
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the mechanistic details of metal binding to mammalian metallothioneins from stoichiometric, kinetic, and binding affinity data
Judith S.
Scheller
,
Gordon W.
Irvine
and
Martin J.
Stillman
 *
*
Department of Chemistry, The University of Western Ontario, London, Ontario N6A 5B7, Canada. E-mail: martin.stillman@uwo.ca; Tel: +1-519-661-3821
First published on 29th January 2018
Abstract
Metallothioneins (MTs) are small, cysteine-rich proteins, found throughout Nature. Their ability to bind a number of different metals with a range of stoichiometric ratios means that this protein family is critically important for essential metal (Zn2+ and Cu+) homeostasis, metal storage, metal donation to nascent metalloenzymes as well as heavy metal detoxification. With its 20 cysteines, metallothionein is also considered to protect cells against oxidative stress. MT has been studied by a large number of researchers over the last 6 decades using a variety of spectroscopic techniques. The lack of distinguishing chromophores for the multitude of binding sites has made the evaluation of stoichiometric properties for different metals challenging. Initially, only 113Cd-NMR spectroscopy could provide strong evidence for the proposed cluster formation of Cd-MT. The extraordinary development of electrospray ionization mass spectrometry (ESI-MS), where all coexisting species in solution are observed, revolutionized MT research. Prior to the use of ESI-MS data, a range of “magic numbers” representing metal-to-MT molar ratios were reported from optical spectroscopic studies. The availability of ESI mass spectral data led to (i) the confirmation of cluster formation, (ii) a conceptual understanding of the cooperativity involved in multiple metal binding events, (iii) the presence of domain specificity between regions of the protein and (iv) mechanistic details involving both binding affinities and rate constants. The kinetic experiments identified the presence of multiple individual binding sites, each with a unique rate constant and an analogous binding affinity. The almost linear trend in rate constants as a function of bound As3+ provided a unique insight that became a critical step in the complete understanding of the mechanistic details of the metalation of MT. To fully define the biological function of this sulfur-rich protein it is necessary to determine kinetic rate constants and binding affinities for the essential metals. Recently, Zn2+ competition experiments between both of the isolated fragments (α and β) and the full-length protein (βα-MT 1a) as well as Zn2+ competition between βα-MT 1a and carbonic anhydrase were reported. From these data, the trend in binding affinities and the values of the Kf of the 7 bimolecular reactions involved in metalation were determined. From the analysis of ESI-MS data for Cu+ binding to βα-MT 1a at different pH-values, a trend in the 20 binding affinities for the complete metalation mechanism was reported. This review details a personal view of the historical development of the determination of stoichiometry for metal binding, the structure of the binding sites, the rates of the metalation reactions and the underlying binding affinities for each metalation step. We have attempted to summarize the experimental developments that led to the publication in May 2017 of the experimental determination of the 20 binding constants for the 20 sequential bimolecular reactions for Cu+ binding to the 20 Cys of apoMT as a function of pH that show the appearance and disappearance of clusters. We report both published data and in a series of tables an assembly of stoichiometries, and equilibrium constants for Zn2+ and Cu+ for many different metallothioneins.
 Judith S. Scheller | Ms Judith Scheller was born in Wurzburg, Germany, and received her degree in Food Chemistry from the Karlsruhe Institute of Technology (KIT), Germany, in 2016. As part of a student exchange between Prof. Dr Martin Stillman and Prof. Dr Andrea Hartwig (KIT) she conducted research in the Stillman Bioinorganic group at the University of Western Ontario, focusing on copper(I) binding to apo-metallothionein using mainly electrospray ionization mass spectrometry. She is now studying for her PhD in biochemistry at Ulm University, Ulm, Germany. |
 Gordon W. Irvine | Dr Gordon Irvine was born in beautiful St. Catharines, Ontario. He graduated with a PhD from Martin Stillman's group at the University of Western Ontario in 2017. He holds a BSc (Hons.) with a double major in Biology and Chemistry from Brock University where he received the distinguished graduating student award for both the Biology and Chemistry departments. He held an NSERC Canada Graduate Scholarship throughout his PhD, which focused on the structural and metalation properties of apo-metallothioneins. Gordon's current research interests involve developing novel mass spectrometry-based techniques for the characterization of disordered proteins. |
 Martin J. Stillman | Martin Stillman received his B.Sc. and Ph.D. from the University of East Anglia, Norwich, UK under the supervised by Prof. Andrew Thomson, FRS. He worked as a postdoctoral fellow at the University of Alberta, Canada. In 1975, he moved to the University of Western Ontario, where since 1986 he has been Professor of Bioinorganic Chemistry. He is currently Bioinorganic Section Editor-in-Chief with the International Journal of Molecular Sciences. Martin Stillman is a Fellow of the Canadian Institute of Chemistry. He is the founding Chair of the CanBIC series of Bioinorganic Conferences held in Parry Sound, Ontario every 2 years beginning in 2007. The 7th conference will take place in May 2019. Three areas of research are of current focus: (i) the metal binding mechanism and structural properties in the human protein metallothionein, (ii) the electronic properties of tetrapyrroles and (iii) the mechanism of action of the Isd heme transport proteins in the human pathogenic Staphylococcus aureus bacterium. Key tools included ESI-mass spectrometry, CD, magnetic CD, and emission spectroscopy, together with the theoretical methods required to assist interpretation, including molecular dynamics for proteins, and DFT-based techniques for the tetrapyrroles. He has published over 250 papers on these topics. His work is funded by the Natural Sciences and Engineering Research Council of Canada. |
1. Introduction
The family of mammalian metallothioneins (MTs) is characterized by small molecular weight (6–8 kDa), high cysteine content (∼30%), and the absence of disulfide bonds as well as the general lack of aromatic amino acids. MT1–5 was first isolated in 1957 as a curious cadmium-containing protein in the horse kidney cortex.6 MTs have now been isolated from a very large number of different organisms throughout Nature.2,7 While mammalian MTs are the best studied MTs, plant MTs have also become a focus of great interest in recent years.8–10 There are 4 major mammalian MT isoforms, MT1–MT4, and a number of sub-isoforms with very similar sequences. The expressions of MT1 and MT2 can be induced by a variety of factors, especially heavy metals;11–13 MT1 is primarily found in the kidneys14 and MT2 in the liver, but both are constitutively expressed.15,16 The other two isoforms, MT3 and MT4, are less inducible and are found in the central nervous system17 and the squamous epithelial tissue,18 respectively. All four isoforms contain 20 cysteine residues, which share Cys-X-Cys, Cys-Cys or Cys-X-X-Cys metal binding motifs.19 MT can bind a vast array of metals, essential as well as toxic, such as Zn2+, Cu+, Cd2+, Hg2+ and As3+ to name a few. Depending on the electron configuration of the metal, stoichiometries from M7-MT to MT20-MT have been reported.4 Its ability to bind a variety of metals, especially d10 metals, resulted in suggested roles in metal homeostasis and heavy metal detoxification within the cell.20–25 It is also a proposed facilitator chaperoning metals to metal-dependent enzymes and metalloproteins,26–28 and finally it has been suggested to be active in protecting cells against oxidative stress.29–31 Recently, its role in intercepting metallodrugs32 and in controlling metals in Alzheimer's disease33 has suggested wider roles in vitro.We have consulted many published resources and have referenced as many as we felt reasonable in this review to provide a general detailed insight into the complex biologically important chemistry of the metallothioneins. These reviews and research reports stretch back to the 1960 paper by Kägi and Vallee34,35 and include the series of monographs from the Metallothionein Conferences, numerous reviews and many thousands of research publications. Here we cite a small sample that will provide the reader with guidance on both the historical developments and the current state of knowledge. The listed papers are just a small sample of those published and omission from our reference list is based on space rather than significance and we apologise to those authors of material we have omitted based simply on space restrictions. While the majority of published reviews mostly discussed function,11,36–38 structure2,3,5,39–43 and/or stoichiometry,4,44–47 we will in this review concentrate on the efforts that have been made to elucidate the metalation mechanism of MT. Recently, a long series of papers from our group have described metalation reaction rate constants and determined the individual binding constants for different metals and characterized the pH dependence of these constants.48–55 This review concerns not only our own perspective on the determination of the individual parameters which are necessary to fully understand the binding mechanism of different metals, but also recent work, particularly concerning metalation pathways of Zn2+ and Cu+ to metallothionein. We have focused in our work on extracting binding constants from ESI-MS data. We have also extracted conformational information, especially on the partially metalated proteins, from, initially, molecular dynamics studies, and later from cysteine modification studies, where we have associated the non-normal distribution of the stepwise modification reactions as measured by ESI-MS methods with the differential shielding of cysteines as a result of folding.43,56,57 Finally, we have over the last 20 years attempted to understand how these quite remarkable natural molecules work. While the down to earth parameters we have reported are vital parameters, as this is a Perspective review, allow me to stand in awe at these natural molecules that can twist and turn to absorb so many different metals into structures and that in many cases exceed inorganic chemistry in their complexity.
2. Metalation of metallothionein
2.1. Experimental tools
Metallothionein is an unusual protein in that it does not contain aromatic amino acids and, as a result, there is no absorption above 230 nm in the apoprotein. In addition, the lack of secondary and tertiary structures in the metal-free protein means that circular dichroism (CD) spectroscopy is not particularly informative about the apoprotein, and finally because the apoprotein is so dynamic with little formal structure, NMR analysis is precluded.58 This lack of quantifiable structure has made the establishment of the initial state in the MT metalation pathway difficult. Recently, various mass spectrometric techniques have been employed to overcome the difficulties in studying the apo-MT structure including ion mobility,59 MS/MS60 and differential cysteine modification.57 We now know that at neutral pH the apoprotein adopts a more compact structure, its native structure, under nearly physiological conditions that facilitate faster Cd2+ binding kinetics56 compared to low pH. However, MS data typically do not give detailed information on metal coordination geometry; coordination numbers are left to EXAFS, NMR and X-ray diffraction studies.Metals commonly associated with MT, including Zn2+, Cd2+, Cu+, Hg2+, and As3+, are coordinated solely by cysteinyl thiols in either digonal, trigonal or tetrahedral coordination geometries. The process of metal binding dramatically changes the structure of this highly dynamic protein. Metal binding introduces new chromophores that facilitate spectroscopic characterization. Most commonly used is the UV-visible absorption spectrum that includes bands in the 220–320 nm region arising from ligand-to-metal charge transfer (LMCT). Also, NMR spectroscopy, both proton and metal based, becomes useful as the metalated protein is far less dynamic than the apo-form. One major issue with a protein that binds multiple metals is that the chromophoric identities of each metal overlap so that the spectral data are the average of all species and are difficult to separate into a stepwise sequence that is necessary for mechanistic analysis. As we will show in this review, ESI-MS data are invaluable in mechanistic studies because all species in the reaction between apo-MT and metals are observed simultaneously. Significantly, the relative concentration can be calculated and used in a traditional manner to obtain binding and rate constants. This means that we are using ESI-mass spectral data in a semi-quantitative manner where calibration anchors the quantitative values. This is much like the use of atomic absorption spectrometry and its reliance on standards.
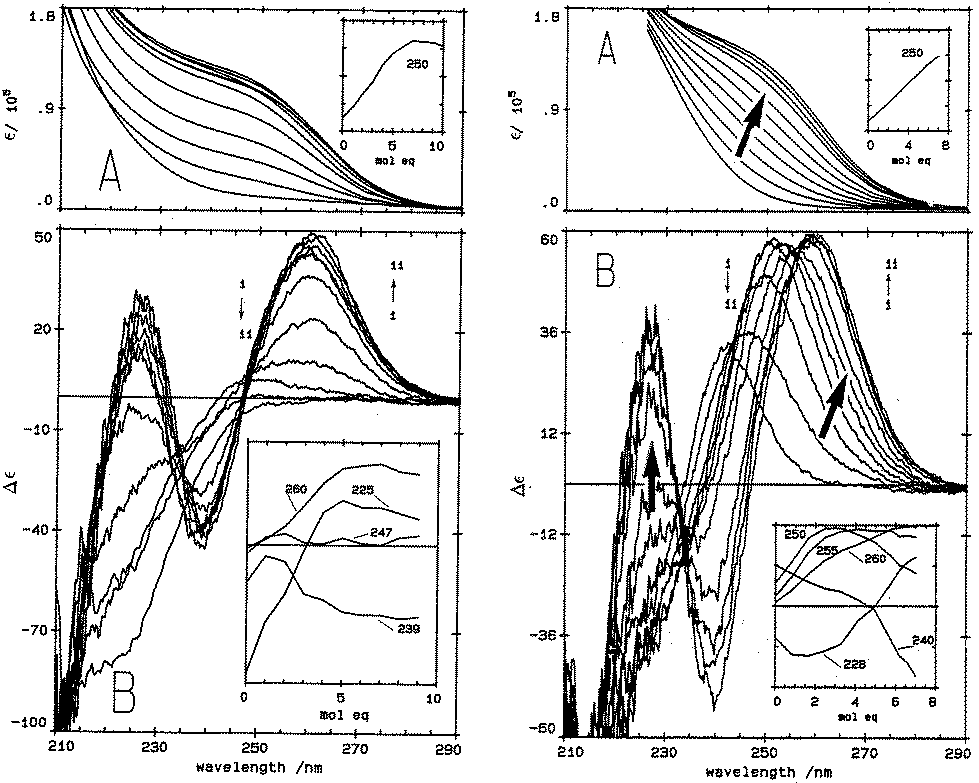 | ||
| Fig. 1 Absorption (A) and CD (B) spectral data for Cd2+ binding to rabbit liver apo-MT 2a and Zn7-MT 2A at pH 7. (LEFT) Absorption (A) and CD (B) spectral data for Cd2+ binding to rabbit liver apo-MT 2a at pH 7. The insets show the change in intensity at each of the wavelengths as a function of Cd2+ added. (Right) Absorption (A) and CD (B) spectral data for Cd2+ binding to rabbit liver Zn7-MT 2a at pH 7. The insets show the change in intensity at each of the wavelengths as a function of Cd2+ added. The difference in CD spectral patterns is due to the prior presence of Zn2+ in the same sites to be occupied by the incoming Cd2+. The distribution of Cd2+ between the two domains is reported as a function of relative binding constants in Fig. 20 below. Reproduced from ref. 61 with the permission of the American Society for Biochemistry and Molecular Biology. | ||
CD spectral data provide detailed information during metal titrations of the protein, with the appearance of maxima, minima and isodiochroic points associated with the formation of specific species. The change in protein structure changes the CD spectral intensity whereas the actual chromophore changes the band maximum. Clearly in the data shown in Fig. 1 for Cd2+ binding to both apo-MT 2A and the Zn7-MT 2A, the absorbance at 250 nm simply increases to give a shoulder (on the edge of the peptide absorption of the protein). The CD spectrum on the other hand grows to a single envelope for the apo-MT 2A as discussed above. Fig. 2 shows what happens when Cd2+ replaces Zn2+ in Zn7-MT using the 3D view to track the spectral changes seen in Fig. 1 (right). Now there are a series of species that form as the Cd2+ replaces Zn2+ more or less isostructurally and the band maximum shifts due to the difference in LMCT energy between the two metals. While these data were recorded at room temperature, we have reported that there is a significant temperature dependence when Cd2+ is added to Zn7-MT 2A that we assigned to the thermally-induced rearrangement in the peptide chain as a function of the change in cluster dimensions.61
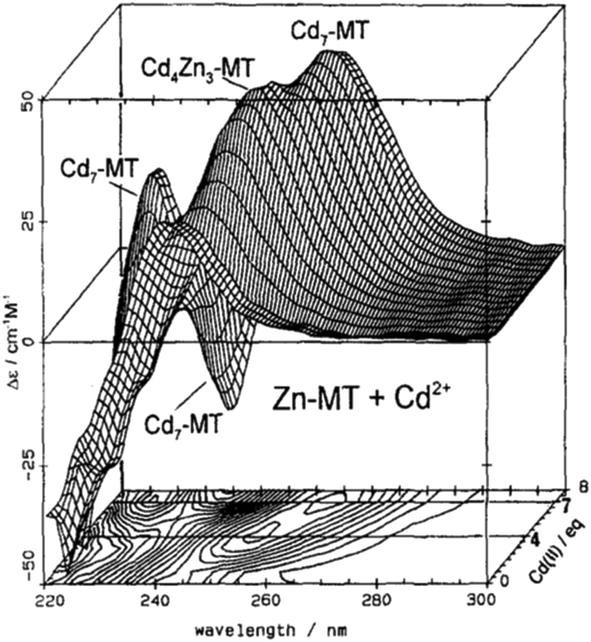 | ||
| Fig. 2 CD spectra recorded during a titration of rabbit liver Zn7MT 2a with Cd2+ at room temperature and pH 7. The CD spectral data reach a maximum of 4 and 7 Cd2+ as indicated by the contour map. Reproduced from ref. 65 with the permission of Wiley and Sons. | ||
These changes in overall structure are far more pronounced when metals that can change their coordination number, and hence geometry, bind to the 20 cysteines of MT. For example, the titration of Cu+ into Zn-MT showed the presence of a number of maxima that were associated with the apparent formation of specific stoichiometric minima for Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu:SCYS in the forming Cu, Zn-MT, Fig. 3. The metalation is highly temperature dependent, with increasing temperatures allowing fast rearrangement to the thermodynamically favorable structures (D).
:Cu:SCYS in the forming Cu, Zn-MT, Fig. 3. The metalation is highly temperature dependent, with increasing temperatures allowing fast rearrangement to the thermodynamically favorable structures (D).
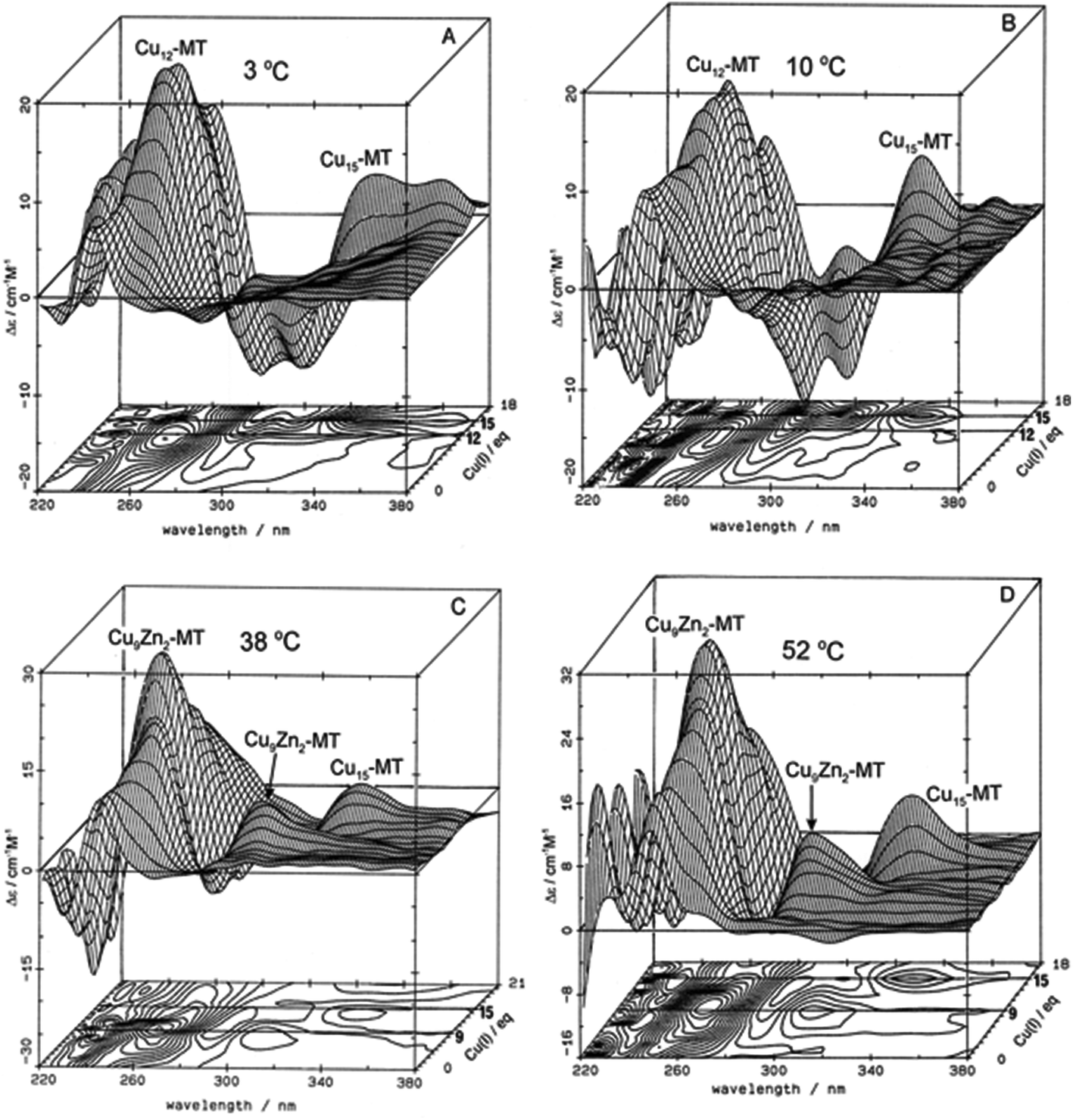 | ||
| Fig. 3 Temperature dependence of Cu+-thiolate cluster formation in rabbit liver Zn7-MT 2A. 3-Dimensional projections and the corresponding contour plots of CD spectra recorded during replacement of Zn2+ in Zn7-MT 2a solutions with Cu1+ at (A) 3 °C, (B) 10 °C, (C) 38 °C and (D) 52 °C, showing the formation of a range of specific species associated with changes in the coordination number of the existing Cu+ as the number of bound metals increases towards 20. Reproduced from ref. 65 with the permission of Wiley and Sons. | ||
Similar large changes in the CD spectra as a function of environment are seen with Hg2+, Fig. 4, under 4 different conditions. (A) Hg2+ was added to apo-MT 2 at pH 7, showing clear formation of Hg7-MT 2A, but this (presumably) clustered species collapses with the addition of further Hg2+, forming possibly a stoichiometrically-relevant species with 11 Hg2+. The spectra in (B) confirm the formation of both the 7 and 11 Hg:MT species when Hg2+ was added to the Zn7MT 2A. Here “0” shows the CD spectrum of the Zn7-MT 2A with a maximum near 230 nm. Like the data shown in Fig. 1, the prearranged or prealigned structure of the MT peptide significantly alters the thermodynamically and kinetically accessible binding sites for the incoming metal. The ionic strength plays an important role as is shown in (C), where at low pH in the presence of high chloride ion concentrations a protein species forms with 18 Hg2+ characterized by a very strong CD spectrum that we assigned as being supermetalated. Much more recently, Rigby-Duncan et al.77 and Sutherland et al.48,78 using CD, ESI-MS and 113Cd NMR methods characterized Cd5-α MT 1A and Cd8-MT 1A as supermetallated species. Finally, the solvent also influences MT metalation: in (D) the solvent was changed to 50% glycol and now the Hg2+ was added to Zn7-MT as shown by its strong positive CD spectrum labelled “0” at 230 nm. The Hg2+ forms only the Hg11-MT species before the protein unfolds and all structure is lost.62–64
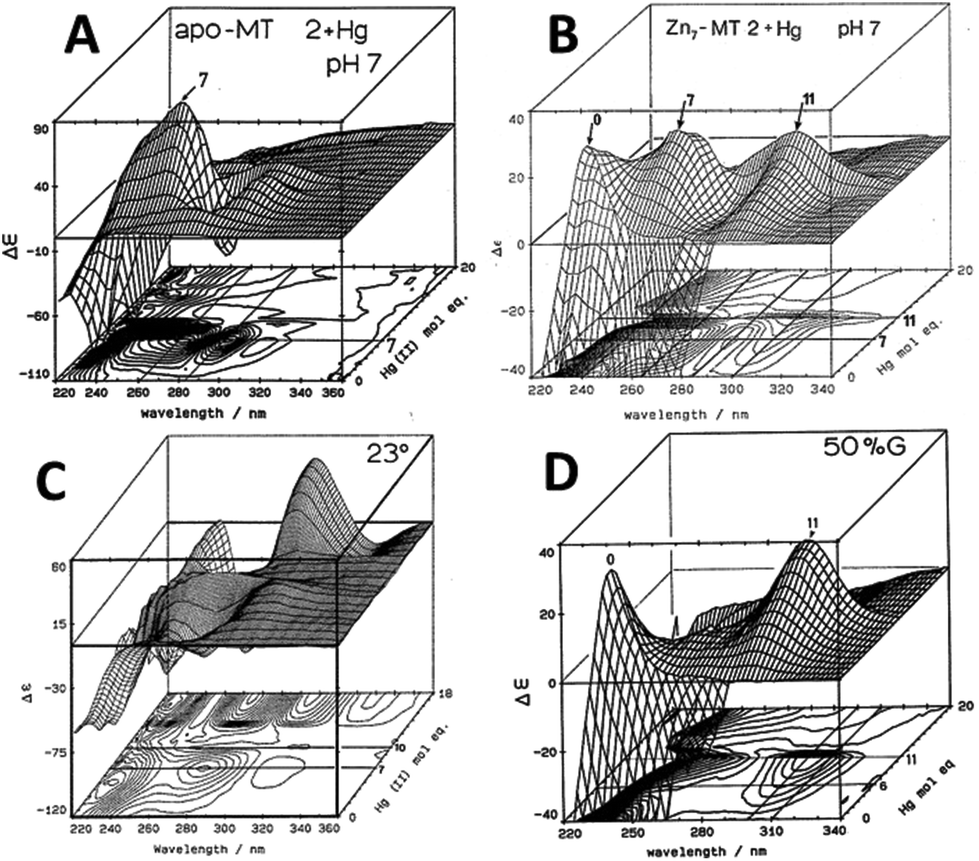 | ||
| Fig. 4 CD spectral changes recorded during titrations of Hg2+ into solutions of rabbit liver apo- and Zn7-MT 2a (rl-MT 2a) showing the formation of different clustered species. (A) A 3-dimensional representation of the formation of Hg18-rlMT 2a using CD spectra recorded during a titration of a single sample of apo-rlMT 2a with Hg2+ at pH 2 and 23 °C in the presence of chloride anions.64 (B) CD spectra recorded during a titration of Zn7-rlMT 2a with Hg2+ at room temperature and at pH 7. The CD spectra show the loss of Zn7-rlMT 2a labeled as “0” followed by the formation of Hg7-rlMT 2a and Hg11-rlMT 2a. (C) A 3-dimensional representation of the formation of Hg18-rlMT 2a using CD spectra recorded during a titration of a single sample of apo-rlMT 2a with Hg2+ at pH 2 and 23 °C in the presence of chloride anions.64 (D) CD spectra recorded during a titration of Zn7-rlMT 2a with Hg2+ at room temperature and at pH 7 in a 50% H2O/ethylene glycol solvent. The CD spectra show the loss of Zn7-rlMT 2a labeled “0” followed by the formation of only Hg11-rlMT 2a. Adapted and reproduced from ref. 61 and 63, with the permission of the American Chemical Society. | ||
The CD spectral data illustrate the complexity of metalation and the difference between the absorption and CD traces illustrates the effect that metal binding has on the orientation of the peptide chain. While the absorption spectrum is dependent on the LMCT band and increases as a function of metal loading, it cannot report on major changes in the structure of the binding site(s). However, CD spectroscopy is sensitive to changes in the wrapping of the peptide around the bound metal – the metal-induced folding described for MT by Duncan & Stillman in 2006.66 The CD spectral intensity arises from the chirality of the peptide as dictated by the selection of cysteines required to bind each metal with the wavelength dependent on the origin of the electronic transition. For the data reported in Fig. 3, the optical transitions are charge transfer in origin, between the Cu+ and the thiolate of the cysteines. The selection of structures is thermodynamically controlled and changes as a function of metal loading as shown in Fig. 3 and 4. Despite assigning at that time specific metal-to-protein ratios with spectral features, results reported much more recently based on detailed, stepwise metal loading experiments monitored by ESI-MS show that for each specific metal:protein ratio, a range of ratios centered on the average metal added exist. The spectral properties are then dominated by the species with the strongest signal, which can distort the analysis. The data described at the end of this review allow more specific assignments to be made. In the end, everything depends on the relative magnitudes of the equilibrium binding constants (where cooperativity requires a larger value compared with the adjacent values) and the spectroscopic motif intensity for each structure formed.
The CD spectral data shown for Cd2+ binding (Fig. 1 and 2), Cu+ binding (Fig. 3) and Hg2+ binding (Fig. 4) clearly demonstrate how the respective metalation reactions with the 20 cysteines of MT 2A involve both different structural motifs (between the beads of isolated metal-thiolate units; see the description of Zn2+ and Cd2+ binding52–54) and clusters. The wavelengths of the band maxima change as a function of the coordination environments of the metal, particularly the coordination number. So, the CD data for the Cd2+-metalation in Fig. 1 and 2 are relatively straightforward, the coordination geometry for beads and clusters remains tetrahedral, but as mentioned above, the CD spectrum is dominated by the electronic exciton coupling between pairs of coordinated Cd2+ in the [Cd4(SCYS)11]3− cluster in the α-domain. On the other hand, the coordination geometry for Cu+ with thiolates is well known to change, with tetrahedral, trigonal and diagonal geometries all reported. Each of these geometries are characterized by charge transfer transitions of different energies, resulting in the CD data of Fig. 3 where the data red shift with the formation of the Cu15-MT species. This clearly (and also logically) indicates the change in coordination geometry from the trigonal expected for Cu12-MT to diagonal. Finally, in the CD data for Hg-MT to be described in Fig. 4, we see an even more dramatic effect of the change in coordination geometry with the Hg:MT ratios of 7, 11 and 18 each resulting in specific structures with individual band maxima and CD motifs. As the number of Hg2+ bound increases past 11 Hg:MT (Fig. 4B and D), analysis of the CD spectral data suggests that the size of the formed clusters exceeds the stable conformation, meaning that the structure collapses but still binds Hg2+ but not in the well-defined clusters of those “magic numbers”,4 so the chirality is not well-defined either and the CD spectral intensity is, therefore, negligible. However, at an Hg:MT ratio approaching 18 (Fig. 4C), another structure forms with a different band maximum, suggesting another change in coordination geometry with the 18 Hg2+ coordinated in a well-defined structure to the 20 cysteines.
We shift now to other spectroscopic methods: first to the use of emission spectroscopy, then NMR, and finally, for the remaining examples, to the remarkable detail of ESI-mass spectral data.
Common use of emission spectroscopy has been limited to Cu-MTs, although there have been reports from Ag-, Pt- and Au-containing MTs.3,67 The first report by Beltramini and Lerch68,69 identified the presence of an emission band near 600 nm when a Cu+ containing MT was excited at 280 nm. This remarkably wide Stokes shift arises from the intersystem crossing populating metal based atomic orbitals. The general nature of the emission properties of Cu-MT was established by many groups and allowed the stoichiometry of Cu:MT to be determined during Cu+ titrations.65,70 The emission intensity did not simply increase as a function of Cu+ loading; rather it increased in a non-linear fashion to a maximum, and then decreased upon further addition of Cu+. This unusual pattern can be explained by the initial formation of Cu–S clusters that exclude water and, therefore, enhance the long lived excited state population, followed by protein unfolding at high Cu:MT ratios exposing Cu+ to water. The lifetime of the Cun-MT excited state at 600 nm was found to be about 5 microseconds.71 This short phosphorescence lifetime points to the presence of Cu+ atomic orbitals as the origin of the emission.72 It is noteworthy that the emission intensity provides a direct indication of the extent of cluster formation in Cu-MTs. Emission spectroscopy can be used as an indirect measure of the structure since compact cluster structures shielded from the solvent have a high emission intensity compared with the unclustered and solvent exposed conformations. This also implies that each metal must have an individual binding constant (Kf) that represents the incremental stepwise reaction of CunMT + Cu → Cun+1MT. However, a more precise method is needed to resolve individual species present in solution and determine the value of these individual Kf's and this is described below using ESI-mass spectral data for these same titrations.
Fig. 5 illustrates the spectral changes that take place with titrations at 2 temperatures.70 In (A) at 10 °C, a maximum at 12 Cu+ added to the Zn7-MT 2 was reached through a steadily increasing signal. However, at 40 °C, the first 6 Cu+ resulted in very little signal. This was interpreted in the paper as being due to the increased mobility of the Cu+ so that at low temperatures the Cu+ was essentially trapped in the kinetic product, whereas at the higher temperatures the Cu+ could rearrange and that thermodynamic product was not emissive. Exploiting the change in emission intensity, Salgado et al.67 probed the structural properties of Cu+ in the presence of a competing metal Ag+. The key to these kinetic studies was that at room temperature the emission recorded is from the Cu–S coordination alone as Ag-SN only emits at 77 K. The 77 K spectra therefore, were a mixture. These titrations have recently been reported using ESI-MS in parallel to follow the speciation of Cu+ when added to the 20 cysteines in apo-MT at room temperature.49
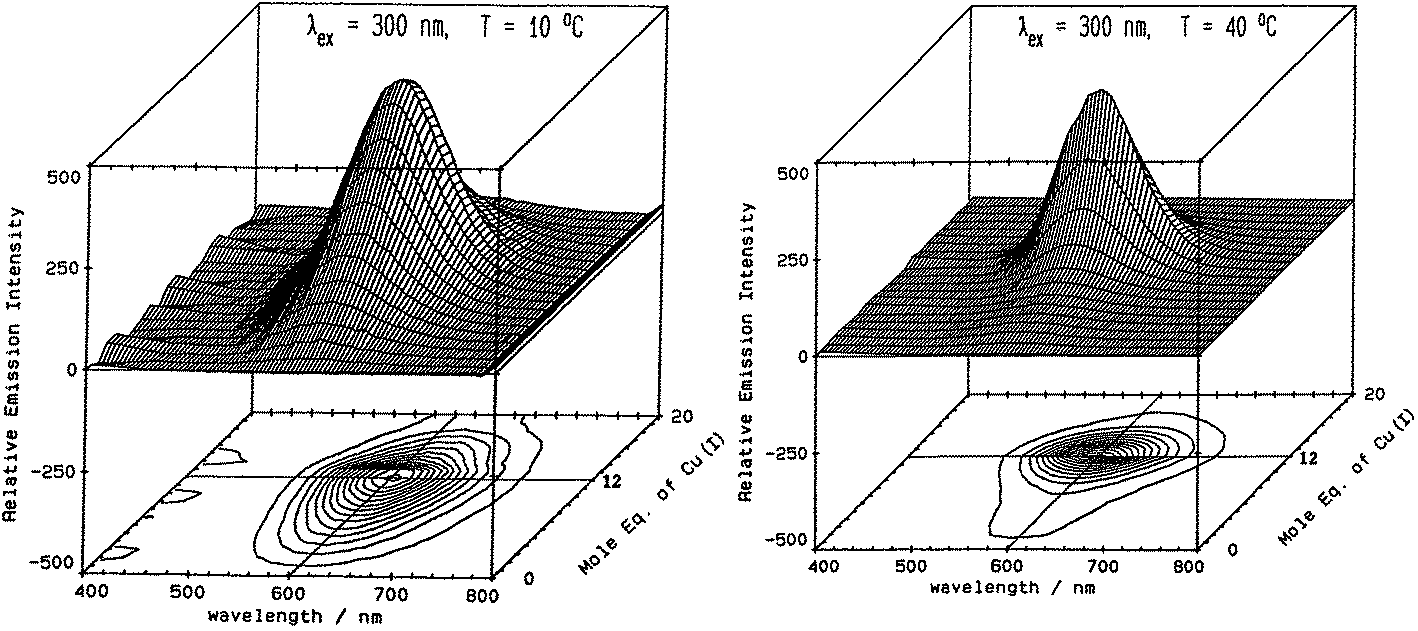 | ||
| Fig. 5 Emission spectra recorded in the 600 nm region for a titration of rabbit liver Zn7-MT 2a with Cu+. (A) Titration at 10 °C shows the gradual intensification of the emission at 600 nm to a maximum at a Cu:MT molar ratio of 12. Addition of Cu+ results in a loss of intensity. (B) Titration at 40 °C shows a very low emission intensity up to a Cu:MT molar ratio of 6 followed by a rapid increase of intensity to a ratio of 12 following which the intensity drops towards 20 Cu+ added. Reproduced from ref. 70 with the permission of the American Chemical Society. | ||
We feel it necessary to emphasize that the clustered domains only exist in MT when the metal loading is high. It appears from experimental data reported to date that there is no mechanism specifically driving metals to any particular coordinating cysteine until clustering is required to accommodate the additional incoming metals.50 We mean by this that there are thermodynamically favored ratios of metal-to-cysteine that are surprisingly flexible in their values compared with typical metal-binding sites in proteins. Only when the number of metals available to bind to the MT cysteines exceeds the isolated structures possible with cysteines do structures form with different arrangements of bridging cysteinyl thiolates because even then the equilibrium binding constants are still very high. MS–MS data are likely the route to determining if there is a single cysteine as the first in apo-MT bound to an incoming metal.
The property of coordinating an additional Cd2+ was first reported by Duncan et al. for the supermetalated Cd5-α domain fragment77 and later by Sutherland et al.78 for the supermetallated Cd4-β domain fragment emphasizing the flexibility of the MT peptide. The initial confirmation of these NMR reports came from the CD spectrum of the Cd5-α-MT77 because the 5th Cd2+ was shown not to be a simple adduct on the outside of the [Cd4(SCYS)11]3− cluster because the CD spectral motif for the exciton split bands centered on 250 nm was replaced by a strong CD motif that was itself centered on the 250 nm band maximum of the LMCT band. The CD spectrum for the first few Cd2+ in Fig. 1B also identifies the presence of this non-clustered Cd2+. 113Cd-NMR studies coupled with CD spectral data showed that new clusters had formed in the α and β domain fragments. The 113Cd-NMR spectrum of the “supermetalated” Cd8MT, Fig. 6, illustrates how the protein can rearrange to accommodate an 8th Cd2+, forming a single super cluster under conditions of excess cadmium.48 Detailed ESI-mass spectral data established that the Cd8-MT 1A was the dominant species. For each of these new species, molecular modeling provided reasonable structures.
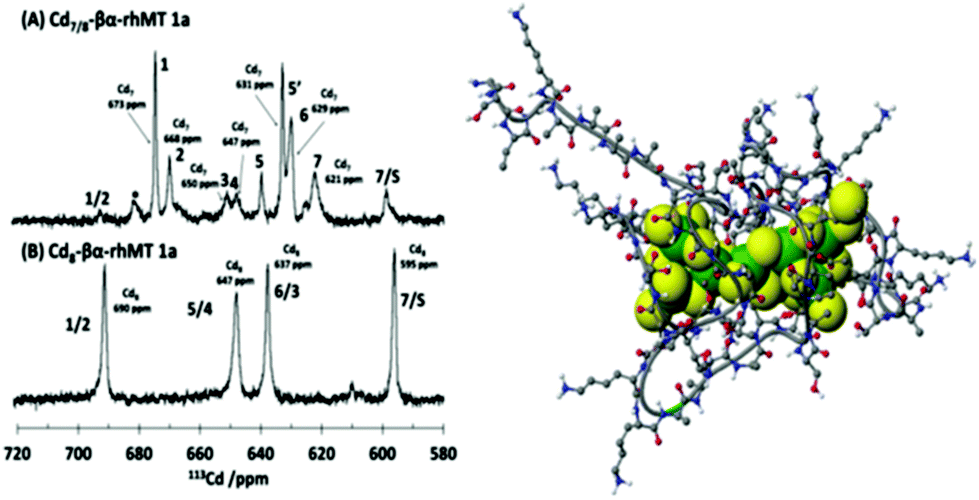 | ||
| Fig. 6 113Cd-NMR spectra recorded for (A) Cd7-rhMT 1a showing the presence of 7 unique sites and (B) Cd8-rhMT 1a showing the presence of only 4 sites with unique chemical shifts. (C) A proposed structure for the Cd8-rhMT 1a using a molecular model. Reproduced from ref. 48 with the permission of the American Chemical Society. | ||
The optical and NMR techniques provided a wealth of information about the metalated MTs and their metalation and demetalation reactions; however, it was clear that these spectroscopic methods report on averages, that is, the spectral data are the sum of the spectral data for all individual species present. To some extent worse, each species will have its own signal intensity so the averaged observed spectra may not and probably do not represent the weighted average of the fractions of the species present. This ambiguity greatly affects the determination of thermodynamic properties that depend on a specific M:protein ratio. So, the CD spectra recorded during the stepwise addition of Cd2+, Cu+, and Hg2+ to Zn7-MT in Fig. 2–5 are averages. The problem is that we do not have any guide as to the breadth of the averages, so although the overall changes are unambiguous and the final saturated products are understandable, the paths to the final products are not as usable as we would like. Particularly, these data do not allow the determination of the relative individual step binding constants with accuracy or precision. The data described from now on using ESI-MS techniques resolved this problem leading to both kinetic rate constants and equilibrium binding constants. The kinetic data for As3+ binding to apo-MT 1A79,80 firmly established that ESI-MS spectral data could be used in a semiquantitative manner to extract precise binding constants and from there, precise but relative equilibrium binding constants. The extreme of those data are the 20 binding constants for Cu+ binding to apo-MT 1A described below, which clearly shows the presence of the cooperatively formed clusters during the stepwise addition of the Cu+.49
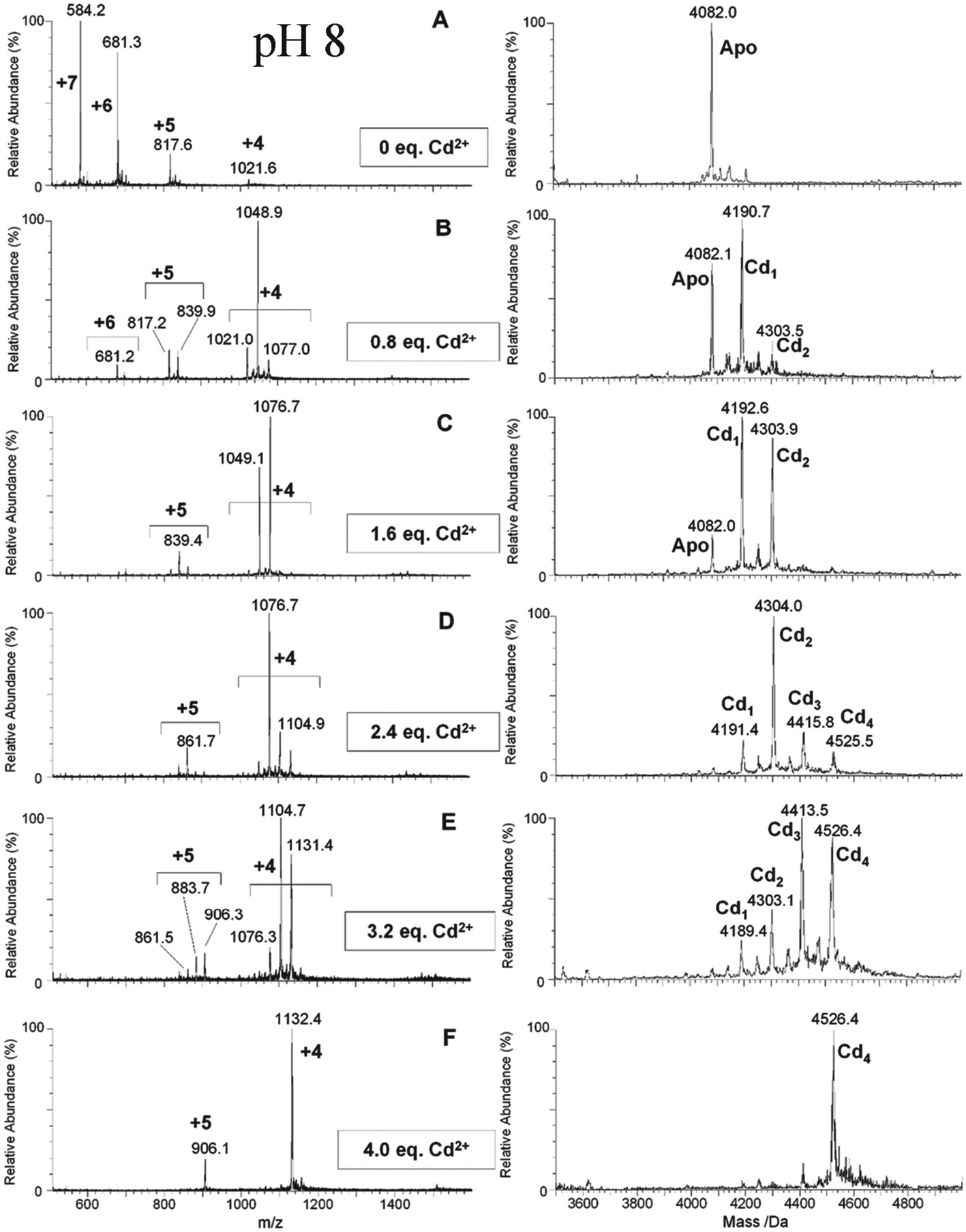 | ||
| Fig. 7 ESI-MS data recorded during a Cd2+ titration of apo-α-rhMT 1a at pH 8. The sequence of A to F shows the change in charge state and deconvoluted parent protein as a function of metalation. At this pH, non-cooperative speciation leads to the formation of Cd1, Cd2, Cd3, and Cd4. This is particularly clear at this pH in D and E. Reproduced and reprinted from ref. 81 with the permission of John Wiley & Sons, Inc. | ||
The Fenselau group were among the first to apply ESI-MS to metallothioneins in the early 1990s and showed that the method could be used semi-quantitatively.82 These studies established the use of ESI-MS for the qualitative monitoring of metal exchange and determination of kinetic parameters.83–85 Thus, the groundwork was laid for a technique that would revolutionize our understanding of the mechanistic details of metal binding by metallothionein.
2.2. Stoichiometry
The original metallated-MTs that were studied in detail were Cd-MTs and the NMR data clearly showed that the stoichiometric ratio was 7 M2+:1MT. These divalent metals were tetrahedrally coordinated solely by cysteinyl thiols so that the two domains were written as M3S9 for the β-domain and M4S11 for the α-domain. The situation was not as clear for Cu+ binding because up to 20 Cu+ could be seen spectroscopically. Overall, Cu12MT was accepted as the equivalent to the M7MT of the divalent metals with more than 12 Cu+ considered by some to be “supersaturated”.38 The same stoichiometry was assigned to Ag-MT.86 However, spectroscopic titrations for the last 30 years have suggested that a number of M:MT ratios are possible. Fig. 8 shows the two familiar structures for the Cd3S9 and Cd4S11 clusters in the β- and α-domains, respectively, together with the proposed structures found during the titration of Cu+ into apo-MT 1,49 the first formed being Cu6S9 in the β-domain, followed by Cu4S6 in the α-domain when 10 Cu+ had bound, a further 10 Cu+ binding finally formed Cu20-MT an unwound loosely folded structure.49
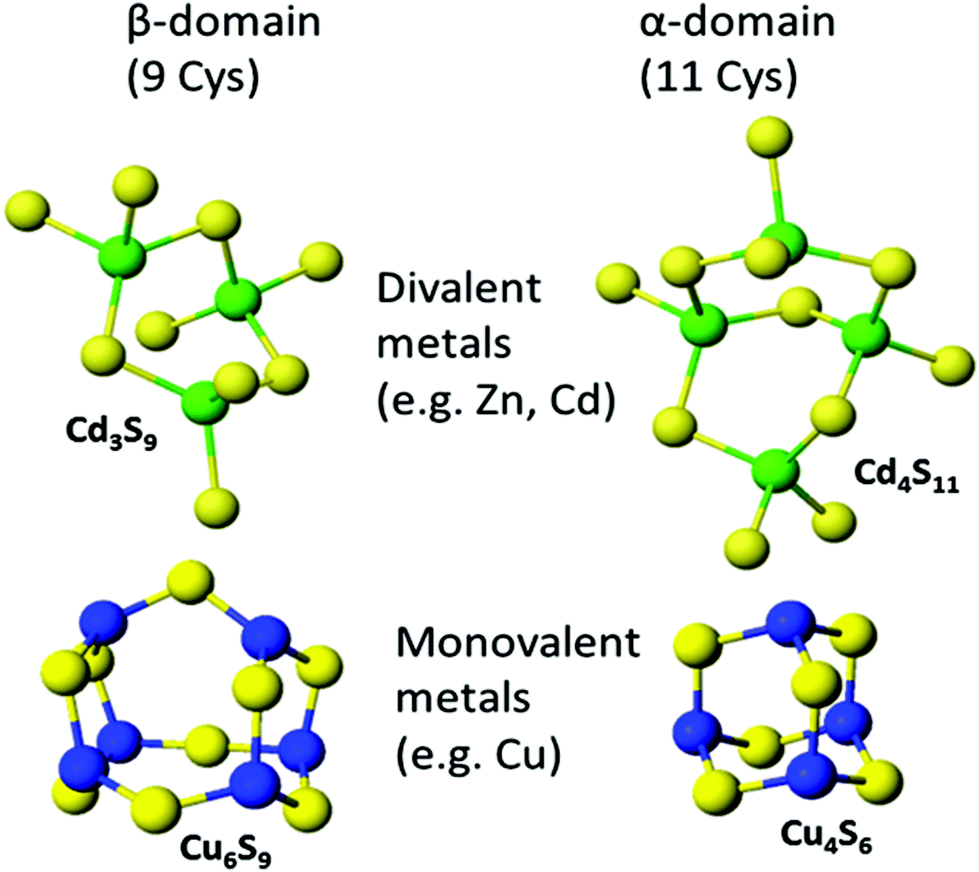 | ||
| Fig. 8 Well established structures of Cd3S9 and Cd4S11 and proposed structures of Cu6S9 and Cu4S6. Adapted from molecular models based on the X-ray and NMR structures90,91 and reproduced from ref. 49 with the permission of the Royal Society of Chemistry. | ||
In the CD data described above for Hg2+, Fig. 4, we showed that at least 3 different clustered structures can form when Hg2+ is titrated into the apo-protein. Table 1 summarizes the reported stoichiometric ratios for a range of MT species and metals. While the mammalian proteins have 20 cysteines, MTs from other organisms have varying numbers of cysteines and we have noted this in the table. Also, while the mammalian MTs bind using cysteines, metal binding in plant and bacterial MTs can involve histidine coordination.87–89 It is clear from the data extracted from the published reports that many M:MT ratios are possible and the most convincing are those determined by ESI-MS because exact masses of all species in solution are obtained. Spectroscopic titrations rely on careful sequential addition of metals which can be inaccurate due to precipitation and oxidative reactions. It should also be noted that in optical spectroscopy, solution averages are measured and using analytical techniques such as atomic absorption and ICP-OES only provides a ratio of the total metal content to an estimate of the protein content without direct connection.
2.3. Kinetics
The reports of the kinetic rate constants by our group have established that important quantitative binding data could be determined from time and temperature resolved ESI-mass spectral data.79,80Table 2 shows a summary of the reported rate constants for metal binding to MT.Up until the establishment of the As3+-MT binding kinetics, it was considered by many that the metals bound at a single rate and with a single binding constant (Kf) in a more or less cooperative fashion. This was due in part to experimental limitations of the stopped-flow spectrophotometers where the complete set of metalation reactions was essentially finished within the dead time of the instrument.92 The extreme speed in which spectroscopically active metals (such as Cd2+ or Cu+) bind to MT made the individual binding kinetics difficult to determine.
The Shaw and Petering groups led in attempts to overcome the difficulty of measuring the fast kinetics of the metal binding reaction to the apo-protein by investigating the demetalation reaction with EDTA, monitored by NMR.93 In addition, the slower metal exchange between Zn7-MT and Cd2+ was monitored by stopped flow.94 Furthermore, the kinetics of metal displacement by covalent cysteine modification were also determined.95 More recent studies by the Russell group showed that metalation/demetalation occurs through very similar pathways yielding identical intermediates in the case of Cd2+ binding to human MT 2A.60
The kinetics of slower binding metal complexes such as cisplatin were determined in the early 2000s by HPLC, atomic absorption and UV-visible absorption spectroscopy.96 This paved the way for the study of As3+ because its unusually slow binding kinetics was perfectly suited for kinetic analysis by ESI-MS.
The As3+ binding data (Fig. 9) showed that the As3+ bound sequentially to the full-length protein.79,80,85 The six rate constants followed a statistical trend where they decrease almost linearly as the binding sites are filled. With no rigid structure defining the binding sites, the incoming metal simply selects a thermodynamically reasonable combination of cysteines to bind to the protein. The rate constant trend can be associated directly with a trend in the six equilibrium binding constants, the Kf-, when we assume that the off-rate is similar for each arsenic. When Kf = kon/koff and koff is constant, the trend in kon is the same as that in Kf. In the data reported for As3+, it was clear that the first As3+ bound with a much greater Kf than the last. The distinction between individual binding constants has more important biological significance for zinc and copper where donation to metalloenzymes is governed by the weakest individual binding constant and not the average binding constant of MxMT (x = 7 M2+, 12 M+). The importance of the kinetic results for As3+ with respect to our group's work to determine binding constants cannot be overstated. The individual rate constants fell on a well-defined trend line for the 3 As3+ binding steps to the two isolated domain fragments, for the 6 As3+ binding steps to the full 20 cysteines in MT97,98 and even for the 9 and 10 As3+ binding steps to the 27 and 33 cysteines of triple β- and α-MT,85 respectively. This almost linear trend for the 20 cysteines of the native apo-MT was strong evidence that the apo-MT peptide acted like a multiple-binding site chelator. This meant we should expect, thermodynamically and statistically, that there would be as many binding constants as metals that bound and that these would trend lower unless there were special products that enhanced cooperative behavior. As we show clearly in the next sections, “special” products are indeed formed. When clustered structures are the thermodynamically favored product then the binding constants for those products distort the expected trend to lower values as the number of metals bound increases, or as the number of available sites diminishes. Indeed, the summary of the rate constants in reference85 (Kf-values are listed in the tables below) is extremely interesting. The rate constants represent the reaction at each step when following a linear trend line. They indicate binding in a similar fashion to fewer and fewer sites, and when there is deviation we can deduce conformational changes occurring. As an example, the binding of the first As3+ is associated with a smaller rate constant than subsequent As3+, which we have assessed as being due to the tight wrapping of the metal-free apo-proteins. We have considered this in modelling studies as well.57,66,99–101
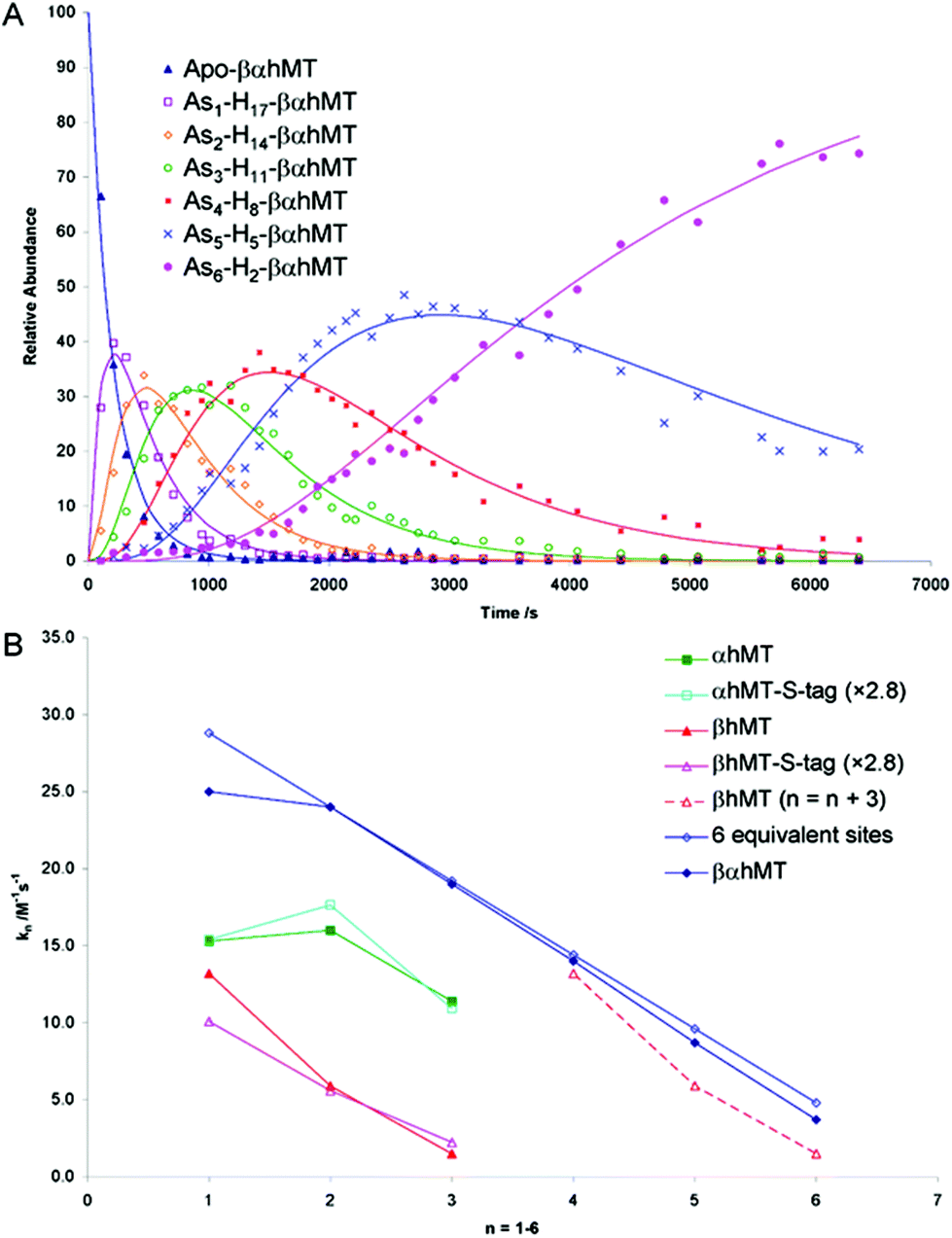 | ||
| Fig. 9 Time dependent ESI-MS data recorded for As3+ binding to rhMT 1a. (A) Time-resolved ESI-MS relative abundances for apo-rhMT 1a following reaction with As3+ at 25 °C and pH 3.5 to form Asn-rhMT (n = 1–6). The reaction was carried out with an As3+:MT stoichiometric ratio of 11:1. The relative abundances of each of the species are shown as the data points in the graph. The lines were calculated by fitting all the data to a series of sequential bimolecular reactions. (B) Comparison of the rate constants calculated from the time-resolved ESI-MS measurements for As3+ metalation of α-rhMT 1a-S-tag, β-rhMT 1a-S-tag, α-rhMT 1a, β rhMT 1a, the full protein βα-rhMT 1a and the trend in rate constant values for 6 equivalent sites where K1 = 28.8 M−1 s−1. The dashed line represents rate constant data for the β-rhMT 1a redrawn with the value of n shifted by three illustrating the similarity to the rate constant trend for the final three As3+ binding to the βα-rhMT 1a. The protein used was recombinant human (rh) MT 1a. Reproduced from Ngu et al. 200879 with permission from the American Chemical Society. | ||
2.4. Equilibrium binding affinities
The experimental data shown in Tables 1–3 highlight the promiscuous nature of metal binding to MT. What is clear is that there is no specific metal binding site in the traditional sense of a well-formed set of coordinating amino acids that typically accept metals with the same geometry. Examples of this would include Zn2+ binding to carbonic anhydrase and the Zn-finger proteins, where the binding site is close to tetrahedral.132,133| Metal | Organism (No. of Cys in sequence) | Isoform | Fragment/protein | Stoichiometry | Geometry | Technique | Reference |
|---|---|---|---|---|---|---|---|
| a Adopted from ref. 10. Abbreviations: CD, circular dichroism spectroscopy; MCD, magnetic circular dichroism spectroscopy; UV-Vis, ultraviolet-visible absorption spectroscopy; AAS, atomic absorption spectrometry; ESI-MS, electrospray ionization-mass spectrometry; MALDI-TOF, matrix-assisted laser deposition ionization-time of flight; FTIR, Fourier transform infrared spectroscopy; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; ICP-AES/OES, inductively coupled plasma–atomic emission spectrometry/optical emission spectroscopy; ESR/EPR, electron spin/paramagnetic resonance spectroscopy; MM/MD, molecular mechanics/molecular dynamics; FPLC, fast protein liquid chromatography; ITC, isothermal titration calorimetry; EXAFS, extended X-ray absorption fine structure; NMR, nuclear magnetic resonance spectroscopy. | |||||||
| Ag | Rabbit (20) | MT2 | βα | Ag18MT | CD | 86 | |
| As | Human (20) | MT1a | α | As3MT | Trigonal | ESI-MS | 80 |
| Human (20) | MT1a | β | As3MT | Trigonal | ESI-MS | 80 | |
| Rabbit (20) | MT2 | βα | As6MT | Trigonal | ESI-MS | 102 | |
| (CH3As)10MT | Diagonal | ||||||
| [(CH3)2As]20MT | Monodentate | ||||||
| Au | Horse kidney (20) | MT | βα | Linear | Chromatographic separation, AAS | 103 | |
| Bi | Not specified | Not specified | Not specified | Bi7MT | Tetrahedral | CD, MCD, absorption | 104 |
| Rabbit (20) | βα | Bi7MT | UV-Vis | 105 | |||
| ICP-AES | |||||||
| Cd | Human (20) | MT1a | α | Cd4MT | Tetrahedral | ESI-MS | 106 |
| Human (20) | MT1a | β | Cd3MT | Tetrahedral | ESI-MS | 106 | |
| Human (20) | MT1a | βα | Cd7MT | Tetrahedral | ESI-MS | 106 | |
| Chickpea (14) | MT2 | Cd5MT | UV-Vis, CD, MCD, ESI-MS, MALDI-TOF | 107 | |||
| Chickpea (12) | MT1 | Cd4MT | UV-Vis, CD, FTIR, ESI-MS | 108 | |||
| Cd5MT | |||||||
| Garden pea (12) | MT1 | Cd3.9MT | AAS, SDS-PAGE | 109 | |||
| Cd5.8MT | |||||||
| Garden pea (12) | MT1 | CdxMT (x = 5.6–6.1) | SDS-PAGE, FPLC | 110 | |||
| Wheat | Ec-1 | Full-length | Cd6Ec-1 | Tetrahedral | UV-Vis, ESI-MS, F-AAS |
111, MS89 |
|
| Wheat | Ec-1 | γ | Cd2γ-Ec-1 | Tetrahedral | ESI-MS, UV-Vis, NMR, F-AAS | 112 | |
| Wheat | Ec-1 | βE | Cd4βE-Ec-1 | Tetrahedral | ESI-MS, F-AAS | 89 | |
| Durum wheat (12) | MT1 | CdxMT (x = 4 ± 1) | UV-Vis, X-ray, MM/MD, SDS-PAGE | 113 | |||
| Cork oak (14) | MT2 | Cd6.3MT | ICP-OES, ESI-MS | 114 | |||
| Grey mangrove (14) | MT2 | Cd3MT | SDS-PAGE, AAS | 115 | |||
| Banana (10) | MT3 | Cd4MT | UV-Vis, AAS, ESI-MS | 116 | |||
| Co | Rabbit (20) | MT1 | βα | Co7MT | Tetrahedral | MCD, ESR | 117 |
| Rabbit (20) | MT2 | βα | Co7MT | Tetrahedral | UV-Vis | 92 | |
| Rabbit (20) | MT2 | βα | Co7MT | Tetrahedral | NMR | 118 | |
| Chickpea (14) | MT2 | Co5MT | UV-Vis | 107 | |||
| Cu | Human (20) | MT3 | βα | Cu10MT | ESI-MS, CD | 119 | |
| Human (20) | MT3 | β | Cu6MT | ESI-MS, CD | 119 | ||
| Human (20) | MT3 | α | Cu4MT | ESI-MS, CD | 119 | ||
| Human (20) | MT1a | βα | Cu20MT | 1Cu:1SCys |
ESI-MS | 49 | |
| Chickpea (12) | MT1 | Cu6MT | UV-Vis, CD | 120 | |||
| Cu9MT | |||||||
| Chickpea (14) | MT2 | Cu6MT | UV-Vis, CD | 120 | |||
| Cu10MT | |||||||
| Chickpea (14) | MT2 | Cu8MT | UV-Vis, CD, MCD, ESI-MS, MALDI-TOF | 107 | |||
| Chickpea (12) | MT1 | Cu6MT | UV-Vis, CD, FTIR, ESI-MS | 108 | |||
| Cu9MT | |||||||
| Garden pea (12) | MT1 | Cu2.3MT | AAS, SDS-PAGE | 109 | |||
| Cu6.2MT | |||||||
| Cork oak (14) | MT2 | Cu5.5MT (+Zn1.7MT) | ICP-OES, ESI-MS | 114 | |||
| Grey mangrove (14) | MT2 | Cu4MT | SDS-PAGE, AAS | 115 | |||
| Wheat | Ec-1 | γ | Cu2γ-Ec-1 | F-AAS, UV-Vis, CD, luminescence | 112 | ||
| Fe | Rabbit (20) | MT1 | βα | Fe7MT | Tetrahedral | MCD, EPR | 121 |
| Hg | Not specified | Not specified | Not specified | Hg7MT | Tetrahedral | CD, MCD, absorption | 104 |
| Rabbit (20) | MT | βα | Hg18MT | CD | 64 | ||
| Ni | Rabbit (20) | MT1 | βα | Ni7MT | Tetrahedral | MCD, ESR | 117 |
| Pb | Not specified | Not specified | Not specified | Pb7MT | Tetrahedral | CD, MCD, absorption | 104 |
| Mouse (20) | MT1 | βα | Pb9MT | ESI-MS | 122 | ||
| Mouse (20) | MT1 | β | Pb4MT | ESI-MS | 122 | ||
| Mouse (20) | MT1 | α | Pb5MT | ESI-MS | 122 | ||
| Human (20) | MT3 | βα | Pb7MT | ITC | 123 | ||
| Pt | Rat liver (20) | MT | βα | Pt10MT | EXAFS | 124 | |
| Rat liver (20) | MT | βα | Pt4.5MT (cis-DDP) | UV-Vis, radioimmunoassay | 125 | ||
| Pt6MT (trans-DDP) | |||||||
| Rat liver (20) | MT | βα | Pt2Zn4MT (cis-DDP) | Chromatographic separation, AAS, covalent Cys modification | 126 | ||
| Rh | Human (20) | MT1a | β | Rh2MT | ESI-MS | 32 | |
| Tc | Rabbit liver (20) | MT1 | βα | Tc7MT | UV-Vis, covalent Cys modification | 127 | |
| Rabbit liver (20) | MT1 | βα | Tc6.2MT | UV-Vis, EXAFS | 128 | ||
| U | Cyanobacteria (9) | SmtA | (UO2)3Zn4MT | Bidentate | ESI-MS, Cd, 1H NMR, SDS-PAGE | 129 | |
| Zn | Human (20) | MT1a | α | Zn4MT | Tetra | ESI-MS | 53 |
| Human (20) | MT1a | β | Zn3MT | Tetra | ESI-MS | 53 | |
| Human (20) | MT1a | βα | Zn7MT | Tetra | ESI-MS | 53 | |
| Wheat | Ec-1 | Full-length | Zn6Ec-1 | Tetrahedral | ESI-MS, F-AAS | 111 | |
| Wheat | Ec-1 | γ | Zn2γ-Ec-1 | Tetrahedral | ESI-MS, F-AAS | 112 | |
| Wheat | Ec-1 | βE | Zn4βE-Ec-1 | Tetrahedral | ESI-MS, F-AAS | 89 | |
| Chickpea (14) | MT2 | Zn5MT | UV-Vis, CD, MCD, ESI-MS, MALDI-TOF | 107 | |||
| Chickpea (12) | MT1 | Zn4MT | UV-Vis, CD, FTIR, ESI-MS | 108 | |||
| Zn5MT | |||||||
| Garden pea (12) | MT1 | Zn5.6 MT | AAS, SDS-PAGE | 109 | |||
| Zn11.5MT | |||||||
| Cork oak (14) | MT2 | Zn3.5 MT | ICP-OES, ESI-MS | 114 | |||
| Grey mangrove (14) | MT2 | Zn3MT | SDS-PAGE, Coomassie blue, AAS | 115 | |||
| Banana (10) | MT3 | Zn3.4 MT | UV-Vis, AAS, ESI-MS | 116 | |||
| Oil palm (10) | MT3A | Zn1.7MT | AAS, SDS-PAGE | 130 | |||
| Metal | Organisma | Isoform | Fragment/protein | Stoichiometry | Technique | Rate constants (k [M−1 s−1]) | Reference |
|---|---|---|---|---|---|---|---|
| a The number of cysteines in the peptide is shown in parentheses. | |||||||
| As | Human (20) | MT1a | α | As3MT | ESI-MS | k 1 5.5 | 80 |
| k 2 6.3 | |||||||
| k 3 3.9 | |||||||
| Human (20) | MT1a | β | As3MT | ESI-MS | k 1 3.6 | 80 | |
| k 2 2.0 | |||||||
| k 3 0.6 | |||||||
| Human (20) | MT1a | βα | As6MT | ESI-MS | k 1 25 | 79 | |
| k 2 24 | |||||||
| k 3 19 | |||||||
| k 4 14 | |||||||
| k 5 8.7 | |||||||
| k 6 3.7 | |||||||
| Bi | Rabbit | MT2 | βα | Bi7MT | UV-Vis, 1H NMR | k 1 5.8 × 10−3 (Cd displacement) | 105 |
| k 2 1.0 × 10−4 (Cd displacement) | |||||||
| k 1 7.2 × 10−3 (Zn displacement) | |||||||
| k 2 5.9 × 10−5 (Zn displacement) | |||||||
| Cd | Human (20) | MT1a | α | Cd4MT | Stopped flow spectrophotometry | k 1–4 60.4 (native) | 56 |
| k 1–4 3.32 (denatured) | |||||||
| Horse kidney (20) | MT | βα | Cd7MT | Absorption spectroscopy | 2.7 × 10−6 (demetalation by EDTA) | 28 | |
| Rabbit (20) | MT2 | βα | Cd7MT | Stopped flow spectrophotometry | pH 4.1 60 ± 10 | 92 | |
| pH 4.6 140 ± 30 | |||||||
| pH 5.1 280 ± 50 | |||||||
| pH 5.4 350 ± 100 | |||||||
| Rabbit (20) | MT2 | α | Cd4MT | Stopped flow spectrophotometry | pH 4.6 170 ± 60 | 92 | |
| pH 5.1 350 ± 100 | |||||||
| pH 5.4 460 ± 200 | |||||||
| Zn | Rabbit (20) | MT2 | βα | Zn7MT | Stopped flow spectrophotometry | pH 4.6 10 ± 1 | 92 |
| pH 5.2 15 ± 1 | |||||||
| pH 5.8 28 ± 2 | |||||||
| pH 6.3 43 ± 7 | |||||||
| pH 6.6 100 ± 15 | |||||||
| pH 7.2 300 ± 100 | |||||||
| Rabbit (20) | MT2 | α | Zn4MT | Stopped flow spectrophotometry | pH 6.6 90 ± 10 | 92 | |
| pH 7.2 230 ± 100 | |||||||
| pH 7.5 690 ± 300 | |||||||
| Pt | Rabbit (20) | MT | βα | Pt7MT | Atomic absorption spectroscopy, UV absorption, HPLC | 0.14 (to apo-MT) | 96 |
| 0.75 to Cd/Zn-MT | |||||||
| 0.53 to Cd7MT | |||||||
| 0.65 to Zn7MT | |||||||
| Zn | Horse kidney (20) | MT | βα | Zn7MT | Absorption | Demetalation by EDTA | 28 |
| k 1 fast | |||||||
| k 2 14.2 × 10−4 | |||||||
| k 3 2.0 × 10−4 | |||||||
| Metal | Isoform | Fragment/Protein | Stoichiometry | Technique | Binding constant value (logK) | Reference |
|---|---|---|---|---|---|---|
| Explanations:a Determined by analysis of the pH dependent titration data.b Binding strength was determined using competitors and a fluorogenic dye.c Binding constant values were determined through competition of zinc binding to carbonic anhydrase. | ||||||
| Cu | MT2 | βα | Cu10MT | ESI-MS, pH 7.5 | K 14.6 | 27 |
| MT1a | βα | Cu20MT | ESI-MSa, pH 7.4 | K 1 15.5 | 49 | |
| K 2 15.0 | ||||||
| K 3 14.6 | ||||||
| K 4 19.3 | ||||||
| K 5 14.6 | ||||||
| K 6 18.9 | ||||||
| K 7 12.9 | ||||||
| K 8 13.1 | ||||||
| K 9 13.7 | ||||||
| K 10 15.5 | ||||||
| K 11 10.7 | ||||||
| K 12 11.2 | ||||||
| K 13 12.9 | ||||||
| K 14 9.5 | ||||||
| K 15 9.5 | ||||||
| K 16 8.6 | ||||||
| K 17 8.1 | ||||||
| K 18 7.0 | ||||||
| K 19 6.1 | ||||||
| K 20 5.0 | ||||||
| Pb | MT3 | βα | Pb7MT | ITC, pH 6.0 | K 1–2 11.7 | 123 |
| K 3–4 10.2 | ||||||
| K 4–7 8.7 | ||||||
| Zn | MT2 | βα | Zn7MT | Fluoresence spectroscopyb, pH 7.4 | K 1–4 11.8 | 131 |
| K 5 10.45 | ||||||
| K 6 9.95 | ||||||
| K 7 7.7 | ||||||
| MT3 | βα | Zn7MT | ITC, pH 6.0 | K 1–4 10.8 | 123 | |
| K 5 10.5 | ||||||
| K 6 9.9 | ||||||
| K 7 7.7 | ||||||
| MT1a | βα | Zn7MT | ESI-MSc | K 1 12.35 | 55 | |
| K 2 12.47 | ||||||
| K 3 12.52 | ||||||
| K 4 12.37 | ||||||
| K 5 12.21 | ||||||
| K 6 12.05 | ||||||
| K 7 11.80 | ||||||
When binding affinities are discussed with respect to MT, it should be clear that the values do not correspond to a specific site but to the binding of the nth metal to Mn−1MT, wherever that may take place. The individual binding event may require backbone rearrangement to position coordinating cysteines, especially during cluster formation. As with the kinetic rate constants, binding affinities tend to decrease statistically as more cysteines are occupied by the already bound metals. Unlike the kinetic constants, there are important exceptions to this general trend. These exceptions tend to be for cluster formation which is a thermodynamically favorable process and is often associated with cooperative binding.
Table 3 contains representative binding constants reported for a range of metals and solution conditions. Only recently have individual constants been reported for each metal bound to MT because the determination of an individual Kf required the measurement of each binding step separately. Only ESI-MS methods provide the resolution required to monitor each step of these multi metal binding reactions. In the case of Zn2+ binding to apo-MT we can summarize the complete reaction in the following Scheme 1.
 | ||
| Scheme 1 A series of 7 single step reactions for Zn2+ binding to apo-MT at pH 7 showing the individual binding constants K1–K7. The values of the individual K's were first reported in ref. 54 and then modified using competition with carbonic anhydrase.55 Scheme adapted from ref. 50 | ||
In this series of reactions, the spectroscopic chromophore of Zn(SCYS)4 is so similar from step to step that we cannot resolve individual Zn2+ binding events. Additionally, the ESI-MS data show that there is a distribution of species that coexist except at the very beginning and end of the titration. This means that the spectroscopic data are simply averages and cannot provide the specific resolution required to obtain each individual binding constant. Individual binding constants for the reactions shown in Scheme 1 were determined by an ESI-MS competition experiment between MT and carbonic anhydrase reported by Pinter et al.55 Carbonic anhydrase was used to calibrate the range of Kf values since its own zinc affinity has been well characterized. Of course, in a competition experiment each protein concentration has to be determined using a method like absorption spectroscopy before mixing since the ionization efficiencies in ESI-MS can differ greatly between peptides.
In the case of Zn2+ binding, ESI-MS competition experiments revealed that the first two Kf values are smaller than subsequent values, where ITC and spectroscopic studies had averaged K1 or 2–4 values and had not shown this counter intuitive phenomenon. The depression of the initial binding constants was interpreted as resulting from the energetically demanding rearrangement of the folded apo-MT to allow the first metals to bind. Covalent modification, ESI-MS studies57 and IM-MS59 confirmed that the apo-protein can exist in a number of conformers, many of which are tightly packed. More ESI-MS results from Irvine et al.52 indicated a further complication in pH dependence of the Kf values, which change drastically around pH 7.0 and 5.0 for Cd2+ and Zn2+ binding, respectively. Cooperativity has been discussed with reference to the metalation mechanism of MT for 40 years or more. Cooperativity manifests itself as a series of increasing Kf values up to the cooperatively formed product. These increasing values are a consequence of more fundamental underlying factors which may include conformational changes and changes in the chemical nature of the ligand as binding proceeds. We can see this effect in the Cu+ binding constants in Table 3 where K4, K6, K10, and K13 are all larger than the surrounding Kf values.
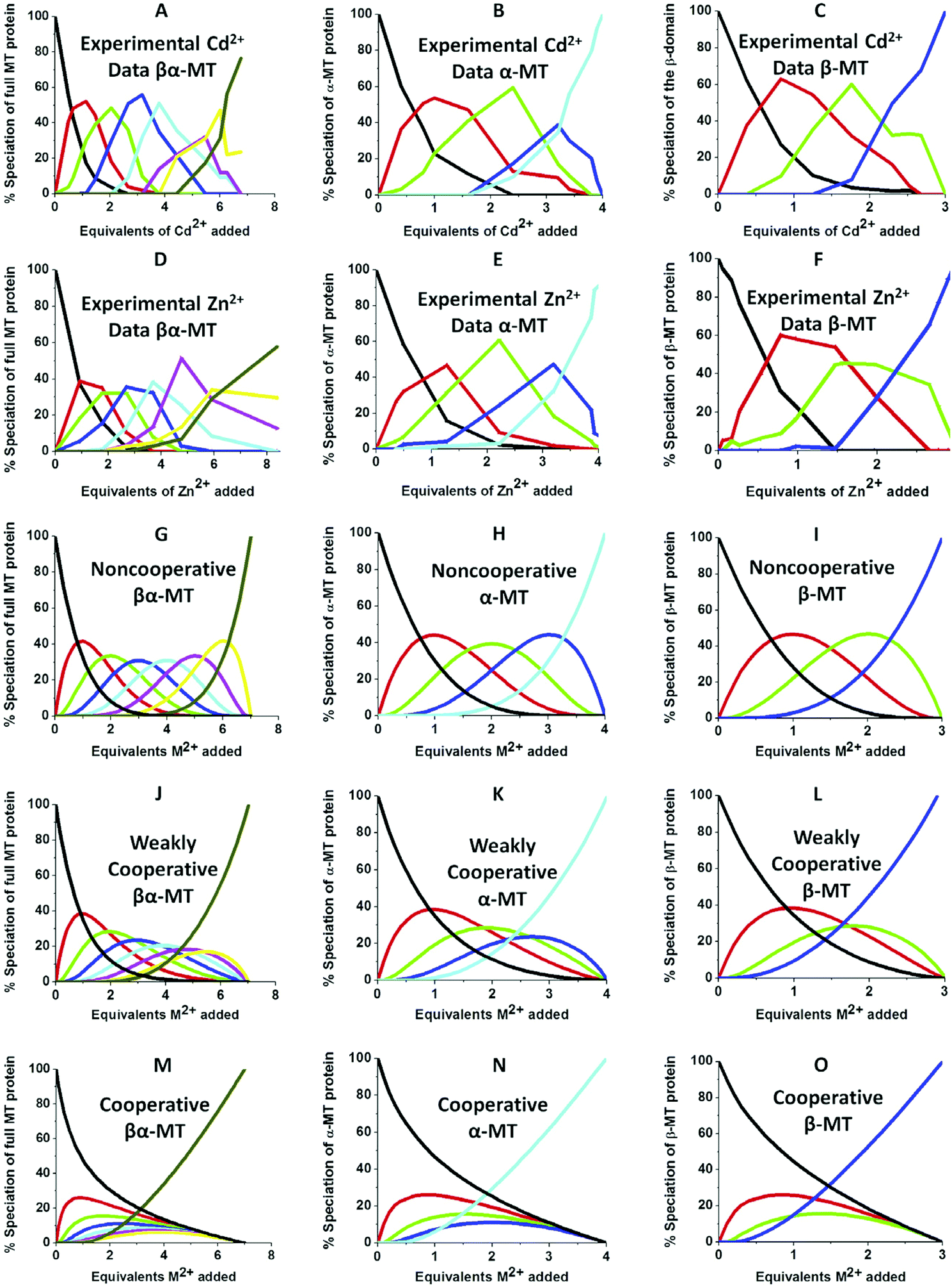 | ||
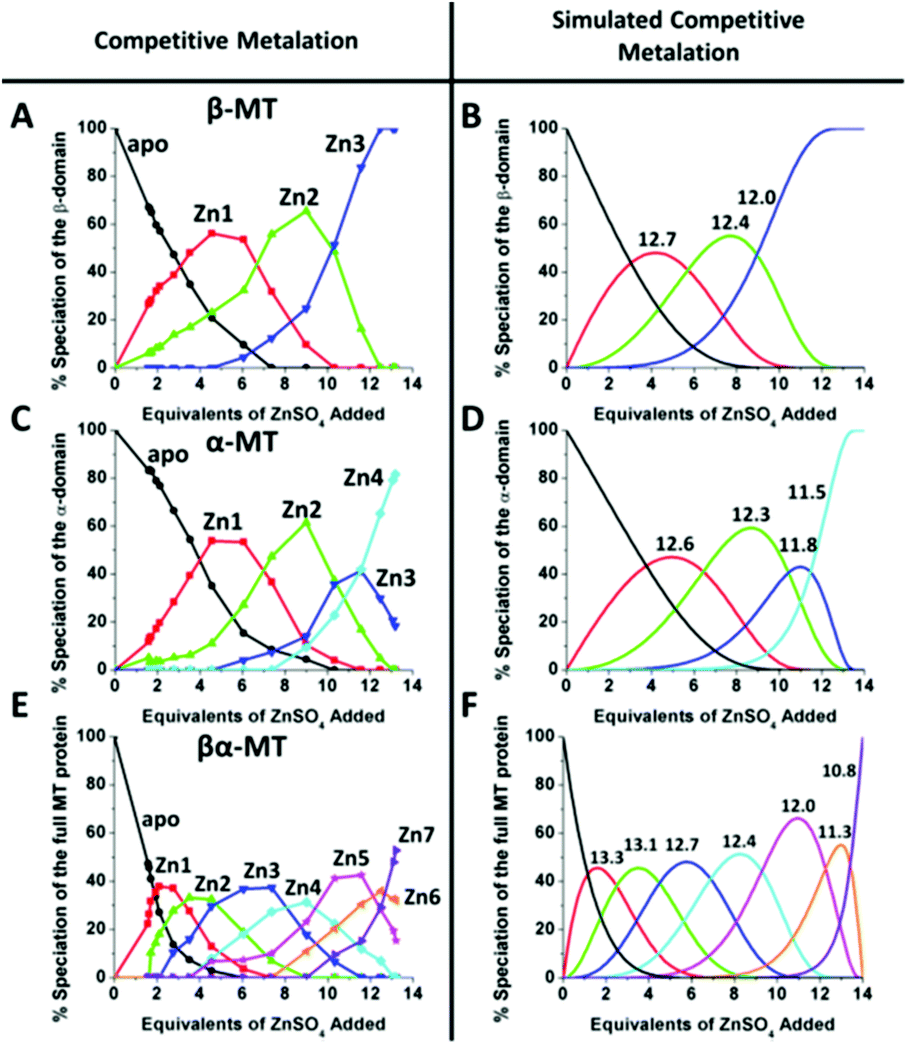
| Fig. 10 Experimental ESI-MS data and simulations of ESI-MS data for the metalation of MT and its isolated domains with Cd2+ and Zn2+. Models and experimental data showing Cd2+ speciation (A–C) and Zn2+ speciation (D–F) during metalation of the full MT protein, and the α- and β-domains. A non-cooperative model (declining Kf's) of ESI-MS data based on a series of linearly decreasing association constants (G–I). A weakly cooperative model (equal Kf's) of ESI-MS data based on a series of equal association constants (J–L). A strongly cooperative model (increasing Kf's) of ESI-MS data based on a series of linearly increasing association constants (M–O). The speciation lines are color coded as follows for each of the full protein (A), the α-MT fragment (B), and the β-fragment (C): BLACK – apo-protein, no metals bound; RED – species with 1 Cd2+ (A–C) or Zn2+ (D–F), or M2+ ion (G–O) bound; GREEN – species with 2 metals bound; BLUE – species with 3 metals bound; PALE BLUE – species with 4 metals bound; MAUVE – species with 5 metals bound; YELLOW – species with 6 metals bound; BROWN – species with 7 metals bound. Reproduced from ref. 51 with the permission of Elsevier. | ||
We can illustrate the spectral characteristics that would be observed for both cooperative and non-cooperative Zn2+ and Cd2+ binding to MT for using ESI-MS metal titration data. First, in Fig. 1051 (A–O), we show experimental and simulated speciation data based on the measurement of Cd2+ (A, B and C) and Zn2+ (D, E and F) binding to the full-length protein and to the isolated α- and β-fragments. The experimental data show that in each of the 6 metalation reactions (A–F) that each, individually-metalated species forms in sequence. This means that 7 metalated species are observed for the full-length protein (A and D), 4 for the α-fragment (B and E) and 3 for the β-fragment (C and F). The simulations of three possible mechanisms, (i) non-cooperative formation of the product (G, H and I), (ii) weakly cooperative formation of the product (J, K and L) and (iii) fully cooperative formation of the product (M, N and O), can be used to assess the type of metalation pathway. Clearly, for MT 1A at this pH the experimentally-determined pathways are all non-cooperative. This led us to ask what controls the choice of pathway during the metalation reaction because other researchers had clearly identified the presence of particularly the M4S11 cluster forming cooperatively.58,134,135 A possible answer was reported by Irvine et al. from pH-dependence of the metalation of apo-MT 1A with Zn2+ and Cd2+. In Fig. 1152 the pH-dependence of the formation of the clustered M4S11 is shown; M4 cluster formation was dominant at low pH (4.5 for Zn2+ and 6.0 for Cd2+). When the pH was increased to >7 the beaded species (M1–M5) were observed, indicating that the M4S11 cluster had been replaced, likely by nMS4 structures (n = 0–4). This pH dependence of the metalation pathways for both Zn2+ and Cd2+ is summarized in Fig. 12.52
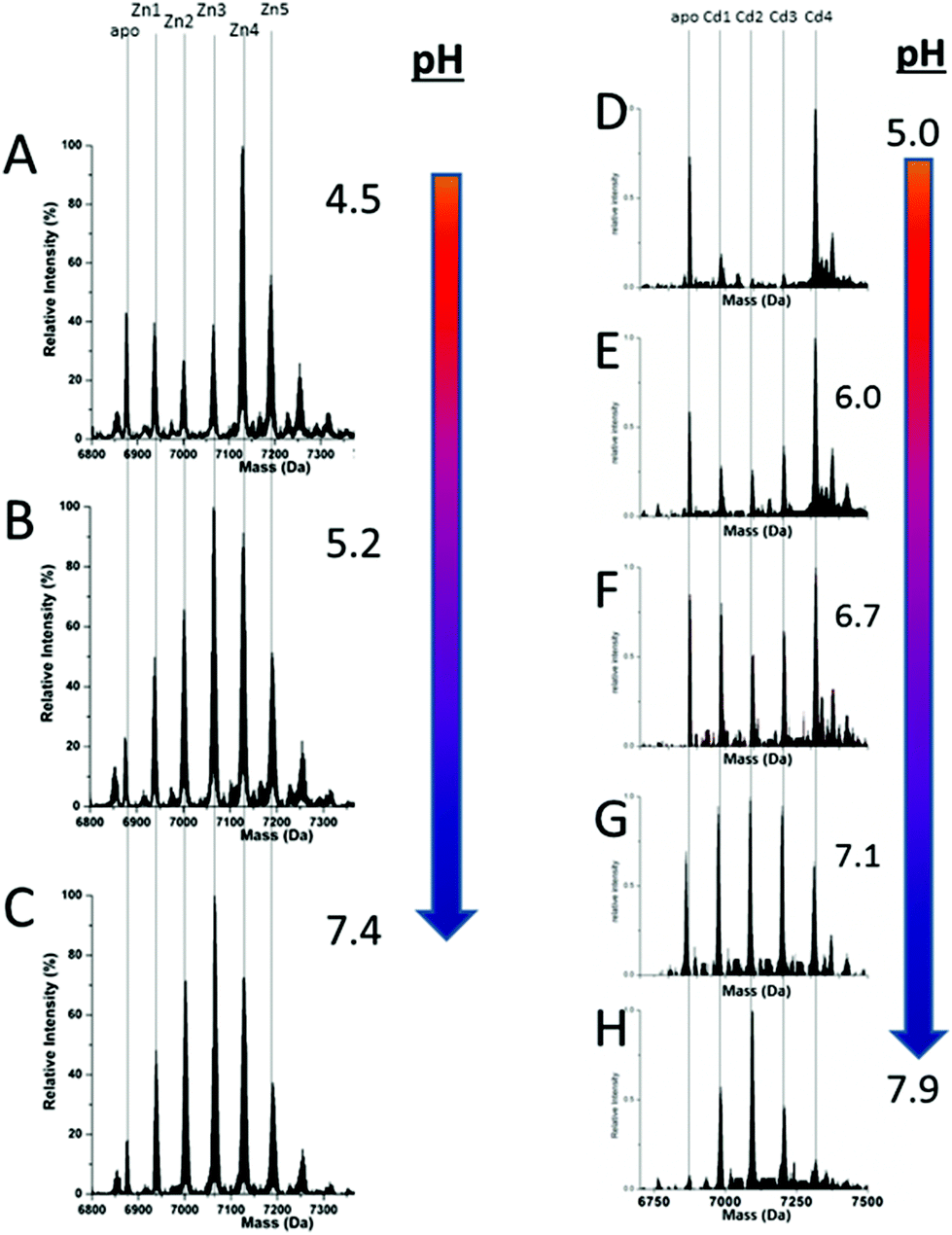 | ||
| Fig. 11 Changes in speciation of Zn2+ and Cd2+ binding to apo-rhMT 1a as a function of pH. (A), (B) and (C) ESI-MS data recorded for a single Zn:MT ratio (≈2.5) at pH 4.5, 5.2 and 7.4 showing the redistribution of Zn2+ from primarily a clustered Zn4 species (with a significant fraction of apo-MT remaining) to a series of distributed, non-cooperative species at pH 7.4. (D, E, F, G and H) ESI-MS data recorded for a single Cd:MT ratio (≈2.5) at pH 5.0, 6.0, 6.7, 7.1, and 7.9 showing the redistribution of Cd2+ from primarily a clustered Cd4 species at pH 5 (with a significant fraction of apo-MT remaining) to a series of distributed, non-cooperative species at pH 7.9. Reproduced from ref. 52 with the permission of the Royal Society of Chemistry. | ||
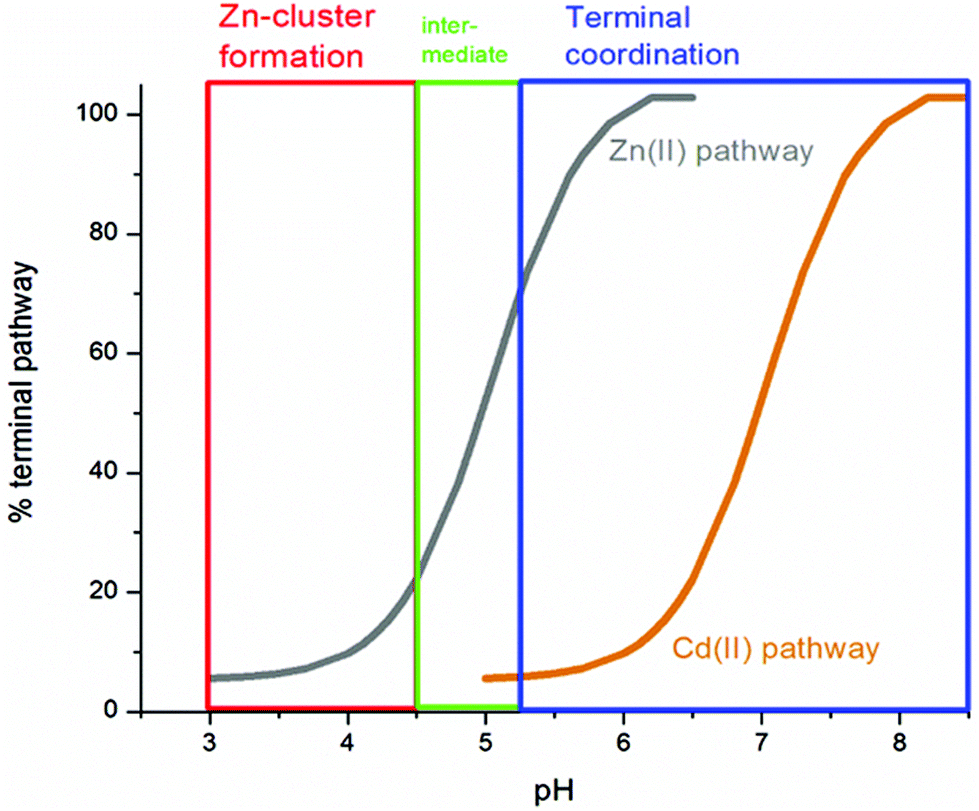 | ||
| Fig. 12 Summary of the trend in M4(SCYS)11 cluster formation as a function of pH for Zn-MT and Cd-MT showing the equilibrium between cooperatively formed clusters at low pH and a distributed, non-cooperatively formed species (“terminal coordination”) at neutral pH. Reproduced from ref. 52 with the permission of the Royal Society of Chemistry. | ||
 | ||
| Fig. 13 Representation of the distribution of metalated Zn-species following the non-cooperative pathway, based on analysis of the abundances in the ESI mass spectral data. (A) Zn2+ titration of apo-α MT, (B) Zn-titration of apo-β MT and (C) Zn2+ titration of apo-MT 1A. The data show the presence of multiple species for every addition of Zn2+. The bar graphs are color coded as follows for each α-MT fragment (A), β-fragment (B) and full protein (C): RED – apo-protein; GREEN – species with 1 Zn2+ bound; PURPLE – species with 2 Zn2+ bound; BLUE – species with 3 Zn2+ bound; PINK – species with 4 Zn2+ bound; ORANGE – species with 5 Zn2+ bound; MAUVE – species with 6 Zn2+ bound; PURPLE – species with 7 Zn2+ bound. Reproduced from ref. 53 with the permission of the American Chemical Society. | ||
This sequential shift in the species distribution as a function of the mol. eq. of Zn2+ added is controlled by the constants of the single step equilibrium reactions (Scheme 1). The solution of this set of 7 reaction equations depends on the initial concentration of the protein, the Zn2+ concentration and the values of the 7 binding constants. Previously, it was not possible to separate the speciation at each titration step into individual metalated species, making it impossible to solve for the 7 Kf values. The mass spectral data provided semi-quantitative estimates of the relative concentrations of each species present in solution (Fig. 13). By fitting the abundances of all species to the set of 7 reaction equations we determined the individual Kf values for each step.54 We then normalized to the average of the values reported by Krezel and Maret.131 A subsequent 3-way competition between the two fragments and the full-length protein allowed the determination of the relative binding constants. The fragments also allowed for the comparison of binding affinities between domains and to quantify the chelation effects of the full-length protein. The quality of the fits can be assessed by a comparison with the simulated speciation based on these same binding constants (Fig. 1454). The important message to take from these experiments is that when competitive chelation occurs, the relative order of the binding constants can be directly determined from the speciation. A very important extra result from this 3-way competition experiment was that the fragment K's exceeded the K's for the full-length protein for the 5th and 6th Zn2+. Our interpretation was that at this point in the stepwise addition of Zn2+ the full protein changed from 5 Zn(SCYS)42− beads to the 2 well-known clusters for the 6th and 7th Zn2+. This massive rearrangement reduced the Kf-values for the full-length protein compared with the isolated fragments. Of course, with different peptides, concentrations must be normalized with a solution-based method, in the case of using the LMCT at 250 nm using Cd2+-remetalation of each peptide and its corresponding extinction coefficient.
 | ||
| Fig. 14 A 3-way competition between the two fragments and the full-length protein allowed the determination of all the relative binding constants. The two fragments and the full protein were present in the same solution with concentration ratios such that the β-MT provided three binding sites, the α-MT provided 4 binding sites and the βα-protein provided 7 binding sites to give a total of 14 sites. Zn2+ was added stepwise and the ESI-MS data were recorded. The ESI-MS data allowed identification of the location of the bound Zn2+ as a function of each of the proteins in the same solution. The simulated data are based on a fit of all data using 14 individual binding constants. Reproduced from ref. 54 with the permission of the American Chemical Society. | ||
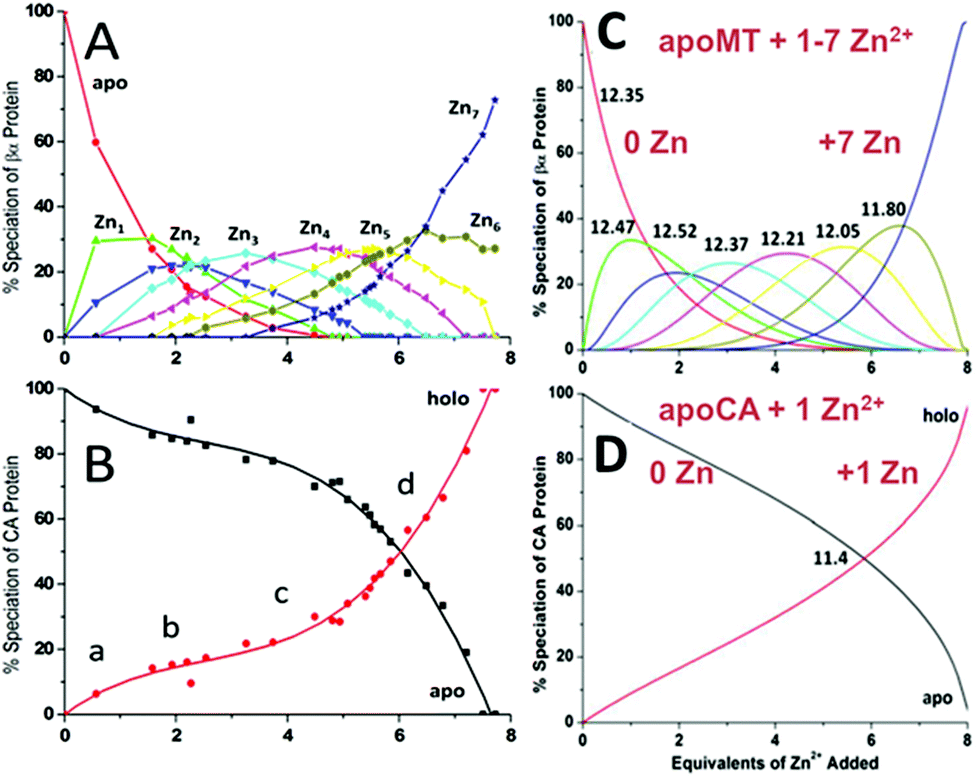 | ||
| Fig. 15 Two-way competition for Zn2+ between apo-rhMT 1a and apo-carbonic anhydrase (CA). ESI mass spectra were recorded as Zn2+ was added to equimolar concentrations of apo-rhMT 1a and CA. (A) Zn-MT speciation and (B) Zn2+ binding to apo-CA from the ESI-MS data showing the appearance of both Zn7MT and Zn1CA (holo-CA) with 8 Zn2+ added. (C) and (D) Calculated binding affinities for Zn2+ for Zn-MT based on the fixed value of 1011.4 for Zn2+ binding to apo-CA. Reproduced from ref. 55 with the permission of the American Chemical Society. | ||
Comparing panels A, C, and E in Fig. 14, it is apparent that βα (full-length protein) binds Zn1 and Zn2 with binding constants greater than the β- or α-fragments. The Kf values determined by the simulation followed the trend established by the As3+ binding kinetics, with a decreasing set of Kf values indicating non-cooperative metalation. However, while the relative Kf values can be determined reliably by ESI-MS and subsequent simulation, the absolute values are more difficult to determine. In this work, we used the average of the 4 values reported by Krezel and Maret131 as a guide for the approximation of absolute Kf values. We note that the speciation shown in Fig. 14 clearly illustrates the problem of assigning spectroscopic signatures to a specific, single number of metals added to MT. We mean, for example, that the speciation data in Fig. 14 E show that taking a stoichiometric ratio of four for Zn:apo-MT does not mean that Zn4-MT is the product, rather a Normal distribution of species each controlled by the relative value of KN. These K values are listed in Table 3.
A more precise method of establishing the absolute Kf values is to use a well-known chelator in direct competition with the protein of interest. Pinter and Stillman55 used the metalation of carbonic anhydrase (CA) with its known log10Kf of 11.4 to compete with βα-MT (Fig. 1555). Once again, the clarity of the speciation observed in the MS data immediately identifies the sequence of Zn2+ metalation for both MT and CA. In Fig. 15A, CA strongly competes for Zn2+ only after Zn5-MT has formed. To illustrate the competitive binding pattern, the Zn2+ contents of the CA and apo-CA were plotted in Fig. 15B. After 6 mol. eq. of Zn2+, CA competes directly with MT for additional Zn2+. Simulations of 8 bimolecular reactions involving 7 sites on MT and one site in CA are shown in Fig. 15C and D. The Kf values determined by this competition experiment are more reliable, as the values are normalized to the log10Kf of CA. Interestingly, while the last few Zn2+ bound have similar Kf values to the data reported by Summers et al. (Fig. 1454) the first Zn2+ bound to MT in the presence of CA binds with a Kf an order of magnitude smaller (1013.3vs. 1012.35). MS data reported by Irvine and Stillman136 showed that following partial metalation of apo-α MT with As3+ both the metalated protein and the metal-free protein adopted a folded conformation even under denaturing conditions. Based on this result, Pinter and Stillman concluded that in the presence of CA, the metallothionein binding constants may be modified due to protein–protein interactions compared with those in the absence of an interacting partner. Our overall conclusion is that the span of the Zn2+Kf-values ranged down one hundred-fold from approximately 1012.5 to 1010.5 and this is the essence of homeostatic control of Zn2+ in the cell. The role of MT in providing Zn2+ for Zn-dependent enzymes, as demonstrated by the competition with CA, is combined with the homeostatic buffering role which requires binding constants that are out of reach of the enzyme. Fig. 14 and 1554 summarize these roles.
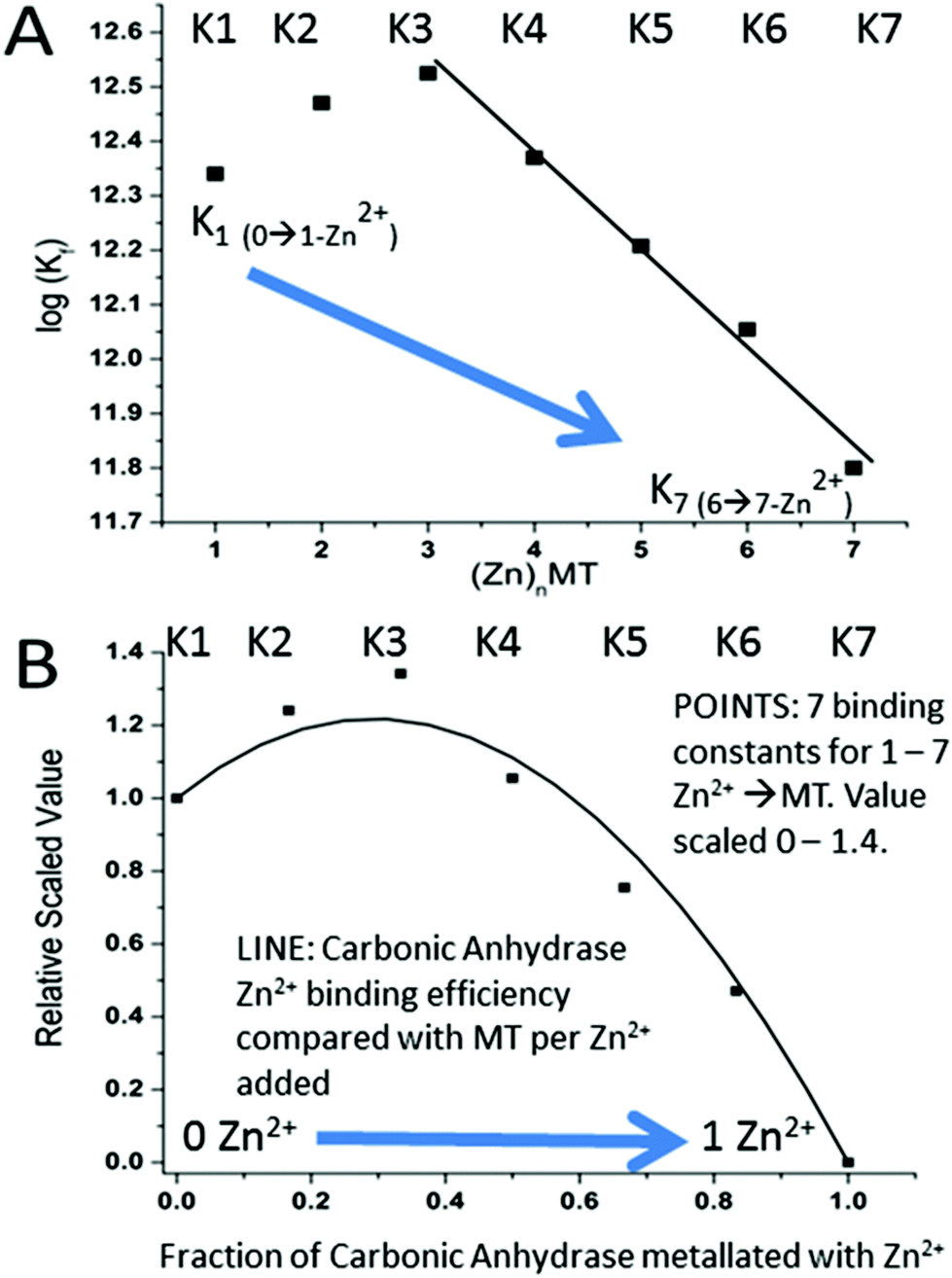 | ||
| Fig. 16 Schematic representation describing how the seven binding constants for the 7 Zn2+ bound to apoMT1a control the competitive uptake of the single Zn2+ by Carbonic Anhydrase (CA) when Zn2+ is added to a mixture of apo-MT 1A and CA. (A) The 7 binding constants calculated from the simulation shown in Fig. 15 as a function of the number of Zn2+'s bound, 1–7. (B) Superposition of (i) (POINTS) the 7 binding constants calculated from the simulation shown in Fig. 15 as a function of the number of Zn2+'s bound, 1–7, and scaled on the x-axis between 0 and 1.4 and on the y-axis between 0 and the complete metalation of CA (y-axis = 1.0). (ii) (SMOOTH LINE) The fraction of Zn2+ bound to apo-MT (y-axis scaled 0–1.4) and to CA (x-axis scaled 0.0–1.0) after each Zn2+ was added in a stepwise fashion. The data show that the first 4 Zn2+ primarily bind to the MT. CA only binds a significant fraction of the added Zn2+ once 5 Zn2+ have bound to the apo-MT 1A. Reproduced from ref. 55 with the permission of the American Chemical Society. | ||
Fig. 16 is a complicated diagram that depicts the control exerted by the 7 Kf's on the competitive Zn2+ binding between apo-MT 1a and CA when both are present in equal concentrations. The Figure shows through the smooth line in (B) how the fractional metalation of CA only finished when close to 5 Zn2+ have bound to the apo-MT 1A. The smooth line shows the fraction of Zn2+ that binds to either the apo-MT 1a (high on the y-axis) or CA (low on the x-axis), where the CA is fully metalated at 1.0 on the x-axis. These properties are summarized in Fig. 17 where the five individually bound Zn2+ exhibit the largest Kf's, and the two final Zn2+ bound result in the 2-domain structure involving bridging cys-thiolates and lower Kf's.
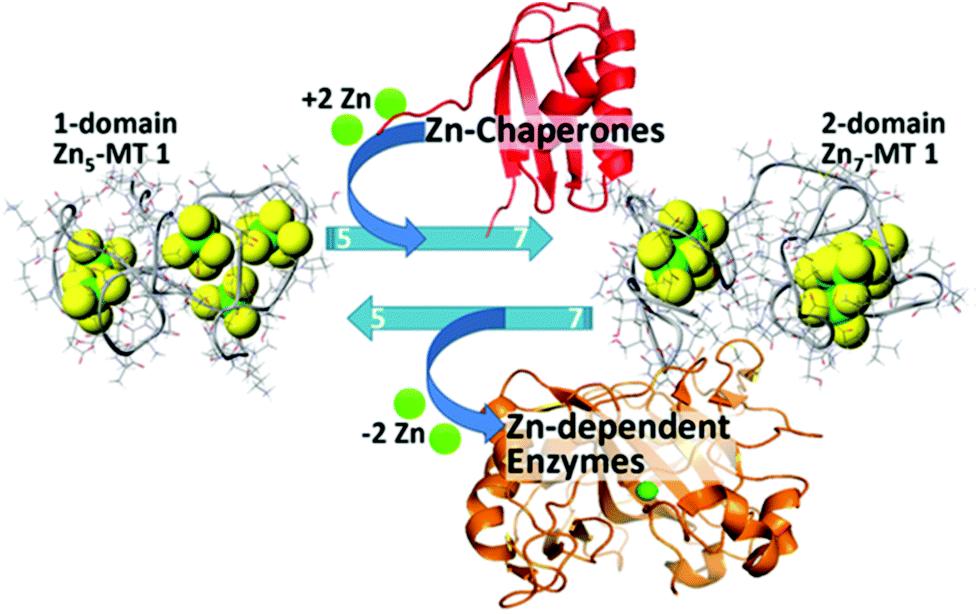 | ||
| Fig. 17 Schematic representation of the speciation in Zn-MT 1a showing the change between 5 individually bound Zn2+ “beads” from the non-cooperative pathway to the two domain structure formed with 7 Zn2+ illustrating the interaction between the Zn-MT and Zn-chaperones and Zn-dependent enzymes at physiological pH. Reproduced from ref. 54 with the permission of the American Chemical Society. | ||
The most detailed mechanistic information has been reported from emission and CD experiments where Cu6MT, Cu9MT, Cu12MT and Cu15MT species resulted in maxima in the spectral data.65,70 However, other groups reported maxima for Cu4 and Cu8MT species141,142 and the cooperative formation of a mixed metal Cd3Cu5MT.143 As we have described above, a major issue during the titration of any metal into MT is the coexistence of multiple species with different numbers of metals bound. In this context, with the addition of 6 mol. eq. Cu+ to MT 1a, species from Cu4 to Cu9 are observed in the mass spectrum.49 That means that spectroscopic methods are not sensitive enough to the specific metal-to-MT stoichiometry and will report intensities based on the average speciation in solution. CD, absorption and emission methods indicate changes in band centers and intensities but the average always blurs the signature of the individual species. The best optical results are obtained when there is a substantial change in chromophore properties. For example, the folding of the protein to form Cu+ clusters results in significant intensification of the emission due to the exclusion of water, which quenches Cu+ emission. When the protein unfolds at large Cu:MT molar ratios, the interaction with water molecules quenches the emission. Similarly, in the CD spectrum, changes in speciation that result in changes in the chiral peptide surrounding the metal centers as a result of the refolding of the peptide around a cluster are the most easily observed transitions.65
The complexity of Cu+ binding is shown in Fig. 1849 where the results of the simulation of the mass spectral data are shown. The mass spectral data for the Cu+ titration were analyzed in terms of 20 consecutive bimolecular reactions (Scheme 2). For a non-cooperative pathway, we would expect a series of species to form with a Normal distribution around the mean mol. eq. of Cu+ added, as seen in Fig. 18A. For a cooperative pathway, we would expect formation of species with specific Cu:MT ratios. The emission and CD data suggested that there would be clustered species formed, likely cooperatively. It was apparent from the mass spectral data that specific species formed with greater abundance, and these species corresponded to clusters previously indicated. However, at higher molar ratios of Cu+, a Normal distribution was observed. Our conclusion was that Cu+ binding to MT involved a mixture of cooperative formation of clusters at lower Cu:MT ratios and non-cooperative binding of Cu+ at higher ratios in isolated sites involving one or two cysteine residues.
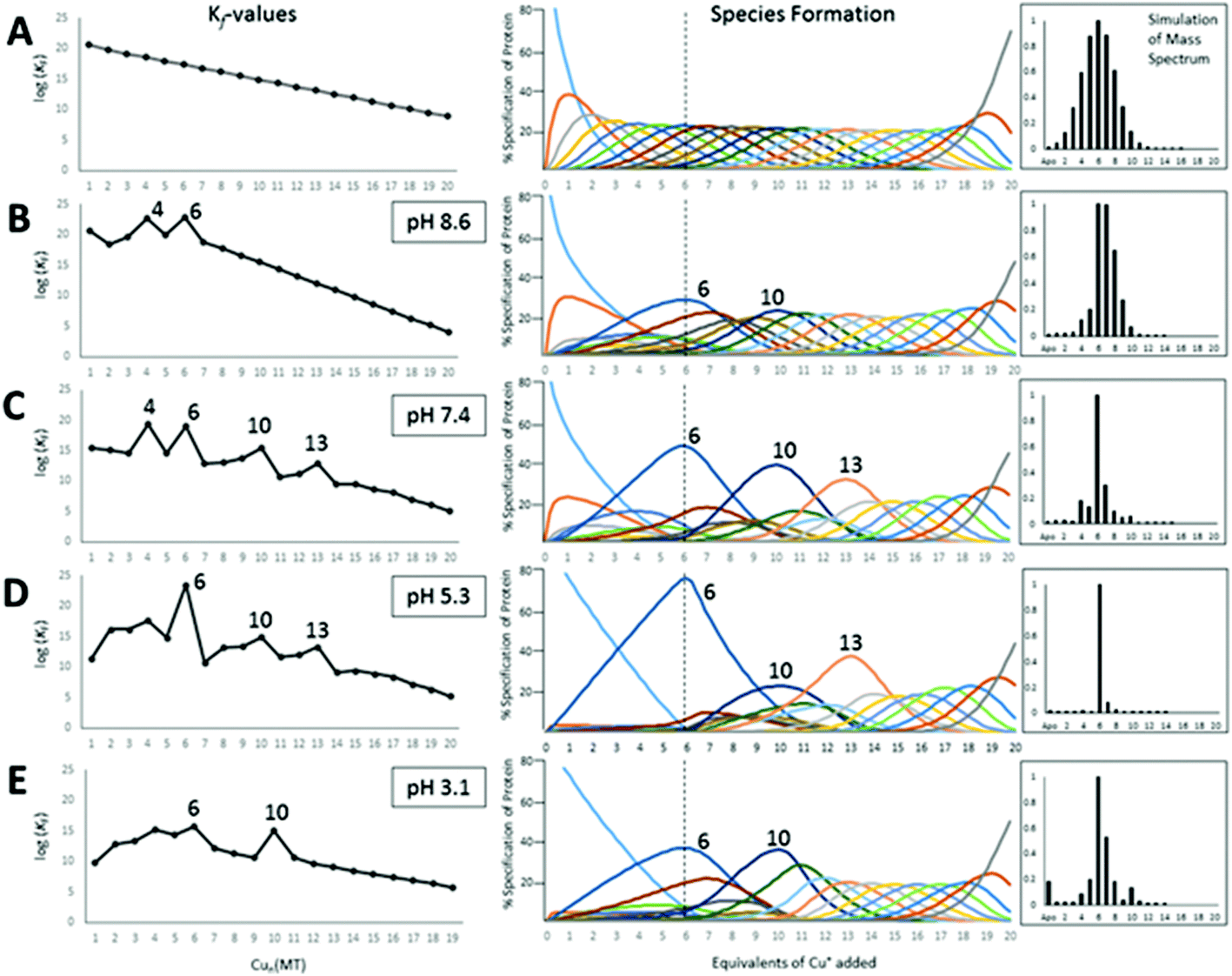 | ||
| Fig. 18 Results of fitting a series of 20 consecutive bimolecular reactions linked by their respective binding constants (Scheme 2) to the experimental ESI-mass spectral data recorded for titrations of apo-rhMT1a with up to 20 Cu+ at pH 8.6, 7.4, 5.3 and 3.1. The left column shows the trend in the individual Kf values as a function of Cu+ loading, the middle column shows individual species formation, and the right column shows a simulated mass spectrum at the titration point of 6 mol. eq. Cu+ added for each pH-value. The top row (A) shows the simulation based on 20 declining Kf values that would be expected when Cu+ binds in a completely non-cooperative manner with no cluster formation. (B)–(E) show the fitted binding constants for the stepwise copper binding reactions at the range of pH's. Cooperative cluster formation changes as a function of the pH. The cluster formation dominates the speciation numbered 6, 10, and 13. Reproduced from ref. 49 with the permission of the Royal Society of Chemistry. | ||
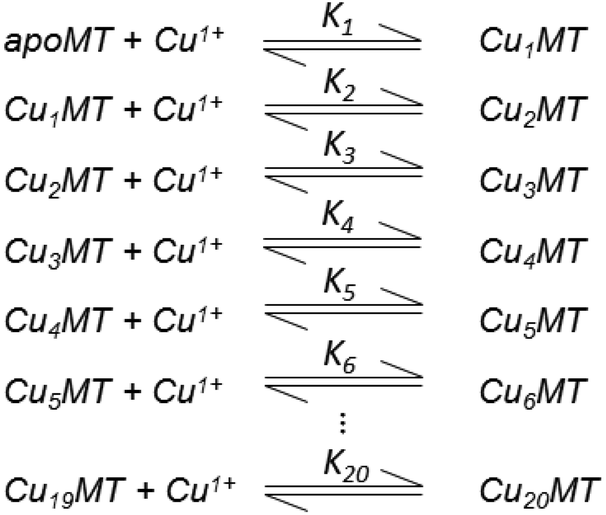 | ||
| Scheme 2 20 bimolecular reactions of Cu+ binding to MT. Adapted from ref. 49. | ||
The HySS simulation program was used to fit the ESI-MS experimental data using 20 Kf values.49Fig. 18 shows that the 4 cooperatively formed species dominate the spectra and simulation (the Cu4, Cu6, Cu10, and Cu13 species). It is likely that first a Cu4S6 cluster is formed, which then forms a Cu6S9 cluster in the β-domain. At 10 mol. eq. of Cu+ two clusters form, Cu6S9 in β and Cu4S6 in α and at 13 mol. eq. the Cu4S6 cluster expands to Cu7S11 (with Cu6S9 still in β). The fitted data clearly show that past the Cu13 point Cu+ is bound non-cooperatively. The simulated species formation in Fig. 18 also accounts for the optical properties previously reported. It should be noted that the oft-cited Cu12 species, which is thought to have 6 Cu in each of the domains, did not feature prominently in the ESI-MS results. It is possible that the previous interpretation of the optical data mistook a mixture of Cu10 and Cu13 species for Cu12 when 12 mol. eq. of Cu was added to the MT. Another possibility includes isoform specific cluster preferences and variation between MTs obtained from different sources (i.e. from different mammals).
