
Sensitive determination of chlorogenic acid in pharmaceutical products based on the decoration of 3D macroporous carbon with Au nanoparticles via polyoxometalates
Tongrui
Zhang
,
Mengjun
Liu
,
Qianqian
Zhang
,
Yeyu
Wang
,
Xiangyi
Kong
,
Lei
Wang
,
Huan
Wang
* and
Yufan
Zhang
 *
*
Key Laboratory of Analytical Science and Technology of Hebei Province, College of Chemistry and Environmental Science, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis, Ministry of Education, Hebei University, 071002 Baoding, P R China. E-mail: zyf@hbu.edu.cn; Fax: +86 0312 5079403; Tel: +86 0312 5079403
First published on 31st May 2017
Abstract
A simple and sensitive electrochemical sensor is constructed for the detection of chlorogenic acid (CGA) based on Au nanoparticles (NPs)/polyoxometalates/3D macroporous carbon (Au-POMs-MPC). Serving as both a reducing and stabilizing agent, the Keggin-type POM, H3PW12O40, is used for the synthesis of stable colloidal Au NPs and then used to link them to MPC at a mild temperature. Because of the unique structural properties and synergetic catalytic effect, Au-POMs-MPC can be developed as an effective sensing platform for the detection of CGA, which showed high activity and excellent analytical performance towards CGA, such as a wide linear range of 2.28 nM–3.24 μM, a high sensitivity of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 554.71 μA mM−1, and a low limit of detection of 2.15 nM. Importantly, the successfully fabricated Au-POMs-MPC device accurately measured the amount of CGA in pharmaceutical samples.
554.71 μA mM−1, and a low limit of detection of 2.15 nM. Importantly, the successfully fabricated Au-POMs-MPC device accurately measured the amount of CGA in pharmaceutical samples.
1. Introduction
CGA is a natural phenolic product metabolized by plants and known as a defence compound against microorganisms and insects.1 It is a prominent polyphenol compound found in considerable amounts in many fruits and vegetables of the human diet (such as coffee, tea, berry fruits, pome fruits, grapes, carrots, tomatoes, potatoes, etc.).2,3 Studies on the health benefits associated with CGA revealed some biological properties such as having anti-oxidant, anti-bacterial and anti-inflammatory activity, being capable of scavenging free radicals and suppressing mutations and having anti-tumor activity.4,5 Additionally, clinical investigations implied that the consumption of CGA can reduce blood sugar levels and potentially exert an anti-diabetic effect, and also have anti-hypertension and anti-obesity effects.6–8 Consequently, the development of reliable methods for the determination of CGA is of particular significance. A number of methods have been employed for the measurement of CGA, such as high performance-liquid chromatography,9–11 capillary electrophoresis,12–14 gas chromatography,15,16 chemiluminescence,17 UV-Vis spectrometry,18,19 infrared spectroscopy with Fourier transformation,20,21 and NMR spectroscopy.22 Commonly, these techniques are costly, complicated, time consuming and operated by highly skilled technicians. In contrast, electrochemical methods can provide simple, rapid, and selective detection with low-cost systems.23–26With the ever increasing demand for electrochemical sensors, the development of efficient and high catalytic activity electrocatalysts has been attracting increasingly strong attention. Gold nanoparticles (Au NPs), a type of noble-metal based electrocatalyst, have attracted increasing interest in bioassays for their unique optical and electrical properties, such as easily controllable size distribution, comparative stability, and friendly biocompatibility with biomolecules.27–34
Recently, the synergetic effect on heterogeneous composite catalysts has gained great attention because the combination of the catalytic component and the support materials can sometimes endow the composite catalysts with unexpectedly improved catalytic properties.35–37 One of the most effective electrocatalysts are noble-metal/carbon composites. Carbon-based materials may offer a platform for supporting noble-metal NPs to form novel hybrid nanostructures with synergetic effects. The combination of noble-metal NPs and carbon materials is of special interest; it is known to show an obviously enhanced electrocatalytic activity.
3D macroporous carbon (MPC) is one of the most promising sources for electrochemical applications among the various types of carbon materials. MPC exhibits a uniform tailored and unique macroporous structure, high specific surface area, large pore volume, excellent conductivity, good thermal stability and chemical inertness, which makes it suitable for application in electrocatalysis and in the design of electrochemical sensors.38–42 In particular, the favourable chemical and electronic interaction between MPC and the nanoparticles leads to a synergistic enhancement of the electrocatalytic activity.
Considering the excellent versatile properties of both Au and MPC, the major objective of this work is to present the synthesis of Au-MPC composite materials and their electrochemical applications. The properties of Au NPs are significantly different compared with those of their bulk materials and can be tuned by their composition, aggregation state, size, shape and local surroundings. A large variety of conditions and protocols were followed for the synthesis of the Au NPs. However, as most Au NPs are synthesized in an organic environment, and require a specific high temperature and long reaction time and the use of the highly hazardous hydrazine as the reductant, the exploration of the preparation of Au NPs under environmentally friendly conditions has been attracting increasing attention.
Polyoxometalates (POMs) are polyatomic anionic ion clusters composed of d-block transition metal-oxides, and they exhibit a wide range of structural, redox, and photochemical characteristics and readily participate in reversible multielectron redox transformations. In reduced forms, due to their electron and proton transfer and/or storage abilities they may act as efficient donors or acceptors of several electrons without structural changes.43,44 POMs also have been shown to serve as reducing and capping agents for metal nanostructures. In general, POM-passivated metal NPs have been prepared through ligand place-exchange reactions, reducing the metal precursors in the presence of the POMs, or reducing the metal precursors using reduced POMs as the reducing agents.45,46
In the current study, relatively uniform Au NPs attached on a MPC surface have been prepared through a facile and green method using POMs as both the reductant and the bridging molecules. The as-prepared novel tri-component Au-POMs-MPC nanohybrids extend the applications of support materials and provide new features regarding the electrocatalytic activities. A new type of Au-POMs-MPC nanocomposite modified electrode has been fabricated and employed as an excellent electrochemical sensor of CGA.
2. Experimental
2.1. Chemical reagents
CGA, HAuCl4·3H2O, the POMs, isopropanol, and N,N′-dimethylformamide (DMF, HPLC grade) were used as purchased from MACKLIN Reagent Co. Ltd. The 0.1 M phosphate buffer solution (PBS pH 7.0), which was made up from NaH2PO4, Na2HPO4, and H3PO4, was employed as a supporting electrolyte. All other reagents were of analytical grade, and all solutions were prepared using double distilled water.2.2. Instrumentation
All of the electrochemical experiments were performed with an Autolab Electrochemistry Workstation (PGSTAT 302N, Metrohm, Switzerland). Electrochemical impedance spectroscopy (EIS) was conducted using the Autolab electrochemical analyzer in a 0.1 M KCl solution containing 5.0 mM K3Fe(CN)6/K4Fe(CN)6, from 0.1 Hz to 10.0 kHz. Scanning electron microscopy (SEM) images were obtained with a Philips XL-30 ESEM, operating at 3.0 kV. Transmission electron microscopy (TEM) images and energy-dispersive X-ray (EDX) spectra were obtained using a JEM-2100F transmission electron microscope JEOL (Japan) operating at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed with a thermo ESCA LAB spectrometer (USA). X-Ray diffraction (XRD) patterns were obtained on an X-ray D/max-2200vpc (Rigaku Corporation, Japan) instrument operated at 40 kV and 20 mA using Cu Kα radiation (k = 0.15406 nm). A conventional three electrode cell was used; the working electrode was a glassy carbon electrode (GCE) or the modified electrode; a platinum electrode was used as the counter electrode whereas an Ag/AgCl (in saturated KCl solution) electrode served as a reference electrode.2.3. Synthesis of tri-component nanohybrids of Au-POMs-MPC
The SiO2 template was prepared by the typical Stöber's method.47 Carbon was introduced into the interstices of the template using the modified method of Jun et al.48 For Au-POMs-MPC, firstly, 1.5 mL of the POMs (10 mg mL−1) was added to a quartz container and mixed with 30 μL of isopropanol; the mixed solution was irradiated under ultra-violet light (500 W) for 30 min and the color of the solution became blue. Secondly, HAuCl4·3H2O (12 μL, 100 mg mL−1) and a MPC suspension (0.2 mL, 5 mg mL−1) were added to the solution of the reduced POMs, with 5 minutes of stirring. Finally, the resulting Au-POMs-MPC nanomaterial was isolated, thoroughly washed with doubly distilled water, and then dried under vacuum overnight. The whole reaction was performed at room temperature and under ambient pressure. An illustration of the preparation of Au-POMs-MPC is presented in Scheme 1.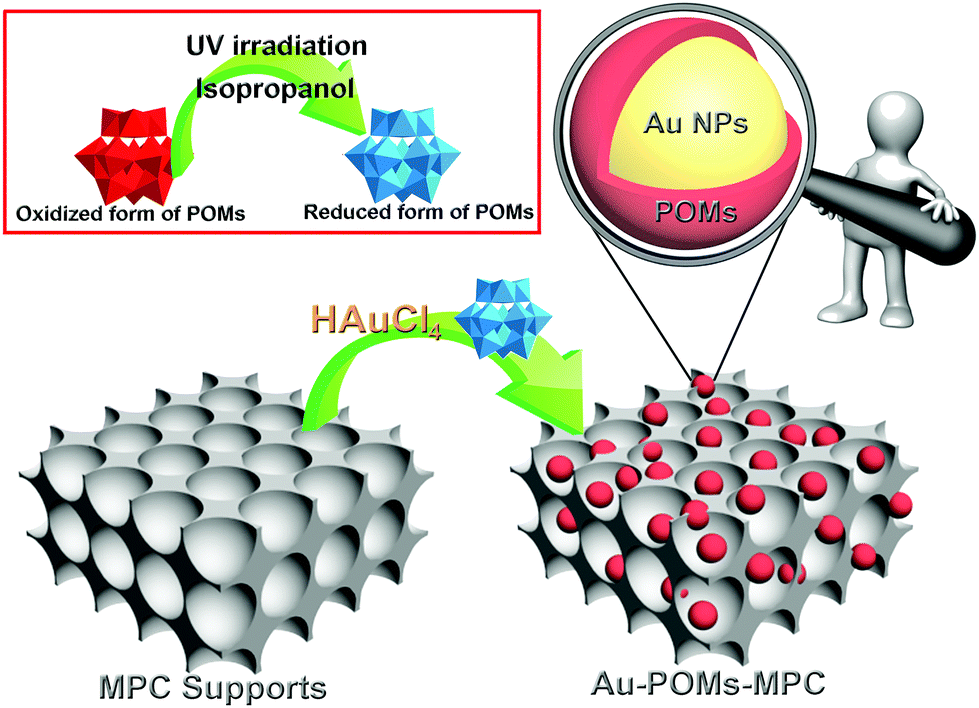 | ||
| Scheme 1 Illustration of the preparation of Au-POMs-MPC. | ||
2.4. Preparation of the modified electrodes
Prior to the modification, the GCE (model CHI104, 3 mm diameter) was polished before each experiment with 1, 0.3 and 0.05 μm alumina powder, respectively, rinsed thoroughly with double distilled water between each polishing step, and then sonicated successively in 1:1 nitric acid, absolute alcohol, and double distilled water. The cleaned electrode was dried with a nitrogen stream for the next modification. To prepare the modified electrodes, 5 mg of the electrode materials was dispersed into 1 mL of DMF to give a homogeneous suspension upon bath sonication. 5 μL of the suspension was dropped onto the GCE and the electrode was then dried at room temperature.
2.5. Pharmaceutical real sample solution preparation
The tablets of CGA were finely powdered in a mortar with a pestle. Calculated amounts of the CGA tablets required were transferred into 25 mL volumetric flasks and were dissolved in absolute alcohol. The solutions were filtered and suitable aliquots of the clear filtrates were collected and stored in the refrigerator for further use.3. Results and discussion
3.1. Characterization of the as-prepared samples
The morphologies of MPC and Au-POMs-MPC were initially characterized by SEM and TEM analysis. The as-prepared pure MPC is seen as a well-defined interconnected macroporous nanostructure (Fig. 1A and B) with a pore size of about 110 nm. A typical TEM image of Au-POMs-MPC is presented in Fig. 1C, which shows that the Au NPs are uniformly dispersed on the surface of MPC. Meanwhile, the lattice fringe shows an interplanar spacing of 0.223 nm for the (111) face-centered cubic structure of the Au NPs by counting from the high-resolution TEM image (Fig. 1D). Fig. 1E illustrates the corresponding histogram of the particle size distribution for the AuNPs by counting from the micrograph from a statistical study of 100 NPs. One can see that the average diameter is about 5.52 nm for Au-POMs-MPC. The composition of the as-synthesized Au-POMs-MPC was confirmed by EDX spectroscopy, as shown in Fig. 1F. It shows the peaks corresponding to the C, O, Au, and W elements (the strong peaks of Cu are from the copper grid), therefore confirming the existence of the Au-POMs in the Au-POMs-MPC nanohybrids.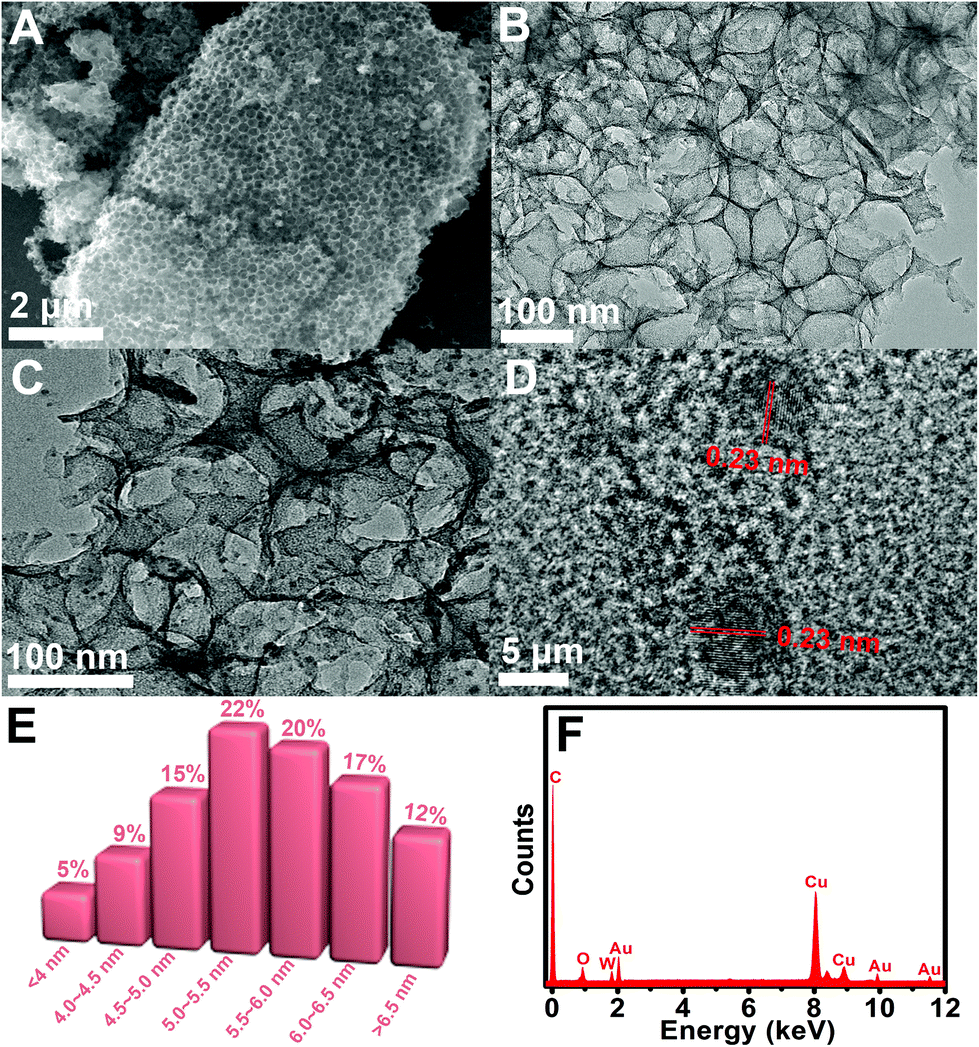 | ||
| Fig. 1 SEM image of MPC (A). TEM images of MPC (B) and Au-POMs-MPC (C). HRTEM image of Au-POMs-MPC (D). Columnar distribution of Au NPs size for Au-POMs-MPC (E). EDX spectra of Au-POMs-MPC (F). | ||
The nanohybrids were further characterized by XPS analysis and XRD patterns. The XPS spectra were used to identify the chemical composition and the binding energy of Au-POMs-MPC (Fig. 2A). The diffraction peak signals of W 4f, Au 4f, W 4d, C 1s, and O 1s confirmed the formation of the Au-POMs-MPC nanocomposite. The presence of W was also detected by XPS, despite the thorough washing of the samples. As shown in Fig. 2B, there is a W 4f5/2 and a W 4f7/2 doublet with the binding energies of 36.3 and 38.6 eV, respectively, which indicates that W is in its full oxidation form in the POMs when assembling in the nanohybrids. With the charge effect corrected by fixing the photoelectric 1s peak of carbon at 284.5 eV, the Au 4f7/2 level is located at 83.7 eV and the Au 4f5/2 level at 87.4 eV (Fig. 2C). These values suggest unambiguously that Au is present only in the metallic form, indicating the formation of the Au NPs on the surface of MPC.30,49 As shown in the XRD spectra of Au-POMs-MPC (Fig. 2D), the characteristic diffraction peaks at 38.5°, 44.5°, 77.4° and 82.8° can be indexed to the (111), (200), (311) and (222) planes of face-centered cubic crystalline Au, respectively. Moreover, diffraction peaks from the POMs were also observed. Their presence shows the unquestionable evidence of the formation of the tri-component Au-POMs-MPC nanohybrids.
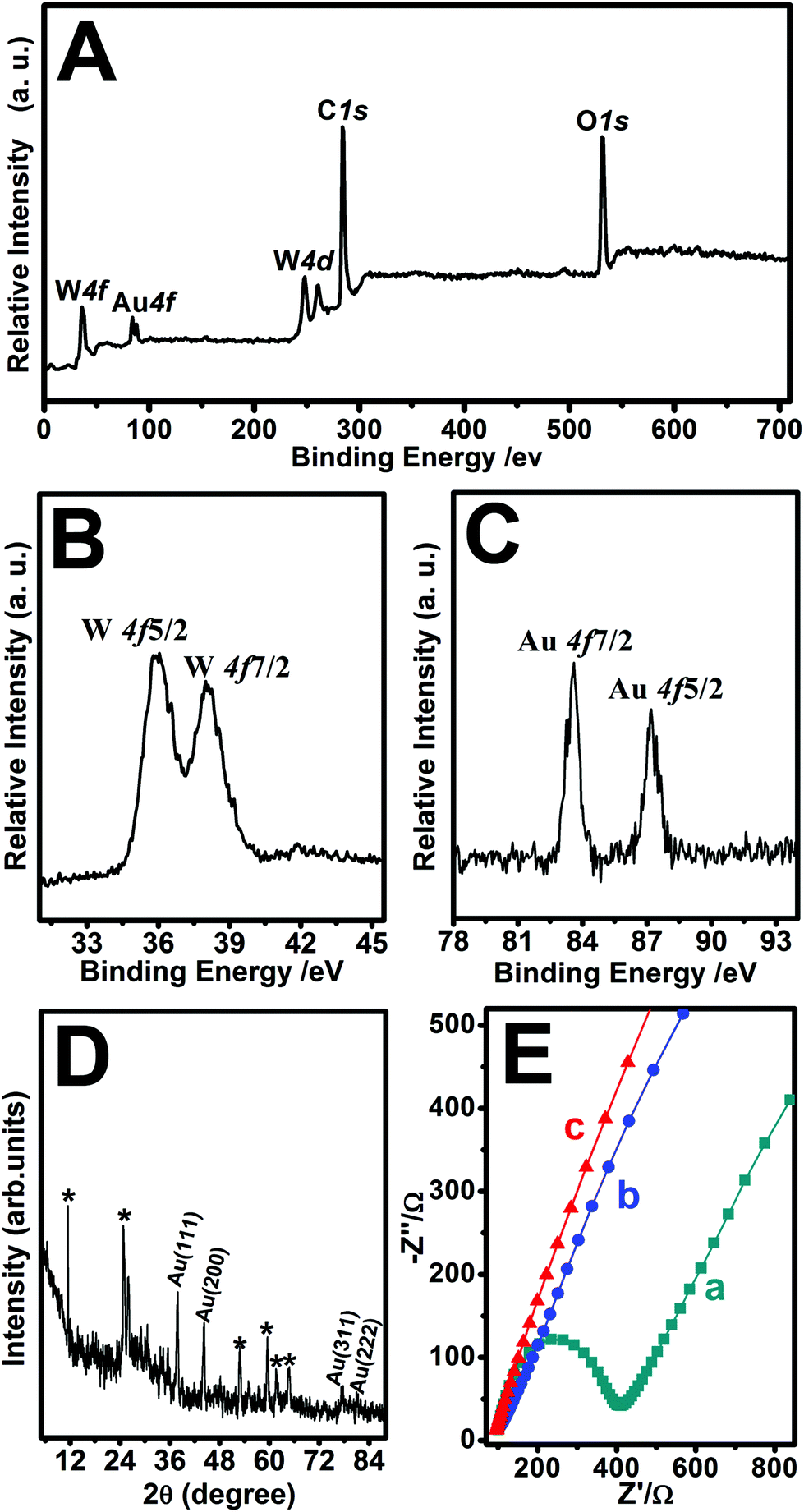 | ||
| Fig. 2 The wide XPS survey spectrum of Au-POMs-MPC (A). High-resolution XPS spectra of W 4f (B) and Au 4f (C) of Au-POMs-MPC. XRD patterns of Au-POMs-MPC (D). EIS of the bare GCE (a), MPC–GCE (b), and Au-POMs-MPC–GCE (c) in a 0.1 M KCl solution containing 0.5 mM K3Fe(CN)6/K4Fe(CN)6 from 0.1 Hz to 10.0 kHz (E). | ||
3.2. Electrochemical behaviors of CGA and its detection
EIS experiments are usually preferred for the investigation of the changes on the electrode surface. Fig. 2E shows the Nyquist diagrams of the EIS for the bare GCE, MPC–GCE, and Au-POMs-MPC–GCE. The bare GCE exhibits a semicircle part at high frequency, which indicates the highest resistance. After being modified with MPC or Au-POMs-MPC, the diameter of the semicircle markedly decreases, indicating that MPC can form good electron pathways between the electrode and electrolyte and can be expected to be a good electrochemical platform.The electrocatalytic activity of different electrodes toward CGA oxidation was studied using typical CV experiments. For the bare GCE (Fig. 3A), there is a small current increase after the addition of a 50 μM CGA solution, reflecting its limited electrocatalytic activity for CGA oxidation. The MPC–GCE exhibits an increase in the catalytic current for the oxidation of CGA (Fig. 3B) compared with the bare GCE. However, the highest oxidation current increase and the lowest onset potential are observed for the Au-POMs-MPC–GCE (Fig. 3C), demonstrating its superior electrocatalytic activity and kinetics toward CGA oxidation compared with the other electrodes. The reproducibility of the catalytic current (Fig. 3D) and onset potential (Fig. 3E) for CGA at different electrodes was measured.
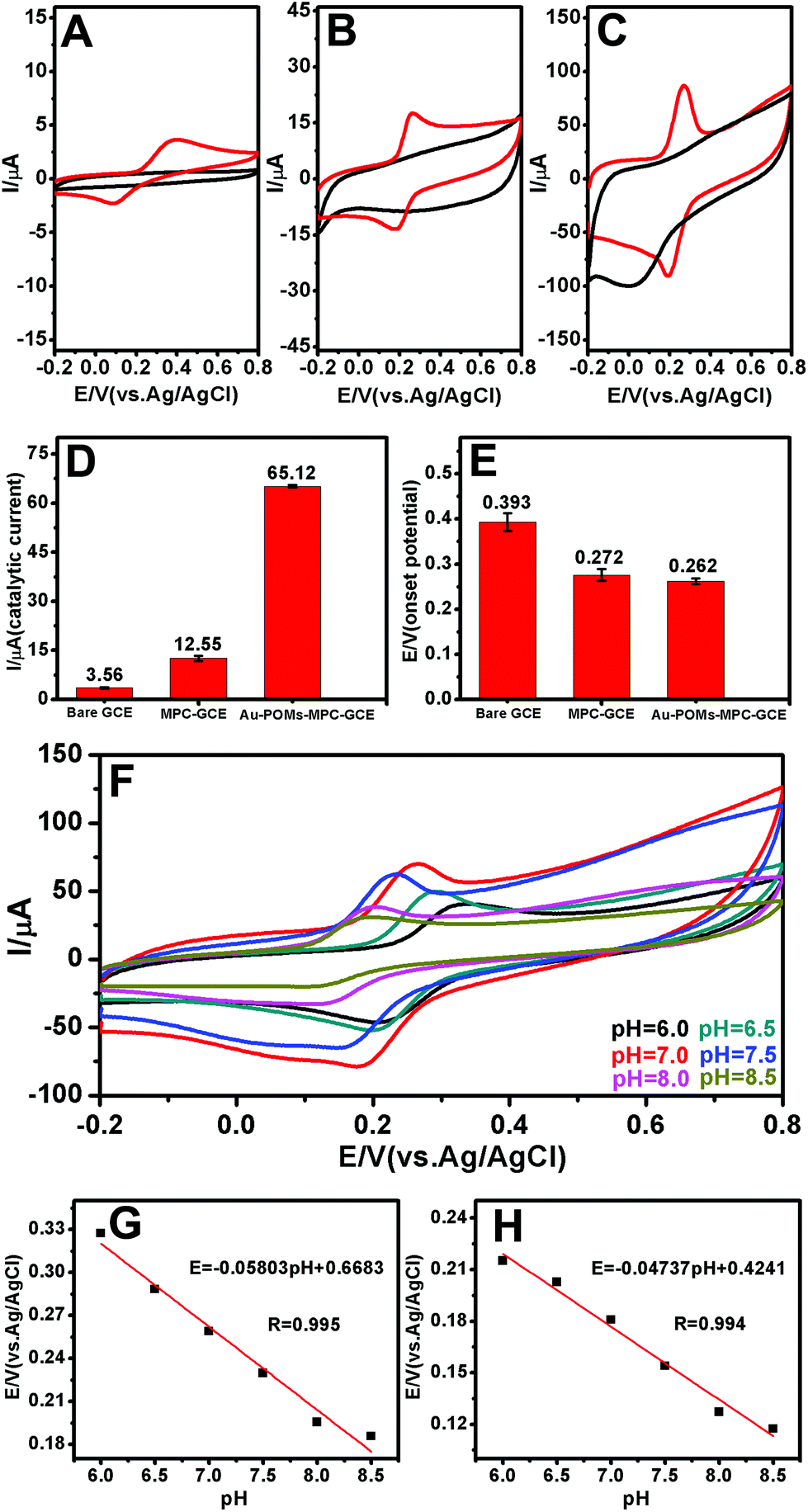 | ||
| Fig. 3 CVs of the bare GCE (A), MPC–GCE (B), and Au-POMs-MPC–GCE (C) in the absence (black line) and presence (red line) of CGA. Scan rate: 50 mV s−1; pH = 7.0. Reproducibility of the catalytic current (D) and onset potential (E) for CGA with different electrodes. CVs obtained for the Au-POMs-MPC–GCE in the presence of CGA at various pH values from 6.0 to 8.5 (F). Effect of pH values on the anodic (G) and cathode (H) peak potential for CGA. | ||
The effect of the pH values on the peak potential of CGA was also examined (Fig. 3F). The peak potential of CGA shifts towards the negative direction with the pH values varying from 6.0 to 8.5. Fig. 3G and H show the plots of the anode and cathode peak potential against the solution pH, respectively. The linear equation for the anodic peak was Epa (V) = −0.05803 pH + 0.6683 and that for the cathode peak was Epc (V) = −0.04737 pH + 0.4241. The values of ΔE/ΔpH are 58.03 and 47.37 mV, which are close to the theoretical slope of a Nernst plot (−59 mV per pH unit at 25 °C). The results indicate that the numbers of the transferred protons and electrons in the redox processes of the Au-POMs-MPC–GCE are equal (2e−/2H+), a finding which is in agreement with those reported in the literature.49–52
The electrochemical behaviors of CGA on the Au-POMs-MPC–GCE with different scan rates were examined using CV (Fig. 4A). A pair of redox waves was observed for CGA, and the oxidation (Fig. 4B) and reduction (Fig. 4C) peak currents increased linearly with the square root of the scan rate in the range of 5–300 mV s−1, implying that the electrochemical kinetics is a diffusion-controlled process.
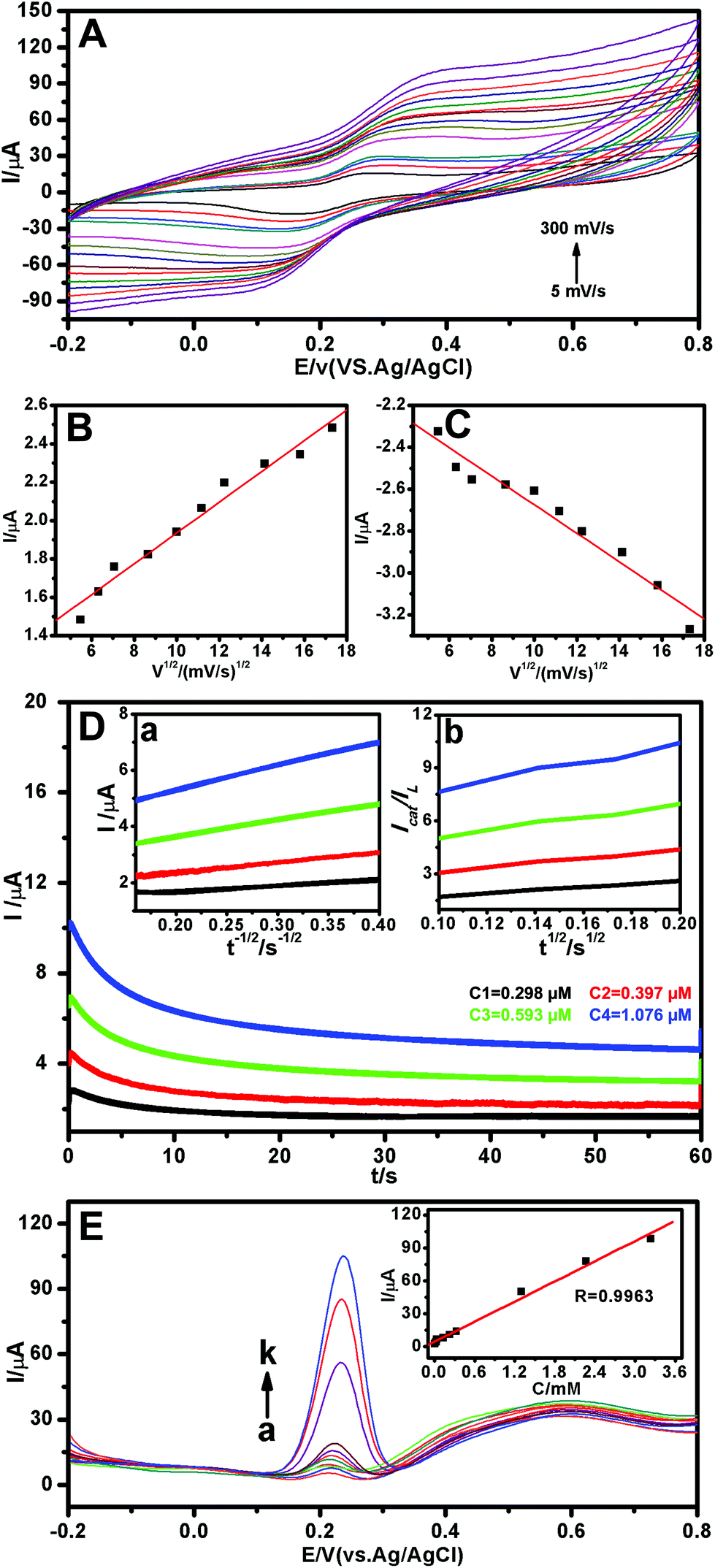 | ||
| Fig. 4 CVs of the Au-POMs-MPC–GCE in 0.1 M PBS (pH 7.0) in the presence of CGA at different scan rates: 5, 10, 15, 20, 30, 40, 50, 75, 100, 125, 150, 200, 250 and 300 mV s−1 (A). The linear relationship between the anodic (B) and cathode (C) peak currents and the square roots of the scan rates. Chronoamperograms of Au-POMs-MPC–GCE in 0.1 M PBS (pH 7.0) solution in the presence (curve C1→C4) of various concentrations of CGA (D). The plots of I versus t−1/2 (inset a) and the slope of the Icat/ILversus t1/2 plots (inset b) obtained from the chronoamperograms, respectively. DPV curves of CGA in PBS (0.1 M, pH 7.0) with the Au-POMs-MPC–GCE (E). The linear dependence of the current response with different concentrations of CGA (inset of E). | ||
The catalytic oxidation of CGA by the Au-POMs-MPC–GCE was also studied by chronoamperometry. The chronoamperograms were obtained for a series of CGA solutions with various concentrations, as shown in Fig. 4D. Inset (a) of Fig. 4D shows the linear relationship of the experimental plots I versus t−1/2 for different concentrations of CGA at intermediate times. The catalytic current is dominated by the rate of the electrocatalytic oxidation of CGA. The diffusion coefficient D could be obtained according to the Cottrell equation:
| I = nFAD1/2cπ−1/2t−1/2 |
where IL is the diffusion limited current in the absence of CGA, Icat is the catalytic current in the presence of CGA, c is the bulk concentration and t is time elapsed (s). From the slope of the Icat/ILversus t1/2 plot, as shown in inset (b) of Fig. 4D, the kcat value was calculated to be 8.11 × 104 M−1 s−1 for CGA.
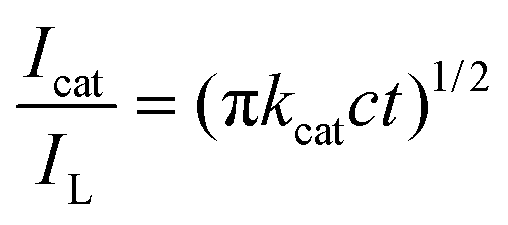
The feasibility of the proposed electrochemical sensor for target CGA detection was verified by the differential pulse voltammetry (DPV) method. Clearly, a series of the DPV curves were obtained from different concentrations of CGA (Fig. 4E). A plot of I versus concentration of CGA with a linear relationship is shown in the inset of Fig. 4E. The CGA sensor displays a linear range of 2.28 nM–3.24 μM (R = 0.9963) with a sensitivity of 30554.71 μA mM−1. The limit of detection is calculated to be 2.15 nM (3N/S), where N is equal to the standard deviation of the background solution without target molecules and S is equal to the sensitivity. The reproducibility of the sensor was also investigated by the DPV method. The RSD of the current signal for 0.5 μM CGA was less than 0.3% for five measurements using the same electrode. After being stored at 4 °C for two weeks, a 4.2% current loss with the Au-POMs-MPC–GCE was obtained by the DPV response of 0.5 μM CGA. A detailed comparison of the CGA detection performance using different CGA electrochemical sensors is summarized in Table 1.
| Working electrode | Potential (V) | Linear range (μM) | Sensitivity (μA mM−1) | Limit of detection (nM) | Ref. |
|---|---|---|---|---|---|
| a A molecularly imprinted siloxane (MIS) film modified Au bare electrode. b Pencil graphite electrodes. c Poly(aminosulfonic acid) modified glassy carbon electrode. d Boron-doped diamond electrode. e Boron-doped diamond electrode. f One of the carbon working electrodes was modified with platinum nanoparticles, reduced graphene oxide and laccase. g Glassy carbon electrode. h Saturated calomel electrode. | |||||
| MIS/Aua | 0.430 (SCEh) | 0.50–14.00 | 46.56 | 148.00 | 1 |
| PGEb | 0.550 (Ag/AgCl) | 0.10–500.00 | 48.94 | 71.40 | 2 |
| PASA/GCEc | 0.480 (Ag/AgCl) | 0.40–1.20 | 2.27 | 40.00 | 6 |
| BDDd | 0.530 (Ag/AgCl) | 5.63–146.25 | 7.95 | 1260.00 | 53 |
| BDDe | 0.490 (Ag/AgCl) | 0.70–11.25 | 8.15 | 137.80 | 54 |
| C-SPE/Pt-NPs/RGO/laccf | — | 2.91–26.47 | 20.00 | 2670.00 | 55 |
| GCEg | 0.187 (Ag/AgCl) | 5.00–50.00 | 49.08 | 1200.00 | 56 |
| Au-POMs-MPC–GCE | 0.262 (Ag/AgCl) | 2.28 × 10−3–3.24 | 30554.71 |
2.15 | This work |
The proposed method was successfully applied to the determination of CGA in a tablet. The samples were treated according to the procedures described in section 2.5. The concentrations of CGA were measured by employing the calibration curve obtained under the same experimental conditions. The results given in Table 2 indicate that the novel electrode is very useful for the rapid and accurate determination of CGA in pharmaceutical samples.
| Samples | UV-vis spectra (μg mL−1) | DPV (μg mL−1) |
|---|---|---|
| Tablet 1 | 1.51 ± 0.001 | 1.51 ± 0.001 |
| Tablet 2 | 1.36 ± 0.002 | 1.22 ± 0.002 |
4. Conclusions
In conclusion, MPC decorated with Au NPs relying on the presence of POMs has been reported. The preparation of the novel Au-POMs-MPC composite used a facile, green, and one-pot synthesis method. The POMs acted as a propelling agent for the NPs toward MPC at a mild temperature with no need for another contributing agent. This strategy led to the strong sticking of the NPs to MPC in a well-distributed manner. Au-POMs-MPC could offer a favourable microenvironment for transferring species in solution, and would also be beneficial for accelerating electron transfer between the electrode and species in solution. A sensitive electrochemical sensor for CGA was developed based on Au-POMs-MPC, which showed a wide linear range, low detection limit, high sensitivity, and good stability. In fact, the successful fabrication of Au-POMs-MPC holds great promise for the design of electrochemical sensors, and is a promising way to promote the development of new electrode materials.Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (No. 21575021 and No. 21505031), the Natural Science Foundation of Hebei Province (No. B2016201018), and the science technology research and development guidance programme project of Baoding City (No. 15ZG006 and 16ZG033).References
- W. d. J. R. Santos, M. Santhiago, I. V. P. Yoshida and L. T. Kubota, Anal. Chim. Acta, 2011, 695, 44–50 CrossRef CAS PubMed.
- I. G. David, D. E. Popa, M. Buleandra, Z. Moldovan, E. E. Iorgulescu and I. A. Badea, Anal. Methods, 2016, 8, 6537–6544 RSC.
- M. N. Clifford, J. Sci. Food Agric., 1999, 79, 362–372 CrossRef CAS.
- K. Koirala, F. B. Sevilla III and J. H. Santos, Sens. Actuators, B, 2016, 222, 391–396 CrossRef CAS.
- M. Lepelley, G. Cheminade, N. Tremillon, A. Simkin, V. Caillet and J. McCarthy, Plant Sci., 2007, 172, 978–996 CrossRef CAS.
- M. Chao and X. Ma, J. Food Drug Anal., 2014, 22, 512–519 CrossRef CAS.
- A.-S. Cho, S.-M. Jeon, M.-J. Kim, J. Yeo, K.-I. Seo, M.-S. Choi and M.-K. Lee, Food Chem. Toxicol., 2010, 48, 937–943 CrossRef CAS PubMed.
- Y. Zhao, J. Wang, O. Ballevre, H. Luo and W. Zhang, Hypertens. Res., 2012, 35, 370–374 CrossRef CAS PubMed.
- I. N. Urakova, O. N. Pozharitskaya, A. N. Shikov, V. M. Kosman and V. G. Makarov, J. Sep. Sci., 2008, 31, 237–241 CrossRef CAS PubMed.
- L. Yang, Q. Yan, H. Mo, X. Li and Q. Wang, J. Chil. Chem. Soc., 2012, 57, 1361–1363 CrossRef CAS.
- L. V. de Melo and A. C. H. F. Sawaya, Rev. Bras. Farmacogn., 2015, 25, 105–110 CrossRef CAS.
- H.-L. Jiang, Y.-Z. He, H.-Z. Zhao and Y.-Y. Hu, Anal. Chim. Acta, 2004, 512, 111–119 CrossRef CAS.
- Z. Li, D. Huang, Z. Tang, C. Deng and X. Zhang, Talanta, 2010, 82, 1181–1185 CrossRef CAS PubMed.
- X. Yao and G. Chen, Anal. Bioanal. Chem., 2007, 388, 475–481 CrossRef CAS PubMed.
- Z. Füzfai and I. Molnár-Perl, J. Chromatogr., A, 2007, 1149, 88–101 CrossRef PubMed.
- K. Shrivas and H.-F. Wu, J. Chromatogr., A, 2007, 1170, 9–14 CrossRef CAS PubMed.
- X. Wang, J. Wang and N. Yang, Food Chem., 2007, 102, 422–426 CrossRef CAS.
- A. Belay, K. Ture, M. Redi and A. Asfaw, Food Chem., 2008, 108, 310–315 CrossRef CAS.
- I. Hečimović, A. Belščak-Cvitanović, D. Horžić and D. Komes, Food Chem., 2011, 129, 991–1000 CrossRef PubMed.
- Z. Wu, B. Xu, M. Du, C. Sui, X. Shi and Y. Qiao, J. Pharm. Biomed. Anal., 2012, 62, 1–6 CrossRef CAS PubMed.
- G. Shi, L. Rao, Q. Xie, J. Li, B. Li and X. Xiong, Vib. Spectrosc., 2010, 53, 289–295 CrossRef CAS.
- G. del Campo, I. Berregi, R. Caracena and J. Zuriarrain, Talanta, 2010, 81, 367–371 CrossRef CAS PubMed.
- X. Du, D. Jiang, N. Hao, J. Qian, L. Dai, L. Zhou, J. Hu and K. Wang, Anal. Chem., 2016, 88, 9622–9629 CrossRef CAS PubMed.
- X. Du, L. Dai, D. Jiang, H. Li, N. Hao, T. You, H. Mao and K. Wang, Biosens. Bioelectron., 2017, 91, 706–713 CrossRef CAS PubMed.
- J. Qian, K. Wang, Y. Jin, X. Yang, L. Jiang, Y. Yan, X. Dong, H. Li and B. Qiu, Biosens. Bioelectron., 2014, 57, 149–156 CrossRef CAS PubMed.
- L. Jiang, J. Qian, X. Yang, Y. Yan, Q. Liu, K. Wang and K. Wang, Anal. Chim. Acta, 2014, 806, 128–135 CrossRef CAS PubMed.
- A. N. Shipway, M. Lahav and I. Willner, Adv. Mater., 2000, 12, 993–998 CrossRef CAS.
- G. Lai, H. Zhang, J. Yong and A. Yu, Biosens. Bioelectron., 2013, 47, 178–183 CrossRef CAS PubMed.
- J.-W. Wu, C.-H. Wang, Y.-C. Wang and J.-K. Chang, Biosens. Bioelectron., 2013, 46, 30–36 CrossRef CAS PubMed.
- Y. Zhang, X. Bo, A. Nsabimana, A. Munyentwali, C. Han, M. Li and L. Guo, Biosens. Bioelectron., 2015, 66, 191–197 CrossRef CAS PubMed.
- H. Wang, S. Yao, Y. Liu, S. Wei, J. Su and G. Hu, Biosens. Bioelectron., 2017, 87, 417–421 CrossRef CAS PubMed.
- H. Fu, L. Zhang, Y. Wang, S. Chen and Y. Wan, J. Catal., 2016, 344, 313–324 CrossRef CAS.
- W. Lin, Y. Lu and Y. Hsu, J. Colloid Interface Sci., 2014, 418, 87–94 CrossRef CAS PubMed.
- L. Bi, Y. Rao, Q. Tao, J. Dong, T. Su, F. Liu and W. Qian, Biosens. Bioelectron., 2013, 43, 193–199 CrossRef CAS PubMed.
- H.-Q. Dong, Y.-Y. Chen, M. Han, S.-L. Li, J. Zhang, J.-S. Li, Y.-Q. Lan, Z.-H. Dai and J.-C. Bao, J. Mater. Chem. A, 2014, 2, 1272–1276 CAS.
- Z. Li, S. Ji, B. G. Pollet and P. K. Shen, Chem. Commun., 2014, 50, 566–568 RSC.
- X. Xie, Y. Xue, L. Li, S. Chen, Y. Nie, W. Ding and Z. Wei, Nanoscale, 2014, 6, 11035–11040 RSC.
- Y. Zhang, X. Bo, A. Nsabimana, C. Han, M. Li and L. Guo, J. Mater. Chem. A, 2015, 3, 732–738 CAS.
- Y. Zhang, H. Wang, Q. Yao, F. Yan, C. Cui, M. Sun and H. Zhang, RSC Adv., 2016, 6, 39618–39626 RSC.
- Y. Song, X. Lu, Y. Li, Q. Guo, S. Chen, L. Mao, H. Hou and L. Wang, Anal. Chem., 2016, 88, 1371–1377 CrossRef CAS PubMed.
- L. M. Jin, F. He, W. L. Cai, J. X. Huang, B. H. Liu and Z. P. Li, J. Power Sources, 2016, 328, 536–542 CrossRef CAS.
- C. Yang, W. Li, Z. Yang, L. Gu and Y. Yu, Nano Energy, 2015, 18, 12–19 CrossRef CAS.
- S. Li, X. Yu, G. Zhang, Y. Ma, J. Yao and P. de Oliveira, Carbon, 2011, 49, 1906–1911 CrossRef CAS.
- R. Liu, S. Li, X. Yu, G. Zhang, S. Zhang, J. Yao, B. Keita, L. Nadjo and L. Zhi, Small, 2012, 8, 1398–1406 CrossRef CAS PubMed.
- D. Kim, J. H. Seog, M. Kim, M. Yang, E. Gillette, S. B. Lee and S. W. Han, Chem. – Eur. J., 2015, 21, 5387–5394 CrossRef CAS PubMed.
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- S. Guo, L. Xu, B. Xu, Z. Sun and L. Wang, Analyst, 2015, 140, 820–826 RSC.
- N. Karikalan, R. Karthik, S. Chen, M. Velmurugan and C. Karuppiah, J. Colloid Interface Sci., 2016, 483, 109–117 CrossRef CAS PubMed.
- E. Haghshenas, T. Madrakian and A. Afkhami, Mater. Sci. Eng., C, 2015, 57, 205–214 CrossRef CAS PubMed.
- T. Li, J. Xu, L. Zhao, S. Shen, M. Yuan, W. Liu, Q. Tu, R. Yu and J. Wang, Talanta, 2016, 159, 356–364 CrossRef CAS PubMed.
- Y. Yardım, E. Keskin and Z. Şentürk, Talanta, 2013, 116, 1010–1017 CrossRef PubMed.
- Y. Yardım, J. Food Sci., 2012, 77, C408–C413 CrossRef PubMed.
- I. Vasilescu, S. A. V. Eremia, R. Penu, C. Albu, A. Radoi, S. C. Litescu and G.-L. Radu, RSC Adv., 2015, 5, 261–268 RSC.
- I. Tomac and M. Seruga, J. Electrochem. Sci., 2016, 11, 2854–2876 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |