in one set of experiments. Student's t-test (p < 0.01) revealed a significant difference among all the uncoated samples, and no significant difference for all coated samples. | ||
To understand whether the observed bacterial adhesion profile is derived from biological characteristics, abiotic microbeads (PS-COOH, 1 μm diameter) were tested on the PDMS surfaces. A similar adhesion trend to bacteria was obtained (Fig. 5G and 6C). The number of adhered microbeads on uncoated PDMS increased from 1070 to 3320 beads mm−2 on PDMS 5:1 and 40:1, respectively, showing an increasing number of adhered microbeads with the decrease of crosslinking degree. On the contrary, the number of adhered microbeads on all coated PDMS species was found to be similar in a very narrow range of 194–230 beads mm−2. The uncoated PDMS 40:1, as noted, permitted 3.1 times higher microbead adhesion than for PDMS 5:1, whereas the coated PDMS 40:1 only attracted 1.2 times the number of microbeads as on coated PDMS 5:1.
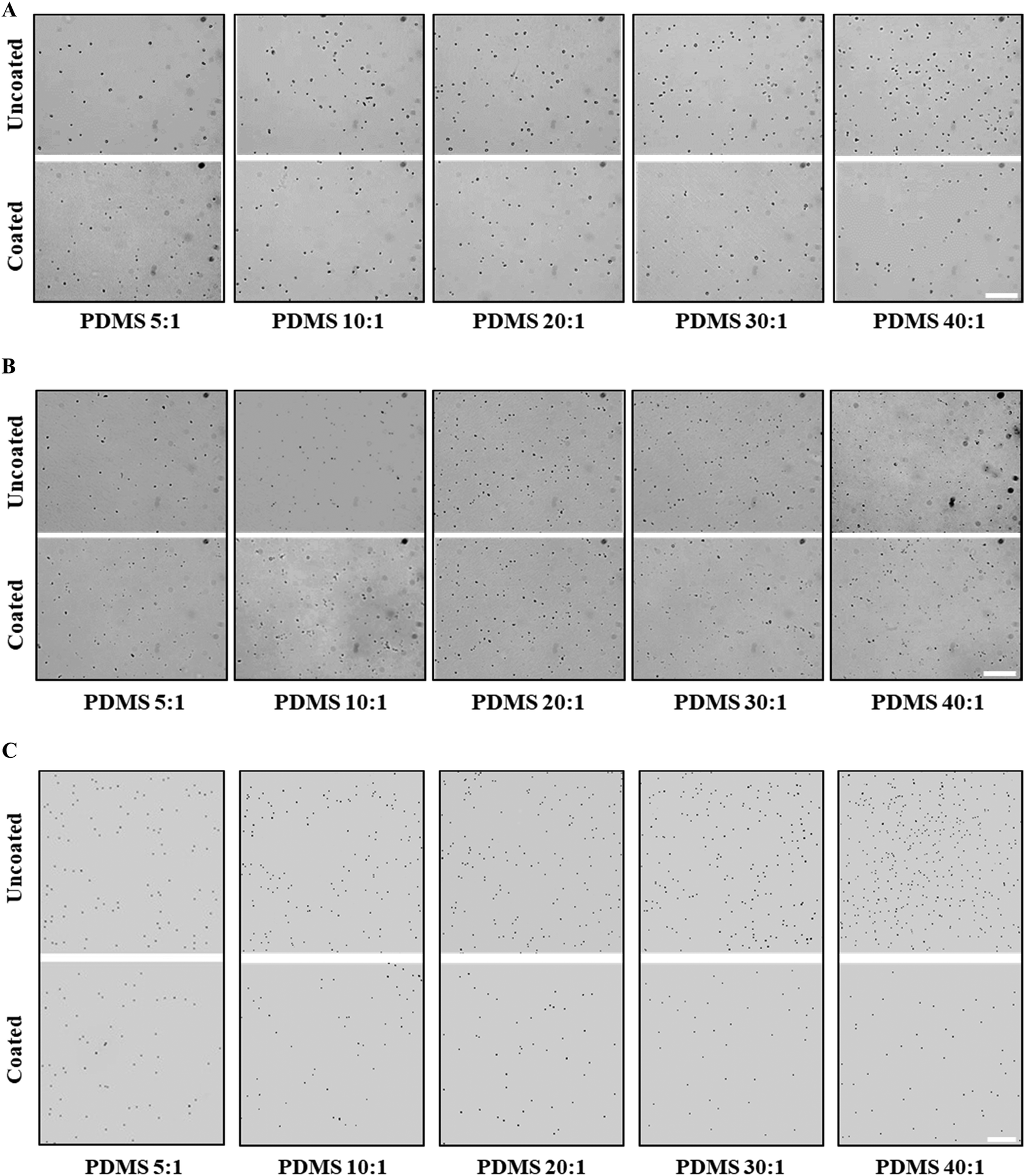 | ||
| Fig. 6 Representative micrographs of adhered S. epidermidis (A), P. aeruginosa (B) and abiotic beads (C) on uncoated and coated PDMS samples. The bacteria and beads were incubated with PDMS samples for 2 hours in PBS under static conditions at 37 °C, followed by careful removal of the suspensions. Subsequently all samples were rinsed with fresh PBS twice to remove unattached bacteria or beads. Adhered bacteria were imaged by optical microscopy and beads were imaged by fluorescent microscopy. Scale bar: 50 μm. | ||
It is noteworthy that the particularly strong increase in bacterial and microbead adhesion for uncoated PDMS 40:1 species correlates well with the distinct increase in the fraction of uncrosslinked PDMS chains for this sample (Fig. 4C) indicating that molecular bridging of the uncrosslinked polymer chains supports bacteria and microbead adhesion, while this mechanism is effectively blocked by the nanolayer deposition.
Based on previous theoretical research about the length of polymer chain ends47 and hydrophobicity affected by the length of the polymer chain,49 the PDMS containing lower crosslinker content is expected to display longer chain ends. In addition, more free, uncrosslinked PDMS chains were derived both from uncoated and coated PDMS samples having lower crosslinker content (Fig. 4C). After plasma coating, the interfacial PDMS chain ends and free/uncrosslinked PDMS chains, however, were blocked as discussed above. Moreover, not only the bulk mechanical property but also the interfacial Young's modulus remained comparable before and after coating. Therefore, the interfacial adhesion force rather than interfacial deformation (Young's modulus) is likely to be the main influential factor for the observed adhesion profile of bacteria and abiotic beads on uncoated and coated PDMS. The interfacial adhesion energy was mainly determined by the length of the polymer chain ends50 and amount of free/uncrosslinked PDMS polymer chains.51 Here, as we hypothesized (Fig. 1, hypothesis II), the polymer chain ends and free/uncrosslinked PDMS polymer chains could not only work as “tentacles” but also contribute to the interfacial adhesion force to influence the nonspecific bacteria adhesion on different PDMS surfaces due to molecular bridging.
