
Palladium-catalyzed C(sp3)–H arylation of lactic acid: efficient synthesis of chiral β-aryl-α-hydroxy acids†
Kai
Chen‡
ab,
Xin
Li‡
a,
Shuo-Qing
Zhang
a and
Bing-Feng
Shi
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: bfshi@zju.edu.cn
bDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA
First published on 7th December 2015
Abstract
A Pd-catalyzed arylation of lactic acid employing 8-aminoquinoline as the directing group has been reported. The protocol is found to be compatible with a broad range of synthetically useful functional groups, thus providing a practical route to chiral β-aryl-α-hydroxy acids. Further, the new reaction has also been applied to the synthesis of pharmaceutically important α-hydroxy acids, such as LY519818 and tesaglitazar.
 Bing-Feng Shi | Bing-Feng Shi received his B.S. degree from Nankai University in 2001 and a PhD degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. Following time as a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is on transition-metal-catalyzed C(sp3)–H functionalization and its application in the synthesis of biologically important small molecules. |
α-Hydroxy acids (AHAs) belong to an important class of α-substituted aliphatic acids and play a significant role as building blocks in conventional chemical synthesis.1 Besides, AHAs, especially the AHAs containing aryl groups at the β-position, are found to be significant structural motifs in numerous bioactive natural products and pharmaceutical molecules (Fig. 1). For example, Danshensu ((R)-2-hydroxy-3-(3,4-dihydroxyphenyl)propionic acid) is a ubiquitous structural motif in many bioactive natural compounds, such as (+)-lithospermic acid.2 Several β-aryl-AHA-based structures, including tesaglitazar, aleglitazar and saroglitazar, have been intensively investigated as potential agonists against peroxisome proliferator-activated receptors (PPARs) for the treatment of type II diabetes.3 Consequently, numerous methods, including asymmetric hydrogenation of enol ethers, asymmetric reduction or alkylation of α-ketoesters, asymmetric α-alkylation of glycolic acid derivatives, enantioselective Passerini reactions, and α-hydroxylation of esters, have been developed for the enantioselective synthesis of AHAs.4 However, these methods still have limitations, such as the employment of expensive chiral ligands or consumption of stoichiometric chiral auxiliaries, and the lack of generality for substrate scopes. Thus, the development of a generalizable and straightforward strategy to access chiral β-aryl-AHAs is highly desirable and remains a great challenge.
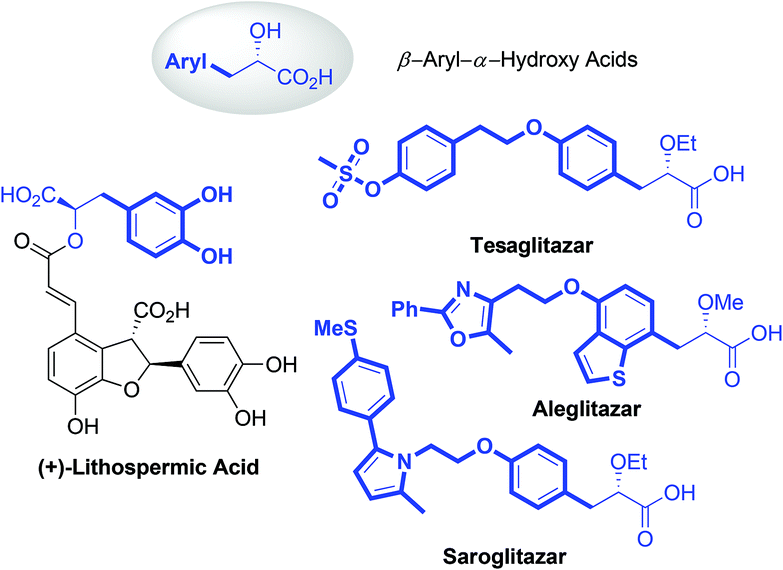 | ||
| Fig. 1 Natural products and pharmaceutical molecules containing α-hydroxy acids. | ||
The last decade has witnessed significant advances in Pd-catalyzed direct functionalization of unactivated C(sp3)–H bonds.5 Basically, two kinds of strategies have been developed and widely used for the efficient activation of C(sp3)–H bonds: (a) the strong coordinated bidentate auxiliaries first introduced by Daugulis in 2005,6,7 which have inspired the discovery of various bidentate auxiliaries;8 and (b) the weakly coordinating N-arylamide (CONHAr or CONHOMe) directing groups promoted by external ligands, which were first developed by Yu.9 In particular, these strategies have found wide applications in the functionalization of naturally occurring α-amino acids (AAs), one of the most abundant biomass-derived chemicals, providing powerful and straightforward methods for the synthesis of unnatural α-AAs.5a,b,e–g,9–11 However, the analogous functionalization of lactic acid remained undeveloped, despite its tremendous potential for synthetic and biological applications.12 So far, only one isolated example of C(sp3)–H arylation of lactic acid directed by 2-methylthioaniline auxiliary has been reported by Daugulis in 2010.6b More recently, the Baudoin group reported a Pd-catalyzed arylation of lactic acid using 2-pyridinylisopropyl (PIP) bidentate auxiliary in the total synthesis of aeruginosin marine natural products.13a We have also achieved the Pd-catalyzed alkylation of lactic acid with alkyl iodides.13b Based on these considerations and our previous work on Pd-catalyzed C(sp3)–H functionalization,14 we were eager to carry out a systematic investigation of the Pd-catalyzed C(sp3)–H arylation of lactic acid. Herein, we describe a Pd-catalyzed arylation of lactic acid for the synthesis of β-aryl-AHAs by employing 8-aminoquinoline (AQ) as the directing group. The protocol possesses good tolerance to various useful functional groups and the reaction can also be easily scaled up, which demonstrates the potential utility of the protocol. Moreover, these reactions also provide a practical access to different biologically important molecules, such as LY519818 and tesaglitazar, potential agonists against peroxisome proliferator-activated receptors (PPARs) for the treatment of type II diabetes.3
It can be expected that the subtle difference of α-substitutions between α-amino acids and α-hydroxy acids might lead to dramatic inconsistence in reactivity due to the following reasons: (a) the N-phthaloyl group that has been widely used in C–H functionalization of α-AAs is more likely to facilitate the desired cyclopalladation via the Thorpe–Ingold effect;14c,15 while the α-oxygen atom on lactic acid would probably have an adverse effect for the desired transformation, largely because of the tendency to trap the palladium catalyst by competitive coordination.16 (b) The α-hydroxyl group is known to be unstable under the catalytic system, and is susceptible to β-oxygen elimination to give the corresponding α,β-unsaturated amides after cyclopalladation. Therefore, the reaction conditions have to be carefully tuned to establish a new catalytic system to address these challenges. We first tested the feasibility of the arylation protocol for an alanine derivative using AgBF4 as the additive.14f However, the arylation of 1a with aryl iodide 2a only resulted in the elimination of MeOH, probably due to the strong Lewis acidity of AgBF4. Therefore, we turned our attention to the screening of various inorganic bases as additives; however, no satisfactory result was obtained (Table 1, entries 1–3). To maintain the high reactivity and suppress the side reaction of elimination, we reexamined other silver salts with weak Lewis acidity. After extensive screening, we found that the use of AgF afforded the desired product in 65% yield (entry 8). Further investigation of different solvents indicated that polar solvents, such as t-AmylOH, MeOH and DMF, were more suitable to this transformation (entries 9–12). To our delight, simply increasing the loading of AgF to 3 equivalents could significantly improve the yield (entry 13, 81%). Interestingly, no diarylation product has been observed in all the cases.
|
|
|||
|---|---|---|---|
| Entry | Additive (equiv.) | Solvent | Yield 3aa (%) |
| a Yields determined by 1H NMR using CH2Br2 as the internal standard. b Isolated yield. c 12 h. | |||
| 1 | Na2CO3 (1.5) | t-AmylOH | Trace |
| 2 | K2CO3 (1.5) | t-AmylOH | 17 |
| 3 | Cs2CO3 (1.5) | t-AmylOH | 18 |
| 4 | Ag2CO3 (1.5) | t-AmylOH | 25 |
| 5 | AgOAc (1.5) | t-AmylOH | 16 |
| 6 | Ag2O (1.5) | t-AmylOH | 50 |
| 7 | AgTFA (1.5) | t-AmylOH | Trace |
| 8 | AgF (1.5) | t-AmylOH | 65 |
| 9 | AgF (1.5) | MeOH | 64 |
| 10 | AgF (1.5) | DCE | 51 |
| 11 | AgF (1.5) | PhMe | 14 |
| 12 | AgF (1.5) | DMF | 60 |
| 13 | AgF (3.0) | t-AmylOH | 81 |
Under the optimized conditions for the arylation of lactic acid-derived amide 1a, we next examined the scope of aryl iodides (Scheme 1). A variety of aryl iodides were found to be compatible with this protocol and afforded the desired arylated products in good yields. Different functional groups, including alkyl (3b–3d, 3h and 3r), alkoxy (3e–3g, 3t and 3w), fluoro (3i), chloro (3j), bromo (3k), acetyl (3l), nitro (3m), trifluoromethyl (3o), methoxycarbonyl (3p), and hydroxyl (3x) groups, were well tolerated under the reaction conditions. When strong electron-deficient 4-iodobenzonitrile was used as the arylation reagent, the modification of the standard reaction conditions (DMF and Ag2O instead of tAmylOH and AgF) could give the desired product 3n in 49% yield. Steric hindrance adjacent to the iodide had a tremendous impact on the reactivity. For example, 2-methyliodobenzene completely shuts down the reaction, and even a less bulky methoxyl group at the ortho-position only gave a moderate yield (3g). Arylation could also proceed smoothly with disubstituted aryl iodides and the desired products were obtained in good yields (3q–3u), which would make this process synthetically attractive. It is worth noting that the arylated product 3r could be converted to the biologically important Danshensu,2b simply by the removal of the AQ auxiliary and protecting group. Additionally, the heterocyclic coupling partners, such as 2-iodothiophene (2w) and 3-iodoindole (2x), were also compatible with this protocol, giving the desired products 3w and 3x in moderate yields (60% and 59%, respectively).
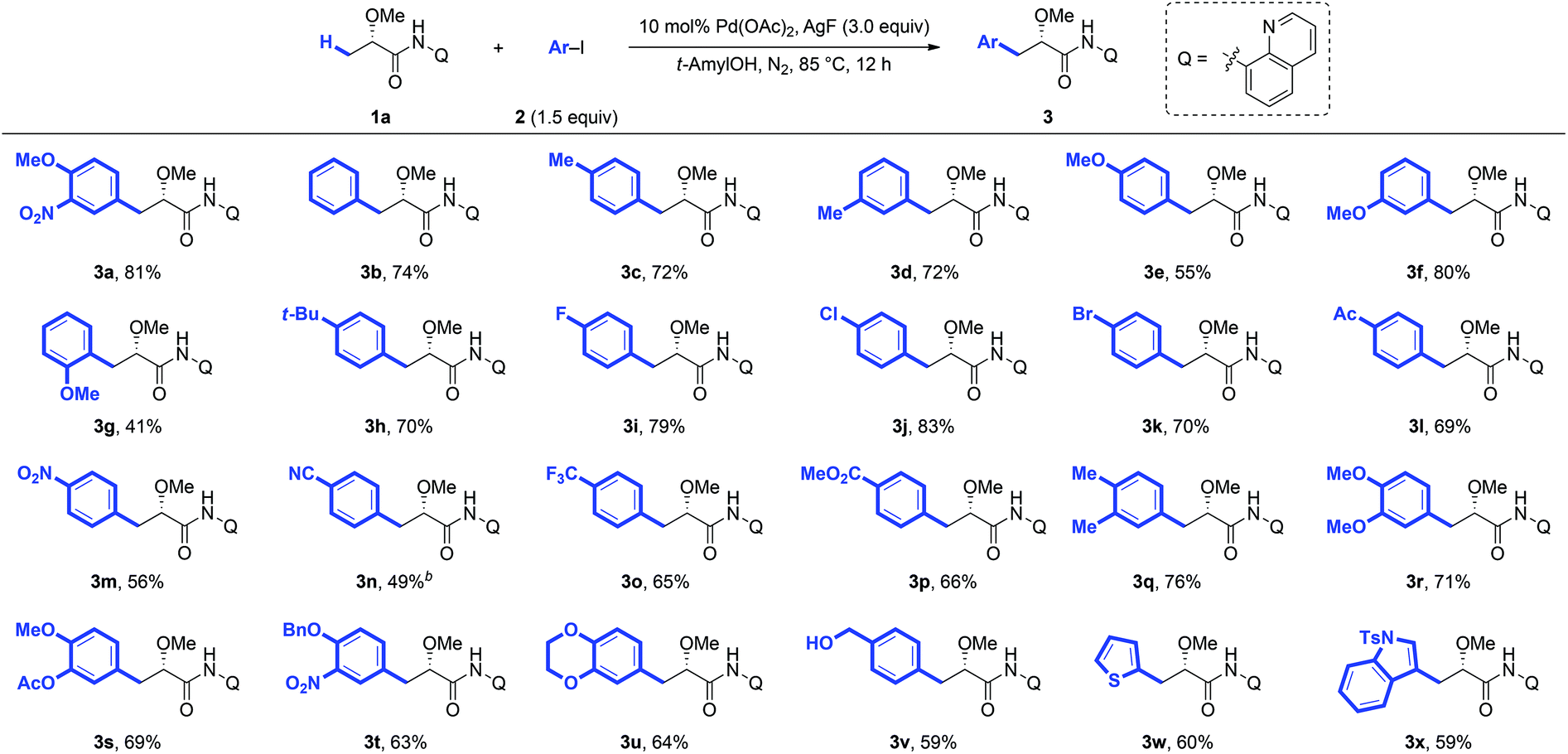 | ||
| Scheme 1 Substrate scope of arylation of lactic acid. | ||
Additionally, the arylation protocol was also compatible with more complicated coupling partners. The coupling of 1a with glucose-derived aryl iodide (2y) under the standard conditions of arylation afforded the corresponding product 3y in 67% yield (Scheme 2A), which suggested the further synthetic potentials in late-stage modification of complex molecules. The reaction could also be scaled up to 5 mmol giving a synthetically useful yield (Scheme 2B). Further the AQ auxiliary could be easily removed in good yield under mild conditions.11e As shown in Scheme 2C, the protection of the secondary amide of 3a with Boc2O, followed by treatment of the resulting tertiary amide with LiOH/H2O2, gave the corresponding α-methoxy carboxylic acid 4a in 76% yield. The BocNHQ 4b could be recovered in quantitative yield.
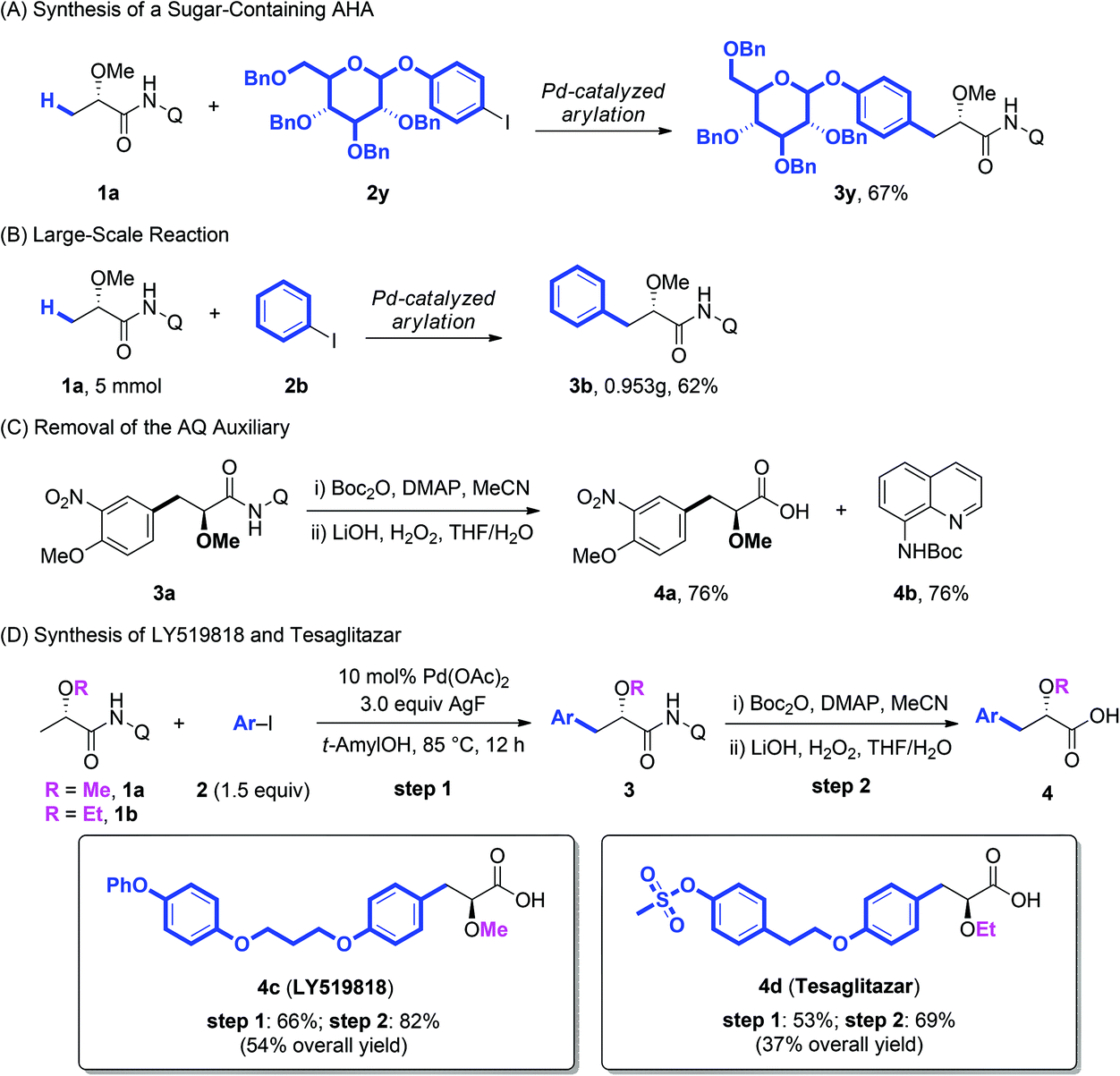 | ||
| Scheme 2 Synthetic application of the Pd-catalyzed arylation of lactic acid. | ||
LY519818 (4c) and tesaglitazar (4d) have been recognized as potential agonists against peroxisome proliferator-activated receptors (PPARs) for the treatment of type II diabetes.3 To demonstrate the practical potential of our protocols, the synthesis of these compounds was conducted with the Pd-catalyzed arylation of lactic acid as the key step. As demonstrated in Scheme 2D, starting from substrates 1a and 1b, LY519818 (4c) and tesaglitazar (4d) were obtained in 54% and 37% overall yield, respectively.
In conclusion, we have developed a palladium-catalyzed AQ-directed arylation of lactic acid derivatives for the synthesis of chiral β-aryl-α-hydroxy acids. Despite the intensive study on the arylation of fatty acids and α-amino acids, the reaction here provides a practical alternative to chiral β-aryl-AHAs. A wide range of aryl and heteroaryl iodides bearing various functional groups underwent the arylation successfully. The synthetic importance of these novel protocols were further demonstrated by the synthesis of two biologically important AHAs, LY519818 and tesaglitazar, potential agonists against peroxisome proliferator-activated receptors (PPARs) for the treatment of type II diabetes.
Acknowledgements
Financial support from the National Basic Research Program of China (2015CB856600) and the NSFC (21572201, 21422206, 21272206) is gratefully acknowledged.Notes and references
- (a) G. M. Coppola and H. F. Schuster, α-Hydroxy Acids in Enantioselective Synthesis, VCH, Weinheim, 1997 CrossRef; (b) S. Hanessian, Total Synthesis of Natural Products: The Chiron Approach, Pergamon, New York, 1983 Search PubMed.
- (a) M. Kendel, G. Barnathan, J. Fleurence, V. Rabesaotra and G. Wielgosz-Collin, Lipids, 2013, 48, 535 CrossRef CAS; (b) Y.-B. Wu, Z.-Y. Ni, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu and B. Cong, Chem. Rev., 2012, 112, 5967 CrossRef CAS; (c) Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441 CrossRef PubMed.
- (a) S. Ebdrup, I. Pettersson, H. B. Rasmussen, H.-J. Deussen, A. F. Jensen, S. B. Mortensen, J. Fleckner, L. Pridal, L. Nygaard and P. Sauerberg, J. Med. Chem., 2003, 46, 1306 CrossRef CAS PubMed; (b) J. A. Martín, D. A. Brooks, L. Prieto, R. González, A. Torrado, I. Rojo, B. L. Uralde, C. Lamas, R. Ferritto, M. D. Martín-Ortega, J. Agejas, F. Parra, J. R. Rizzo, G. A. Rhodes, R. L. Robey, C. A. Alt, S. R. Wendel, T. Y. Zhang, A. Reifel-Miller, C. Montrose-Rafizadeh, J. T. Brozinick, E. Hawkins, E. A. Misener, D. A. Briere, R. Ardecky, J. D. Fraser and A. M. Warshawsky, Bioorg. Med. Chem. Lett., 2005, 15, 51 CrossRef PubMed.
- For selected reviews and examples, see: (a) A. Mori and S. Inoue, Cyanation of Carbonyl and Imino Groups, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Heidelberg, 1999, ch. 28 Search PubMed; (b) M. J. Burk, Acc. Chem. Res., 2000, 33, 363 CrossRef PubMed; (c) M. J. Burk, C. S. Kalberg and A. Pizzano, J. Am. Chem. Soc., 1998, 120, 4345 CrossRef; (d) E. Díez, D. J. Dixon and S. V. Ley, Angew. Chem., Int. Ed., 2001, 40, 2906 CrossRef; (e) S. Li, S.-F. Zhu, J.-H. Xie, S. Song, C.-M. Zhang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 1172 CrossRef PubMed; (f) Y. Zhang, X. Liu, L. Zhou, W. Wu, T. Huang, Y. Liao, L. Lin and X. Feng, Chem. – Eur. J., 2014, 20, 15884 CrossRef CAS PubMed . And references therein.
- For recent reviews on C(sp3)–H activation, see: (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (c) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (f) L. Yang and H. Huang, Catal. Sci. Technol., 2012, 2, 1099 RSC; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (h) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459 CrossRef CAS PubMed; (i) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- For selected examples on the use of 8-AQ auxiliary, see: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef PubMed; (c) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC.
- For recent reviews on the functionalization of C–H bonds utilizing bidentate-chelation assistance, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (b) B. Zhang, H.-X. Guan, B. Liu and B.-F. Shi, Chin. J. Org. Chem., 2014, 34, 1487 CrossRef CAS; (c) L. C. Misal Castro and N. Chatani, Chem. Lett., 2015, 44, 410 CrossRef; (d) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (e) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef.
- (a) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070 CrossRef CAS PubMed; (b) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem. – Eur. J., 2012, 18, 5541 CrossRef CAS PubMed; (c) M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152 CrossRef CAS PubMed; (d) G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed; (e) N. Rodríguez, J. A. Romero-Revilla, M. Á. Fernández-Ibáñez and J. C. Carretero, Chem. Sci., 2013, 4, 175 RSC; (f) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712 RSC; (g) Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868 CrossRef CAS PubMed; (h) H. H. Al Mamari, E. Diers and L. Ackermann, Chem. – Eur. J., 2014, 20, 9739 CrossRef CAS PubMed.
- (a) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (b) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed; (c) R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194 CrossRef CAS PubMed.
- For recent reviews on the synthesis of α-amino acids via C–H functionalization: (a) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed; (b) Z. Huang and G. Dong, Tetrahedron Lett., 2014, 55, 5869 CrossRef CAS; (c) Q. Zhang, K. Chen and B.-F. Shi, Synlett, 2014, 1941 CAS.
- (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (b) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (c) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS PubMed; (d) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS PubMed; (e) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef PubMed; (f) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3952 RSC.
- (a) For an isolated example of arylation of lactic acid using 2-methylthioaniline auxiliary, see: ref. 6b; (b) For an isolated example of methoxylation of lactic acid using 2-pyridinylisopropyl auxiliary, see: ref. 6c.
- (a) D. Dailler, G. Danoun and O. Baudoin, Angew. Chem., Int. Ed., 2015, 54, 4919 CrossRef CAS PubMed; (b) K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Chem. Commun. Search PubMed , under revision.
- (a) Q. Zhang, K. Chen, W.-H. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (b) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC; (c) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC; (d) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed; (e) K. Chen, S.-Q. Zhang, H.-Z. Jiang, J.-W. Xu and B.-F. Shi, Chem. – Eur. J., 2015, 21, 3264 CrossRef CAS PubMed; (f) K. Chen, S.-Q. Zhang, J.-W. Xu, F. Hu and B.-F. Shi, Chem. Commun., 2014, 50, 13924 RSC; (g) Q. Zhang, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, J. Am. Chem. Soc., 2015, 137, 8219 CrossRef CAS PubMed.
- For the Thorpe–Ingold effect in cyclometallation, see: B. L. Shaw, J. Am. Chem. Soc., 1975, 97, 3856 CrossRef CAS.
- For selected examples of ethers as the directing group, see: (a) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed; (b) Á. Iglesias, R. Álvarez, Á. R. Lera and K. Muñiz, Angew. Chem., Int. Ed., 2012, 51, 2225 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00319a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |