
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic MnIII–FeII hybrid complexes formed by a functionalized MnIII Anderson polyoxometalate coordinated to FeII: observation of a field-induced slow relaxation of magnetization in the MnIII centres and a photoinduced spin-crossover in the FeII centres†
Alexandre
Abhervé
a,
Mario
Palacios-Corella
a,
Juan Modesto
Clemente-Juan
a,
Raphael
Marx
b,
Petr
Neugebauer
b,
Joris
van Slageren
b,
Miguel
Clemente-León
*a and
Eugenio
Coronado
*a
aInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, Paterna, 46980, Spain. E-mail: miguel.clemente@uv.es; eugenio.coronado@uv.es; Fax: +34 963543273; Tel: +34 963544405
bInstitut für Physikalische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
First published on 27th May 2015
Abstract
The synthesis and crystal structure of an Anderson POM functionalized with two 2,6-di(pyrazol-1-yl)-pyridine (1-bpp) ligands are reported (compound 1). High-frequency electron paramagnetic resonance (HF-EPR) and magnetic measurements show that it presents a significant negative axial zero-field splitting and field-induced slow relaxation of magnetization due to the presence of isolated MnIII anisotropic magnetic ions. Complexation of 1 with FeII gives rise to a 2D cationic network formed by Anderson POMs coordinated to two FeII ions through the two tridentate 1-bpp ligands and to other two FeII ions through two oxo ligands in compound 2, and to an anionic polymeric network formed by Anderson POMs coordinated through the 1-bpp ligands to two FeII, which are coordinated to two 1-bpp ligands from two neighbouring POMs, in compound 3. The crystal structure of 2 has been solved. Magnetic properties show that the FeII atoms of 3 remain in the low-spin state, while those of 2 remain in the high-spin state due to coordination to oxygen atoms from a neighbouring POM and dimethylformamide and water solvent molecules. Irradiation of 3 at 10 K with green light induces a spin-crossover (LIESST effect) with a small but significant photoconversion (∼8%). Finally, AC susceptibility measurements of 2, 3 and (C16H36N)3[MnMo6O18{(OCH2)3CNH2}2] (4) confirm field-induced slow relaxation of magnetization of MnIII Anderson POMs.
Introduction
Polyoxometalates (POMs) constitute a family of molecular-metal oxides with unique electronic and structural properties and a variety of applications in areas like catalysis, medicine and material science.1,2 An interesting possibility for these polyanions is that they can be functionalized with organic ligands,3 affording rationally designed, predictable and consistent POM-based hybrid structures.4 One of the most successful strategies uses tris-alkoxo-amide tripods as anchoring ligands. This approach has already been used for the incorporation of a large variety of organic ligands into Lindqvist, Anderson and Dawson–Wells structures.4–9 For instance, discrete or polymeric complexes have been obtained from tris-alkoxo-pyridyl ligands of various denticity (pyridyl,10 bipyridyl11,12 and terpyridine13).The preparation and characterization of magnetic POMs following this strategy remains largely unexplored.13 Still, complex magnetic functionalities could be expected if the appropriate functionalization is chosen. An attractive example can result from the association of a magnetic POM molecule with a spin-crossover (SCO) complex. In SCO systems, low-spin (LS) to high-spin (HS) transitions can be triggered through a variety of external stimuli (temperature, pressure or electromagnetic radiation). They constitute one of the most spectacular examples of molecular bistability. To the best of our knowledge, there are no previous reports of POMs showing SCO behaviour. In this work, we will explore this topic through the incorporation of the tridentate ligand, 2,6-bis(pyrazol-1-yl)pyridine (1-bpp) (Scheme 1), into a MnIII Anderson POM. This ligand has been chosen because FeII complexes of 1-bpp usually present very abrupt spin transitions with thermal hysteresis close to room temperature.14,15 Furthermore, they often exhibit spin-crossover induced by irradiation (light-induced excited spin state trapping effect, LIESST) with relatively long lifetimes of the photoinduced metastable states.15b To reach this goal we first prepared the 1-bpp functionalized Anderson POM (C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5 (1) and subsequently the compounds [Fe(H2O)(C3H7NO)]2[MnMo6O24(C16H15N6O)2](OH)·(H2O)·(C3H7NO)1.5 (2) and (C16H36N)[Fe(MnMo6O24(C16H15N6O)2)]·(H2O)4 (3), formed by the reaction of 1 with Fe2+.
 | ||
| Scheme 1 Molecular structure of 1-bpp (left) and 4 (right). | ||
Interestingly, during the magnetic characterization of 1, we have found that the magnetically anisotropic MnIII ion behaves as a single-molecule magnet (SMM) showing field-induced slow relaxation of the magnetization. This behaviour is rare in POMs.1c In fact, it was only observed for the first time in 2008 in mononuclear complexes based on lanthanoids ([Ln(W5O18)2]9− POM series)16a and in magnetic clusters based on the {[XW9O34]2[MnIII4MnII2O4(H2O)4]}12− (X = Si, Ge) POM.17 Very recently, in 2015, this behaviour has been observed for mononuclear complexes based on d metal ions ([M(SiW9O34)2]17−/18− (M = FeIII, CoII and MnIII)).18 Owing to the current interest raised by mononuclear SMMs based on MnIII,19 we have studied in this work how general this behaviour is in the Anderson structures containing this transition metal ion. Thus, the magnetic properties of 1, 2 and 3 will be compared with those of a functionalized MnIII Anderson POM reported in the literature, (C16H36N)3[MnMo6O18{(OCH2)3CNH2}2] (4), (Scheme 1).20 In the second part of this work, we will also study how light affects the SCO behaviour in the hybrid MnIII–FeII compound 3.
Experimental
General remarks
(C16H36N)4α-[Mo8O26],21 bppCOOEt22 and (C16H36N)3[MnMo6O18{(OCH2)3CNH2}2] (4)20 were synthesized according to the literature methods. All other materials and solvents were commercially available and used without further purification.Synthesis of TRIS-bpp
Under nitrogen atmosphere, bppCOOEt (181 mg, 0.64 mmol), (HOCH2)3CNH2 (77 mg, 0.64 mmol) and K2CO3 (88 mg, 0.64 mmol) were suspended in dry dimethyl sulfoxide (DMSO) (5 mL) and stirred at room temperature for 18 h. The reaction mixture was then filtered, and the solvent was removed by vacuum. The residue was dissolved in EtOH (3 mL) and the product precipitated by slowly adding H2O (15 mL). The white precipitate was filtered, washed with diethyl ether, and dried to give pure TRIS-bpp (87 mg, 38%). 1H NMR (d6-DMSO, 300 MHz): 8.99 (dd, J = 3, 0.75 Hz, 2H, HIm1), 8.12 (s, 2H, HPyr), 7.92 (dd, J = 2, 0.75 Hz, 2H, HIm2), 7.82 (br, 1H, HNH), 6.67 (dd, J = 3, 2 Hz, 2H, HIm3), 4.69 (t, J = 6 Hz, 3H, HOH), 3.74 (d, J = 6 Hz, 6H, HCH2).Synthesis of (C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5(1)
(C16H36N)4α-[Mo8O26] (150 mg, 0.07 mmol) and Mn(CH3COO)3·2H2O (27 mg, 0.10 mmol) were dissolved in dry dimethylacetamide (DMAc, 8 mL). Then, a solution of TRIS-bpp (87 mg, 0.24 mmol) in dry DMAc (3 mL) was added, and the mixture was heated at 80 °C for 18 h. The obtained orange solution was allowed to cool down. After two days, yellow cubic crystals of 1 were obtained (177 mg, 98%). 1H NMR (d6-DMSO, 300 MHz): 8.97 (d, J = 2.7 Hz, 4H, H Im1), 8.09 (br, 4H, HPyr), 7.91 (d, J = 1.2 Hz, 4H, HIm2), 6.65 (dd, J = 2.7, 1.2 Hz, 4H, HIm3), 3.16 (m, 24H, HC1), 1.56 (q, J = 7.5 Hz, 24H, HC2), 1.31 (sx, J = 7.5 Hz, 24H, HC3), 0.93 (t, J = 7.5 Hz, 36H, HC4) (presence of 2 equiv. of DMAc confirmed by peaks at 1.96, 2.78 and 2.94 ppm). IR (KBr pellet, cm−1): 2960 (ν C–H, s), 2935 (ν C–H, s), 2874 (ν C–H, s), 1670 (m), 1618 (m), 1570 (m), 1552 (sh), 1524 (m), 1483 (sh), 1462 (s), 1396 (ν C–H, s), 1362 (w), 1320 (w), 1288 (w), 1259 (w), 1207 (w), 1151 (w), 1111 (sh), 1097 (ν C–O, w), 1047 (m), 1036 (ν C–O, sh), 1030 (sh), 941 (ν Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, vs), 922 (ν MoO, vs), 903 (ν MoO, vs), 789 (m), 760 (m), 667 (ν Mo–O–Mo, vs), 565 (m), 464 (m). Anal. calcd for (C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5: C, 41.0; H, 6.3; N, 9.2%. Found: C, 40.9; H, 5.8; N, 9.1%.
O, vs), 922 (ν MoO, vs), 903 (ν MoO, vs), 789 (m), 760 (m), 667 (ν Mo–O–Mo, vs), 565 (m), 464 (m). Anal. calcd for (C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5: C, 41.0; H, 6.3; N, 9.2%. Found: C, 40.9; H, 5.8; N, 9.1%.
Synthesis of [Fe(H2O)(C3H7NO)]2[MnMo6O24(C16H15N6O)2](OH)·(H2O)·(C3H7NO)1.5(2)
(C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5 (1) (25.8 mg, 0.01 mmol) was dissolved in acetonitrile (3 mL). A solution of Fe(ClO4)2·xH2O (5.1 mg, 0.02 mmol) in acetonitrile (3 mL) was added slowly, and the resulted mixture was stirred for 30 min at room temperature. The orange precipitate was filtered and recrystallized in dimethylformamide (10 mL). After three days, red crystals were obtained (0.8 mg, 4%) IR (KBr pellet, cm−1): 2922 (ν C–H, s), 2875 (ν C–H, s), 1654 (m), 1648 (m), 1628 (m), 1572 (m), 1528 (m), 1500 (m), 1460 (s), 1405 (ν C–H, s), 1327 (w), 1295 (w), 1274 (w), 1211 (s), 1173 (s), 1156 (w), 1098 (ν C–O, s), 1055 (ν C–O, sh), 1025 (sh), 972 (s), 946 (ν MoO, vs), 925 (ν MoO, vs), 912 (ν MoO, vs), 796 (s), 765 (m), 668 (ν Mo–O–Mo, vs), 569 (m), 466 (m). Anal. calcd for [Fe(H2O)(C3H7NO)]2[MnMo6O24(C16H15N6O)2]·(OH)·(H2O): C, 23.3; H, 2.6; N, 10.0%. Found: C, 21.59; H, 3.62; N, 8.99%.
Synthesis of (C16H36N)[Fe(MnMo6O24(C16H15N6O)2)]·(H2O)4(3)
(C16H36N)3[MnMo6O24(C16H15N6O)2]·(C4H9NO)2·(H2O)2.5 (1) (12.9 mg, 0.005 mmol) was dissolved in dry acetonitrile (5 mL). A solution of Fe(ClO4)2·xH2O (0.005 mmol) in dry acetonitrile (500 μL) was added slowly, and the resulted mixture was stirred for 10 min at room temperature. The orange precipitate was centrifuged, washed with dry acetonitrile (5 mL), and dried under vacuum (4 mg, 40%). IR (KBr pellet, cm−1): 2958 (ν C–H, s), 2923 (ν C–H, s), 2872 (ν C–H, s), 1670 (m), 1624 (m), 1570 (m), 1527 (m), 1499 (sh), 1459 (s), 1400 (ν C–H, s), 1390 (w), 1363 (w), 1323 (w), 1264 (w), 1209 (w), 1169 (w), 1096 (ν C–O, w), 1052 (ν C–O, sh), 1026 (sh), 972 (s), 945 (ν MoO, vs), 922 (ν MoO, vs), 903 (ν MoO, vs), 795 (m), 764 (m), 665 (ν Mo–O–Mo, vs), 567 (m), 462 (m). Anal. calcd for (C16H36N)[Fe(MnMo6O24(C16H15N6O)2)]·(H2O)4: C, 28.8; H, 3.7; N, 9.1%. Found: C, 27.5; H, 2.5; N, 9.1%. The bands at 1624, 1390, 1169 and 972 cm−1 supported the coordination of FeII to 1-bpp.
Physical measurements
Infrared (IR) spectra were recorded in the solid state (KBr pellets) using a Nicolet Avatar 320 FTIR spectrometer in the 400–4000 cm−1 range. C, H and N elemental analyses were done using a CE Instruments EA 1110 CHNS Elemental analyser. The Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo and Fe:Mn:Mo ratios were measured using a Philips ESEM X230 scanning electron microscope equipped with an EDAX DX-4 microprobe. 1H NMR spectra were acquired using a Bruker AVANCE DRX 300 spectrometer.
:Mo and Fe:Mn:Mo ratios were measured using a Philips ESEM X230 scanning electron microscope equipped with an EDAX DX-4 microprobe. 1H NMR spectra were acquired using a Bruker AVANCE DRX 300 spectrometer.
Single crystals of all compounds were mounted on glass fibres using a viscous hydrocarbon oil to coat the crystal and then were transferred directly to the cold nitrogen stream for data collection. All reflection data were collected at 120 K for 1 and 180 K for 2 using a Supernova diffractometer (1) and using a Supernova Atlas Dual Source diffractometer (2) equipped with a graphite-monochromated Enhance (Mo) X-ray Source (λ = 0.7107 Å). The CrysAlisPro program, Oxford Diffraction Ltd., was used for unit cell determinations and data reduction. Empirical absorption correction was performed using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. Crystal structures were solved by direct methods with the SIR97 program23 and refined against all F2 values with the SHELXL-2013 program24 using the WinGX graphical user interface.25 All non-hydrogen atoms were refined anisotropically except as noted and hydrogen atoms were placed in calculated positions and refined isotropically with a riding model. The structure of 2 showed a weak diffraction due to the presence of disordered solvent molecules in the structure. Due to this, it was not possible to refine anisotropically C and N atoms. Initial refinements revealed the presence of a substantial volume of unresolvable solvent (DMF) molecules in 2. The subroutine SQUEEZE from PLATON26 was used to remove the diffracting component of disordered solvents resulting in a void of ca. 741.5 Å3 and 142 electrons per cell omitted. This corresponds to ca. 3 DMF molecules per unit cell. Crystallographic data are summarized in Table S1, ESI.† CCDC 1058519 and 1058520. 0.5 mm glass capillaries were filled with polycrystalline samples of compound 1 and mounted and aligned using a Empyrean PANalytical powder diffractometer, using CuKα radiation (λ = 1.54177 Å). A total of 3 scans were collected at room temperature in the 2θ range 5°–40°.
A Q-TOF Premier mass spectrometer with an orthogonal Z-spray electrospray source (Waters, Manchester, UK) was used for electrospray ionization mass spectrometry (ESI-MS). The temperature of the source block was set to 100 °C and the desolvation temperature to 120 °C. A capillary voltage of 3.3 kV was used in the negative scan mode, and the cone voltage was set to 5 V to control the extent of fragmentation of the identified species. TOF mass spectra were acquired in the W-mode operating at a resolution of ca. 15000 (fwhm). Mass calibration was performed using a solution of sodium iodide in isopropanol/water (50:50) from m/z 50 to 3000. Acetonitrile sample solutions were infused via syringe pump directly connected to the ESI source at a flow rate of 10 μL min−1. The observed isotopic pattern of each compound perfectly matched the theoretical isotope pattern calculated from their elemental composition using the MassLynx 4.1 program.
Magnetic measurements were performed with Quantum Design MPMS-XL-5 SQUID and PPMS-9 magnetometers on powdered polycrystalline samples. Photomagnetic measurements were performed with irradiation from a Diode Pumped Solid State Laser DPSS-532-20 from Chylas coupled via an optical fibre to the cavity of the SQUID magnetometer. The optical power at the sample surface was adjusted to 3.4 mW cm−2, and it was verified that it resulted in no significant change in magnetic response due to heating of the sample. The photomagnetic samples consisted of a thin layer of compound whose weight was obtained by comparison with the magnetic measurement of a more accurately weighted sample of the same compound. High-frequency EPR (HF-EPR) spectra (100–370 GHz) were recorded using a home-built spectrometer. Its microwave source is a 8–20 GHz signal generator (VDI) in combination with an amplifier–multiplier chain (VDI) to obtain the required frequencies. It features a quasi-optical bridge (Thomas Keating) and induction mode detection. The detector is a QMC magnetically tuned InSb hot electron bolometer. The sample is located in an Oxford Instruments 15/17 T cryomagnet equipped with a variable temperature insert (1.5–300 K). The sample was measured as a 5 mm pressed pellet, which was mixed with eicosane (ratio 1:1, 25 mg each). Spectral simulations were performed using the EasySpin 4.5.3 simulation software. A modulation amplitude of 80 mA (80 G) was used to modulate the magnetic field. Two temperature sensors allowed monitoring of the sample temperature with high accuracy. The sample was investigated at different frequencies and temperatures (Table S2, ESI†). A linewidth of 120 mT (FWHM) was used. The powder spectrum is obtained using 91 orientations.
Results and discussion
Syntheses
1-bpp-functionalized Anderson POM (1) was synthesized in several steps following adapted literature procedures (Scheme 2). The starting material for the preparation of the functionalized POM was tris-(hydroxymethyl)-functionalized 1-bpp (TRIS-bpp), which was obtained from 1-bpp-4′-carboxyethylester. The ester was obtained by esterification with ethanol of the carboxylate 1-bpp derivative (bppCOOH). The functionalization of the POM was performed in dry dimethylacetamide (DMAc). It can also be performed with similar conditions in dry acetonitrile but this gives rise to a less pure product in a lower yield. Single crystals were obtained by slow evaporation of the DMAc solution of the compound. 1H NMR spectra confirm the purity of TRIS-bpp and 1 and the grafting of 1-bpp to the POM in 1 (Fig. S1, ESI†). As observed previously in terpyridine-functionalized Anderson POM,13 the electronic influence of the cluster (paramagnetic MnIII) induces changes in the chemical shifts of the methylene protons of the ligand but not in those of the aromatic ones. Further characterization of 1 by elemental analysis, IR spectroscopy (Fig. S2, ESI†), microanalysis and electrospray ionization mass spectrometry (ESI-MS) is consistent with the bifunctionalization of the POM. Microanalysis shows a Mn:Mo ratio close to 1:6. Fig. S3, ESI,† shows the ESI-MS (negative mode) analysis of a solution of 1 in acetonitrile. The three most intense peaks appear at m/z values of 543.2, 814.8 and 935.9, which correspond to [MnMo6O24(C16H15N6O)2]3− ([1]3−), [MnMo6O24(C16H15N6O)2]2− (H+ + [1]3−) and [(C16H36N)MnMo6O24(C16H15N6O)2]2− (tetrabutylammonium (TBA)+ + [1]3−) species. The charge of the species present in the spectrum has been unambiguously characterized by single ion recording (SIR) at the highest resolution of the spectrometer with monoisotopic peaks separated by 1/z. Fig. S4, ESI†, shows the isotopic distributions of the most intense peaks. As these peaks arise from species in which the POM remains intact, we can conclude that the structure of the polyanion is preserved in solution.
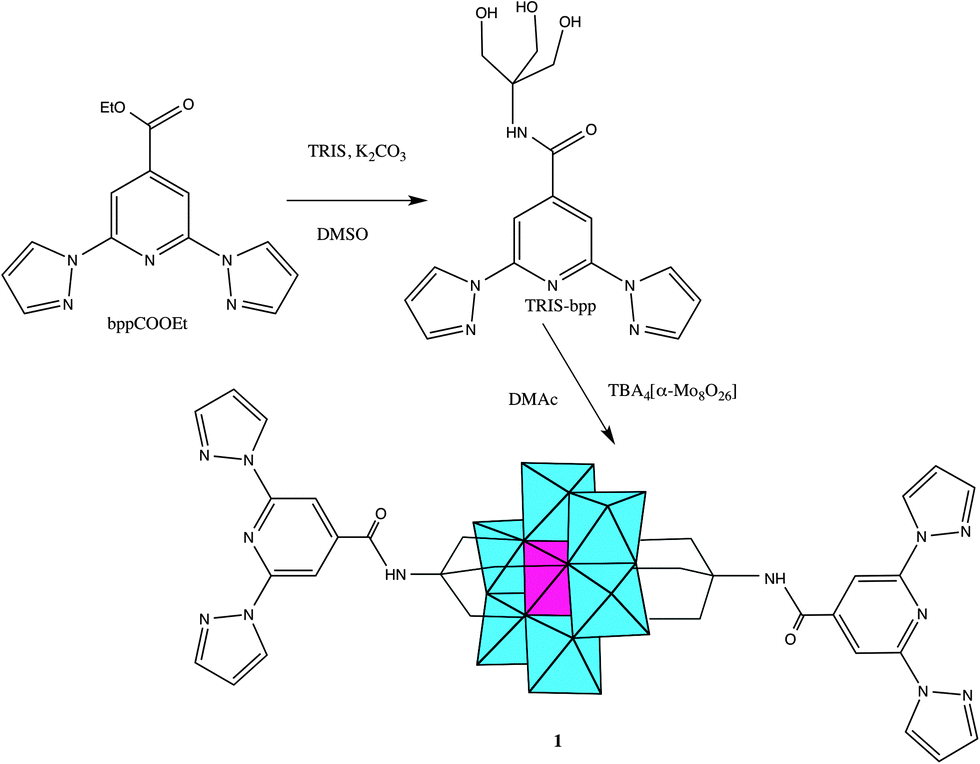 | ||
| Scheme 2 Synthesis of TRIS-bpp and 1. | ||
When 1 was reacted with Fe2+ in acetonitrile, a precipitate immediately formed, as observed in Lindqvist POM functionalized with terpyridine.27 The precipitation takes place after the addition of one equivalent of Fe2+ to the POM suggesting the formation of a polymeric compound in which every POM is coordinated to two FeII, which, at the same time, are coordinated to two 1-bpp from two POMs (Scheme 3). Elemental analysis of this precipitate is consistent with the formula (C16H36N)[Fe(MnMo6O24(C16H15N6O)2)]·(H2O)4 (3). Furthermore, microanalysis shows a Fe:Mn:Mo ratio close to 1:1:6 and the IR spectrum (Fig. S2, ESI†) and magnetic properties (below) are consistent with coordination of two 1-bpp to FeII. Unfortunately, it was not possible to get single crystals of this compound to solve the structure. If two equivalents of Fe2+ are added, an orange precipitate is obtained with a Fe:Mn:Mo ratio close to 2:1:6. This could indicate that FeII are either coordinated to 1-bpp from the POM or act as counterions. This precipitate was partially soluble in polar aprotic solvents such as DMSO, DMAc and dimethylformamide (DMF). This dissolution may involve dissociation of the 1-bpp-metal coordination bond as observed in pyridyl-functionalized hexavanadates.5 Indeed, recrystallization in DMF of the compound gave rise to compound 2, in which octahedral coordination around FeII is completed with DMF and water solvent molecules and oxo groups from neighbouring POM (see below). The presence of DMF and water molecules coordinated to the MII (Mn, Co, Ni, Zn) metals has also been observed in pyridyl-functionalized hexavanadates.5
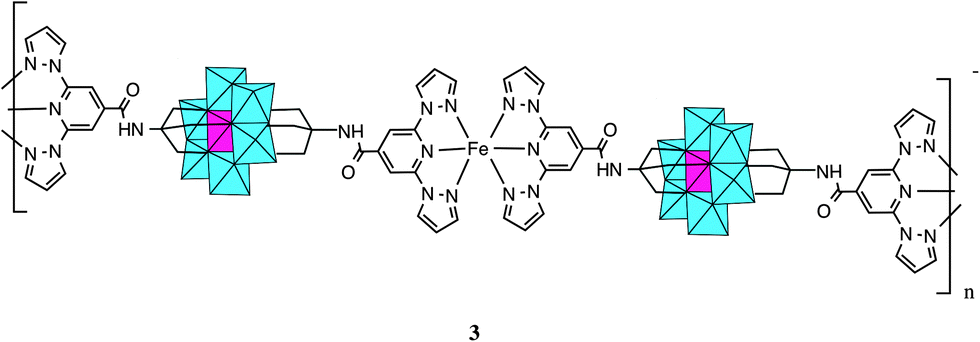 | ||
| Scheme 3 Proposed structure for the polymeric network of 3. | ||
Structure
1 crystallizes in the monoclinic space group P21/n. The asymmetric unit is composed of two half crystallographically independent anions, three TBA+ cations, two DMAc solvent molecules and water molecules that present some disorder. The two crystallographically independent anions contain an inversion centre placed in the Mn. They present the common Anderson POM structure with six MoO6 octahedral edge-sharing units forming a hexagon around the central MnO6 octahedron. As both alkoxo ligands from the bpp-ligand are directly linked to the MnIII ion, this corresponds to the δ isomer of the Anderson structure (Fig. 1).28 All metal atoms essentially lie in a common plane, with a maximum deviation of 0.003 Å from the best least-squares plane. The octahedral coordination geometry of the central MnIII ion is quite regular, with three Mn–O distances of 1.961(3), 1.977(3) and 2.014(3) Å for Mn1 and 1.913(3), 2.019(3) and 2.022(3) Å for Mn2, and cis-O–Mn–O bond angles between 87.00(10)° and 93.00(11)° and trans-O–Mn–O angles of 180° due to the presence of an inversion center in Mn. In contrast to previous MnIII complexes exhibiting a field-induced slow relaxation of magnetization, the coordination sphere around MnIII does not exhibit a marked tetragonal distortion.18,19 For example, distances and angles in 1 are considerably closer to a perfect octahedron than those of TBA7H10[MnIII(SiW9O34)2]·3H2O (Mn–O distances of 1.928(5), 1.939(5) and 2.332(5) Å, cis-O–Mn–O bond angles of 77.75(19)° and 88.2(2)° and trans-O–Mn–O angle of 101.36(19)°).18 It should be noted that the rigidity of the POM framework prevents the MnIII ion from undergoing significant Jahn–Teller distortions. Due to this, the coordination octahedron is only very slightly elongated (Mn1) or compressed (Mn2). The small distortions observed correspond to a slight compression of the octahedron, bringing the two faces capped by the organic ligands closer together, as in other MnIII Anderson POMs.20,28 Indeed, distances between these two faces of the octahedron (2.186–2.189 Å) are slightly shorter than those between other faces (2.312–2.338 Å). The two crystallographically independent POMs present a different orientation. They are surrounded by TBA+ cations and solvent molecules (Fig. S5, ESI†). The shortest distance between MnIII belonging to different POMs is 14.317 Å. Hydrogen bonds are observed between the terminal oxo groups of the POM and water molecules. Furthermore, the NH groups of the two POMs form hydrogen bonds with a DMAc molecule and a water molecule. Powder X-ray diffraction of 1 confirms the structure of the compound (Fig. S6, ESI†).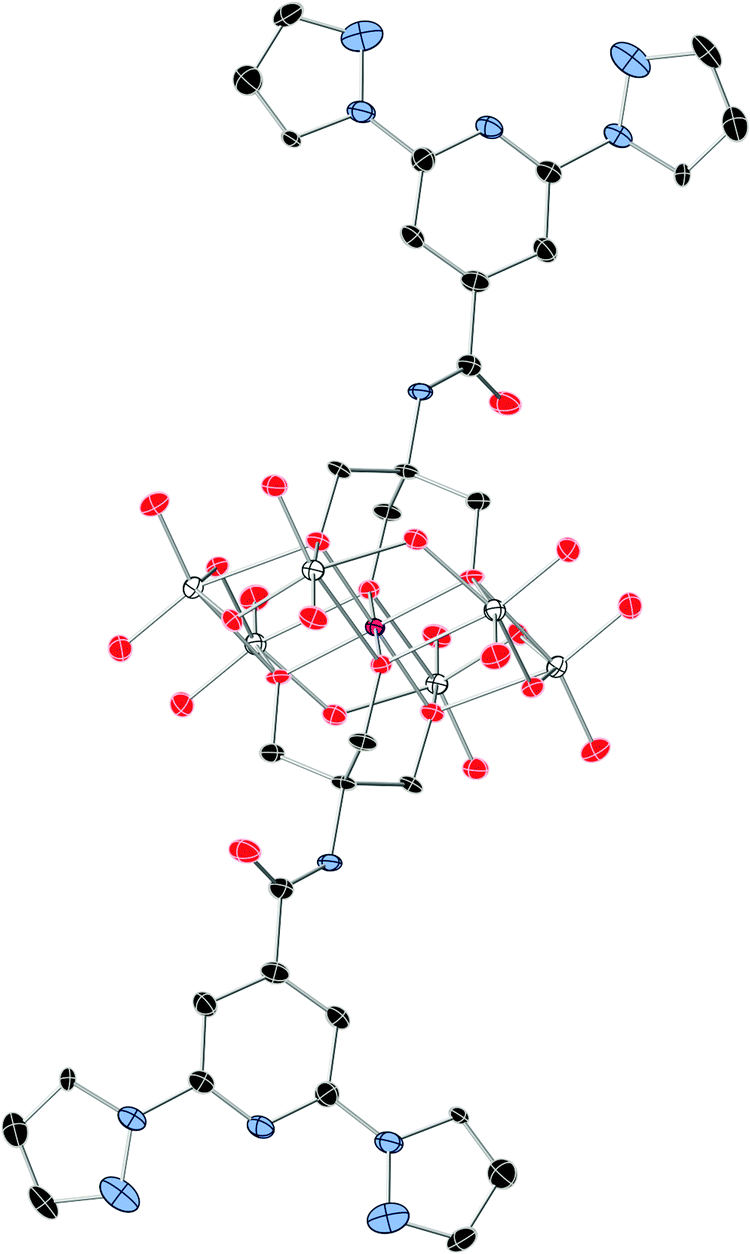 | ||
| Fig. 1 The structure of the functionalized-POM in compound 1. (Mn (pink), Mo (white), C (black), N (blue), and O (red)). Hydrogen atoms have been omitted for clarity. | ||
2 crystallizes in the monoclinic space group C2/c. The asymmetric unit is composed of half a crystallographically independent anion and one crystallographically independent Fe coordinated to a DMF and a water molecule. Furthermore, it contains half crystallographically independent OH− and water molecules. The structure of the anion is the same as that of the 1-bpp-functionalized Anderson POM found in 1 with an inversion centre placed in Mn, but, in contrast to 1, it presents a two dimensional (2D) polymeric structure (Fig. 2). Thus, each functionalized Anderson POM is coordinated to two FeII ions through the two tridentate 1-bpp ligands and to other two FeII ions through two oxo ligands linked to two Mo ions (Mo2). The octahedral coordination around FeII is completed with one DMF and water solvent molecules and the oxo ligand from a neighbouring POM, mentioned above. This gives rise to a 2D network in the bc plane formed by interconnected [FeII(H2O)(C3H7NO)]2[MnIIIMo6O24(C16H15N6O)2]+ units (Fig. 2). The octahedral coordination geometry of the central MnIII presents three Mn–O distances of 1.906(11), 1.998(11) and 2.034(12) Å, cis-O–Mn–O bond angles between 87.1(5)° and 92.9(5)° and trans-O–Mn–O angles of 180° due to the presence of the inversion centre in Mn. FeII presents a more distorted octahedral coordination geometry. The shortest distance is that with the O atom from DMF (1.985(19) Å). Fe–O distances to the water molecule and oxo ligand from POM are intermediate (2.128(16) and 2.206(13) Å), while Fe–N distances to the 1-bpp ligand range from 2.182(16) to 2.224(16) Å. These distances indicate that FeII is in the HS state. A lateral view of two neighbouring layers, shown in Fig. S7, ESI,† allows us to distinguish the microporous channels, which are formed along the crystallographic c-axis. These pores are occupied by three disordered DMF solvent molecules (see above). In addition to these DMF molecules, the space between the cationic layers is occupied by water solvent molecules and OH− groups, which are connected through hydrogen bond interactions with NH groups and POM oxo groups from the layers. Hydrogen bond formation agrees with the presence of half crystallographically independent OH− anions (O200 in Fig. S7, ESI†), which counterbalances the positive charge of the 2D layer. Powder X-ray diffraction of 2 could not be performed due to the small amount of sample available.
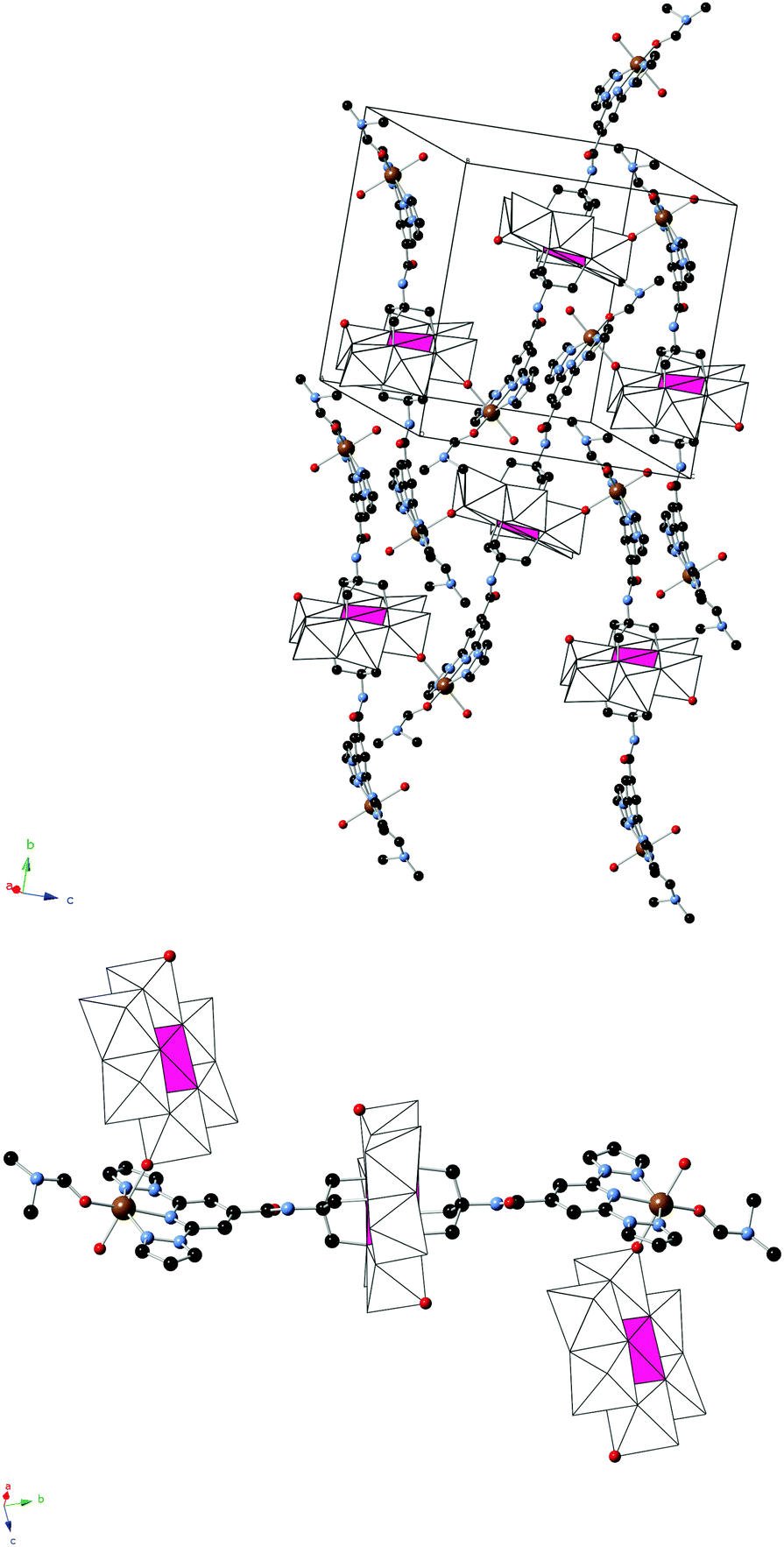 | ||
| Fig. 2 Illustration of a layer of functionalized-POM linked through Fe2+ ions in the structure of 2 (top) and of the repeating trimeric unit with two coordinated POMs (bottom) (Fe (yellow), Mn (pink), Mo (white), C (black), N (blue), and O (red)). Red oxygen atoms from the POM are those coordinated to Fe2+ ions. Hydrogen atoms have been omitted for clarity. | ||
Magnetic properties
Temperature dependence of the product of the molar magnetic susceptibility times the temperature (χmT) of a pressed pellet of 1 in eicosane is shown in Fig. 3. The χmT value at room temperature (2.9 cm3 mol−1 K) is consistent with an isolated MnIII with S = 2 and g = 2.0. Upon cooling, the χmT value remains constant until 40 K. Below this temperature, there is an abrupt decrease which indicates that there is an appreciable zero-field splitting as observed in other MnIII mononuclear complexes. This is further confirmed by the isothermal magnetization (M) curves of 1 in the temperature range of 2–10 K, which cannot be superposed at high H/T values (Fig. 4). This indicates that there is a strong magnetic anisotropy of the ground state of MnIII. From simultaneous fitting of susceptibility and magnetization data using the Magpack program (Fig. 4 and Fig. S8, ESI†),29 a D value = −5.75 cm−1, a E value = 0.01 cm−1 and a g value = 2 have been obtained. A more precise value of the negative axial anisotropy and a rhombic term presence was determined by high-frequency electron paramagnetic resonance (HF-EPR).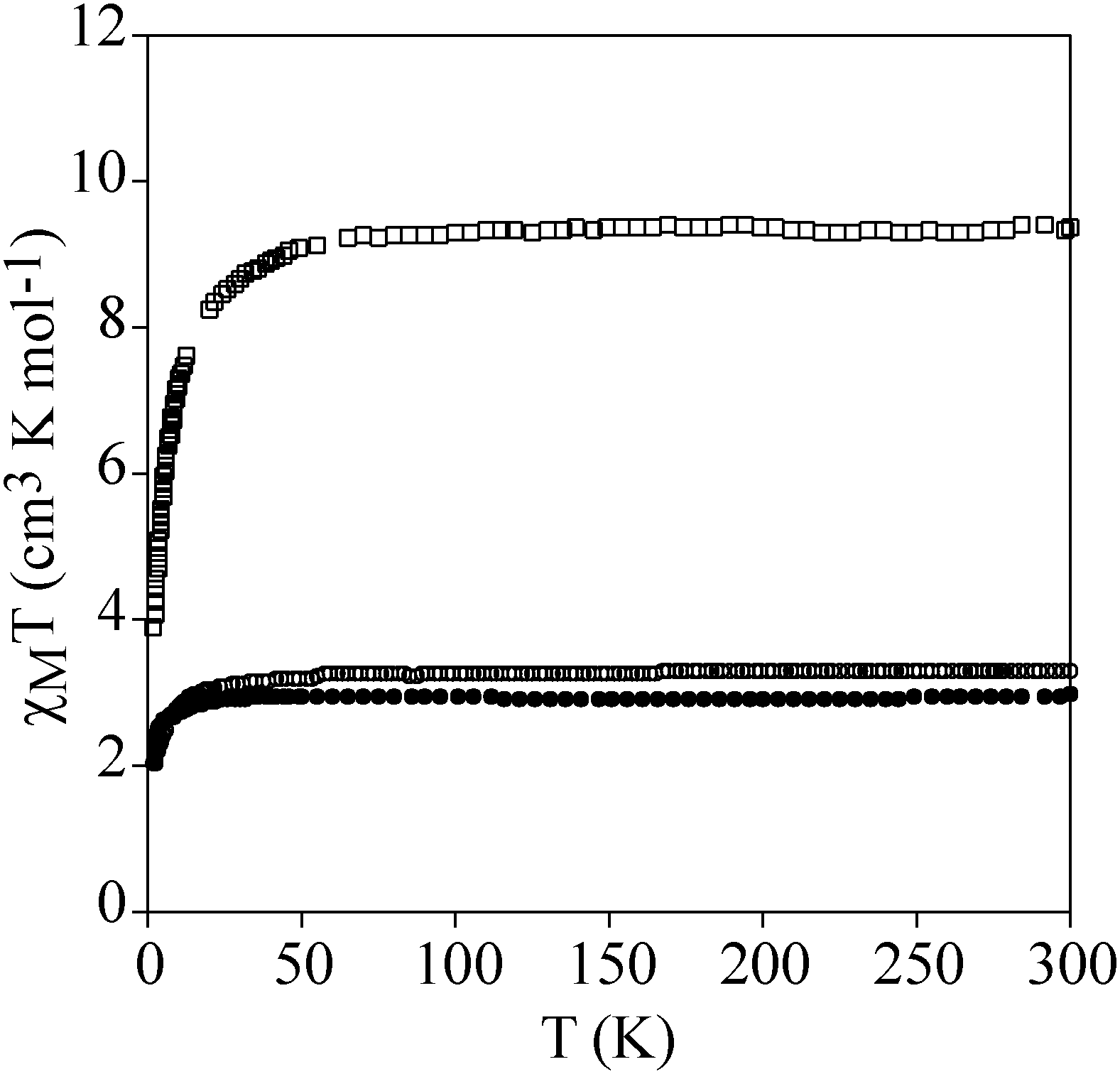 | ||
| Fig. 3 Temperature dependence of χmT of 1 (full circles), 2 (empty squares) and 3 (empty circles). | ||
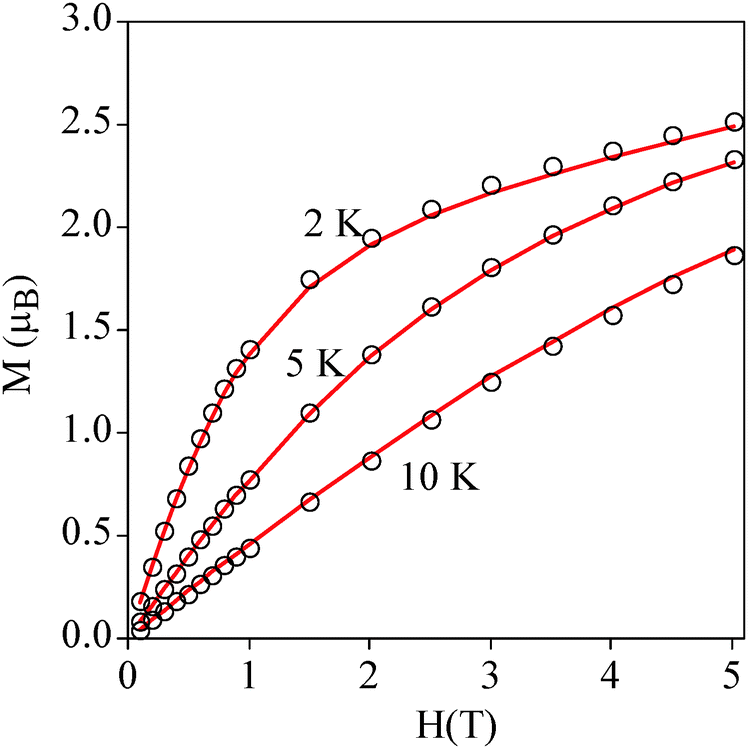 | ||
| Fig. 4 Isothermal magnetization of 1 at 2, 5 and 10 K. The continuous line corresponds to the fit (see the text for details). | ||
HF-EPR is a useful technique to study mononuclear MnIII complexes.30 HF-EPR spectra of a pressed pellet of 1 in eicosane at different temperatures and frequencies (see Table S2, ESI†) are shown in Fig. 5; Fig. S9 and S10, ESI.† Simulations of these spectra using the EasySpin simulation software31 clearly confirm the negative sign of D. All simulations were done using the following set of parameters: D = −5.24 cm−1, E = 0.39 cm−1 and giso = 1.98. Interestingly, the slightly distorted octahedral geometry of MnIII in the Anderson POM gives rise to a higher axial zero-field splitting parameter than values found in literature for other MnIII complexes presenting a clear tetragonal elongation of the coordination sphere of MnIII,30 including those showing a field-induced slow relaxation of magnetization (D ranging from −3.2 to −4.55 cm−1).19 On the other hand, this D value is closer to that of TBA7H10[MnIII(SiW9O34)2]·3H2O (D = −5.28 cm−1), which also presents a clear tetragonal distortion.18 Finally, the rhombic E-term is slightly lower than most of these complexes (E ∼ 0.5–0.7 cm−1)19b,c,e but higher than that found for Na5[Mn(L-tart)2]·12H2O (E = 0.032 cm−1)19d and TBA7H10[MnIII(SiW9O34)2]·3H2O (E = 0.00119 cm−1).18
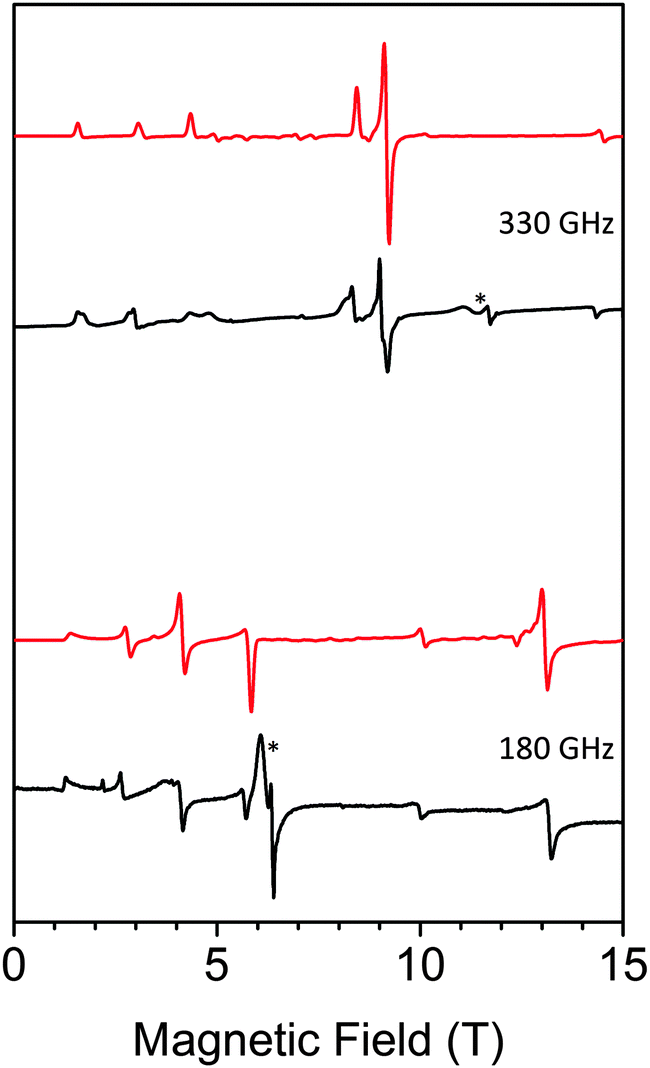 | ||
| Fig. 5 HF-EPR spectra of 1 (black: experiment, red: simulation) at 330 GHz (top) and 180 GHz (bottom) and 10 K. * denotes small impurities in the sample. | ||
The relaxation properties of 1 were studied by susceptibility measurements performed with an alternating magnetic field (AC susceptibility). In the absence of a magnetic field, no signal in the out of phase molar susceptibility (χm′′) is observed. When magnetic fields of 0.2 or 0.5 T are applied, strong frequency-dependent peaks in both the in phase molar susceptibility (χm′) and χm′′ appear with clear maxima of χm′′ below 3 K (Fig. 6 and Fig. S8, ESI†). This is a clear indication that 1 presents a field-induced slow relaxation of magnetization.
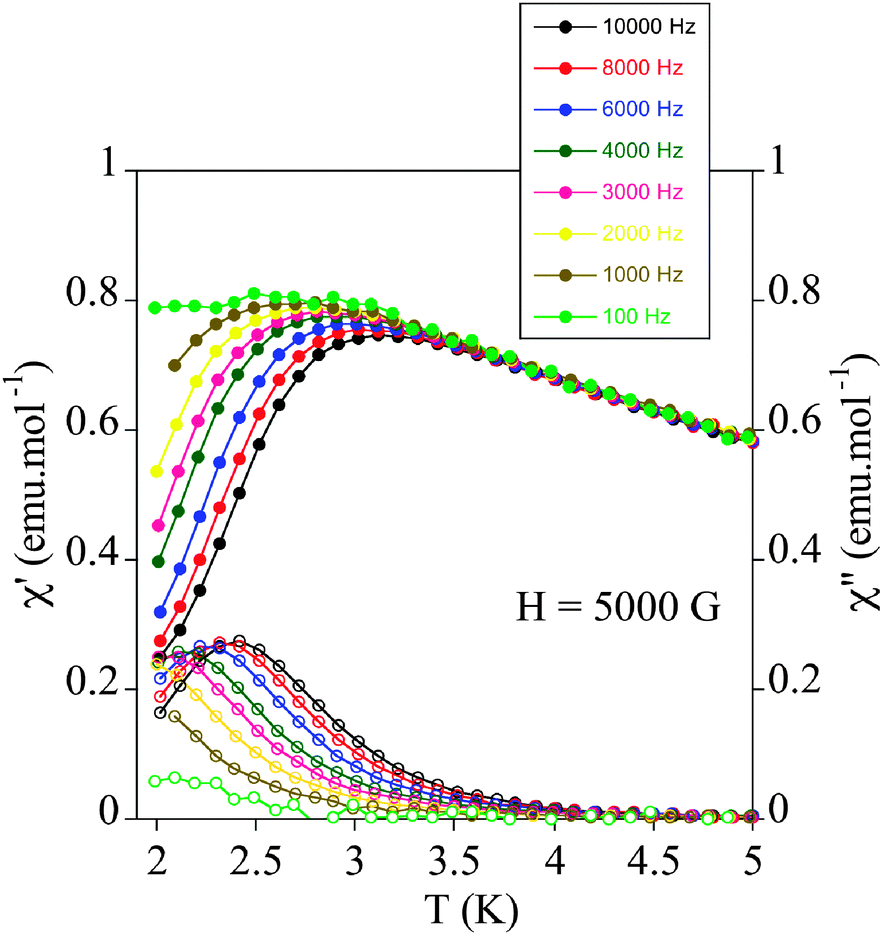 | ||
| Fig. 6 Temperature dependence of the in-phase AC susceptibility (χm′) (filled symbols) and the out-of-phase AC susceptibility (χm′′) (empty symbols) of 1 under an applied field of 0.5 T. | ||
Notice that only a few mononuclear MnIII complexes, reported very recently, have shown this behaviour.19 They are Ph4P[MnIII(opbaCl2)(py)2] (H4opbaCl2 = N,N′-3,4-dichloro-o-phenylenebis(oxamic acid), py = pyridine, and Ph4P+ = tetraphenylphosphonium cation),19a [MnIII(5-TMAM(R)-salmen)(H2O)CoIII(CN)6]·7H2O·MeCN (5-TMAM(R)-salmen = (R)-N,N-(1-methylethylene)bis(5-trimethylammoniomethylsalicylideneiminate)),19b [MnIII{(OPPh2)2N}3],19c Na5[Mn(L-tart)2]·12H2O (L-tart = L-tartrate),19d MnIII(dbm)3 (dbm− = dibenzoylmethanido), [MnIII(dbm)2(L)2](ClO4) (L = dimethyl sulfoxide or pyridine),19e and the TBA7H10[MnIII(SiW9O34)2]·3H2O POM mentioned above.18 The maxima of χm′′ in 1 appear at lower temperature (2.4 K) than those of compounds Ph4P[MnIII(opbaCl2)(py)2]19a (∼3.6 K) or [MnIII(dbm)2(L)2](ClO4) (L = pyridine)19e (∼2.6 K) but higher than those of compounds [MnIII(5-TMAM(R)-salmen)(H2O)CoIII(CN)6]·7H2O·MeCN,19b [MnIII{(OPPh2)2N}3],19c [MnIII(dbm)3]19e [MnIII(dbm)2(L)2](ClO4) (L = dimethyl sulfoxide)19e and TBA7H10[MnIII(SiW9O34)2]·3H2O18 (lower than 2.3 K). On the other hand, the values of the relaxation time, which are calculated from the maximum of χm′′ at a given frequency (τ = 1/2πν), follow the Arrhenius law characteristic of a thermally activated mechanism (τ = τ0exp(Ea/kBT)) (Fig. S8, ESI†). The calculated values of the pre-exponential factor and the activation energy (τ0 = 9 ± 2 × 10−9 s and Ea = 12.6 ± 0.3 cm−1 at 0.2 T and τ0 = 7 ± 1 × 10−9 s and Ea = 13.1 ± 0.4 cm−1 at 0.5 T) are consistent with those of the other MnIII complexes showing this behaviour.19 In these compounds, τ0 and Ea are field dependent. The extrapolated zero-field Ea value (12 ± 1 cm−1) is close to the gap between the ground and first excited states obtained from HF-EPR data (14.5 cm−1). This could indicate that an Orbach process of magnetic relaxation is operative via the first excited mS state as observed in lanthanoid complexes.32 On the other hand, the Cole–Cole plots of 1 at 2.1 K and applied fields of 0.2 and 0.5 T give almost perfect semicircles, which can be fitted by the generalized Debye model (Fig. S11, ESI†).33 The calculated low values of the α parameter (α = 0.11 at 0.2 T and α = 0.13 at 0.5 T) support a single relaxation process (α = 0 for a Debye model). These values are similar to those found in Ph4P[MnIII(opbaCl2)(py)2] (0.089–0.216).19a
Temperature dependence of χmT of powdered samples of 2 and 3 is shown in Fig. 3. χmT values at room temperature (9.4 cm3 mol−1 K for 2 and 3.3 cm3 mol−1 K for 3) are close to the expected contributions for an isolated MnIII with S = 2 and g = 2.0 plus two FeII in the HS state for 2. These data are consistent with metal-ligand distances in the structure of 2 that indicate that FeII is in the HS state. This is in agreement with the crystal field splitting caused by the coordination of FeII with O atoms (N3O3 coordination sphere), which is weaker than that caused by the N atoms in 3. In fact, in 3, FeII is coordinated to two 1-bpp ligands leading to a LS state of FeII. As χmT values for 2 remain constant upon cooling, we can conclude that there is no magnetic interaction between the MnIII centre (S = 2) and the two HS FeII centres (S = 2). This is due to the magnetic isolation between MnIII and the HS FeII, which is provided by the relatively long TRIS-bpp bridging ligand or MoO6 units. Indeed, minimum distances between Fe and Mn are close to 6.4 Å in 2. Finally, as spin-crossover of other 1-bpp derivatives has been observed at temperatures well above 300 K,34χmT of 3 has been measured up to 400 K. Unfortunately, χmT remains close to LS values indicating that no spin-crossover is taking place in this polymer. To see if it was possible to photoinduce the spin-crossover, 3 was irradiated with green light (λ = 532 nm, optical power 3.4 mW cm−2) at 10 K. A small but significant increase of the magnetic signal was observed. After three hours, the irradiation was switched off and the temperature was then increased at the standard rate of 0.3 K min−1. The χmT product firstly increases upon warming from 10 K due to zero-field splitting of the HS FeII and reaches a maximum near 40 K (Fig. 7). At higher temperatures, χmT decreases to reach similar values to those obtained before irradiation above 70 K. The maximum difference between the two curves (∼0.25 cm3 mol−1 K) indicates a photoconversion close to 8%. This low photoconversion is similar to that observed in other FeII compounds showing a disordered structure and high T1/2.35 Further studies are needed to understand the photomagnetic behaviour of this compound (spectroscopic studies and relaxation kinetics of the photo-induced metastable state).
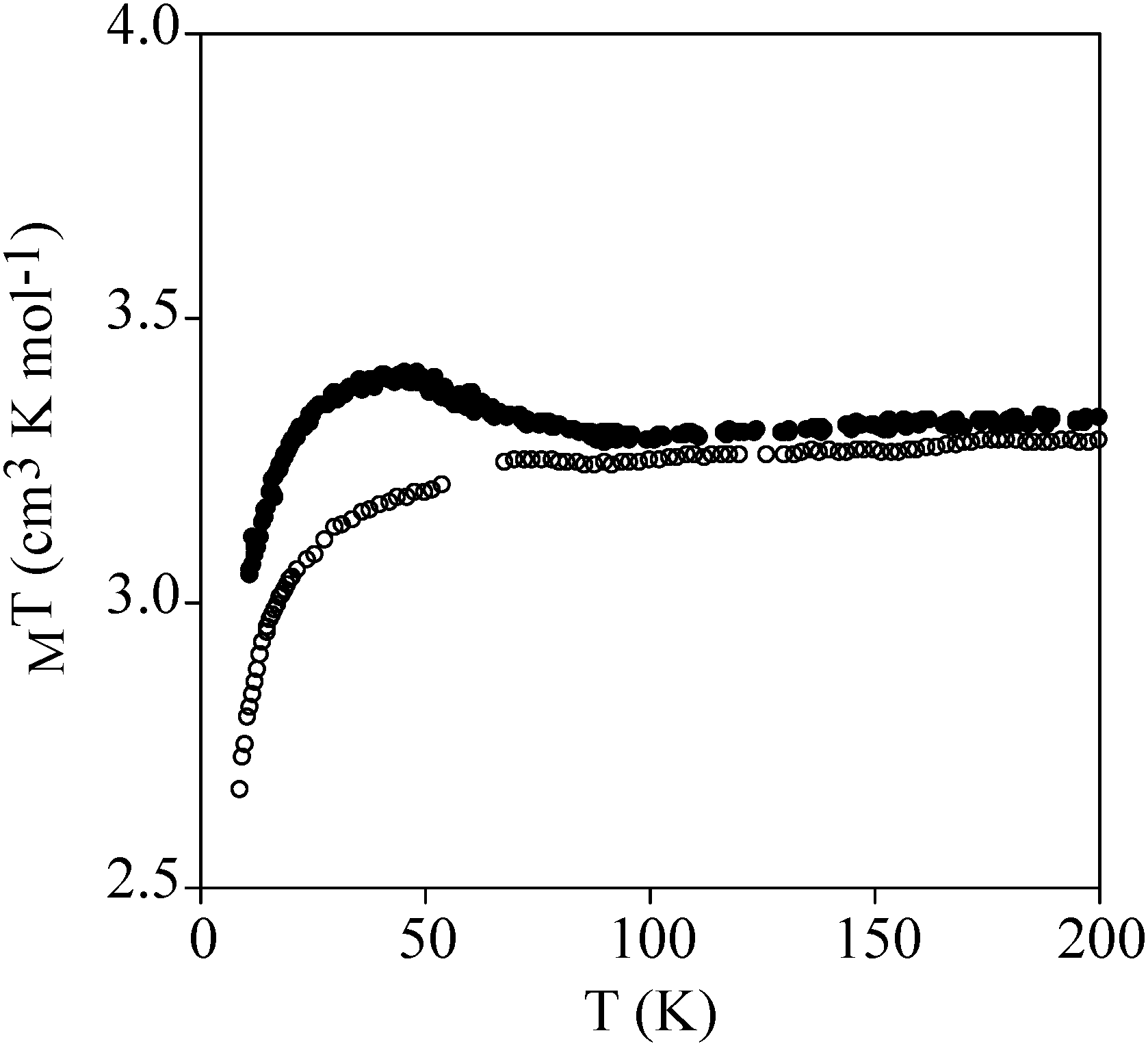 | ||
| Fig. 7 Temperature dependence of χmT of 3. Empty circles: data recorded without irradiation; full circles: data recorded after irradiation at 10 K. | ||
To test if SMM behaviour is general for this type of structure, we have measured the relaxation properties of 2 and 3 by AC susceptibility measurements. Furthermore, we have studied for the first time the magnetic properties of 4 (Scheme 1), one of the simplest Anderson POM with MnIII reported in the literature.20 We have to take into account that, while unfunctionalized Anderson-type polyoxomolybdates with MnII, FeIII, NiII, and ZnII are known in the literature, all MnIII Anderson POM structures reported to date correspond to functionalized POMs.202, 3 and 4 show strong frequency-dependent χm′ and χm′′ peaks under an applied magnetic field of 0.5 T below 3 K as in 1. This is a clear indication that field-induced slow relaxation of magnetization is a common feature for this type of structure (Fig. 8 and Fig. S12, ESI†). The calculated values of τ0 and Ea of these compounds are similar to those of 1 and the other MnIII complexes showing this behaviour (Fig. S12 and Table S3, ESI†).19
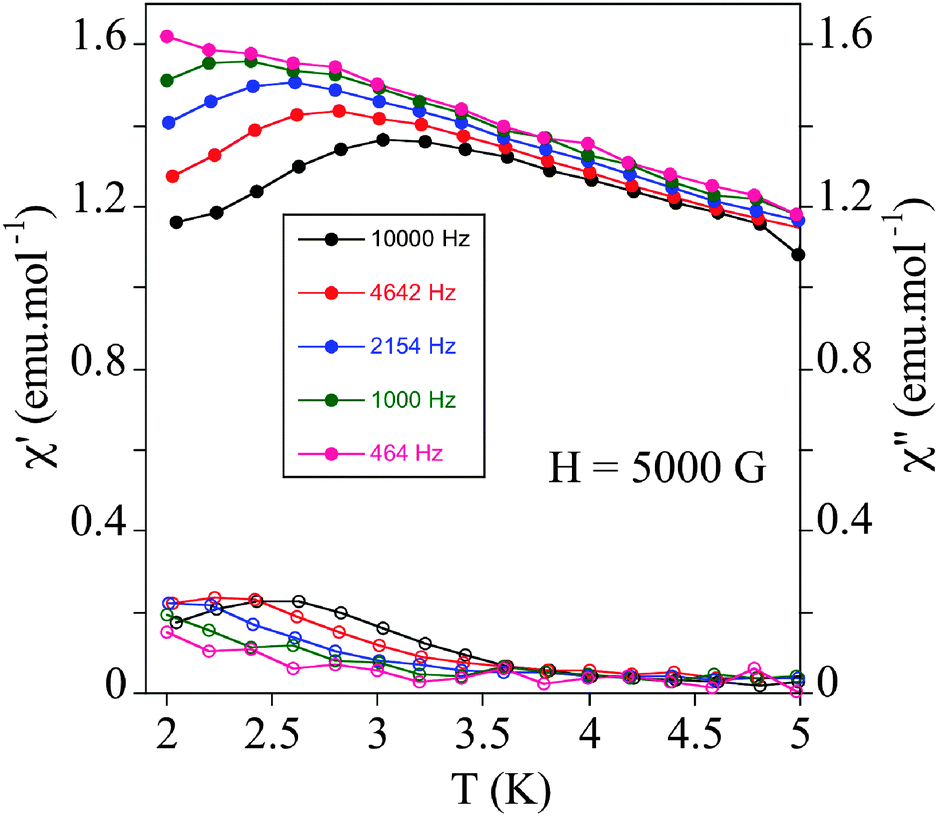 | ||
| Fig. 8 Temperature dependence of the in-phase AC susceptibility (χm′) (filled symbols) and the out-of-phase AC susceptibility (χm′′) (empty symbols) of 2 under an applied field of 0.5 T. | ||
Conclusions
In this work, two tridentate 1-bpp ligands have been incorporated into an Anderson POM in compound 1 using the tris-alkoxo-amide tripodal functionalization as shown by single crystal X-ray diffraction. Direct reaction of 1 in a 1:1 Fe2+:POM ratio gives rise to a 1D polymer in compound 3, whereas a 2:1 Fe2+:POM ratio leads to a precipitate partially soluble in DMF, which leads to 2 after recrystallisation. These results confirm the versatility of the coordination chemistry of tris-alkoxo-amide functionalized POMs to obtain a great variety of structures ranging from a 2D cationic network in compound 2 or an anionic polymer in compound 3. Two conclusions can be extracted from the structure of 2: (i) the excess of metal leads to coordination with the oxo groups of the POM and (ii) recrystallization in polar aprotic solvents such as DMF involves dissociation of the 1-bpp-metal coordination bond. Similar behaviour has been observed in other functionalized POMs such as pyridyl-functionalized hexavanadates.5
The magnetic properties of 1 have shown that it presents a field-induced slow relaxation of magnetization due to magnetic anisotropy of MnIII, as observed in other mononuclear MnIII complexes reported very recently. This is the second example of d-metal POM exhibiting this behaviour reported in the literature. Until very recently, slow relaxation of magnetization in POMs had only been found in POMs containing lanthanoids.16 The similar behaviour of the reference compound 4, which is one of the simplest functionalized Anderson POM reported to date, and 2 and 3 confirms that this type of behaviour is general for this type of structure. Furthermore, it shows that a high Jahn–Teller tetrahedral distortion as that of those previous MnIII complexes showing that field-induced slow relaxation of magnetization is not needed to obtain such behaviour. This result opens the way for the preparation of hybrid POMs combining this property with other magnetic properties of interest. Thus, spin-crossover behaviour could be expected if two 1-bpp ligands were coordinated to FeII, as observed in compound 3. The magnetic properties indicate that, although FeII complexes remain in the LS state in all the temperature range, it is possible to induce spin-crossover by light irradiation (LIESST effect). However, the LS to HS photoconversion is limited (∼8%). Possible strategies to improve these results are the use of other counterions or solvents as the spin transition of this type of complexes is very sensitive to the changes of packing and intermolecular interactions resulting from different counterions or solvent molecules. Another possibility is to decrease the ligand field by the introduction of other substituents in the 1-bpp derivative.
Acknowledgements
Financial support from the EU (SPINMOL ERC Adv. Grant), the Spanish MINECO (CTQ-2011-26507, MAT2011-22785 and MAT2014-56143), DFG (INST 41/863-1, SPP1601) and the Generalitat Valenciana (Prometeo and ISIC-Nano programs) is gratefully acknowledged. J. M. Martínez-Agudo from the University of Valencia is gratefully acknowledged for magnetic measurements. Gabriel Peris-Pérez and Cristian Vicent-Barrera from Universitat Jaume I are gratefully acknowledged for single crystal X-ray diffraction measurements of 2 and ESI-MS measurements of 1.Notes and references
- (a) M. T. Pope, Comprehensive Coordination Chemistry II, 4, Elsevier Ltd, Oxford, UK, 2004, p. 635 Search PubMed; (b) C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS PubMed (Special issue: polyoxometalates); (c) J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- C. Bosch-Navarro, B. Matt, G. Izzet, C. Romero-Nieto, K. Dirian, A. Raya, S. I. Molina, A. Proust, D. M. Guldi, C. Martí-Gastaldo and E. Coronado, Chem. Sci., 2014, 5, 4346 RSC.
- (a) Y. Zhu, P. Yin, F. Xiao, D. Li, E. Bitterlich, Z. Xiao, J. Zhang, J. Hao, T. Liu, Y. Wang and Y. Wei, J. Am. Chem. Soc., 2013, 135, 17155 CrossRef CAS PubMed; (b) I. Bar-Nahum, H. Cohen and R. Neumann, Inorg. Chem., 2003, 42, 3677 CrossRef CAS PubMed.
- M. P. Santoni, G. S. Hanan and B. Hasenknopf, Coord. Chem. Rev., 2014, 281, 64 CrossRef CAS.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2598 CrossRef CAS.
- J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094 CrossRef CAS PubMed.
- C. Allain, S. Favette, L. M. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 3433 CrossRef CAS.
- C. Allain, D. Schaming, N. Karakostas, M. Erard, J.-P. Gisselbrecht, S. Sorgues, I. Lampre, L. Ruhlmann and B. Hasenknopf, Dalton Trans., 2013, 42, 2745 RSC.
- I. Ahmed, R. Farha, Z. Huo, C. Allain, X. Wang, H. Xug, M. Goldmann, B. Hasenknopf and L. Ruhlmann, Electrochim. Acta, 2014, 110, 726 CrossRef.
- M. P. Santoni, A. K. Pal, G. S. Hanan, M. C. Tang, K. Venne, A. Furtos, P. Ménard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48, 200 RSC.
- M. P. Santoni, A. K. Pal, G. S. Hanan and B. Hasenknopf, Dalton Trans., 2014, 43, 6990 RSC.
- P. Yin, T. Li, R. S. Forgan, C. Lydon, X. Zuo, Z. N. Zheng, B. Lee, D. L. Long, L. Cronin and T. Liu, J. Am. Chem. Soc., 2013, 135, 13425 CrossRef CAS PubMed.
- M. P. Santoni, A. K. Pal, G. S. Hanan, A. Proust and B. Hasenknopf, Inorg. Chem., 2011, 50, 6737 CrossRef CAS PubMed.
- (a) M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS; (b) M. A. Halcrow, New J. Chem., 2014, 38, 1868 RSC.
- (a) T. Buchen, P. Gütlich, K. H. Sugiyarto and H. A. Goodwin, Chem. – Eur. J., 1996, 2, 1134 CrossRef CAS; (b) S. Marcen, L. Lecren, L. Capes, H. A. Goodwin and J. F. Létard, Chem. Phys. Lett., 2002, 358, 87 CrossRef CAS; (c) G. Chastanet, C. A. Tovee, G. Hyett, M. A. Halcrow and J. F. Létard, Dalton Trans., 2012, 41, 4896 RSC.
- (a) M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874 CrossRef CAS PubMed; (b) M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467 CrossRef CAS PubMed; (c) M. J. Martínez-Pérez, S. Cardona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-García, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Ariño, J. Sesé, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 CrossRef; (d) S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Camon, M. Evangelisti, F. Luis, M. J. Martínez-Pérez and J. Sesé, J. Am. Chem. Soc., 2012, 134, 14982 CrossRef CAS PubMed.
- C. Ritchie, A. Ferguson, H. Nojiri, H. N. Miras, Y. F. Song, D. L. Long, E. Burkholder, M. Murrie, P. Kögerler, E. K. Brechin and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 5609 CrossRef CAS PubMed.
- R. Sato, K. Suzuki, T. Minato, M. Shinoe, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 4081 RSC.
- (a) J. Vallejo, A. Pascual-Álvarez, J. Cano, I. Castro, M. Julve, F. Lloret, J. Krzystek, G. De Munno, D. Armentano, W. Wernsdorfer, R. Ruiz-García and E. Pardo, Angew. Chem., Int. Ed., 2013, 52, 14075 CrossRef CAS PubMed; (b) R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300 CrossRef CAS PubMed; (c) A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869 CrossRef CAS PubMed; (d) G. A. Graig, J. J. Marbey, S. Hill, O. Roubeau, S. Parsons and M. Murrie, Inorg. Chem., 2015, 54, 13 CrossRef PubMed; (e) L. Chen, J. Wang, Y.-Z. Liu, Y. Song, X.-T. Chen, Y.-Q. Zhang and Z.-L. Xue, Eur. J. Inorg. Chem., 2015, 271 CrossRef CAS.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406 CrossRef CAS.
- M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Inorg. Chem., 1979, 18, 93 CrossRef CAS.
- T. Vermonden, D. Branowska, A. T. M. Marcelis and E. J. R. Sudhölter, Tetrahedron, 2003, 59, 5039 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14 CrossRef CAS PubMed.
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081 CrossRef CAS.
- J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985 CrossRef.
- (a) J. Limburg, J. S. Vrettos, R. H. Crabtree, G. W. Brudvig, J. C. de Paula, A. Hassan, A. L. Barra, C. Duboc-Toia and M. N. Collomb, Inorg. Chem., 2001, 40, 1698 CrossRef CAS PubMed; (b) C. Mantel, A. K. Hassan, J. Pécaut, A. Deronzier, M. N. Collomb and C. Duboc-Toia, J. Am. Chem. Soc., 2003, 125, 12337 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- R. Marx, F. Moro, M. Dörfel, L. Ungur, M. Waters, S. D. Jiang, M. Orlita, J. Taylor, W. Frey, L. F. Chibotaru and J. van Slageren, Chem. Sci., 2014, 5, 3287 RSC.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS.
- A. Abhervé, M. Clemente-León, E. Coronado, C. J. Gómez-Garcia and M. López-Jordà, Dalton Trans., 2014, 43, 9406 RSC.
- V. Mishra, H. Mishra, R. Mukherjee, E. Codjovi, J. Linares, J. F. Létard, C. Desplanches, C. Baldé, C. Enachescu and F. Varret, Dalton Trans., 2009, 7462 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic table, 1H NMR of TRIS-bpp and 1, IR spectra of 1 and 3, ESI-MS of 1, structural views of 1 and 2, X-ray powder diffraction pattern of 1, AC susceptibility data of 1, 3 and 4 and HF-EPR spectra of 1. CCDC 1058519 and 1058520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc01089f |
| This journal is © The Royal Society of Chemistry 2015 |