Conclusions
Based on a systematic study of uncoated and coated PDMS that was plasma-coated with a HMDSO-derived nanolayer, similar bulk mechanical properties (deformability, Young's modulus and shear modulus) were found for the same PDMS species before and after plasma coating. The results of water contact angle and interfacial adhesion force analysis lead to the conclusion that uncoated PDMS with low Young's modulus contained more free polymer chains and longer chain ends on the surface. These free PDMS polymer chains and longer PDMS polymer chain ends could adsorb more bacteria and abiotic microbeads, leading to higher bacterial adhesion and polystyrene microbeads on uncoated PDMS with reducing crosslinking content. The free polymer chains and polymer chain ends on PDMS surfaces were successfully blocked by a 2 nm plasma-polymerized HMDSO layer after plasma coating, leading to similar adhesion ability of E. coli, E. coli type 1 fimbriae mutants, P. aeruginosa, S. epidermidis and polystyrene microbeads on all PDMS species. This work provides the first evidence that bacterial adhesion on the tested PDMS substrates could be largely attributed to the available free PDMS polymer chains and PDMS polymer chain ends available indicated by interfacial adhesion force for molecular bridging on the sample surfaces.Methods
Chemical and reagents
All chemicals and reagents were purchased from Sigma-Aldrich (Buchs, Switzerland) and applied as received unless otherwise noted. Fluorescent carboxylate modified polystyrene beads with average diameter of 1 μm and composition of 2.5% (L4655, Sigma-Aldrich, Switzerland) were also used as received. Phosphate-buffered saline at pH 7.4 was prepared as follows: 8 g L−1 NaCl, 0.2 g L−1 KH2PO4 and 1.44 g L−1 Na2PO4 in distilled water. Bacterial growth medium (LB broth) was prepared as follows: 10 g L−1 tryptone, 5 g L−1 yeast extract and 5 g L−1 NaCl in distilled water.PDMS substrate preparation
PDMS substrates were prepared as reported using a Sylgard184 silicone elastomer kit (Dow corning Inc., USA) with different weight ratios to curing agent of 5:1, 10:1, 20:1, 30:1 and 40:1.25,26 After thorough mixing, the mixtures were degassed under vacuum for 30 min and subsequently 15 mL of each mixture were poured into plastic Petri dishes (Greiner Bio-One GmbH, Austria, diameter of 9.4 cm) to reach PDMS samples with ∼1.8 mm thickness. Then all the Petri dishes were placed on horizontally levelled racks inside a vacuum drying oven (SalvisLab Vacucenter, Switzerland) under vacuum for 30 min and later incubated at 60 °C for 24 hours after vacuum release. All samples unless otherwise noted were immersed in 70% ethanol for 20 min and vacuum dried before further usage.
Plasma polymerization coating on PDMS substrates
For plasma treatment a pilot-scale reactor was used with an electrode area of 21 × 70 cm2, where 13.56 MHz radiofrequency was applied (capacitively coupled) for plasma excitation in a volume of ∼100 L.38 Prepared PDMS substrates varying in crosslinking content were directly placed on this electrode to be treated in the same run and the chamber was evacuated down to 10−3 Pa. The samples were first activated by Ar plasma (gas flow rate of 80 sccm, power input of 100 W, at 10 Pa pressure) for 10 s to enhance adhesion (by avoiding “harsh” plasma interaction conditions) to the subsequently deposited nanolayer. The ∼2 nm thick PDMS-like plasma polymer film (film density of 1.2 ± 0.05 g cm−3) was deposited using a gaseous mixture of hexamethyldisiloxane (HMDSO) in Ar (4 sccm/20 sccm, 50 W, 7 Pa) requiring a deposition time of 8 s according to the previously determined deposition rate on PDMS samples (Fig. S1, ESI†), which can be well controlled and reproduced.Water contact angle measurement
A Drop Shape Analyzer DSA25E (Krüss GmbH, Germany) was used to measure water contact angle on coated and uncoated PDMS samples immediately after coating (punched discs with a diameter of 20 mm).Atomic force microscope (AFM) analysis
AFM characterization was performed on coated and uncoated PDMS immediately after coating (punched discs with a diameter of 20 mm) in PBS by a Flex Bio-AFM (Nanosurf, Switzerland). Surface topography and force curve measurement were performed with a cantilever PointProbe® Plus Non-Contact/Soft Tapping Mode – Au Coating (PPP-NCST-Au, Nanosensor, Switzerland). Young's modulus was analysed based on elastic deformation of all samples from AFM-force spectroscopy by AtomicJ converting raw deflection vs. sample displacement plots into force vs. deformation plots according to the probe signal sensitivity (nm V−1) and spring constant (N m−1).52–54Gel fraction determination
The gel fraction was calculated as reported.30 The mass of coated and uncoated PDMS samples (punched discs with a diameter of 20 mm) was measured immediately after coating before (W1) and after (W2) extraction in pure ethanol for one week. During ethanol extraction, the ethanol was changed 6 times to immerse samples. Later on, all samples were dried in a vacuum oven at 55 °C for 24 h. The gel fraction was calculated as follows:| Gel fraction = W2/W1 × 100% |
Rheology characterization
Rheological properties of coated and uncoated PDMS immediately after coating (all samples were cut into discs with a diameter of 20 mm) were measured using an in situ plate-to-plate rheometer (Anton-Paar 301, Graz). Samples were placed on the bottom plate and a tool master was located above the sample surface in a furnace of 37 °C. (1) Dynamic strain-sweep measurement was conducted to determine linear viscoelastic range at a fixed angular frequency of 10 rad s−1 and strain of 0.01–500%. (2) Time-sweep measurements were performed at constant angular frequency of 10 rad s−1 and strain of 0.5% for uncoated and coated PDMS samples. Shear complex modulus (G*) was calculated as:G* is shear complex modulus, G′ is storage modulus and G′′ is loss modulus.
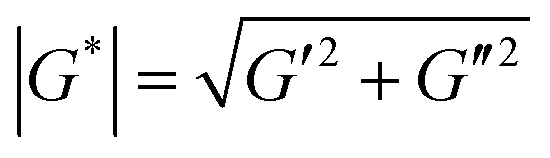
(3) Dynamic frequency-sweep measurement was conducted at constant strain of 0.5% and angular frequency of 1–300 rad s−1. Three independent measurements were conducted and one set result is displayed in Fig. S3 (ESI†). (4) For adhesion force measurement, the samples were located at the center of the bottom plate and the rheometer upper plate was lowered to the samples till physical contact. Every sample was pressed by the upper plate at a constant normal force (FN) of 10 N for 5 seconds. Later, the upper plate was programmed to move upwards at a speed of 5 mm s−1 until a gap distance (h) of 30 mm was reached. The measured normal forces were recorded against gap distance. FN against gap distance was integrated by Origin 2018 resulting in adhesion energy of every sample with the upper plate. (5) The length of the PDMS chain ends was estimated by theoretical calculation and Monte Carlo simulation.47 The length of the PDMS chain ends was estimated according to:
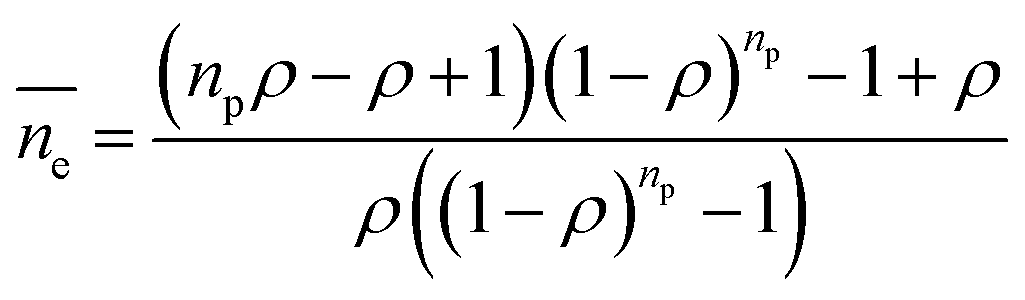
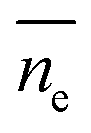 is the average length of the chain ends; np is the length of the uniform polymer of p monomers and ρ is the density of branching points of every polymer chain.
is the average length of the chain ends; np is the length of the uniform polymer of p monomers and ρ is the density of branching points of every polymer chain.
Thickness measurement of the HMDSO coating layer
Two methods were used to measure the thickness. On the one hand, ellipsometry (Imaging Ellipsometer nanofilm_ep4, ACCURION, Germany) was applied to determine the HMDSO modification layer on PDMS substrates immediately after coating and EP4Modle software was used to fit the thickness of the HMDSO modification layer.55 On the other hand, thicker films (>20 nm) on Si wafer and PDMS samples were assessed by profilometry (Dektak 150, Veeco) and weighing of the substrates before and after deposition (using a microbalance, Mettler AE200) in order to obtain a curve of thickness vs. deposition time, which can be linearly fitted to adjust a required deposition time for the intended film thickness in the nanometer range (Fig. S1, ESI†). Moreover, the film density was derived from thickness and mass.Elemental composition of sample surfaces
The chemical compositions on coated and uncoated PDMS were determined immediately after coating by X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe II instrument with a monochromatic AlKα X-ray source, USA). The atomic concentrations were calculated by the software of CasaXPS Version 2.3.12 with instrument specific sensitivity factors.Bacteria culture
E. coli BW25113, a strain widely utilized for biofilm research,56,57 and its isogenic FimH knockout mutant JW4283-3, the type 1 fimbriae null E. coli mutant AAEC191A and its fimbriae complemented recombinant AAEC191A (pSH2),58S. epidermidis ATCC 155 and P. aeruginosa DSM 1117 were used in this study. Bacteria colonies were picked from an agar plate, incubated in 10 mL bacterial growth medium LB in 50 mL Falcon tubes and cultivated in an orbital shaker at 160 rpm at 37 °C overnight. The growth medium of the type 1 fimbriae null E. coli mutant AAEC191A contained 100 μg mL−1 Ampicillin and the one for the fimbriae complemented recombinant AAEC191A (pSH2) contained 100 μg mL−1 Ampicillin and 10 μg mL−1 Chloramphenicol.Adhesion assay
The bacterial adhesion assays were conducted as reported.29 Uncoated and coated PDMS samples were cut into discs with a diameter of 20 mm and placed in 12 well plates immediately after coating (TPP, Switzerland). All freshly punched samples (discs with a diameter of 20 mm) were sterilized with 2 mL 70% ethanol for 20 min and subsequently immersed in 2 mL PBS for 2 h before the adhesion assay. The overnight cultures of bacteria in LB were centrifuged at 7500 rpm for 5 min at 4 °C and washed with 10 mL PBS three times. The bacterial cells were suspended in PBS again to reach 2 × 106 colony forming units (CFU) mL−1. A volume of 1.5 mL of bacteria suspension was added to every well and all samples were subsequently incubated at 37 °C for 2 h without shaking. After incubation, the bacteria suspension was removed and all samples were gently washed with fresh 2 mL PBS twice to remove non-attached cells. The assay with fluorescent polystyrene beads was applied with the exact same method. The stock suspension of the fluorescent polystyrene beads was diluted 8000X and suspended again with PBS to reach the same CFU mL−1 of bacteria suspension.29Adhered bacteria and bead quantification
All samples immersed in bacteria suspension were first treated with bacterial fixation solution (4% paraformaldehyde and 2.5% glutaraldehyde) for 30 min, and later with surface passivation solution (0.1% bovine serum albumin) for 5 min. The bacteria adhered on surfaces were analysed by optical microscopy and the beads were analysed by fluorescent microscopy (excitation 485/20 nm and emission 528/20 nm). An inverted microscope Eclipse Ti2E (Nikon, Japan) with a 40X objective lens was utilized for quantification of adhered bacteria and beads.Statistics
Statistical analyses were performed by utilizing unpaired and two-tailed Student's t-tests for comparison between two groups. Statistical significances were indicated by asterisks in the figures (*p < 0.01).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Kongchang Wei and Dr Xiao-Hua Qin for technical support with rheology measurement, and Dr Mahsa Zabara and Jianing Li for introduction of the ellipsometer. We also thank Hervé Straub for help with using optical microscopy and Jules Valentin with techniques for quantification of adhered bacteria. The authors thank Prof. Madeleine Ramstedt and Dr Martin Held for providing the E.coli strains BW25113 and JW4283-3. The type 1 fimbriae null E.coli strain AAEC191A was provided by Prof. Evgeni Sokurenko (University of Washington, Seattle, USA) and the pSH2 plasmid containing the entire fim cluster was kindly provided by Prof. Paul Orndorff (NCSU, USA). The authors greatly acknowledge Prof. Manfred Heuberger for giving valuable scientific input to the manuscript.References
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623 CrossRef CAS.
- C. R. Armbruster and M. R. Parsek, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4317–4319 CrossRef CAS.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS.
- P. K. Singh, M. R. Parsek, E. P. Greenberg and M. J. Welsh, Nature, 2002, 417, 552 CrossRef CAS.
- D. McDougald, S. A. Rice, N. Barraud, P. D. Steinberg and S. Kjelleberg, Nat. Rev. Microbiol., 2012, 10, 39 CrossRef CAS.
- G. O'Toole, H. B. Kaplan and R. Kolter, Annu. Rev. Microbiol., 2000, 54, 49–79 CrossRef.
- S. Hou, E. A. Burton, R. L. Wu, Y.-Y. Luk and D. Ren, Chem. Commun., 2009, 1207–1209 RSC.
- H. Cao, K. Tang and X. Liu, Mater. Horiz., 2018, 5, 264–267 RSC.
- J. Li, W. Liu, D. Kilian, X. Zhang, M. Gelinsky and P. K. Chu, Mater. Horiz., 2019, 6, 1271–1282 RSC.
- L.-L. Li, H.-W. An, B. Peng, R. Zheng and H. Wang, Mater. Horiz., 2019 10.1039/C8MH01670D.
- A. V. Singh, V. Vyas, R. Patil, V. Sharma, P. E. Scopelliti, G. Bongiorno, A. Podesta, C. Lenardi, W. N. Gade and P. Milani, PLoS One, 2011, 6, e25029 CrossRef CAS.
- C. Díaz, M. C. Cortizo, P. L. Schilardi, S. G. Gómez de Saravia and M. A. F. Lorenzo de Mele, Mater. Res., 2007, 10, 11–14 CrossRef.
- S. Wu, S. Altenried, A. Zogg, F. Zuber, K. Maniura-Weber and Q. Ren, ACS Omega, 2018, 3, 6456–6464 CrossRef CAS.
- T. R. Scheuerman, A. K. Camper and M. A. Hamilton, J. Colloid Interface Sci., 1998, 208, 23–33 CrossRef CAS.
- S. Perni and P. Prokopovich, Soft Matter, 2013, 9, 1844–1851 RSC.
- S. Wu, F. Zuber, K. Maniura-Weber, J. Brugger and Q. Ren, J. Nanobiotechnol., 2018, 16, 20 CrossRef.
- N. Saha, C. Monge, V. Dulong, C. Picart and K. Glinel, Biomacromolecules, 2013, 14, 520–528 CrossRef CAS.
- J. A. Lichter, M. T. Thompson, M. Delgadillo, T. Nishikawa, M. F. Rubner and K. J. Van Vliet, Biomacromolecules, 2008, 9, 1571–1578 CrossRef CAS.
- D. P. Bakker, F. M. Huijs, J. de Vries, J. W. Klijnstra, H. J. Busscher and H. C. van der Mei, Colloids Surf., B, 2003, 32, 179–190 CrossRef CAS.
- C. Guégan, J. Garderes, G. Le Pennec, F. Gaillard, F. Fay, I. Linossier, J.-M. Herry, M.-N. B. Fontaine and K. V. Réhel, Colloids Surf., B, 2014, 114, 193–200 CrossRef.
- K. W. Kolewe, S. R. Peyton and J. D. Schiffman, ACS Appl. Mater. Interfaces, 2015, 7, 19562–19569 CrossRef CAS.
- K. W. Kolewe, J. Zhu, N. R. Mako, S. S. Nonnenmann and J. D. Schiffman, ACS Appl. Mater. Interfaces, 2018, 10, 2275–2281 CrossRef CAS.
- Y. Wang, A. Guan, I. Isayeva, K. Vorvolakos, S. Das, Z. Li and K. S. Phillips, Biomaterials, 2016, 95, 74–85 CrossRef CAS.
- S. Pinto, P. Alves, C. Matos, A. Santos, L. Rodrigues, J. Teixeira and M. Gil, Colloids Surf., B, 2010, 81, 20–26 CrossRef CAS.
- F. Song and D. Ren, Langmuir, 2014, 30, 10354–10362 CrossRef CAS.
- F. Song, M. E. Brasch, H. Wang, J. H. Henderson, K. Sauer and D. Ren, ACS Appl. Mater. Interfaces, 2017, 9, 22176–22184 CrossRef CAS PubMed.
- Y. H. An and R. J. Friedman, J. Biomed. Mater. Res., 1998, 43, 338–348 CrossRef CAS.
- B. Pidhatika, J. Möller, E. M. Benetti, R. Konradi, E. Rakhmatullina, A. Mühlebach, R. Zimmermann, C. Werner, V. Vogel and M. Textor, Biomaterials, 2010, 31, 9462–9472 CrossRef CAS PubMed.
- H. Straub, C. M. Bigger, J. Valentin, D. Abt, X.-H. Qin, L. Eberl, K. Maniura-Weber and Q. Ren, Adv. Healthcare Mater., 2019, 1801323 CrossRef PubMed.
- J. D. P. Valentin, X.-H. Qin, C. Fessele, H. Straub, H. C. van der Mei, M. T. Buhmann, K. Maniura-Weber and Q. Ren, J. Colloid Interface Sci., 2019, 552, 247–257 CrossRef CAS PubMed.
- S. Sahli, Y. Segui, S. Ramdani and Z. Takkouk, Thin Solid Films, 1994, 250, 206–212 CrossRef CAS.
- D. Hegemann, B. Nisol, S. Watson and M. R. Wertheimer, Plasma Chem. Plasma Process., 2017, 37, 257–271 CrossRef CAS.
- D. Hegemann, N. Hocquard and M. Heuberger, Sci. Rep., 2017, 7, 17852 CrossRef PubMed.
- R. Rochotzki, M. Arzt, F. Blaschta, E. Kreyβig and H. Poll, Thin Solid Films, 1993, 234, 463–467 CrossRef CAS.
- Z. Cai, W. Qiu, G. Shao and W. Wang, Sens. Actuators, A, 2013, 204, 44–47 CrossRef CAS.
- H. Hillborg, J. Ankner, U. W. Gedde, G. Smith, H. Yasuda and K. Wikström, Polymer, 2000, 41, 6851–6863 CrossRef CAS.
- M. J. Owen and P. J. Smith, J. Adhes. Sci. Technol., 1994, 8, 1063–1075 CrossRef CAS.
- N. E. Blanchard, B. Hanselmann, J. Drosten, M. Heuberger and D. Hegemann, Plasma Processes Polym., 2015, 12, 32–41 CrossRef CAS.
- S. Forster and S. McArthur, Biomicrofluidics, 2012, 6, 036504 CrossRef CAS.
- V. Barbier, M. Tatoulian, H. Li, F. Arefi-Khonsari, A. Ajdari and P. Tabeling, Langmuir, 2006, 22, 5230–5232 CrossRef CAS PubMed.
- A. Karkhaneh, H. Mirzadeh and A. Ghaffariyeh, J. Appl. Polym. Sci., 2007, 105, 2208–2217 CrossRef CAS.
- J. Juárez-Moreno, A. Ávila-Ortega, A. Oliva, F. Avilés and J. Cauich-Rodríguez, Appl. Surf. Sci., 2015, 349, 763–773 CrossRef.
- M. Quirynen and C. Bollen, J. Clin. Periodontol., 1995, 22, 1–14 CrossRef CAS.
- C. M. Bollenl, P. Lambrechts and M. Quirynen, Dent. Mater., 1997, 13, 258–269 CrossRef.
- H. Tang, T. Cao, X. Liang, A. Wang, S. O. Salley, J. McAllister and K. S. Ng, J. Biomed. Mater. Res., Part A, 2009, 88, 454–463 CrossRef.
- B. Cappella and G. Dietler, Surf. Sci. Rep., 1999, 34, 1–104 CrossRef CAS.
- M. Lang, D. Göritz and S. Kreitmeier, Macromolecules, 2003, 36, 4646–4658 CrossRef CAS.
- J. Möller, T. Lühmann, M. Chabria, H. Hall and V. Vogel, Sci. Rep., 2013, 3, 2884 CrossRef.
- V. Purcar, O. Cinteza, M. Ghiurea, A. Balan, S. Caprarescu and D. Donescu, Bull. Mater. Sci., 2014, 37, 107–115 CrossRef CAS.
- J. J. Lin, J. A. Silas, H. Bermudez, V. T. Milam, F. S. Bates and D. A. Hammer, Langmuir, 2004, 20, 5493–5500 CrossRef CAS PubMed.
- J. J. Sahlin and N. A. Peppas, J. Biomater. Sci., Polym. Ed., 1997, 8, 421–436 CrossRef CAS.
- P. Hermanowicz, M. Sarna, K. Burda and H. Gabryś, Rev. Sci. Instrum., 2014, 85, 063703 CrossRef PubMed.
- K. Gotlib-Vainshtein, O. Girshevitz, C. N. Sukenik, D. Barlam, E. Kalfon-Cohen and S. R. Cohen, J. Phys. Chem. C, 2013, 117, 22232–22239 CrossRef CAS.
- A. C. Chang and B. H. Liu, Mech. Mater., 2018, 118, 17–21 CrossRef.
- R. Synowicki, Phys. Status Solidi C, 2008, 5, 1085–1088 CrossRef CAS.
- T. K. Wood, Environ. Microbiol., 2009, 11, 1–15 CrossRef CAS.
- J. Zhao, Q. Wang, M. Li, B. D. Heijstra, S. Wang, Q. Liang and Q. Qi, Microbiology, 2013, 159, 633–640 CrossRef CAS.
- R. Nakao, M. Ramstedt, S. N. Wai and B. E. Uhlin, PLoS One, 2012, 7, e51241 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh01191a |
| This journal is © The Royal Society of Chemistry 2020 |