We do recognize that the data in Fig. 18 represent a biologically unlikely set of reactions. It is not suggested that in vivo 20 Cu+ bind to apo-MT, rather the significance in the data shown in Fig. 18 and described fully in ref. 49 is clearly showing how the cooperative formation of clusters is dependent on the pH and that the structural properties are fluxional – different pathways are followed depending on the environment (pH), temperature, and metalation status. Dogmatically assigning cluster formation to MT is not correct; clusters form when it is a thermodynamic product. As the Zn2+-binding studies showed, bead-formation takes place at physiological pH up to 5 Zn2+ added, only then do the 6th and 7th Zn2+ added result in the rearrangement to the two-domain, clusters of the X-ray and NMR structures. In other words, the 2 last sites only exist because those metals that can use bridging thiolates can increase the metal loading in that way. The supermetalated Cd8-MT is just the next phase as the 2-cluster structure is lost and a single structure forms.48,77
50 shows the analysis of the mass spectral data in terms of selectivity between the two fragments. Fig. 19A and C indicate that at pH 7.4 there is little domain specificity with the first metal binding fractionally more to the β-domain than the α-domain. Between 2 and 5 metals added, the α-domain is slightly preferred because the α-domain binds 4 metals compared to 3 for the β-domain. The conclusion reached was that at pH 7.4 for the MT 1A protein there would be minimal domain specificity with a non-cooperative metalation pathway. The situation is quite different at lower pH (5.8); thus, following initial occupancy of the β-domain, the α-domain dominates as a result of the cooperative formation of the M4S11 cluster. The conclusion is that at lower pH there would be domain specificity for the full-length apoprotein with the α-domain dominating due to cooperative cluster formation.
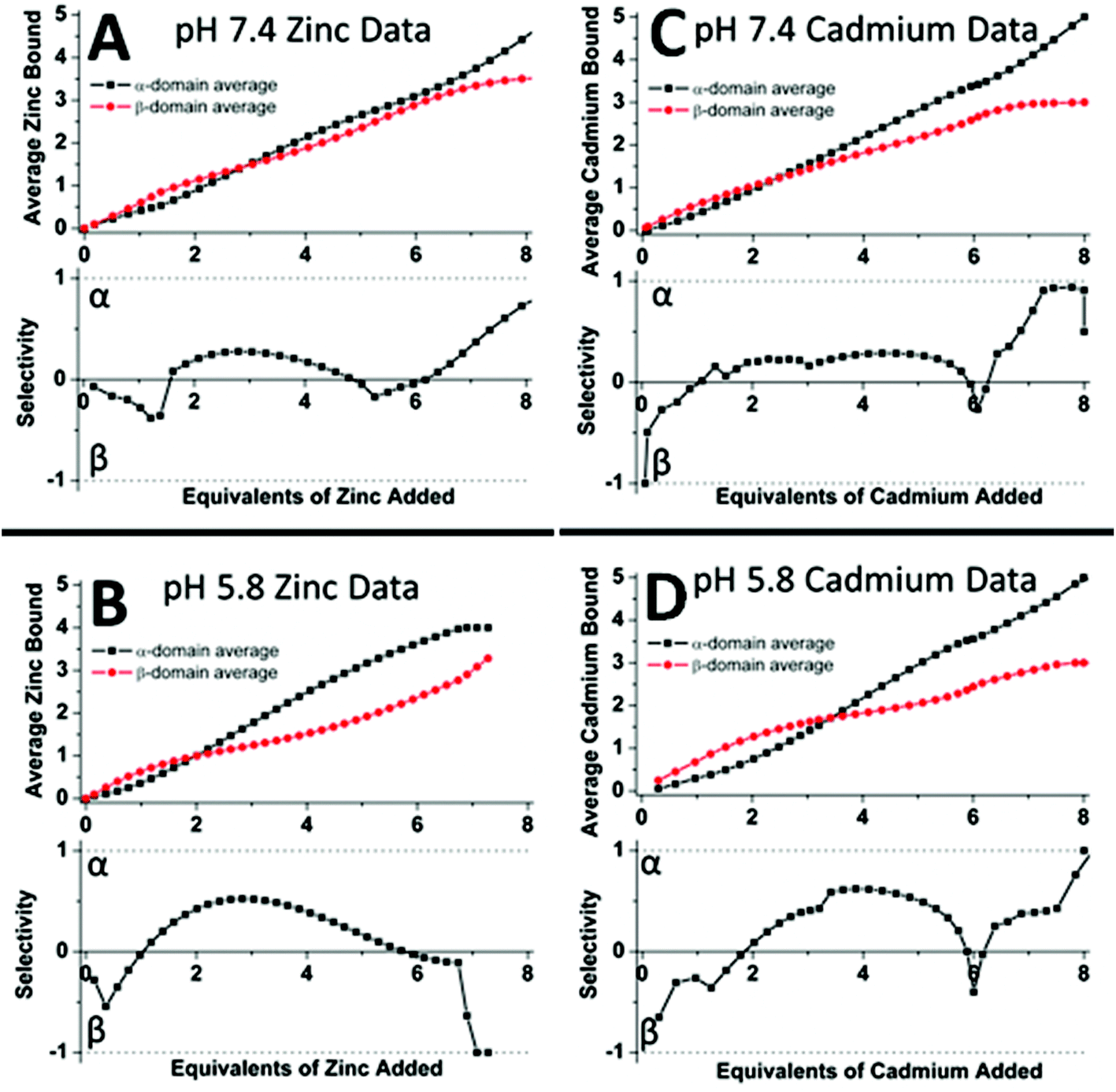 | ||
| Fig. 19 Analysis of the experimental ESI-MS data for Zn2+ (A, B) and Cd2+ (C, D) titrations of equimolar solutions of the α- and β-domain fragments showing the distribution of the Zn2+ and Cd2+ between the two domain fragments as a function of pH. The average Zn2+ bound graphs (top in A and B) and the average Cd2+ bound graphs (top in C and D) show the weighted distribution of the metal in the specified fragment (domain). The selectivity of each stepwise-added Zn2+/Cd2+ as a function of the pH is shown in the lower panels where if the data are negative this means that the Zn2+/Cd2+ are preferentially located in the β-fragment whereas if the data are positive the Zn2+/Cd2+ are preferentially located in the α-fragment. The graphs show that fragment (domain) selectivity is minimal at pH 7.4 (the dotted line is close to the “0” line until the 4th metal that has to bind to the α-domain fragment) but α is preferred at pH 5.8 for both metals (the dotted line is clearly above the “0” line for the 2–4 metal-added stages). Reproduced from ref. 50 with the permission of the American Chemical Society. | ||
50 outlines the different intertwined pathways for complete replacement of Zn2+ by Cd2+, for the full-length protein. Pathway 2a represents the cooperative, domain specific formation of Cd4S11. Pathway 3a shows the non-cooperative, non-domain specific displacement of 4 Zn2+ by Cd2+. The ESI mass spectral data recorded at pH 5.8 show that neither Zn2+ nor Cd2+ binding follows a domain selective binding mechanism when Cd2+ is added to the Zn2+ saturated domain fragments. This means that the binding affinity constants for the two metals span both fragments in their magnitude, resulting in a distribution of metals as indicated in Pathway 3a.
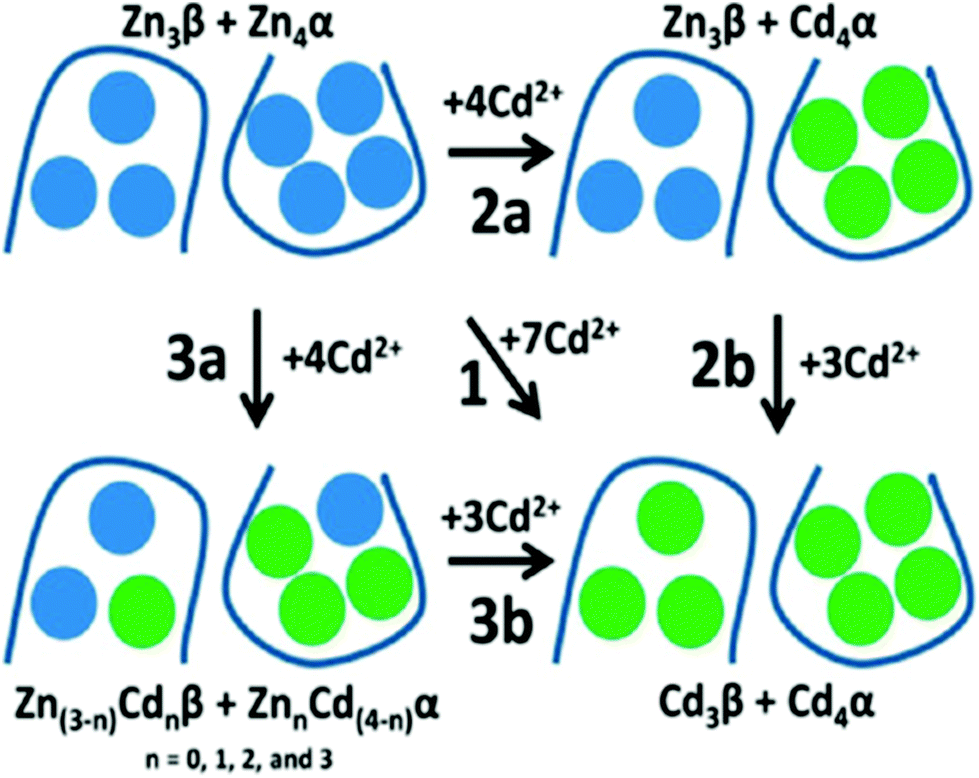 | ||
| Fig. 20 Schematic representation of the domain choices in the binding site for Cd2+ when binding to Zn7-βα rhMT1a. Pathway 1 implies complete replacement of Zn2+ by Cd2+ with no stepwise placement identified; pathway 2a shows cooperative formation of [Cd4S11]3− in the α-domain; pathway 3a shows a distributed binding of Cd2+ forming mixed Zn2+/Cd2+ clusters in both domains, followed by Pathway 3b with saturation of both domains. The associated binding constant data supported Pathways 3a and 3b: that is, non-cooperative displacement of Zn2+. Reproduced from ref. 50 with the permission of the American Chemical Society. | ||
3. Conclusions
Our goal in this very personal review was to summarize our laboratory's decades-long progress towards an understanding of the metal binding mechanism of mammalian MTs and the subsequent internal rearrangements. First, we should restate that the major property of the majority of MTs is that they bind multiple metals into well-defined binding sites when saturated with metals. However, the lack of specificity in both metal identity, binding site location and even geometry has complicated the determination of mechanistic details. This ambiguity has arisen from the overlap in metalation status and the lack of significant and individual differences in the chromophoric properties of each of the several possible metals when bound. The clearest data were reported by X-ray diffraction studies and 113Cd-NMR and 1H-NMR results. However, these data were from the metal saturated proteins which meant that the stoichiometry, cluster formation and coordination geometry were well defined, but that the mechanism involved in the stepwise binding into those sites was not available.The many spectroscopic studies involving a variety of optical methods indicated that metalation is accompanied by changes in the peptide, but the overlap of chromophores blurred the determination of specific speciation during stepwise binding. Clarity on the stepwise binding mechanism was delivered by ESI mass spectral data. The report on the kinetics of As3+ binding outlined each metalation step and the effect of the declining availability of cysteines on the rate constants was understood. For the purposes of determining binding affinities it was necessary to separate the speciation such that each equilibrium reaction could also be defined. The As3+-binding kinetics provided evidence that the binding affinities should decline as a function of metal loading. This then led to the determination of the effect of cooperativity on the binding affinities. The cooperative formation of clusters was well known, but the effect on the remaining metalation reactions up to saturation was not well described. The ESI mass spectral data provided the details necessary to incorporate both cooperative, cluster-driven pathways and non-cooperative, distributed pathways into the overall mechanistic description of MT metalation. These developments have led us to the complete analysis of Cu+ binding to apo-MT with its 20 binding constants. In a sense, the unraveling of the complex metalation pathways of metallothionein and its role in cellular chemistry has really only just begun. We look forward to detailed mechanistic details of the major metals involved in vivo and an understanding of the environmental factors controlling the metalation status and, therefore, the homeostatic and redox roles of metallothioneins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the NSERC of Canada through Discovery and Research Tools and Instruments Grants (to M.J.S.) and a Canada Graduate Scholarship (CGS-D) to G.W.I. We also thank Prof. Andrea Hartwig (KIT, Germany) for a highly productive collaboration that allowed Ms Scheller to work in M.J.S.'s laboratory in 2016 and 2017. M.J.S. thanks the many students who have worked on and have maintained their interest in the metallothionein projects for the last 20 years; without your dedication and perseverance the latest results described here would not have been possible. We thank Ms. A. Zhang for the Table of Contents graphics that, we hope, shows the properties of metallothionein gradually coming into focus since the first reports of 1957. We wish to also acknowledge Mr Doug Hairsine, who has continued to provide excellent technical assistance with the mass spectrometer for many years. Our work could not be carried out without the continued excellence of the electronic, computational and mechanical support of all our instruments over many years from Mr John Vanstone and the team members of the Department of Chemistry Electronics Shop. We thank Dr Eva Freisinger for data on the wheat proteins. We also acknowledge and thank the anonymous reviewers who provided many insightful and valuable comments.References
- C. A. Blindauer, Chem. Commun., 2015, 51, 4544–4563 RSC.
- C. A. Blindauer and O. I. Leszczyszyn, Nat. Prod. Rep., 2010, 27, 720–741 RSC.
- M. J. Stillman, Coord. Chem. Rev., 1995, 144, 461–511 CrossRef CAS.
- D. E. Sutherland and M. J. Stillman, Metallomics, 2011, 3, 444–463 RSC.
- D. E. Sutherland and M. J. Stillman, Metallomics, 2014, 6, 702–728 RSC.
- M. Margoshes and B. L. Vallee, J. Am. Chem. Soc., 1957, 79, 4813–4814 CrossRef CAS.
- L. Vergani, M. Grattarola, C. Borghi, F. Dondero and A. Viarengo, FEBS J., 2005, 272, 6014–6023 CrossRef CAS PubMed.
- O. I. Leszczyszyn, H. T. Imam and C. A. Blindauer, Metallomics, 2013, 5, 1146–1169 RSC.
- E. Freisinger, Dalton Trans., 2008, 6663–6675 RSC.
- E. Freisinger, J. Biol. Inorg. Chem., 2011, 16, 1035–1045 CrossRef CAS PubMed.
- C. D. Klaassen and J. Liu, Drug Metab. Rev., 1997, 29, 79–102 CrossRef CAS PubMed.
- G. K. Andrews, Biochem. Pharmacol., 2000, 59, 95–104 CrossRef CAS PubMed.
- E. Tokuda, S.-I. Ono, K. Ishige, A. Naganuma, Y. Ito and T. Suzuki, Toxicology, 2007, 229, 33–41 CrossRef CAS PubMed.
- N. O. Nartey, J. V. Frei and M. G. Cherian, Lab. Invest., 1987, 57, 397–401 CAS.
- S. Somji, M. A. Sens, D. L. Lamm, S. H. Garrett and D. A. Sens, Cancer Detect. Prev., 2000, 25, 62–75 Search PubMed.
- A. Miles, G. Hawksworth, J. Beattie and V. Rodilla, Crit. Rev. Biochem. Mol. Biol., 2000, 35, 35–70 CrossRef CAS PubMed.
- B. A. Masters, C. J. Quaife, J. C. Erickson, E. J. Kelly, G. J. Froelick, B. P. Zambrowicz, R. L. Brinster and R. D. Palmiter, J. Neurosci., 1994, 14, 5844–5857 CAS.
- C. J. Quaife, S. D. Findley, J. C. Erickson, G. J. Froelick, E. J. Kelly, B. P. Zambrowicz and R. D. Palmiter, Biochemistry, 1994, 33, 7250–7259 CrossRef CAS PubMed.
- Y. Kojima, C. Berger, B. L. Vallee and J. Kägi, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3413–3417 CrossRef CAS.
- K. Balamurugan and W. Schaffner, Biochim. Biophys. Acta, 2006, 1763, 737–746 CrossRef CAS PubMed.
- S. Lutsenko, Curr. Opin. Chem. Biol., 2010, 14, 211–217 CrossRef CAS PubMed.
- C. M. St.Croix, K. J. Wasserloos, K. E. Dineley, I. J. Reynolds, E. S. Levitan and B. R. Pitt, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2002, 282, L185–L192 CrossRef CAS PubMed.
- D. J. Ecker, T. R. Butt, E. J. Sternberg, M. P. Neeper, C. Debouck, J. A. Gorman and S. T. Crooke, J. Biol. Chem., 1986, 261, 16895–16900 CAS.
- G. Isani and E. Carpene, Biomolecules, 2014, 4, 435–457 CrossRef PubMed.
- S. R. Sturzenbaum, O. Georgiev, A. J. Morgan and P. Kille, Environ. Sci. Technol., 2004, 38, 6283–6289 CrossRef CAS PubMed.
- G. Meloni, V. Sonois, T. Delaine, L. Guilloreau, A. Gillet, J. Teissie, P. Faller and M. Vasak, Nat. Chem. Biol., 2008, 4, 366–372 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- T.-Y. Li, A. J. Kraker, C. F. Shaw and D. H. Petering, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6334–6338 CrossRef CAS.
- G. Meloni, P. Faller and M. Vasak, J. Biol. Chem., 2007, 282, 16068–16078 CrossRef CAS PubMed.
- A. Krezel and W. Maret, Biochem. J., 2007, 402, 551–558 CrossRef CAS PubMed.
- B. Ruttkay-Nedecky, L. Nejdl, J. Gumulec, O. Zitka, M. Masarik, T. Eckschlager, M. Stiborova, V. Adam and R. Kizek, Int. J. Mol. Sci., 2013, 14, 6044–6066 CrossRef CAS PubMed.
- D. L. Wong and M. J. Stillman, Chem. Commun., 2016, 52, 5698–5701 RSC.
- E. Atrian-Blasco, A. Santoro, D. L. Pountney, G. Meloni, C. Hureau and P. Faller, Chem. Soc. Rev., 2017, 46, 7683–7693 RSC.
- J. H. Kägi and B. L. Vallee, J. Biol. Chem., 1960, 235, 3460–3465 Search PubMed.
- J. H. Kägi and B. L. Vallee, J. Biol. Chem., 1961, 236, 2435–2442 Search PubMed.
- P. Babula, M. Masarik, V. Adam, T. Eckschlager, M. Stiborova, L. Trnkova, H. Skutkova, I. Provaznik, J. Hubalek and R. Kizek, Metallomics, 2012, 4, 739–750 RSC.
- D. H. Nies, E. Freisinger and G. J. Krauss, Ecological Biochemistry: Environmental and Interspecies Interactions, 2014, pp. 236–257 Search PubMed.
- A. Krężel and W. Maret, Int. J. Mol. Sci., 2017, 18, 1237 CrossRef PubMed.
- M. Vasak, J. Trace Elem. Med. Biol., 2005, 19, 13–17 CAS.
- J. Kägi, M. Vasák, K. Lerch, D. E. Gilg, P. Hunziker, W. R. Bernhard and M. Good, Environ. Health Perspect., 1984, 54, 93 CrossRef.
- M. J. Stillman and A. P. Presta, Molecular biology and toxicology of metals, Taylor & Francis, New York, 2000, pp. 276–299 Search PubMed.
- M. J. Stillman, C. F. Shaw and K. T. Suzuki, Metallothionein: Synthesis, structure, and properties of metallothioneins, phytochelatins, and metal-thiolate complexes, Wiley-VCH, 1992 Search PubMed.
- G. W. Irvine and M. J. Stillman, Int. J. Mol. Sci., 2017, 18, 913 CrossRef PubMed.
- J. Chan, Z. Huang, M. E. Merrifield, M. T. Salgado and M. J. Stillman, Coord. Chem. Rev., 2002, 233, 319–339 CrossRef.
- T. T. Ngu, M. D. Dryden and M. J. Stillman, Biochem. Biophys. Res. Commun., 2010, 401, 69–74 CrossRef CAS PubMed.
- T. T. Ngu, S. Krecisz and M. J. Stillman, Biochem. Biophys. Res. Commun., 2010, 396, 206–212 CrossRef CAS PubMed.
- D. L. Wong, M. E. Merrifield-MacRae and M. J. Stillman, Lead: Its Effects on Environment and Health, 2017, vol. 17, p. 241 Search PubMed.
- D. E. Sutherland, M. J. Willans and M. J. Stillman, J. Am. Chem. Soc., 2012, 134, 3290–3299 CrossRef CAS PubMed.
- J. S. Scheller, G. W. Irvine, D. L. Wong, A. Hartwig and M. J. Stillman, Metallomics, 2017, 9, 447–462 RSC.
- T. B. Pinter, G. W. Irvine and M. J. Stillman, Biochemistry, 2015, 54, 5006–5016 CrossRef CAS PubMed.
- D. E. Sutherland, K. L. Summers and M. J. Stillman, Biochem. Biophys. Res. Commun., 2012, 426, 601–607 CrossRef CAS PubMed.
- G. W. Irvine, T. B. Pinter and M. J. Stillman, Metallomics, 2016, 8, 71–81 RSC.
- D. E. Sutherland, K. L. Summers and M. J. Stillman, Biochemistry, 2012, 51, 6690–6700 CrossRef CAS PubMed.
- K. L. Summers, D. E. Sutherland and M. J. Stillman, Biochemistry, 2013, 52, 2461–2471 CrossRef CAS PubMed.
- T. B. Pinter and M. J. Stillman, Biochemistry, 2014, 53, 6276–6285 CrossRef CAS PubMed.
- G. W. Irvine, K. E. R. Duncan, M. Gullons and M. J. Stillman, Chem. – Eur. J., 2015, 21, 1269–1279 CrossRef CAS PubMed.
- G. W. Irvine, M. Santolini and M. J. Stillman, Protein Sci., 2017, 26, 960–971 CrossRef CAS PubMed.
- M. Good, R. Hollenstein, P. J. Sadler and M. Vasak, Biochemistry, 1988, 27, 7163–7166 CrossRef CAS PubMed.
- S.-H. Chen, L. Chen and D. H. Russell, J. Am. Chem. Soc., 2014, 136, 9499–9508 CrossRef CAS PubMed.
- S.-H. Chen, W. K. Russell and D. H. Russell, Anal. Chem., 2013, 85, 3229–3237 CrossRef CAS PubMed.
- M. J. Stillman, W. Cai and A. J. Zelazowski, J. Biol. Chem., 1987, 262, 4538–4548 CAS.
- W. Cai and M. J. Stillman, J. Am. Chem. Soc., 1988, 110, 7872–7873 CrossRef CAS.
- W. Lu and M. J. Stillman, J. Am. Chem. Soc., 1993, 115, 3291–3299 CrossRef CAS.
- W. Lu, A. J. Zelazowski and M. J. Stillman, Inorg. Chem., 1993, 32, 919–926 CrossRef CAS.
- A. Presta, A. R. Green, A. Zelazowski and M. J. Stillman, Eur. J. Biochem., 1995, 227, 226–240 CrossRef CAS PubMed.
- K. E. R. Duncan and M. J. Stillman, J. Inorg. Biochem., 2006, 100, 2101–2107 CrossRef PubMed.
- M. T. Salgado, K. L. Bacher and M. J. Stillman, J. Biol. Inorg. Chem., 2007, 12, 294–312 CrossRef CAS PubMed.
- M. Beltramini and K. Lerch, FEBS Lett., 1982, 142, 219–222 CrossRef CAS PubMed.
- M. Beltramini and K. Lerch, FEBS Lett., 1981, 127, 201–203 CrossRef CAS PubMed.
- A. R. Green, A. Presta, Z. Gasyna and M. J. Stillman, Inorg. Chem., 1994, 33, 4159–4168 CrossRef CAS.
- M. J. Stillman, Z. Gasyna and A. J. Zelazowski, FEBS Lett., 1989, 257, 283–286 CrossRef CAS PubMed.
- M. J. Stillman and Z. Gasyna, Methods Enzymol., 1991, 205, 540–555 CAS.
- J. D. Otvos and I. M. Armitage, J. Am. Chem. Soc., 1979, 101, 7734–7736 CrossRef CAS.
- B. A. Messerle, A. Schaffer, M. Vasak, J. H. R. Kagi and K. Wuthrich, J. Mol. Biol., 1990, 214, 765–779 CrossRef CAS PubMed.
- A. Robbins, D. McRee, M. Williamson, S. Collett, N. Xuong, W. Furey, B. Wang and C. Stout, J. Mol. Biol., 1991, 221, 1269–1293 CAS.
- S. Hu, B. Ye, X. Yi, Z. Cao, D. Wu, C. Shen and J. Wang, Talanta, 2016, 155, 272–277 CrossRef CAS PubMed.
- K. E. R. Duncan, C. W. Kirby and M. J. Stillman, FEBS J., 2008, 275, 2227–2239 CrossRef CAS PubMed.
- D. E. Sutherland, M. J. Willans and M. J. Stillman, Biochemistry, 2010, 49, 3593–3601 CrossRef CAS PubMed.
- T. T. Ngu, A. Easton and M. J. Stillman, J. Am. Chem. Soc., 2008, 130, 17016–17028 CrossRef CAS PubMed.
- T. T. Ngu and M. J. Stillman, J. Am. Chem. Soc., 2006, 128, 12473–12483 CrossRef CAS PubMed.
- K. E. Rigby-Duncan and M. J. Stillman, FEBS J., 2007, 274, 2253–2261 CrossRef PubMed.
- X. Yu, M. Wojciechowski and C. Fenselau, Anal. Chem., 1993, 65, 1355–1359 CrossRef CAS PubMed.
- J. Zaia, D. Fabris, D. Wei, R. L. Karpel and C. Fenselau, Protein Sci., 1998, 7, 2398–2404 CrossRef CAS PubMed.
- Y. Hathout, D. Fabris and C. Fenselau, Int. J. Mass Spectrom., 2001, 204, 1–6 CrossRef CAS.
- T. T. Ngu, J. A. Lee, T. B. Pinter and M. J. Stillman, J. Inorg. Biochem., 2010, 104, 232–244 CrossRef CAS PubMed.
- A. J. Zelazowski and M. J. Stillman, Inorg. Chem., 1992, 31, 3363–3370 CrossRef CAS.
- C. A. Blindauer, M. T. Razi, D. J. Campopiano and P. J. Sadler, J. Biol. Inorg. Chem., 2007, 12, 393–405 CrossRef CAS PubMed.
- O. I. Leszczyszyn, R. Schmid and C. A. Blindauer, Proteins: Struct., Funct., Genet., 2007, 68, 922–935 CrossRef CAS PubMed.
- E. A. Peroza, R. Schmucki, P. Guntert, E. Freisinger and O. Zerbe, J. Mol. Biol., 2009, 387, 207–218 CrossRef CAS PubMed.
- D. A. Fowle and M. J. Stillman, J. Biomol. Struct. Dyn., 1997, 14, 393–406 CAS.
- A. Presta, D. A. Fowle and M. J. Stillman, J. Chem. Soc., Dalton Trans., 1997, 977–984, 10.1039/A605462E.
- J. Ejnik, J. Robinson, J. Zhu, H. Försterling, C. F. Shaw and D. H. Petering, J. Inorg. Biochem., 2002, 88, 144–152 CrossRef CAS PubMed.
- T. Gan, A. Munoz, C. F. Shaw-III and D. H. Petering, J. Biol. Chem., 1995, 270, 5339–5345 CrossRef CAS PubMed.
- J. Ejnik, C. F. Shaw-III and D. H. Petering, Inorg. Chem., 2010, 49, 6525–6534 CrossRef CAS PubMed.
- C. F. Shaw III, L. He, A. Muñoz, M. M. Savas, S. Chi, C. L. Fink, T. Gan and D. H. Petering, J. Biol. Inorg. Chem., 1997, 2, 65–73 CrossRef.
- D. Hagrman, J. Goodisman, J. C. Dabrowiak and A.-K. Souid, Drug Metab. Dispos., 2003, 31, 916–923 CrossRef CAS PubMed.
- T. T. Ngu and M. J. Stillman, Dalton Trans., 2009, 5425–5433 RSC.
- T. T. Ngu and M. J. Stillman, IUBMB Life, 2009, 61, 438–446 CrossRef CAS PubMed.
- K. E. R. Duncan and M. J. Stillman, FASEB J., 2006, 20, A501–A501 Search PubMed.
- K. E. Rigby, J. Chan, J. Mackie and M. J. Stillman, Proteins: Struct., Funct., Bioinf., 2006, 62, 159–172 CrossRef CAS PubMed.
- K. E. Rigby and M. J. Stillman, Biochem. Biophys. Res. Commun., 2004, 325, 1271–1278 CrossRef CAS PubMed.
- G. Jiang, Z. Gong, X.-F. Li, W. R. Cullen and X. C. Le, Chem. Res. Toxicol., 2003, 16, 873–880 CrossRef CAS PubMed.
- G. Schmitz, D. Minkel, D. Gingrich and C. Shaw, J. Inorg. Biochem., 1980, 12, 293–306 CrossRef CAS PubMed.
- W. Bernhard, M. Good, M. Vašák and J. H. Kägi, Inorg. Chim. Acta, 1983, 79, 154–155 CrossRef.
- H. Sun, H. Li, I. Harvey and P. J. Sadler, J. Biol. Chem., 1999, 274, 29094–29101 CrossRef CAS PubMed.
- D. E. K. Sutherland and M. J. Stillman, Biochem. Biophys. Res. Commun., 2008, 372, 840–844 CrossRef CAS PubMed.
- X. Wan and E. Freisinger, Metallomics, 2009, 1, 489–500 RSC.
- O. Schicht and E. Freisinger, Inorg. Chim. Acta, 2009, 362, 714–724 CrossRef CAS.
- A. M. Tommey, J. Shi, W. P. Lindsay, P. Urwin and N. J. Robinson, FEBS Lett., 1991, 292, 48–52 CrossRef CAS PubMed.
- P. Kille, D. R. Winge, J. L. Harwood and J. Kay, FEBS Lett., 1991, 295, 171–175 CrossRef CAS PubMed.
- E. A. Peroza and E. Freisinger, J. Biol. Inorg. Chem., 2007, 12, 377–391 CrossRef CAS PubMed.
- J. Loebus, E. A. Peroza, N. Blüthgen, T. Fox, W. Meyer-Klaucke, O. Zerbe and E. Freisinger, J. Biol. Inorg. Chem., 2011, 16, 683 CrossRef CAS PubMed.
- K. Bilecen, U. H. Ozturk, A. D. Duru, T. Sutlu, M. V. Petoukhov, D. I. Svergun, M. H. Koch, U. O. Sezerman, I. Cakmak and Z. Sayers, J. Biol. Chem., 2005, 280, 13701–13711 CrossRef CAS PubMed.
- G. Mir, J. Domènech, G. Huguet, W.-J. Guo, P. Goldsbrough, S. Atrian and M. Molinas, J. Exp. Bot., 2004, 55, 2483–2493 CrossRef CAS PubMed.
- G.-Y. Huang and Y.-S. Wang, Aquat. Toxicol., 2010, 99, 86–92 CrossRef CAS PubMed.
- E. Freisinger, Inorg. Chim. Acta, 2007, 360, 369–380 CrossRef CAS.
- M. Vasak, J. H. Kaegi, B. Holmquist and B. L. Vallee, Biochemistry, 1981, 20, 6659–6664 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, L. Messori and M. Vasak, J. Am. Chem. Soc., 1989, 111, 7296–7300 CrossRef CAS.
- E. Artells, O. Palacios, M. Capdevila and S. Atrian, FEBS J., 2014, 281, 1659–1678 CrossRef CAS PubMed.
- X. Wan, O. Schicht and E. Freisinger, Z. Anorg. Allg. Chem., 2013, 639, 1365–1369 CrossRef CAS.
- M. Good and M. Vasak, Biochemistry, 1986, 25, 8353–8356 CrossRef CAS PubMed.
- Ò. Palacios, À. Leiva-Presa, S. Atrian and R. Lobinski, Talanta, 2007, 72, 480–488 CrossRef PubMed.
- M. C. Carpenter, A. S. Shah, S. DeSilva, A. Gleaton, A. Su, B. Goundie, M. L. Croteau, M. J. Stevenson, D. Wilcox and R. N. Austin, Metallomics, 2016, 8, 605–617 RSC.
- A. Pattanaik, G. Bachowski, J. Laib, D. Lemkuil, C. Shaw, D. Petering, A. Hitchcock and L. Saryan, J. Biol. Chem., 1992, 267, 16121–16128 CAS.
- A. J. Żelazowski, J. S. Garvey and J. D. Hoeschele, Arch. Biochem. Biophys., 1984, 229, 246–252 CrossRef.
- A. Kraker, J. Schmidt, S. Krezoski and D. Petering, Biochem. Biophys. Res. Commun., 1985, 130, 786–792 CrossRef CAS PubMed.
- M. Morelock, T. Cormier and G. Tolman, Inorg. Chem., 1988, 27, 3137–3140 CrossRef CAS.
- W. B. Jones, T. E. Elgren, M. M. Morelock, R. Elder and D. E. Wilcox, Inorg. Chem., 1994, 33, 5571–5578 CrossRef CAS.
- C. Acharya and C. A. Blindauer, Inorg. Chem., 2016, 55, 1505–1515 CrossRef CAS PubMed.
- S. N. A. Abdullah, S. Cheah and D. J. Murphy, Plant Physiol. Biochem., 2002, 40, 255–263 CrossRef CAS.
- A. Krezel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS PubMed.
- N. J. Pace and E. Weerapana, Biomolecules, 2014, 4, 419–434 CrossRef PubMed.
- D. W. Christianson and C. A. Fierke, Acc. Chem. Res., 1996, 29, 331–339 CrossRef CAS.
- P. M. Gehrig, C. You, R. Dallinger, C. Gruber, M. Brouwer, J. H. R. Kagi and P. E. Hunziker, Protein Sci., 2000, 9, 395–402 CrossRef CAS PubMed.
- J. Byrd and D. R. Winge, Arch. Biochem. Biophys., 1986, 250, 233–237 CrossRef CAS PubMed.
- G. W. Irvine and M. J. Stillman, Biochem. Biophys. Res. Commun., 2013, 441, 208–213 CrossRef CAS PubMed.
- R. A. Himes, G. Y. Park, A. N. Barry, N. J. Blackburn and K. D. Karlin, J. Am. Chem. Soc., 2007, 129, 5352–5353 CrossRef CAS PubMed.
- R. Pufahl, C. Singer, K. Peariso, S.-J. Lin, P. Schmidt, C. Fahrni, V. C. Culotta, J. Penner-Hahn and T. O'halloran, Science, 1997, 278, 853–856 CrossRef CAS PubMed.
- V. Martin-Diaconescu, K. N. Chacón, M. U. Delgado-Jaime, D. Sokaras, T.-C. Weng, S. DeBeer and N. J. Blackburn, Inorg. Chem., 2016, 55, 3431–3439 CrossRef CAS PubMed.
- E. W. Dahl and N. K. Szymczak, Angew. Chem., Int. Ed., 2016, 55, 3101–3105 CrossRef CAS PubMed.
- D. L. Pountney, I. Schauwecker, J. Zarn and M. Vasak, Biochemistry, 1994, 33, 9699–9705 CrossRef CAS PubMed.
- B. Roschitzki and M. Vašák, Biochemistry, 2003, 42, 9822–9828 CrossRef CAS PubMed.
- M. Vaher, N. Romero-Isart, M. Vašák and P. Palumaa, J. Inorg. Biochem., 2001, 83, 1–6 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |